

Abstract
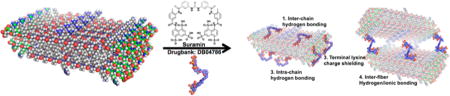
Self-assembly of multidomain peptides (MDP) can be tailored to carry payloads that modulate the extracellular environment. Controlled release of growth factors, cytokines, and small-molecule drugs allows for unique control of in vitro and in vivo responses. In this study, we demonstrate this process of ionic cross-linking of peptides using multivalent drugs to create hydrogels for sustained long-term delivery of drugs. Using phosphate, heparin, clodronate, trypan, and suramin, we demonstrate the utility of this strategy. Although all multivalent anions result in good hydrogel formation, demonstrating the generality of this approach, suramin led to the formation of the best hydrogels per unit concentration and was studied in greater detail. Suramin ionically cross-linked MDP into a fibrous meshwork as determined by scanning and transmission electron microscopy. We measured material storage and loss modulus using rheometry and showed a distinct increase in G′ and G″ as a function of suramin concentration. Release of suramin from scaffolds was determined using UV spectroscopy and showed prolonged release over a 30 day period. Suramin bioavailability and function were demonstrated by attenuated M1 polarization of THP-1 cells compared to positive control. Overall, this design strategy has allowed for the development of a novel class of polymeric delivery vehicles with generally long-term release and, in the case of suramin, cross-linked hydrogels that can modulate cellular phenotype.
INTRODUCTION
Multidomain peptide-based hydrogels present a novel platform for delivery of small-molecule drugs, growth factors, and cells.1–5 Amphiphilic MDPs have demonstrated the ability to self-assemble as a function of amino acid composition and ionic buffers (e.g., Mg2+/Ca2+/PO43−).6,7 The specific MDP used in this study, KSLSLSLRGSLSLSLKGRGDS, which we call SLac, has been previously reported.2 SLac is composed of a central block of alternating hydrophilic serine and hydrophobic leucine residues, flanked on both sides with lysines that aid in its solubilization. Antiparallel β sheet formation creates a facial amphiphile with the side chains of leucine and serine constituting the hydrophobic and hydrophilic faces, respectively. Association of the antiparallel β sheets in a bilayer facilitates fiber formation so that the nonpolar leucine side chains are sequestered away and hydroxyl groups of serine remain exposed to the aqueous medium (Figure 1).
Figure 1.
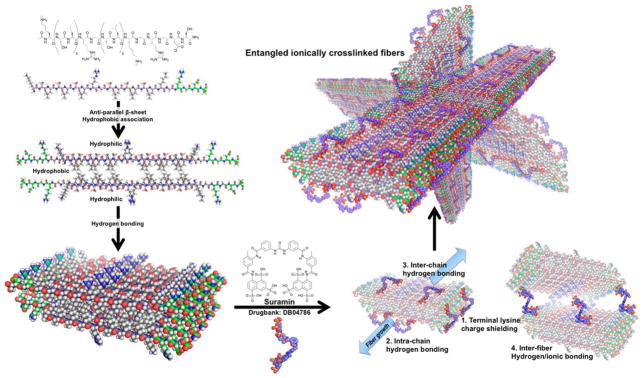
Schematic of molecular self-assembly. Designer peptides with –RGDS terminal sequence. Main SL-repeat regions arrange antiparallel to each other and hydrogen bond into sheets. Polyvalent suramin allows for intra- and intermolecular hydrogen bonding and ionic interaction with terminal lysines. Stabilized short fibers’ terminal charges are screened, allowing fiber growth and polymerization into hydrogel networks.
To augment the peptides’ function, an –LRG– moiety is present to facilitate biodegradation, and an –RGDS sequence to promote cell adhesion. At neutral pH, the protonated amines of the lysine residues resist molecular self-assembly of growing fibers. The resulting molecular frustration limits the extent of fibrillation. The addition of high-ionic-strength buffers or polyvalent anions can specifically aid in relieving molecular frustration, shielding positive or negative charges and allowing formation of long-range microscopic fibers.7,8 Noting the propensity for fiber growth with charge shielding because of polyvalent anions, we hypothesized that the addition of specific drug molecules with polyvalency to MDP hydrogels may aid in sequestration and controlled delivery. Here, we have investigated this notion with the use of several polyvalent drugs. These include heparin, clodronate, trypan, and suramin (Figure 2). All of these molecules resulted in the rapid formation of high-quality hydrogels. Because suramin resulted in the strongest hydrogels per unit concentration used, we focused most of our characterization on suramin–SLac hydrogels.
Figure 2.

Polyanionic drugs used. A variety of polyanionic drugs that are clinically important were chosen for ionic cross-linking with matrices.
The importance of controlling delivery of drugs is underscored by two key facets: (i) the desire to minimize systemic side effects of drugs such as chemotherapeutics while treating local regions of tissue in the immediate vicinity of pathologies with high doses of drug and (ii) tuning the delivery of drugs allows for sustained dosing without reducing the frequency and quantity of drug delivered. For example, treatment options for neoplastic disease typically involve radiation, chemotherapeutics, and noninvasive/minimally invasive treatments if diagnosed in early stages. For late-stage solid tumors, (e.g., glioblastoma multiforme) treatment involves surgical resection, coupled with postoperative radiation and/or chemotherapy.9–11 Systemic chemotherapeutics are typically used in high concentrations so that malignancies in the far reaches of the extracellular space still receive the minimum effective therapeutic dosage at the cost of systemic side effects.10–12 To counteract this, biocompatible polymers with anticancer payloads can be administered during surgical resection.13,14 This release strategy may potentially reduce the immune-compromising chemotherapeutic burden, allow targeted chemotherapy, and reduce localized malignancy.15,16 Further, this strategy with controlled prolonged release can be extended to a variety of other multivalent drug candidates such as heparin (for prolonged anticlotting), trypan (for localized treatment of parasites), and clodronate (for localized osteoporosis treatment).
RESULTS AND DISCUSSION
To determine material–drug interactions, we first noted the propensity for MDP to form robust hydrogels when like charges were shielded by negative ions, such as PO43−, allowing fiber cross-linking (Figure 1).
Extending from this, we noted the potential for a variety of anionic drugs to participate in a similar role, while potentially providing higher mechanical strength because of their conformation, molecular weight, and charge density (Figure 2). To determine which of these strategies would best cross-link MDP, we used parallel-plate rheometry.
Rheological characterization of hydrogel scaffolds when cross-linked with multivalent drugs may help elucidate potential mechanisms for cross-linking. Similar therapeutic doses of drugs were chosen for loading into hydrogels. Therapeutic doses approximate the concentration of low suramin, Figure 3. Low to high suramin loading spanned the range of concentrations that would diffuse to tissue or be bolus-dosed. All multivalent drugs showed a marked increase in G′ and G″ as a function of concentration (p < 0.01) (Figure 3). Given the high G′ and G″, the gels created were easily handleable and manipulable. Uniquely, depending on the cross-linking molecule, distinct rheological properties were noted. Increasing PO43− buffer ionic strength from 5 to 38.5 mM resulted in a significant increase in mechanical strength. Extending this notion of phosphate-based cross-linking, a clinically relevant diphosphonate used for bone healing, clodronate disodium, also resulted in a distinct increase in G′/G″ over that of phosphate alone or saline. The two phosphonate groups of clodronate are capable of shielding a greater number of charges and creating cross-links to multiple MDPs. To test this hypothesis, heparin, a large molecule with several negative charges, was chosen next. Heparin-loaded gels showed a significant increase in mechanical strength over that of PO43− but not that of clodronate alone. Further investigating functionality, a polysulfonate, trypan blue, typically used in cell culture viability assays, cross-linked MDP with a similar distinct increase in G′/G″. However, increases in mechanical responses yield gels of similar strength to those cross-linked with heparin. Finally, suramin, with six sulfonate groups, at a similar concentration, showed a much higher increase in strength (an order of magnitude higher G′/G″) as compared to other drugs used. Although the concentration and total number of charges in heparin gels is the greatest, gels prepared with suramin displayed the highest increase in strength (highest G′ and G″, Figure 3). This suggests that suramin has a conformationally and structurally suitable architecture in the presence of MDP to aid in its shielding of charges and cross-linking. An illustrative movie shows how we hypothesize interactions between suramin and MDP (Movie 1).
Figure 3.
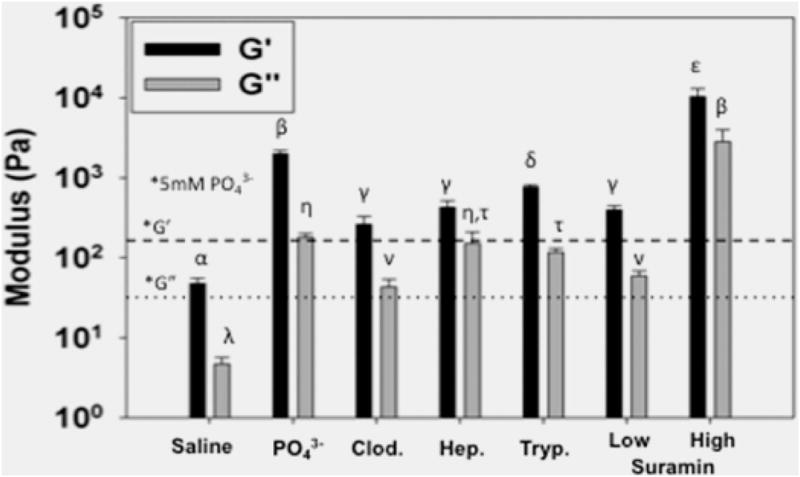
Rheological properties of cross-linked peptide hydrogels. Higher concentrations of negative ion charge shield MDP charges to a greater extent, yielding increased storage moduli (G′) and loss moduli (G″). Significant increases were noted when PO43− concentration was increased, compared to prior values (*, 5 mM PO43− from Galler et al., G′ (dashed line), G″ (dotted line)).2 Increasing the density of PO43− groups, using bisphosphonate clodronate (Clod.), resulted in an increase in mechanical strength. Charge density did not result in significantly increased moduli when comparing heparin (Hep.) and trypan blue (Tryp.) at similar concentrations. Charge density and conformation of suramin resulted in an order of magnitude increase in mechanical strength with increase in concentration. Similar Greek letter indicates no statistically significant difference for each receptor (*, p < 0.01).
Previous reports from our group have demonstrated the cross-linking potential for small multivalent molecules such as PO43− and drugs such as heparin.2,3,5 Basic modeling of the multivalent-drug–SLac interaction suggests that at neutral pH deprotonated charged groups can interact with several positively charged terminal lysine amine groups reducing charge–charge repulsion and promoting intra- and interpeptide cross-linking. Extended to suramin, we hypothesize that negatively charged sulfonate residues cross-link positively charged lysine side chains (Figure 1). FTIR spectroscopy of hydrogels showed a characteristic extended β-sheet hydrogen-bonding peak between 1610–1630 cm−1 and a characteristic antiparallel β-sheet peak at 1695 cm−1 for all peptide mixtures (Figure 4A). Circular dichromism (CD) spectra similarly showed the presence of β-sheet secondary structure, with a minimum around 216 nm and maximum around 195 nm (Figure 4B). Formation of a fibrous structure within hydrogels at high concentrations of suramin/high ionic strength resulted in lower peak magnitudes at the 195 nm maxima. Peptide hydrogels that formed were optically clear and conformed to the shape of the mold they were cast in (Figure 4C). Microstructure of peptide scaffolds, probed using TEM and SEM, showed formation of a nanofibrous matrix (Figure 4D,E).
Figure 4.

Characterization of peptide structure. (A) FTIR spectroscopy showing characteristic peaks for the formation of a β-sheet structure when peptide is mixed with saline, HBSS, high-ionic-strength phosphate, and two different suramin concentrations. (B) CD spectroscopy confirmed formation of β-sheet structure. Note that gels formed aggregates, resulting in lower minima/maxima intensity for higher concentration of suramin. For FTIR and CD, all samples were identically diluted as detailed in the Experimental Methods. (C) Peptide–drug mixtures cast in cylindrical molds create optically transparent gels that maintain their structure. (D) Peptide hydrogels with drug loading create nanofiber matrix seen in negative-stain TEM, scale bar = 50 nm. (E) SEM of dehydrated hydrogel, scale bar = 1 μm.
CONTROLLED RELEASE OF AN IONICALLY SEQUESTERED DRUG
Previous studies have shown the ability to tailor MDP with unique functionality on the basis of peptide sequence.2 Additionally, MDP can be loaded with drugs, growth factors, cytokines, and cellular secretome.1,2,17 As noted, chemical cross-linking of drugs with subsequent controlled release can strongly promote localized tissue responses and obviate systemic side effects. To this end, the potential of suramin as a potent antiangiogenic, antineoplastic, and antimicrobial drug to serve as a chemical cross-linker as demonstrated above was noted. Given suramin’s high IV dosing and frequency of dosing (1 g/3–7 days in adults), delivery of suramin in situ may prove to be advantageous. However, concerns exist over retention of suramin at the delivery site and leakage of the drug into lymphatic circulation. This may be reduced by ionically cross-linked suramin sequestered within hydrogels with slow, steady release (Figure 1, Figure 5). Suramin cross-linked gels showed long-term release at both low and high concentrations, with much of the drug still present in the carrier hydrogel at 30 days (Figure 5). At high suramin loading (1.0 mg of suramin, 2.0 mg of peptide) hydrogel scaffolds showed an initially linear cumulative mass release followed by a tapering off to 57.9 ± 1.1% with the rest of the drug (42.1%) remaining trapped within the hydrogel. At the lower suramin loading (0.1 mg of suramin, 2.0 mg of peptide), hydrogel scaffolds exhibited significantly lower cumulative release, 38.7 ± 3.2%, with the rest of the drug (61.3%) remaining trapped within the hydrogel (Figure 5B,C). Modeling of suramin release from polymers was performed to help determine the mechanism of release, as detailed in the Experimental Methods.
Figure 5.

Drug release from scaffolds. (A) Short- and long-term drug release data overlapped. (B) Maximum cumulative suramin concentrations reach 0.6 mg/mL (for high loading) and 0.04 mg/mL (for low loading). (C) Cumulative percentage mass released from scaffolds was determined, modeled, and (D) first derivative release rates determined.
Release from 10 mg/mL loaded gels was diffusion dependent, t ≈ 0.5, R2 = 0.972.18 Release from 1 mg/mL loaded gels was non-Fickian-diffusion-/case II-transport-dependent, t ≈ 1.0, R2 = 0.993, suggesting erosion of hydrogel ionic cross-links was releasing suramin.18 First derivatives of release profiles showed that release rates were initially faster for both loading concentrations, which then tapered after 2 weeks to a near-linear release rate (Figure 5D). The aggressive design of the release experiment, with release being probed daily, may overestimate release from hydrogel scaffolds because diffusive and convective transport enhances drug release, and the suramin reservoir concentration in the release media is depleted. However, we assayed release aliquots over a shorter time period more frequently (hr vs day) and determined no significant difference in kinetics of release (Figure 5A).19 Given the release data, we hypothesize that the immediate microenvironment will be affected by low concentrations of the drug over the first 30 days, with much of the suramin release being dictated by cellular adhesion to MDP hydrogels, mediated by an –RGDS terminal sequence, and degradation of MDP matrix, mediated by MMP-susceptible –LRG– domain. Sustained and targeted delivery of suramin has been a goal of several groups, liposomal encapsulation of suramin for antiviral applications,20 encapsulation of suramin/paclitaxel into PLLA/PLGA microparticles for cancer treatment,21 and local delivery to inhibit neointimal hyperplasia postangioplasty,22 to name a few. However, of primary concern with any of these techniques is the release, detailed above, and bioavailability, detailed below, of the suramin after loading into the carrier.
PRESERVATION OF BIOLOGICAL ACTIVITY IN IONICALLY CROSS-LINKED GELS
Drug release from polymeric scaffolds for sustained release has been sought after for decades.10,12,15,23–26 Several strategies have been attempted for the capture of drugs including dissolving tablets, micelles for hydrophobic drugs, multiwalled microparticles, polymer wafers, covalent immobilization into carriers or onto surfaces, and ionic layer-by-layer self-assembly, to name a few. Of concern is the unfavorable interaction of polymeric carriers with loaded drugs or surrounding tissue, such as covalent linker addition for conjugation to scaffolds or surfaces that may attenuate activity of functionality of drugs or non-natural degradation products that may elicit an inflammatory response, complicating drug action or the nonconforming nature of solid delivery systems.9,10,12,15,16,25,26 In this article, we have explored the potential for a drug to actively interact with its carrier by cross-linking it. Similar studies by our group and others have shown limited inflammatory responses with synergistic activity of passively loaded cytokines or growth factors.1,3,4,17 As a vital next step, it was important to confirm the maintenance of activity of ionically sequestered drugs after loading into MDP carriers. Because suramin is known to have distinct effects on growth factors/growth factor receptors, we utilized suramin-releasing gels to determine biological activity.27 Suramin-loaded SLac hydrogels were seeded with THP-1 macrophages. THP-1 cells were chosen because of their neoplasticity, monocytic origin, and establishment as an human cell line.28 Lipopolysaccharide (LPS)-activated THP-1 macrophages adhered to drug-laden SLac hydrogels. These cells were exposed to ionically trapped suramin, which decreased M1 polarization with decreased levels of TNF-α and IL-1β compared to M1 cells (Figure 6). This is probably due to known P2Y receptor antagonism, as reported previously.29 Together, this data demonstrates preservation of suramin effector functionality and inhibition of proinflammatory M1 phenotype development in LPS-induced human monocytic leukemia cell line THP-1 cells.
Figure 6.

Macrophage polarization as a function of suramin loading. Suramin loading hydrogels repressed the expression of M1 macrophage marker CC chemokine receptor 7 (CCR7)+ and release of proinflammatory cyotkines (B) tumor necrosis factor alpha (TNF-α) and (C) interleukin 1 β (IL-1β).
MEASURING IN VIVO ACTIVITY
Suramin-cross-linked SLac hydrogels may have potential for localized drug delivery. To demonstrate this, high-concentration suramin was used to cross-link 1 wt % SLac hydrogels. Suramin-cross-linked gels, compared to PO43−-cross-linked ones, showed significantly higher stiffness (G′/G″, Figure 3). However, hydrogels were still injectable, allowing site-delivered subcutaneous implantation. Suramin hydrogels showed a marked decrease in cellular infiltration compared to PO43−-cross-linked SLac hydrogels (Figure 7). This potentially may be due to the increased stiffness of matrices that encumber cellular infiltration.30–32 Furthermore, a significant increase in immunostained M2 macrophages was observed in suramin gels compared to unloaded gels. MDP hydrogels have previously demonstrated the lack of a fibrous capsule and excellent ECM integration.5,33 Suramin-loaded hydrogels show a similar lack of fibrous encapsulation and excellent integration (Figure 7). These results demonstrate the minimal effect implanted gels have on surrounding tissue. Specifically, (i) cellular infiltrate was localized to the implant, (ii) M2 macrophage polarization was localized to within the implant and the immediate vicinity, and (iii) no fibrous tissue walling-off of the implant was observed. Having established these key chemical and biomechanical facets, future studies in sophisticated in vivo cancer models can now be envisioned. These syringe-deliverable constructs may help a variety of strategies that allow localized drug delivery,34 complemented by functional peptide signaling,33 that ultimately offer another tool for targeted therapeutics.
Figure 7.
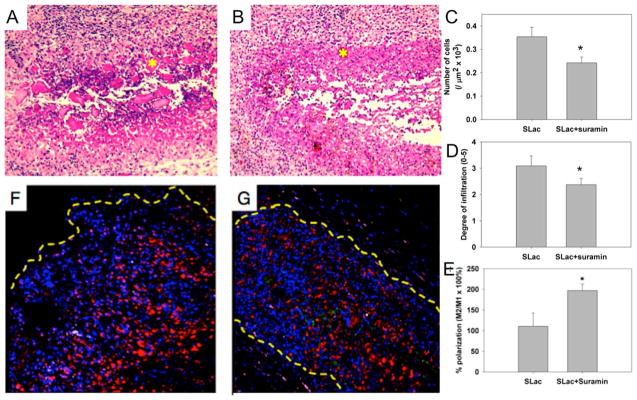
In vivo evaluation of scaffolds. (A) Scaffolds with suramin-loading show large platelets of MDP uninfiltrated (*), compared to (B) a similar region (*) in SLac-only scaffolds. (C) Quantification of infiltration showed a significantly lower number of cells, (D) infiltrating loaded scaffolds to a diminished extent, (E) with a greater M2 macrophage polarization. Representative images of suramin-loaded (F) and -unloaded (G) scaffolds shown. Nuclei, DAPI blue; macrophages, CD68-red; M1, CCR7-green; M2, CD163-purple. (*, p < 0.01).
In this study, we have demonstrated the ability of polyvalent drugs to ionically cross-link multidomain peptides. Specifically, we modeled interactions of peptides with drugs, showing stabilization of an antiparallel β-sheet structure that enhanced long-range nanofibrous meshwork formation and mechanically robust gels. We further demonstrated cross-linking of scaffolds using a variety of clinically relevant polyvalent drugs such as suramin, clodronate, heparin, and trypan blue. Sequestered suramin was shown to slowly release from hydrogel scaffolds, with less than 40–60% releasing over the first 30 days, depending on loading concentration. Preservation of suramin activity on attenuation of M1 phenotype of LPS-stimulated THP-1 monocytic leukemia cells was demonstrated. Subsequent in vivo studies in mammalian subcutaneous tumor-resection models will aim to demonstrate attenuated scaffold infiltration, muted angiogenesis, and limited malignancy as a function of suramin loading.
EXPERIMENTAL METHODS
All chemicals and reagents were purchased from Sigma-Aldrich, unless otherwise noted.
Peptide Design and Characterization
Multidomain peptides were designed on the basis of previously published sequences from our laboratory: SLac, K-(SL)3-(RG)-(SL)3-K-GRGDS.2 All peptides, resin, and coupling reagents were purchased from Aapptec (Louisville, KY). Standard solid-phase peptide synthesis was performed on an Apex Focus XC (Aapptec) apparatus using Rink amide resin with 0.37 mM loading and N-terminal acetylation. After cleavage from resin, crude mass was checked prior to dialysis with 500–1200 MWCO dialysis tubing (Sigma-Aldrich, St. Louis, MO) against Milli-Q water. Peptides were subsequently lyophilized, confirmed for purity using time-of-flight electrospray ionization mass spectrometry, MicroTOF ESI (Bruker Instruments, Billerica, MA), and reconstituted at 20 mg/mL in sterile 298 mM sucrose.
Peptide (Nanofiber)–Drug Interaction
Peptides were modeled in PyMOL Molecular Graphics System, version 1.5.0.5 (Schrödinger, LLC, San Diego, CA) on the basis of previous published work. Previously published work from our lab indicates that the peptide chains form an antiparallel β sheet that sequesters the nonpolar residues away from water. Hydrogen bonding between the sandwiched β sheets facilitates 1D propagation in nanofibers. The antiparallel β-sheet arrangement of peptides in SLac was confirmed using Fouriertansform infrared spectroscopy (FTIR). For modeling in PyMOL, 8–10 peptide chains were arranged adjacently in an antiparallel fashion with hydrophobic/hydrophilic faces (dependent on amino acid R chain) facing the same direction. The two hydrophobic faces were brought in proximity of each other as suggested by previous studies.8 Ioinc interactions between facial amphiphilic fibers were then hypothesized with suramin (RCSB DrugBank ID: DB04786).
Scanning Electron Microscopy and Transmission Electron Microscopy
Microscopic morphology of SLac scaffolds gelled with suramin was determined using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). For SEM, samples were ethanol-dehydrated, critical-point-dried, sputter-coated with 7 nm gold (Denton, Moorestown, NJ), and imaged using an FEI Quanta 400 ESEM (FEI Company, Hillsboro, OR). Fibrillar network for SLac was confirmed using TEM. For TEM, samples were prepared by adding peptide solution directly onto Quantifoil R1.2/1.3 holey carbon mesh on copper grids. Excess peptide was blotted, and the grid was dried prior to negative staining using 2.0% pH 7 phototungstic acid for 8 min. Dried grids were imaged using a JEOL 2010 microscope (120 kV).
Circular Dichroism
All CD experiments were performed on a Jasco J-815 spectropolarimeter equipped with a Peltier temperature-controlled stage. All spectra were collected from 190 to 250 nm at 25 °C. Peptide was dissolved in sucrose at 2 wt %. CD samples were prepared at pH 7.4 by gelling peptide in equal volumes of 0.9% saline, Hank’s balanced salt solution (HBSS), 7.7 mM PO43−, and 1.0 or 0.1 mg/mL suramin. For each experiment, 20 μL aliquots of each sample were gelled in situ on a quartz cell with a path length of 0.01 cm. The molar residual elipticity (MRE) was calculated from the following formula:
where θ is the observed ellipticity in millidegrees, m is the molecular weight in grams per mole (g mol−1), C is the concentration in milligrams per millileter (mg ml−1), l is the path length of the cuvette in centimeters (cm), and nr is the number of amino acids in the peptide.
Attenuated Total Reflectance Infrared Spectroscopy
Aliquots (10 μL each) of peptide gel at pH 7.4 were pipetted onto a Golden Gate diamond window and dried under nitrogen until a thin film of peptide was achieved. IR spectra (64 accumulations) were taken using a Jasco FT/IR-660 spectrometer.
Mechanical Analysis
Peptide solutions were made by dissolving lyophilized SLac in 298 mM sucrose/water at a concentration of 2 wt %, pH 7.4. Hydrogels were constructed by addition of HBSS at a 1:1 ratio. Multivalent drugs were added at similar concentrations, approximating physiological doses, to SLac solutions with final concentrations of 0.4 mM SLac (1 wt %); concentration of positive lysine charges, 0.8 mM; 3.8 mM (0.5 wt %) or 0.38 mM (0.05 wt %) suramin; 3.2 mM trypan blue (0.2 wt %); ~2.78 mM heparin (5 wt %); 3.4 mM clodronate disodium; 38 and 5 mM phosphate buffer; and 0.9% saline. Negatively charged polyvalent ions in buffer solution facilitated intermolecular ionic interactions with lysine residues, cross-linking the hydrogel. Rheological behavior of peptide hydrogels was determined using an 8 mm parallel-plate geometry at a gap of 250 μm. A 50 μL sample of hydrogel was constructed and, within 30 min, placed on stainless-steel plates of a rheometer (AR-G2, TA Instruments). A frequency sweep (0.1–100 Hz, at constant 1% strain), strain sweep (0–1000% strain, at 1 Hz), and shear recovery (1% strain for 30 min, 100% strain for 60 s, 1% strain for 30 min) were performed. We ensured the phase angle δ ≤ 90° to ensure no slipping.
Suramin Drug Loading and Release
Suramin was dissolved in 0.9% saline and loaded into MDP hydrogel (100 μL of SLac dissolved in 298 mM surcose + 100 μL of drug/saline) in microcentrifuge tubes. Release media (1 mL of HBSS) was added to gels placed in a humidified, 5% CO2 cell culture incubator at 37 °C. Aliquots (200 μL each) of release media were assayed daily with replenishment for 30 days. To determine short-term release kinetics, in a separate setup, aliquots were assayed at 1, 2, 4, 8, 12, 24, 48, and 72 h. Concentration of drug was determined using UV spectrophotometry at 313 nm, n = 5. Mass-release data were plotted as a function of cumulative release of drug in Sigmaplot (Systat, Chicago, IL). Drug release was modeled using the Korsmeyer–Peppas equation.35
Monocyte/Macrophage Culture and Differentiation
Human monocytic leukemia cell line THP-1 cells (ATCC, Manassas, VA), were cultured in media (ATCC-formulated RPMI-1640 medium supplemented with 0.05 mM 2-mercaptoethanol, fetal bovine serum (10%), 100 mg/mL penicillin, and 100 mg/mL streptomycin) at a concentration of 200 000 cells/mL. Cells were grown in suspension and diluted when concentration reached 0.8–1.0 million cells/mL. Media was changed every 3 days as necessary. THP-1 monocytes were cultured to M0 macrophages by pulsing with 5 nm phorbol 12-myristate 13-acetate, phorbol 12-myristate 13-acetate (PMA, Sigma-Aldrich) for 5 min. Adherent M0-differentiated cells were incubated with interferon gamma (IFN-γ, 20 ng/mL, Gibco, Life Technologies, Carlsbad, CA) and LPS (20 ng/mL, Sigma-Aldrich) for M1 (R&D Systems) for 24 h at 37 °C. For macrophage plasticity studies, THP-1 cells were differentiated to M0 macrophages as described. An aliquot of cells (0.5 M cells/cm2) were seeded atop 100 μL of SLac hydrogels made with 1 wt % suramin (final = 1 wt % peptide, 0.5 wt % suramin), 0.1 wt % suramin (final = 1 wt % peptide, 0.05 wt % suramin), or PBS (final = 1 wt % peptide), n = 5, in 16-well glass-bottomed glass slides. Media containing IFN-γ (20 ng/mL, Gibco, Life Technologies, Carlsbad, CA) and LPS (20 ng/mL, Sigma-Aldrich) was added atop hydrogels to stimulate M1 phenotype. Control hydrogels were gelled using PBS. M0 control did not receive IFN-γ/LPS; positive control media was supplemented with 0.5 or 0.05 mg of suramin (identical to mass of suramin in gels).29 After 24 h, media aliquots were stored at −80 °C, and proinflammatory markers IL-1β and TNF-α were measured (Biolegend, San Diego, CA). Gels were then fixed in formalin and immunostained for M1 marker CCR7 (Novus, Littleton, CO) and DAPI. The number of CCR7+ cells divided by the number of nuclei gave the M1 polarization ratio, quantified by counting six random image sections at 20× magnification per sample, of five samples per group, using NIH ImageJ.
Subcutaneous Implants
All experiments were approved by the Rice University Institutional Animal Care and Use committee. Female Wisar rats (225–250g, Charles River Laboratories, Wilmington, MA) were anesthetized using isofluorane (2% for induction and 1% for maintenance), dorsal aspect shaved, and sterilely prepped. Scaffolds were loaded in syringes, and 200 μL subcutaneous injections of each SLac and SLac+suramin (100 mg/mL) were made in four different 1.5-inch-spaced randomized sites on the dorsal aspect, on either side, between the lower thoracic and upper lumber vertebrae, n = 4 for each scaffold for 1 week. Rats were then euthanized using an overdose of isoflurane, CO2 asphyxiation, and bilateral thoracic puncture. The dorsal skin was removed around the entire implant, washed with PBS, and fixed in neutral buffered formalin for 24 h prior to processing. Tissue was then processed into paraffin blocks, sectioned at 7 μm, deparraffinized, and stained for cellular infiltrate using hematoxylin and eosin (H&E), and nuclei were counted in three random fields per sample and averaged over all samples from each group using ImageJ (NIH, Bethesda, MD). Infiltration of implants was graded on a 5 point scale: 1 = periphery (<50%, with large parts of scaffold uninfiltrated, center uninfiltrated), 2 = 50–80% (with small regions of scaffold exposed, center uninfiltrated), 3 = center infiltrated (with small regions of scaffold exposed), 4 = few to no scaffold regions visible, and 5 = implant indistinguishable from native tissue except for complete dense cellular repopulation. Cellular infiltrate was phenotyped using immunostaining. Immune cell staining was performed for panmacrophage, rabbit antirat CD68 (Abcam); M1 macrophages, goat antirat CCR7 (Novus); and M2 macrophages, mouse antirat CD163 (AbDSerotec). Secondary antibodies used were (i) AF647 donkey antirabbit, (ii) AF488 donkey antigoat, and (iii) AF555-donkey antimouse (Life Technologies). Nuclei were counterstained with DAPI (Life Technologies). Cellular infiltrate was quantified using NIH ImageJ, and the M1/M2 polarization ratio was determined.
Ionic cross-linking of scaffolds by drugs is a novel approach to the preparation of drug-delivering hydrogels. We have previously shown that heparin, a highly charged proteoglycan, can attenuate drug release from MDP.3,4,17,34 In this study, we show that heparin and a number of other small-molecule drugs act as the hydrogel cross-linking agent and are therefore effectively incorporated into the MDP. These ionically cross-linked MDPs show improved rheological performance because of the charged interaction of drugs and scaffolds (Figure 3). The resulting hydrogel is composed of nanofibrous β sheets and is cross-linked in a fashion similar to our previous PO43−-mediated charge-shielding approach to self-assembly (Figure 1 and Movie 1). Covalent attachment of drugs directly to the scaffold can be an effective method to present drugs and attenuate their release.36 Conventional covalent immobilization and physisorption strategies can result in altered chemical presentation, introduction of non-native polymeric sequences, or systemic side effects.9,10,16,35 Here, we show attenuation of suramin release over a long period, localized delivery via syringe, and maintenance of biological activity. Importantly, because the drug in this case is not simply cargo within the hydrogel but intimately participating in its structure, the release duration is very long (Figure 5). Most hydrogels loaded with small-molecule drugs, and even those loaded with relatively large proteins, result in short-term burst release that has limited their applicability.9,10,16,35 MDP interior hydrophobic domains may provide next generation strategies for solubilization of hydrophobic drugs while allowing aqueous solubility of nanofiber conjugates.
Supplementary Material
Acknowledgments
The authors appreciate help and support from the Shared Equipment Authority (SEA) at Rice University where imaging work was performed. The authors would like to thank the Breast Cancer Pathology Core at the Baylor College of Medicine for assistance with histomorphometry, and Dr. Jorge Wong at Rice University for assistance with WEO Movie 1. The work presented in this manuscript was supported by grants from the Welch Foundation (Grant C1557) NIH for J.D.H. (R01 DE021798) and V.A.K. (F32 DE023696).
Footnotes
A video illustrating self-assembly of MDP with suramin is presented in the HTML version of the paper.
The authors declare no competing financial interest.
References
- 1.Bakota EL, Wang Y, Danesh FR, Hartgerink JD. Biomacromolecules. 2011;12:1651–1657. doi: 10.1021/bm200035r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galler KM, Aulisa L, Regan KR, D’Souza RN, Hartgerink JD. J Am Chem Soc. 2010;132:3217–3223. doi: 10.1021/ja910481t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galler KM, Hartgerink JD, Cavender AC, Schmalz G, D’Souza RN. Tissue Eng, Part A. 2012;18:176–184. doi: 10.1089/ten.tea.2011.0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galler KM, D’Souza RN, Hartgerink JD, Schmalz G. Adv Dent Res. 2011;23:333–339. doi: 10.1177/0022034511405326. [DOI] [PubMed] [Google Scholar]
- 5.Galler KM, Cavender A, Yuwono V, Dong H, Shi S, Schmalz G, Hartgerink JD, D’Souza RN. Tissue Eng, Part A. 2008;14:2051–2058. doi: 10.1089/ten.tea.2007.0413. [DOI] [PubMed] [Google Scholar]
- 6.Aulisa L, Dong H, Hartgerink JD. Biomacromolecules. 2009;10:2694–2698. doi: 10.1021/bm900634x. [DOI] [PubMed] [Google Scholar]
- 7.Dong H, Paramonov SE, Aulisa L, Bakota EL, Hartgerink JD. J Am Chem Soc. 2007;129:12468–12472. doi: 10.1021/ja072536r. [DOI] [PubMed] [Google Scholar]
- 8.Bakota EL, Sensoy O, Ozgur B, Sayar M, Hartgerink JD. Biomacromolecules. 2013;14:1370–1378. doi: 10.1021/bm4000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serwer LP, James CD. Adv Drug Delivery Rev. 2012;64:590–597. doi: 10.1016/j.addr.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Wang PP, Frazier J, Brem H. Adv Drug Delivery Rev. 2002;54:987–1013. doi: 10.1016/s0169-409x(02)00054-6. [DOI] [PubMed] [Google Scholar]
- 11.Guerin C, Olivi A, Weingart JD, Lawson HC, Brem H. Invest New Drugs. 2004;22:27–37. doi: 10.1023/b:drug.0000006172.65135.3e. [DOI] [PubMed] [Google Scholar]
- 12.Wang AZ, Langer R, Farokhzad OC. Annu Rev Med. 2012;63:185–198. doi: 10.1146/annurev-med-040210-162544. [DOI] [PubMed] [Google Scholar]
- 13.Cho K, Wang X, Nie S, Chen ZG, Shin DM. Clin Cancer Res. 2008;14:1310–1316. doi: 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- 14.Ta HT, Dass CR, Dunstan DE. J Controlled Release. 2008;126:205–216. doi: 10.1016/j.jconrel.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 15.Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Adv Drug Delivery Rev. 2014;66:2–25. doi: 10.1016/j.addr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brannon-Peppas L, Blanchette JO. Adv Drug Delivery Rev. 2004;56:1649–1659. doi: 10.1016/j.addr.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Bakota E, Chang BH, Entman M, Hartgerink JD, Danesh FR. J Am Soc Nephrol. 2011;22:704–717. doi: 10.1681/ASN.2010040403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu Y, Kao WJ. Expert Opin Drug Delivery. 2010;7:429–444. doi: 10.1517/17425241003602259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Welter M, Rieger H. PLoS One. 2013;8:e70395. doi: 10.1371/journal.pone.0070395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mastrangelo E, Mazzitelli S, Fabbri J, Rohayem J, Ruokolainen J, Nykanen A, Milani M, Pezzullo M, Nastruzzi C, Bolognesi M. Chem Med Chem. 2014;9:933–939. doi: 10.1002/cmdc.201300563. [DOI] [PubMed] [Google Scholar]
- 21.Nie H, Fu Y, Wang CH. Biomaterials. 2010;31:8732–8740. doi: 10.1016/j.biomaterials.2010.07.080. [DOI] [PubMed] [Google Scholar]
- 22.Rasheed Q, Cacchione JG, Berry J, Richards F, Bryant L, Strony J, Enger C, Hodgson JM. Cathet Cardiovasc Diagn. 1994;31:240–245. doi: 10.1002/ccd.1810310317. [DOI] [PubMed] [Google Scholar]
- 23.Sweet JL, Pillay V, Choonara YE. Drug Delivery. 2007;14:309–318. doi: 10.1080/10717540701203067. [DOI] [PubMed] [Google Scholar]
- 24.Siepmann J, Siepmann F. J Controlled Release. 2012;161:351–362. doi: 10.1016/j.jconrel.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Hines DJ, Kaplan DL. Crit Rev Ther Drug Carrier Syst. 2013;30:257–276. doi: 10.1615/critrevtherdrugcarriersyst.2013006475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hearnden V, Sankar V, Hull K, Juras DV, Greenberg M, Kerr AR, Lockhart PB, Patton LL, Porter S, Thornhill MH. Adv Drug Delivery Rev. 2012;64:16–28. doi: 10.1016/j.addr.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 27.Coffey RJ, Jr, Leof EB, Shipley GD, Moses HL. J Cell Physiol. 1987;132:143–148. doi: 10.1002/jcp.1041320120. [DOI] [PubMed] [Google Scholar]
- 28.Qin Z. Atherosclerosis. 2012;221:2–11. doi: 10.1016/j.atherosclerosis.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 29.Sakaki H, Tsukimoto M, Harada H, Moriyama Y, Kojima S. PLoS One. 2013;8:e59778. doi: 10.1371/journal.pone.0059778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chaudhuri O, Koshy ST, Branco da Cunha C, Shin JW, Verbeke CS, Allison KH, Mooney DJ. Nat Mater. 2014;13:970–978. doi: 10.1038/nmat4009. [DOI] [PubMed] [Google Scholar]
- 31.Branco da Cunha C, Klumpers DD, Li WA, Koshy ST, Weaver JC, Chaudhuri O, Granja PL, Mooney DJ. Biomaterials. 2014;35:8927–8936. doi: 10.1016/j.biomaterials.2014.06.047. [DOI] [PubMed] [Google Scholar]
- 32.Lu P, Weaver VM, Werb Z. J Cell Biol. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar VA, Taylor NL, Shi S, Wang BK, Jalan AA, Kang MK, Wickremasinghe NC, Hartgerink JD. ACS Nano. 2015;9:860–868. doi: 10.1021/nn506544b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wickremasinghe NC, Kumar VA, Hartgerink JD. Biomacromolecules. 2014;15:3587–3595. doi: 10.1021/bm500856c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar VA, Taylor NL, Shi S, Wickremasinghe NC, D’Souza RN, Hartgerink JD. Biomaterials. 2015;52:71–78. doi: 10.1016/j.biomaterials.2015.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang M, Xu D, Jiang L, Zhang L, Dustin D, Lund R, Liu L, Dong H. Chem Commun (Cambridge, UK) 2014;50:4827–4830. doi: 10.1039/c4cc01568a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.