

Abstract
Extracellular shed vesicles, including exosomes and microvesicles, are disseminated throughout the body and represent an important conduit of cell communication. Cancer-cell-derived microvesicles have potential as a cancer biomarker as they help shape the tumor microenvironment to promote the growth of the primary tumor and prime the metastatic niche. It is likely that, in cancer cell cultures, the two constituent extracellular shed vesicle subpopulations, observed in dynamic light scattering, represent an exosome population and a cancer-cell-specific microvesicle population and that extracellular shed vesicle size provides information about provenance and cargo. We have designed and implemented a novel microfluidic technology that separates microvesicles, as a function of diameter, from heterogeneous populations of cancer-cell-derived extracellular shed vesicles. We measured cargo carried by the microvesicle subpopulation processed through this microfluidic platform. Such analyses could enable future investigations to more accurately and reliably determine provenance, functional activity, and mechanisms of transformation in cancer.
Keywords: Microvesicle, Exosome, Biomarker, Cancer, Microfluidic, Deterministic lateral displacement
1 Introduction
Extracellular shed vesicles (ESVs), including exosomes and cancer-cell-derived microvesicles, are disseminated throughout the body (Antonyak et al. 2011; Lee et al. 2011; Li et al. 2006; Bobrie et al. 2011; Muralidharan-Chari et al. 2010). We use the term exosome to refer to ESVs contained within multivesicular bodies (MVBs), that are trafficked to the cell surface, and released via fusion of MVBs with the cell membrane. Exosomes are thought to be generated by both normal and cancerous cells (Johnstone et al. 1987; Peinado et al. 2011). We use the term microvesicle to refer to ESVs that bud from cancer cell surfaces (D’Souza-Schorey and Clancy 2012; Antonyak et al. 2011; Lee et al. 2011). ESVs represent an important conduit of cell communication (Keller et al. 2006; Peinado et al. 2011; van Niel et al. 2006) and have potential as a disease state biomarker (Burgess 2013; Skog et al. 2008; Wang et al. 2013; D’Souza-Schorey and Clancy 2012; Nilsson et al. 2009). ESVs contain membrane-associated, cytosolic, and nuclear molecules including specifically packaged signaling proteins, enzymes, miRNAs, and RNA transcripts (Grange et al. 2011; Skog et al. 2008; Mathivanan and Simpson 2009; Cocucci et al. 2009; Antonyak et al. 2012; Lee et al. 2011; Li et al. 2012; Al-Nedawi et al. 2008; Al-Nedawi et al. 2009; Di Vizio et al. 2012). Recipient cells, upon ESV uptake, can experience a change in their behavior and function (Keller et al. 2006; Peinado et al. 2011; van Niel et al. 2006) due to cargoes in the ESVs. ESVs play a role in many systems, including immune responses (Kim et al. 2006; van Niel et al. 2006; Valenti et al. 2007), reproduction (Mincheva-Nilsson and Baranov 2010; Dragovic et al. 2011), virus proliferation (György et al. 2011; van der Pol et al. 2012; Schorey and Bhatnagar 2008), and cancer progression (Muralidharan-Chari et al. 2010; D’Souza-Schorey and Clancy 2012; Peinado et al. 2011). Cancer-cell-derived ESVs represent a heterogenous population that exhibits a large range of sizes with unique subpopulations (Antonyak et al. 2011; Muralidharan-Chari et al. 2010; van der Pol et al. 2010; Cocucci et al. 2009; Choi et al. 2007; Santana et al. 2014). We have recently demonstrated that cancer-cell-derived ESVs exhibit a bimodal size distribution (Santana et al. 2014). It is likely that the two constituent cancer-cell-derived ESV subpopulations in this size distribution represent an exosome population and a cancer-cell-specific microvesicle population (Santana et al. 2014) and that size correlates with biological properties of interest (van der Pol et al. 2012; D’Souza-Schorey and Clancy 2012). Microvesicles are ubiquitous in populations shed by cancer cells and decorate the surface of these cells (Antonyak et al. 2011; Santana et al. 2014).
ESV characterization is difficult because ESVs are small and exist in a complex biological milieu. The ability to discern chemical, biological, or physical differences among ESV subpopulations emanating from the same cell population is extremely challenging. Current microvesicle harvesting approaches concentrate ESVs by means of ultracentrifugation (Choi et al. 2007; Jorgensen et al. 2013; Wubbolts et al. 2003), filtration (Antonyak et al. 2011; Simpson et al. 2009; Lawrie et al. 2009; Mathivanan et al. 2010), and immunoaffinity (Coren et al. 2008; Tauro et al. 2012; Mathivanan et al. 2010), or some combination thereof. Although centrifugation and immunoaffinity approaches enable measurements reflecting averaged properties of heterogeneous ESV populations, they neither enable subpopulation cargo analysis nor efficiently isolate an intact ESV subpopulation for use in a biological assay. Centrifugation and filtration can concentrate ESVs within a sample, but centrifugation does not separate subpopulations. Filtration can isolate a targeted size population, but, to date, the recovery efficiency and purity have not been quantified. Furthermore, pressure drops across filters may damage the isolated ESV subpopulation.
To address these limitions, we have designed and implemented a novel microfluidic technology that separates microvesicles, as a function of diameter, from heterogeneous populations of cancer-cell-derived extracellular shed vesicles by using the principles of deterministic lateral displacement (DLD) (Inglis et al. 2006; Huang et al. 2004). Microfluidic devices can be designed to control particle trajectories as a function of their properties (Pamme 2007; Smith et al. 2012; Gleghorn et al. 2013; Heller and Bruus 2008; Loutherback et al. 2010; Huang et al. 2004; Pratt et al. 2011; Li et al. 2007; Hawkins et al. 2007). Microfluidic obstacle arrays for controlling particle trajectories have broad utility in medicine and biology (Huang et al. 2004; Green et al. 2009; Holm et al. 2011; Inglis et al. 2006). These technologies tend to separate particles with diameter between approximately 1–10μm (Inglis et al. 2010; Gleghorn et al. 2010); as such, convection dominates transport within these systems, given fluid velocities, target particle diameters, and obstacle array dimensions. Unique considerations must be taken into account when designing devices to isolate microvesicles from extracellular shed vesicles which have diameters between approximately 10nm–1μm, as diffusional transport always plays a significant role for small-diameter species (Huang et al. 2004; Heller and Bruus 2008).
2 Materials and methods
2.1 Device fabrication
The device geometry was drawn using L-Edit® (Tanner Research, Inc., Monrovia, CA). A chrome-plated quartz mask was fabricated using a DWL2000 (Heidelberg Instruments Mikrotechnik, GmbH, Heidelberg, Germany) and stripped of excess resist and chrome on a Hamatech-Steag Mask Processor (Süss MicroTec, Sunnyvale, CA). 100-mm silicon wafers were aggressively descummed with an oxygen plasma, dry-stripping process using an Aura 1000 Resist Strip (Gasonic Instruments, Inc., Calgary, Alberta). All wafers were primed under vacuum with hexamethyldisilazane (HMDS) using a YES-LP III Vapor Prime Oven (Yield Engineering Systems, Inc., Livermore, CA). This process yields an HMDS monolayer that promotes adhesion of the photoresist polymer to the wafer. To deposit the photoresist, Megaposit® SPR®220-3.0 (Shipley Company, L.L.C., Marlborough, MA), 3mL were deposited onto the primed wafer. Following deposition, the wafer was spun at 4000 r· min for 30 seconds to yield an approximately 3–μm layer. To cure the photoresist, wafers were soft baked at 115 °C for 90 seconds. Wafers were patterned with the mask bearing the image of the device geometry on a High Resolution Mask Aligner (ABM, Inc., Silicon Valley, CA) by exposing for 6 seconds with 365nm light at 11.2mW cm−2. Patterned wafers were subsequently hard baked at 115 °C for 90 seconds. Wafers were developed for 120 seconds in AZ® 726 MIF Developer (AZ Electronic Materials USA, Corp., Somerville, NJ) on a Hamatech-Steag Wafer Processor (Süss MicroTec, Sunnyvale, CA). To etch channels, wafers were Bosch-etched on a Unaxis 770 Deep Silicon Etcher (Oerlikon, Pfäffikon, Switzerland) to a depth of approximately 100μm, as measured on a NewView™ 7300 3D Optical Surface Profilometer (Zygo Corp., Middlefield, CT). To remove the remaining photoresist, wafers were piranha cleaned using a Hamatech Hot Piranha (Süss MicroTec, Sunnyvale, CA) and dry-stripped using an Aura 1000 Resist Strip, as previously described. For storage, prior to use, wafers were coated with uncured SPR®220-3.0 and diced on a K&S® 7100 Dicing Saw (Kulicke & Soffa Industries, Inc., Fort Washington, PA). All stages of device fabrication were completed in the Cornell Nanoscale Science and Facility (CNF).
2.2 Device construction
Polydimethylsiloxane (PDMS) gaskets were made in a 5:1 elastomer base-curing agent ratio, using a Sylgard® 184 silicone elastomer kit (Dow Corning®, Midland, MI). PDMS gaskets were cured at 60° for 4 hours. After curing, gaskets were cut to size and cleaned with acetone, isopropanol, and deionized water; gaskets were dried with compressed nitrogen gas. Devices were stripped of photoresist as described in Section 2.1. Immediately following stripping, PDMS gaskets and cleaned devices were primed for bonding in a Glen 1000 Plasma Strip (Yield Engineering Systems, Inc., Livermore, CA) with an oxygen plasma at 0.4torr at 100W for 180 seconds. Silicon devices and PDMS gaskets were manually aligned and bonded and subsequently baked at 60 °C for 1 hour to secure the bond between the gasket and the device. Device inlets and outlets were fitted with 50-μm inner diameter Tygon® tubes (Saint-Gobain Performance Plastics, Corp., Akron, OH); devices were manually primed with ethanol.
2.3 Surface modification
Devices were functionalized with 5000-Da silanated polyethylene glycol (silanated-PEG, Nanocs, Inc., New York, NY) by incubating the sealed device with 1mg mL−1 silanated-PEG in 95 % ethanol, 5 % deionized water (vol./vol.) for 120 minutes. To remove the PEG solution, devices were rinsed with ethanol, purged with deionized water, and then with 3 % (m/v) bovine serum albumin (BSA) in phosphate buffered saline (PBS) (3 % BSA buffer). Devices were stored in 3 % BSA buffer, at room temperature, for up to 4 hours prior to use.
2.4 Cell culture
The BxPC-3 (CRL-1687™, pancreatic adenocarcinoma) cell line was obtained from the American Type Culture Collection (ATCC®, Manassas, Virginia) and cultured in Roswell Park Memorial Institute (RPMI-1640; Lonza, Walkersville, MD) cell medium containing 10 % fetal bovine serum (FBS; Gemini BioProducts, West Sacramento, CA). Cells were cultured under standard conditions (37 °C, humidified, 5 % carbon dioxide environment). The culture medium was exchanged regularly according to standard sterile techniques. The cells were maintained in 25-cm−2 rectangular cell culture flasks and 150-mm cell culture dishes.
2.5 Sample preparation
Fluorescent polystryene microspheres, (1 % (vol./vol.); Bangs Laboratories, Inc., Fishers, IN), of diameters 51 nm, 190 nm, and 2.01μm, were used to characterize microdevice performance. These beads were suspended in 3 % BSA buffer by mixing 10, 30, and 50μL of stock bead solution, respectively, in 1mL of the 3 % BSA buffer solution.
ESVs were harvested from BxPC-3 cells by collecting the conditioned medium from 30e6 cells subjected to a 12-hour serum starvation. Following medium collection, intact cells and cell debris were removed by centrifugation at 300 × g for 10 minutes and 12000 × g for 20 minutes. Partially clarified conditioned medium was collected for subsequent processing. To analyze MV content, the supernatant sample was filtered through a 0.22μm Steriflip® filter (EMD Millipore, Billerica, MA) and rinsed with 5mL of PBS. Microvesicles retained on the filter surface were then lysed with 2mL of cell lysis buffer (25mM Tris, 100mM NaCl, 1 % Triton X-100, 1mM EDTA, 1mM DTT, 1mM NaVO4, 1mM β-glycerol phosphate, 1μg mL−1 aprotinin, 1μg mL−1 leupeptin). 200μL supernatant samples were extracted for each ELISA measurement. For the conditioned media controls, 200-μL of supernatant was collected. For microdevice processing, 170-μL conditioned media supernatant samples were added to 30μL of fluorescent bead samples, for flow verification purposes, and coprocessed with a 3 % BSA buffer sheath flow. Following microdevice processing, 200μL of device outputs δ, undeflected (heterogeneous ESV) output, and ε, deflected (microvesicle) output, were analyzed (see Fig. 1 for output locations).
Fig. 1.
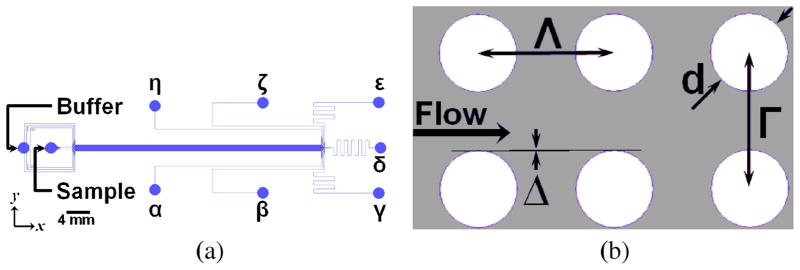
Microfluidic MV Separation Device Design. a Device Schematic. Note that the MV sample is centered on the input and is surrounded by a sheath flow. Particles (microspheres and ESVs) experience a symmetric and effectively infinite flow field. The volumetric flow rate of each input channel, buffer and sample inputs, has been matched to ensure uniform input velocity. The mean flow is in the x-direction; the mean fluid flow in the y-direction is zero. In this system, output ε contains the displaced MV population and output δ contains the heterogeneous ESV population. b Post Array Design. The geometric parameters are designed to yield a threshold diameter of 250nm. The center-to-center spacing are indicated by Λ and Γ. The obstacle diameter is indicated by d. The offset is indicated by Δ. the shoulder-to-shoulder gap is 6μm and the offset angle is 0.16°
2.6 Experimental visualization and setup
All experiments were conducted on the stage of a Nikon® LVUDM100 upright microscope (Nikon Instruments, Inc., Melville, NY). An X-cite® fluorescence illumination source (Lumen Dynamics Group, Inc., Mississauga, ON) excited and visualized fluorescent nano- and microspheres through a fluorescein isothiocyanate (FITC), Texas Red/Cy3.5, and 4′,6-diamidino-2-phenylindole (DAPI) cube (Chroma Technology Corp., Bellows Falls, VT). All images were collected with Q-Capture Pro 7™(Quantitative Imaging, Corp., Surrey, BC) and an EXi Blue™ fluorescence microscopy camera (Quantitative Imaging). All image intensity datasets were extracted from a 500-frame, 60-fps averaged camera feed with ImageJ.
The buffer solution and prepared samples were delivered through the device by syringe pumps (Chemyx, Inc., Stafford, TX) at a rate of 3.74mL h−1 and 0.15mL h−1, respectively. The effluent of each outlet was collected in 1.5mL tubes (Eppendorf, AG, Hamburg, Germany).
2.7 ELISA
Vascular endothelial growth factor (VEGF) content was measured at the output ports of interest with the Human VEGF Quantikine ELISA Kit (R&D Systems, Inc., Minneapolis, MN) according to the manufacturer’s instructions. Concentration readings were scaled and adjusted to account for diluents (3 % BSA, lysis buffer, PBS, and serum-free RPMI).
2.8 Data analysis
All statistics were extracted using Matlab®. Reported errors represent standard error of the mean for six measurements, unless noted otherwise. To eliminate background noise from the output readings, the integral of total pixel intensity for all regions of interest was used to adjust the intensity reading of each channel. To calculate output channel purity, the product of the output channel composition and the volume integral of ESVs produced by BxPC-3 cells was calculated.
3 Results and discussion
A microfluidic device, Fig. 1, was designed to separate microvesicles from samples containing heterogeneous extracellular shed vesicle populations by use of deterministic lateral displacement (Huang et al. 2004). The threshold diameter, i.e. the diameter above which particles experience deterministic lateral displacement resulting from interactions with the obstacle array, is 250μm, as seen in Fig. 2a. The threshold diameter is dictated by the array properties, see Fig. 1b. This threshold diameter is designed to lie in the natural gap existing between the two cancer-cell-derived ESVs subpopulations shed by BxPC-3 cells (Santana et al. 2014). The array geometry could be tuned to efficiently separate species with unique threshold diameters (Smith et al. 2012; Gleghorn et al. 2013). In this system, the Peclet number (Pe), that is, the ratio of convective to diffusional transport rates, is significantly greater than unity (Pe = 𝓞(107 – 109)) and is calculated as , where Λ is the center-to-center obstacle spacing in the direction of flow (see Fig. 1), 〈Uflow〉 is the average fluid velocity, and D is the particle diffusivity dictated by the Stokes-Einstein equation . As such, flow-associated transport is dominated by convection. In the direction orthogonal to the mean direction of flow, transport is mediated by deterministic lateral displacement (DLD) (Huang et al. 2004; Inglis et al. 2006) and by diffusion, to varying degrees, as a function of the particle diameter (dparticle). The relative transport contributions of these phenomenon can be directly compared in the ratio of the characteristic diffusional transport length (ldiffusion) and the characteristic DLD transport length (lDLD), that is , where the characteristic time scale, t̃, is derived from microfluidic array properties and the flow rate , and UDLD is the transverse velocity of the particles resulting from deterministic lateral displacement. Figure 2b shows the value of this transport ratio as a function of ESV diameter. In the case of small-diameter ESVs (i.e. exosomes), which are on the order of 10–100nm, diffusional transport plays a significant role. As such, diffusion may limit a particle’s ability to enter a collision-mode trajectory, as mediated by DLD, and decrease the device’s separation efficiency (Heller and Bruus 2008).
Fig. 2.
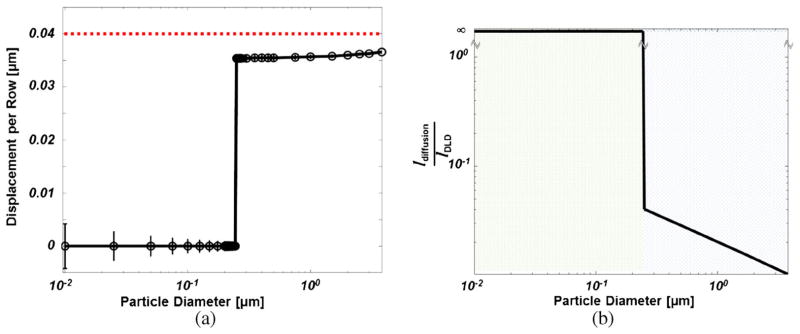
Device Performance and Transport. a Calculated Displacement. The total calculated displacement per obstacle as a function of ESV diameter was determined by FEM (COMSOL). The markers represent calculated displacement accounting for particle–obstacle interactions; the vertical bars represent the maximum possible displacement resulting from diffusional transport over the timescale between obstacle interactions. This diffusional transport is a function of particle diameter ( , where Γ is the center-to-center spacing in the direction transverse to flow, and Reflow is the Reynolds number of the flow). The curvature in the displacement curve for ESV diameters above the threshold diameter is a consequence of diameter-dependent particle migration resulting from obstacle interactions. The dashed line indicates the maximum theoretical displacement per row. b Transport Length Ratio. The ratio of the characteristic diffusional transport length to the characteristic deterministic lateral displacement (DLD) transport length as a function of particle diameter demonstrates that multiple transport phenomena must be considered in a separation device. For the device described in this work, small-diameter ESVs (below the threshold diameter) move laterally by diffusion. Large-diameter ESVs (MVs with diameters above the threshold diameter) move laterally by DLD-dominated transport, and the role played by diffusion decreases with particle size
The post array and device geometry were designed in an incremental process. Approximate particle trajectories in this system were predicted by means of a ballistic model (Gleghorn et al. 2013); this approach predicts trajectories for dilute particles treated as Lagrangian tracers experiencing inelastic obstacle collisions (Gleghorn et al. 2013). The ballistic model provides a baseline obstacle array geometry from which subsequent designs are based. Subsequently, a finite element modeling (FEM) approach (Gleghorn et al. 2013) was used to predict the threshold diameter and resultant particle trajectories and displacements. Informed by the FEM data, iterative changes were made to the array’s geometric parameters, accounting for device length, pressure drops, and diffusional transport, until the target threshold diameter, 250nm, and total displacement across the device length, 𝓞(100μm), were achieved. The total displacement is designed to yield a spatial separation that overcomes small-diameter particles’ diffusion across the device length. The displacements resulting from the final array, as shown in Fig. 1b, are shown in Fig. 2a.
On-device microvesicle separation performance was evaluated by processing three sets of fluorescent microspheres whose diameters represent ESV diameters of interest, one whose diameter is below the threshold diameter (51nm), one whose diameter is less than but near the threshold diameter (190nm), and one above the threshold diameter (2.01μm). As shown in Fig. 3, microspheres with diameters less than the 250-nm threshold diameter were negligibly displaced whereas microspheres with diameters above the threshold diameter were displaced, by means of deterministic lateral displacement, into the adjacent output (output ε). For the 190nm-diameter population, the population with a diameter near the threshold, there is some degree of device-mediated displacement in excess of diffusion, as indicated by the composition of this population in output ε. In comparing output ε to output δ, there is a significant concentration of microvesicle-sized species in output ε, in accordance with the device design. The MV-concentrated output, output ε, is 98.5 ± 31.6 % pure, by volume, with a recovery efficiency of 39.3 ± 12.4 %.
Fig. 3.

In polystyrene bead separation experiments, the microfluidic obstacle array preferentially deflects large-diameter particles in the target output (ε). Particles with diameters below the threshold diameter demonstrate minimal deflection from the input stream across the length of the device. Particles with diameters near the threshold diameter indicate a minimal degree of device-mediated deflection into output ε. There is a statistically significant difference between the composition of large- and intermediate-diameter particles in output ε (p < 0.05). Note: “< Threshold Diameter” indicates 51nm particles; “≲ Threshold Diameter” indicates 190nm particles; “> Threshold Diameter” indicates 2.01μm particles
Following validation of this novel microfluidic platform, by means of polystyrene nano- and microsphere separation, we processed ESVs harvested from BxPC-3 cells through the microfluidic device. As ESVs are not readily visualized during separation experiments, given their size and the similarity of their optical properties to those of the buffer solution, ESV-containing conditioned media samples were supplemented with fluorescent microspheres to verify flow separation. The fidelity of this approach for microvesicle chemical analysis was determined by quantifying the amount of VEGF present within the effluent of output δ, the heterogeneous-ESV containing output, and output ε, the microvesicle-containing output. We selected VEGF detection as an exemplary chemical readout, as VEGF (a) stimulates angiogenesis, (b) is found in cancer-cell-derived ESVs (Skog et al. 2008; Taraboletti et al. 2006; Lee et al. 2011), and (c) promotes metastasis (Skobe et al. 2001; Stacker et al. 2001; Ishigami et al. 1998).
As shown in Fig. 4, output ε has higher VEGF content than output δ; the statistically significant difference between the VEGF content of output δ and output ε is likely attributable to the preferential concentration of MVs in output ε. Soluble VEGF contained within the conditioned medium primarily contributes to the VEGF readout in output δ, as compared to that of output ε, given the limited diffusion of this species across the device length (radius of gyration, ; ldiffusion ≈ 10μm).
Fig. 4.
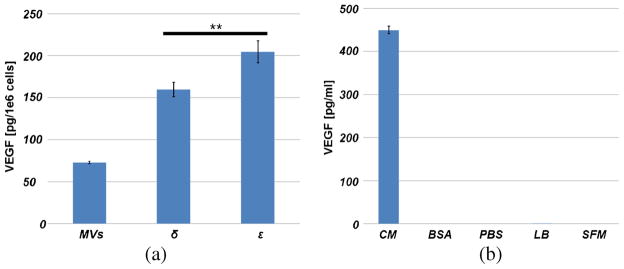
a VEGF Readings. An ELISA assay was used to measure the total VEGF content in concentrated MVs harvested from 35mL of BxPC-3 conditioned medium, and from 200μL samples collected from output δ and ∊ of the microfluidic device, all measurements are scaled by total cell numbers and volume harvested, and are adjusted by the volume of diluent added. These measurements show that processing conditioned medium directly in the microdevice affords sufficient sample for immunodetection. The statistically significant difference (p < 0.05) in results between the VEGF content of the unique outputs likely results from the preferential concentration of MVs in output ε as compared to δ. b Controls. VEGF measurements were conducted on the following controls: unlysed conditioned medium (UCM), BSA, PBS, lysis buffer (LB), and serum-free medium (SFM). BSA, PBS, LB, and SFM are diluents in various measurements, the controls demonstrate that none but soluble VEGF in conditioned medium contribute additional VEGF to output readings. Note: n = 2 for all controls, except the conditioned medium
Current extracellular shed vesicle and microvesicle harvesting approaches rely on centrifugation, filtration, and immunoaffinity. ESVs isolated by means of centrifugation, in which samples are pelleted (Jorgensen et al. 2013; Wubbolts et al. 2003; Yuan et al. 2009; Tauro et al. 2012) or enriched (Choi et al. 2007), and immobilization (Coren et al. 2008; Tauro et al. 2012), in which ESVs are chemically-bound to an immunocoated surface, yield samples consisting of all species (i.e. exosomes and microvesicles) within the sample. By comparison, our microfluidic device yields a pure sample of intact microvesicles. As such, chemical analyses from our system, as compared to centrifugation or immobilization, represent components only expressed by microvesicles, instead of those expressed by all constituent ESVs. Filtration, in which off-the-shelf, low-protein-absorption 0.22μm filters are typically used (Antonyak et al. 2011; Mathivanan et al. 2010; Simpson et al. 2009; Lawrie et al. 2009), benefits from the serendipitous fact that the pore size aligns well with the natural gap (Santana et al. 2014) between small- and large-diameter ESV subpopulations. An attractive aspect of filtration is that it recovers and concentrates the resultant sample (Antonyak et al. 2011). Increasing pressure drops across filters, resulting from pores being filled by microvesicles, may damage the harvested population; to date, this phenomenon has not been quantified but a direct comparison between filter-concentrated MVs and microfluidic-concentrated MVs in future studies may clarify this. The current microfluidic device’s most appealing aspects are its geometric tunability of threshold diameter and gentle treatment of ESVs, ensuring no ESV destruction or consolidation, and the demonstrated purity of the isolated MVs, and the confirmed difference in protein content of separated ESV subpopulations. As this device does not concentrate vesicles, it has not demonstrated, to date, the sample volumes achieved via filtration and centrifugation (Antonyak et al. 2011; Jorgensen et al. 2013) for use in cell transformation assays or Western blots. Next-generation devices may address this by multiplying throughput.
4 Conclusions
Cancer-cell-derived ESVs, specifically microvesicles, are believed to play important roles in cancer progression including changing the behaviors of cells that make-up the tumor microenvironment in ways that drive primary tumor growth. ESVs, which as also shed into circulation, are believed to contribute to the priming of the metastatic niche. As cargo is believed to be correlated with size, it is important to isolate the large-diameter microvesicle population to identify unique cargoes and further establish microvesicles’ role as a cancer biomarker. In this work, we developed a deterministic lateral displacement microfluidic device for the isolation of cancer-cell-derived microvesicles from conditioned medium containing a heterogeneous cancer-cell-derived extracellular shed vesicle population. We highlighted an exemplary case of chemical detection by measuring VEGF within each ESV-containing microfluidic output and showed a statistically-significant difference between the outputs, which likely results from MV deflection to and concentration in the target output (ε). Additional chemical analyses could be conducted to measure other cargo of interest in on-going efforts to establish cancer-cell-derived MVs as cancer biomarkers and their role in cancer progression. The microfluidic device we designed and tested demonstrated a microvesicle-sized particle recovery efficiency of 39 % with a corresponding purity of 98.5 % in target output. The primary limitation of efficiency is attributable to the role of diffusion in the transport of such small particles. This technology enables separation of microvesicle-sized particles and can be used to identify cargo carried by the microvesicle subpopulation. Such analyses could enable future investigations revealing microvesicles’ provenance, functional activity, and mechanisms of transformation in cancer.
Acknowledgments
This work was supported by the National Cancer Institutes under Award Number U54CA143876 and the Alfred P. Sloan Foundation.
Contributor Information
Steven M. Santana, Sibley School of Mechanical and Aerospace Engineering, Cornell University, 238 Upson Hall, Ithaca, NY 14853, USA
Marc A. Antonyak, Department of Molecular Medicine, Vet Medical Center, Room C3, Cornell University, Ithaca, NY 14853, USA
Richard A. Cerione, Department of Molecular Medicine, Vet Medical Center, Room C3, Cornell University, Ithaca, NY 14853, USA
Brian J. Kirby, Email: kirby@cornell.edu, Sibley School of Mechanical and Aerospace Engineering, Cornell University, 238 Upson Hall, Ithaca, NY 14853, USA
References
- Antonyak MA, Bo L, Lindsey K, Johnson JL, Druso JE, Bryant KL, Holowka DA, Cerione RA, Boroughs LK. Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Pro Natl Acad Sci. 2011;108(42):17569–17569. doi: 10.1073/pnas.1017667108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TH, D’Asti E, Magnus N, Al-Nedawi K, Meehan B, Rak J. Microvesicles as mediators of intercellular communication in cancer–the emerging science of cellular ’debris’. Semin Immunopathol. 2011;33(5):455–67. doi: 10.1007/s00281-011-0250-3. [DOI] [PubMed] [Google Scholar]
- Li XB, Zhang ZR, Schluesener HJ, Xu SQ. Role of exosomes in immune regulation. J Cell Mol Med. 2006;10(2):364–375. doi: 10.1111/j.1582-4934.2006.tb00405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobrie A, Colombo M, Raposo G, Théry C. Exosome secretion: molecular mechanisms and roles in immune responses. Traffic (Copenhagen, Denmark) 2011;12(12):1659–68. doi: 10.1111/j.1600-0854.2011.01225.x. [DOI] [PubMed] [Google Scholar]
- Muralidharan-Chari V, Clancy JW, Sedgwick A0, D’Souza-Schorey C. Microvesicles: mediators of extracellular communication during cancer progression. J Cell Sci. 2010;123(Pt 10):1603–11. doi: 10.1242/jcs.064386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes) J Biol Chem. 1987;262(19):9412–9420. [PubMed] [Google Scholar]
- Peinado H, Lavotshkin S, Lyden D. The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol. 2011;21(2):139–46. doi: 10.1016/j.semcancer.2011.01.002. [DOI] [PubMed] [Google Scholar]
- D’Souza-Schorey C, Clancy JW. Tumor-derived microvesicles: shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev. 2012;26(12):1287–99. doi: 10.1101/gad.192351.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller S, Sanderson MP, Stoeck A, Altevogt P. Exosomes: from biogenesis and secretion to biological function. Immunol Lett. 2006;107:102–108. doi: 10.1016/j.imlet.2006.09.005. [DOI] [PubMed] [Google Scholar]
- van Niel G, Porto-Carreiro I, Simoes S, Raposo G. Exosomes: a common pathway for a specialized function. J Biochem. 2006;140(1):13–21. doi: 10.1093/jb/mvj128. [DOI] [PubMed] [Google Scholar]
- Burgess DJ. Glioblastoma: Microvesicles as major biomarkers. Nat Rev Cancer. 2013;13(1):8. doi: 10.1038/nrc3424. [DOI] [PubMed] [Google Scholar]
- Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT, Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10(12):1470–6. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Li H, Yan Z, Shenghua J. Peripheral blood microvesicles are potential biomarkers for hepatocellular carcinoma - Cancer Biomarkers - Volume 13, Number 5/2013 - IOS Press. Cancer Biomarkers. 2013;13(5):351–357. doi: 10.3233/CBM-130370. [DOI] [PubMed] [Google Scholar]
- Nilsson J, Skog J, Nordstrand A, Baranov V, Mincheva-Nilsson L, Breakefield XO, Widmark A. Prostate cancer-derived urine exosomes: a novel approach to biomarkers for prostate cancer. Br J Cancer. 2009;100(10):1603–7. doi: 10.1038/sj.bjc.6605058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grange C, Tapparo M, Collino F, Vitillo L, Damasco C, Deregibus MC, Tetta C, Bussolati B, Camussi G. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res. 2011;71(15):5346–56. doi: 10.1158/0008-5472.CAN-11-0241. [DOI] [PubMed] [Google Scholar]
- Mathivanan S, Simpson RJ. ExoCarta: a compendium of exosomal proteins and RNA. Proteomics. 2009;9(21):4997–5000. doi: 10.1002/pmic.200900351. [DOI] [PubMed] [Google Scholar]
- Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2009;19(2):43–51. doi: 10.1016/j.tcb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Antonyak MA, Wilson KF, Cerione RA. R(h)oads to microvesicles. Small GTPases. 2012;3(4):219–24. doi: 10.4161/sgtp.20755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Antonyak MA, Zhang J, Cerione RA. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene. 2012;31(45):4740–9. doi: 10.1038/onc.2011.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, Rak J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008;10(5):619–24. doi: 10.1038/ncb1725. [DOI] [PubMed] [Google Scholar]
- Al-Nedawi K, Meehan B, Rak J. Microvesicles: messengers and mediators of tumor progression. Cell Cycle. 2009;8:2014–2018. doi: 10.4161/cc.8.13.8988. [DOI] [PubMed] [Google Scholar]
- Di Vizio D, Morello M, Dudley AC, Schow PW, Adam RM, Morley S, Mulholland D, Rotinen M, Hager MH, Insabato L, Moses MA, Demichelis F, Lisanti MP, Hong W, Klagsbrun M, Bhowmick NA, Rubin MA, D’Souza-Schorey C, Freeman MR. Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. Am J Pathol. 2012;181(5):1573–84. doi: 10.1016/j.ajpath.2012.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Bianco N, Menon R, Lechman ER, Shufesky WJ, Morelli AE, Robbins PD. Exosomes derived from genetically modified DC expressing FasL are anti-inflammatory and immunosuppressive. Molecular therapy: The Journal of the American Society of Gene Therapy. 2006;13(2):289–300. doi: 10.1016/j.ymthe.2005.09.015. [DOI] [PubMed] [Google Scholar]
- Valenti R, Huber V, Iero M, Filipazzi P, Parmiani G, Rivoltini L. Tumor-released microvesicles as vehicles of immunosuppression. Cancer Res. 2007;67(7):2912–2915. doi: 10.1158/0008-5472.CAN-07-0520. [DOI] [PubMed] [Google Scholar]
- Mincheva-Nilsson L, Baranov V. The role of placental exosomes in reproduction. Am J Reprod Immunol (New York, NY: 1989) 2010;63(6):520–33. doi: 10.1111/j.1600-0897.2010.00822.x. [DOI] [PubMed] [Google Scholar]
- Dragovic RA, Gardiner C, Brooks AS, Tannetta DS, Ferguson DJP, Hole P, Carr B, Redman CWG, Harris AL, Dobson PJ, Harrison P, Sargent IL. Sizing and phenotyping of cellular vesicles using nanoparticle tracking analysis. Nanomedicine: Nanotechnology, Biology and Medicine. 2011;7(6):780–8. doi: 10.1016/j.nano.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- György B, Szabó TG, Pásztói M, Pál Z, Misják P, Aradi B, László V, Pállinger E, Pap E, Kittel A, Nagy G, Falus A, Buzás EI. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci: CMLS. 2011;68(16):2667–88. doi: 10.1007/s00018-011-0689-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Pol E, Böing AN, Harrison P, Sturk A, Nieuwland R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev. 2012;64(3):676–705. doi: 10.1124/pr.112.005983. [DOI] [PubMed] [Google Scholar]
- Schorey JS, Bhatnagar S. Exosome function: from tumor immunology to pathogen biology. Traffic. 2008;9:871–881. doi: 10.1111/j.1600-0854.2008.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Pol E, Hoekstra AG, Sturk A, Otto C, van Leeuwen TG, Nieuwland R. Optical and non-optical methods for detection and characterization of microparticles and exosomes. J Thromb Haemost: JTH. 2010;8(12):2596–607. doi: 10.1111/j.1538-7836.2010.04074.x. [DOI] [PubMed] [Google Scholar]
- Choi DS, Lee JM, Park GW, Lim HW, Bang JY, Kim YK, Kwon KH, Ho JK, Kim KP, Gho YS. Proteomic analysis of microvesicles derived from human colorectal cancer cells. J Proteome Res. 2007;6(12):4646–55. doi: 10.1021/pr070192y. [DOI] [PubMed] [Google Scholar]
- Santana SM, Antonyak MA, Cerione RA, Kirby BJ. Cancerous epithelial cell lines shed extracellular vesicles with a bimodal size distribution that is sensitive to glutamine inhibition. Phys Biol. 2014 doi: 10.1088/1478-3975/11/6/065001. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen M, Baek R, Pedersen S, Sondergaard EKL, Kristensen SR, Varming K. Extracellular vesicle (EV) array: microarray capturing of exosomes and other extracellular vesicles for multiplexed phenotyping. Journal Extracellular Vesicles. 2013:2. doi: 10.3402/jev.v2i0.20920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wubbolts R, Rachel SL, Veenhuizen PTM, Schwarzmann G, Mobius W, Hoernschemeyer J, Slot JW, Geuze HJ, Stoorvogel W. Proteomic and biochemical analyses of human b cell-derived exosomes: potential implications for their function and multivesicular body formation. J Biol Chem. 2003;278(13):10963–10972. doi: 10.1074/jbc.M207550200. [DOI] [PubMed] [Google Scholar]
- Simpson RJ, Lim JWE, Moritz RL, Mathivanan S. Exosomes: proteomic insights and diagnostic potential. Expert Review of Proteomics. 2009;6(3):267–83. doi: 10.1586/epr.09.17. [DOI] [PubMed] [Google Scholar]
- Lawrie AS, Albanyan A, Cardigan RA, Mackie IJ, Harrison P. Microparticle sizing by dynamic light scattering in fresh-frozen plasma. Vox Sanguinis. 2009;96(3):206–12. doi: 10.1111/j.1423-0410.2008.01151.x. [DOI] [PubMed] [Google Scholar]
- Mathivanan S, Lim JWE, Tauro BJ, Ji H, Moritz RL, Simpson RJ. Proteomics analysis of A33 immunoaffinity-purified exosomes released from the human colon tumor cell line LIM1215 reveals a tissue-specific protein signature. Molecular & cellular Proteomics: MCP. 2010;9(2):197–208. doi: 10.1074/mcp.M900152-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coren LV, Shatzer T, Ott DE. CD45 immunoaffinity depletion of vesicles from Jurkat T cells demonstrates that exosomes contain CD45: no evidence for a distinct exosome/HIV-1 budding pathway. Retrovirology. 2008;5(1):64. doi: 10.1186/1742-4690-5-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauro BJ, Greening DW, Mathias RA, Ji H, Mathivanan S, Scott AM, Simpson RJ. Comparison of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for isolating human colon cancer cell line LIM1863-derived exosomes. Methods (San Diego, Calif) 2012;56(2):293–304. doi: 10.1016/j.ymeth.2012.01.002. [DOI] [PubMed] [Google Scholar]
- Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteome. 2010;73:1907–1920. doi: 10.1016/j.jprot.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Inglis DW, Davis JA, Austin RH, Sturm JC. Critical particle size for fractionation by deterministic lateral displacement. Lab on a Chip. 2006;6(5):655–8. doi: 10.1039/b515371a. [DOI] [PubMed] [Google Scholar]
- Huang LR, Cox EC, Austin RH, Sturm JC. Continuous particle separation through deterministic lateral displacement. Science. 2004;304:987–990. doi: 10.1126/science.1094567. [DOI] [PubMed] [Google Scholar]
- Pamme N. Continuous flow separations in microfluidic devices. Lab on a Chip. 2007;7(12):1644. doi: 10.1039/b712784g. [DOI] [PubMed] [Google Scholar]
- Smith JP, Barbati AC, Santana SM, Gleghorn JP, Kirby BJ. Microfluidic transport in microdevices for rare cell capture. Electrophoresis. 2012;33(21):3133–3142. doi: 10.1002/elps.201200263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleghorn JP, Smith JP, Kirby BJ. Transport and collision dynamics in periodic asymmetric obstacle arrays: Rational design of microfluidic rare-cell immunocapture devices. Phys Rev E. 2013;88(3):032136. doi: 10.1103/PhysRevE.88.032136. [DOI] [PubMed] [Google Scholar]
- Heller M, Bruus H. A theoretical analysis of the resolution due to diffusion and size dispersion of particles in deterministic lateral displacement devices. J Micromech Microeng. 2008;18(7):075030. [Google Scholar]
- Loutherback K, Chou KS, Newman J, Puchalla J, Austin RH, Sturm JC. Improved performance of deterministic lateral displacement arrays with triangular posts. Microfluid Nanofluid. 2010;9(6):1143–1149. [Google Scholar]
- Pratt ED, Huang C, Hawkins BG, Gleghorn JP, Kirby BJ. Rare cell capture in microfluidic devices. Chem Eng Sci. 2011;66:1508–1522. doi: 10.1016/j.ces.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Dalton C, Crabtree JH, Nilsson G, Kaler KV. Continuous dielectrophoretic cell separation microfluidic device. Lab on a chip. 2007;7(2):239–48. doi: 10.1039/b613344d. [DOI] [PubMed] [Google Scholar]
- Hawkins BG, Smith AE, Syed YA, Kirby BJ. Continuous-flow particle separation by 3D insulative dielectrophoresis using coherently shaped, dc-biased, ac electric fields. Anal Chem. 2007;79:7291–7300. doi: 10.1021/ac0707277. [DOI] [PubMed] [Google Scholar]
- Green JV, Radisic M, Murthy SK. Deterministic lateral displacement as a means to enrich large cells for tissue engineering. Anal Chem. 2009;81(21):9178–82. doi: 10.1021/ac9018395. [DOI] [PubMed] [Google Scholar]
- Holm SH, Beech JP, Barrett MP, Tegenfeldt JO. Separation of parasites from human blood using deterministic lateral displacement. Lab on a chip. 2011;11(7):1326–32. doi: 10.1039/c0lc00560f. [DOI] [PubMed] [Google Scholar]
- Inglis DW, Herman N, Vesey G. Highly accurate deterministic lateral displacement device and its application to purification of fungal spores. Biomicrofluidics. 2010;4(2):024109. doi: 10.1063/1.3430553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleghorn JP, Pratt ED, Denning D, Liu H, Bander NH, Tagawa ST, Nanus DM, Giannakakou PA, Kirby BJ. Capture of circulating tumor cells from whole blood of prostate cancer patients using geometrically enhanced differential immunocapture (GEDI) and a prostate-specific antibody. Lab on a chip. 2010;10:27–29. doi: 10.1039/b917959c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraboletti G, D’Ascenzo S, Giusti I, Marchetti D, Borsotti P, Millimaggi D, Giavazzi R, Pavan A, Dolo V. Bioavailability of VEGF in tumor-shed vesicles depends on vesicle burst induced by acidic pH. Neoplasia (New York, NY) 2006;8(2):96–103. doi: 10.1593/neo.05583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, Riccardi L, Alitalo K, Claffey K, Detmar M. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med. 2001;7(2):192–8. doi: 10.1038/84643. [DOI] [PubMed] [Google Scholar]
- Stacker SA, Caesar C, Baldwin ME, Thornton GE, Williams RA, Prevo R, Jackson DG, Nishikawa S, Kubo H, Achen MG. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med. 2001;7(2):186–91. doi: 10.1038/84635. [DOI] [PubMed] [Google Scholar]
- Ishigami SI, Arii S, Furutani M, Niwano M, Harada T, Mizumoto M, Mori A, Onodera H, Imamura M. Predictive value of vascular endothelial growth factor (VEGF) in metastasis and prognosis of human colorectal cancer. Br J Cancer. 1998;78(10):1379–1384. doi: 10.1038/bjc.1998.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Farber EL, Rapoport AL, Tejada D, Deniskin R, Akhmedov NB, Farber DB. Transfer of microRNAs by embryonic stem cell microvesicles. PloS one. 2009;4(3):4722. doi: 10.1371/journal.pone.0004722. [DOI] [PMC free article] [PubMed] [Google Scholar]