

Summary
Rothmund-Thomson syndrome (RTS) is an autosomal recessive hereditary disorder associated with mutation in RECQL4 gene, a member of the human RecQ helicases. The disease is characterized by genomic instability, skeletal abnormalities and predisposition to malignant tumors, especially osteosarcomas. The precise role of RECQL4 in cellular pathways is largely unknown, however recent evidence suggest its involvement in multiple DNA metabolic pathways. This study investigates the roles of RECQL4 in DNA double strand break (DSB) repair. The results show that RECQL4-deficient fibroblasts are moderately sensitive to γ-irradiation and accumulate more γH2AX and 53BP1 foci than control fibroblasts. This is suggestive of defects in efficient repair of DSB’s in the RECQL4 deficient fibroblasts. Real time imaging of live cells using laser confocal microscopy show that RECQL4 is recruited early to laser induced DSBs and remains for a shorter duration than WRN and BLM indicating its distinct role in repair of DSBs. Endogenous RECQL4 also colocalizes with γH2AX at the site of DSBs. The RECQL4 domain responsible for its DNA damage localization has been mapped to the unique N-terminus domain between amino acids 363–492, which shares no homology to recruitment domains of WRN and BLM to the DSBs. Further, the recruitment of RECQL4 to laser induced DNA damage is independent of functional WRN, BLM or ATM proteins. These results suggest distinct cellular dynamics for RECQL4 protein at the site of laser induced DSB and that it might play important roles in efficient repair of DSB’s.
Keywords: RecQ helicase, Rothmund-Thomson syndrome (RTS), Werner syndrome (WRN), Bloom syndrome (BLM), Double strand break repair, Premature aging
Introduction
The RecQ helicase family is a group of evolutionarily conserved DNA unwinding proteins that play diverse roles in multiple DNA metabolic and repair processes. Five RecQ homologues have been identified in humans and mice: RECQL1, BLM, WRN, RECQL4 and RECQL5. Defects in human RecQ helicases are associated with chromosomal and developmental abnormalities, cancer susceptibility and premature aging (Brosh, Jr. & Bohr, 2007), and three loci encoding human RecQ helicases are causally linked to recessive hereditary diseases characterized by genomic instability and cancer predisposition (Bohr, 2008; Hickson, 2003). Werner syndrome (WS) and Bloom syndrome (BS) are caused by defects in WRN and BLM helicase, respectively. Defects in human RECQL4 are linked to Rothmund Thomson (RTS) Type 2, RAPADILINO and Baller-Gerold syndromes (Kitao et al., 1999; Siitonen et al., 2003; Van Maldergem et al., 2006). Although human WRN and BLM helicases are well characterized, relatively little is known about the biochemistry, cellular biology and function of human RECQL4.
RTS is a rare, autosomal recessive disorder characterized by poikiloderma, growth deficiency, juvenile cataracts, premature aging and predisposition to malignant tumors especially osteosarcomas (Wang et al., 2003;Vennos et al., 1992; Stinco et al., 2008). Approximately two-thirds of RTS patients have mutations in RECQL4 which are predicted to result in a truncated protein due to premature termination of protein synthesis (Lindor et al., 2000; Wang et al., 2003). These patients are referred to as RTS Type 2 and show strong predisposition to osteosarcoma (Wang et al., 2003). Cells from RTS patients show genomic instability and chromosomal abnormalities such as trisomy, aneuploidy and chromosomal rearrangements (Vennos et al., 1992; Der Kaloustian, V et al., 1990; Orstavik et al., 1994; Durand et al., 2002; Anbari et al., 2000).
RECQL4 is a 1208 amino acid protein (133 kDa) with a central helicase domain, which is characteristic for RecQ helicases (Kitao et al., 1998). However, RECQL4 does not share homology to two other conserved RecQ motifs, RQC and HRDC (helicase and RNaseD C-terminal), which are present in human BLM and WRN helicases. RECQL4 also possesses two N-terminal nuclear localization signal (NLS) sequences (Burks et al., 2007). RECQL4 has an intrinsic ATPase activity and single-strand DNA annealing activity (Yin et al., 2004; Macris et al., 2006). Recent studies have shown that the RECQL4 also has helicase activity in vitro (Xu & Liu, 2009; Capp et al., 2009; Suzuki et al., 2009).
The biological functions of RECQL4 are not yet known. However, it has been proposed that the RecQL4 N-terminal region plays a role in recruiting DNA polymerase α to nascent DNA replication forks in Xenopus egg extract (Sangrithi et al., 2005; Matsuno et al., 2006). Recently, it has been shown that human RECQL4 is a part of the repliosome complex and interacts with MCM10, MCM2-7, CDC45 and GINS (Xu et al., 2009). Another study indicates that the assembly of Cdc45-MCM2-7-GINS (CMG) complex at the replication fork requires RECQL4 (Im et al., 2009). These findings suggest an active involvement of RECQL4 in the assembly of the pre-replication complex and initiation of DNA replication.
Recent findings also indicate the involvement of RECQL4 in some DNA repair pathways. Fibroblasts from RTS patients are sensitive to replication-blocking DNA damaging agents including hydroxyurea, camptothecin and doxorubicin (Jin et al., 2008). RTS cells showed sensitivity to H2O2 and accumulate more H2O2-induced DNA strand breaks than control cells, suggesting that RECQL4 may stimulate repair of H2O2-induced DNA damage (Werner et al. 2006; Schurman et al., 2009). Biochemically, RECQL4 modulates the intrinsic activities of APE1, FEN1 and DNA polβ, indicating its role in base excision repair (BER) pathway (Schurman et al., 2009). RECQL4 also interacts with poly (ADP-ribose) polymerase 1 (PARP-1), which is implicated in DNA recombination, DNA repair, and transcriptional regulation (Woo et al., 2006). RECQL4 colocalizes with the Xeroderma Pigmentosum Group A (XPA) protein in human cells treated with UV, suggesting that RECQL4 might facilitate nucleotide excision repair (NER)-mediated repair of UV-induced DNA damage in human cells (Fan & Luo, 2008). Furthermore, RECQL4 and Rad51 colocalize in the nuclei of cells treated with etoposide, suggesting that RECQL4 might play a role in homologous recombination (HR)-mediated repair of DSBs (Petkovic et al., 2005). But the mechanism of this interaction is not known. There is also RECQL4 involvement in the repair of DSBs in Xenopus egg extracts (Kumata et al., 2007). Moreover, there are inconsistent reports about the sensitivity of RTS cells from patients and from MEFs derived from the RECQL4-deficient mice towards γ-irradiation, which predominantly forms DSB in the cells (Vennos et al., 1992; Cabral et al., 2008; Jin et al., 2008; Hoki et al., 2003).
In this work, we studied the cellular dynamics of RECQL4 in live cells at the site of laser induced DSBs using confocal laser scanning microscopy as well as sensitivity assays. First, we show that the RTS-deficient primary fibroblasts are moderately sensitive to γ-irradiation and accumulate more γ-irradiation induced γ-H2AX and 53BP1 foci that do not get efficiently resolve than control fibroblasts; this suggests that there are DNA repair defects in RTS cells compared to control cells. RECQL4 is recruited early to the laser-induced DNA damage site. Recruitment of RECQL4 to DNA damage requires an N-terminal domain which does not share homology to WRN and BLM proteins. Furthermore, RECQL4 shows distinct cellular dynamics at the site of laser induced DSBs compared to WRN and BLM.
Results
RTS cells are moderately sensitive to γ-irradiation
The importance of RECQL4 in DNA double strand break (DSB) repair was examined by measuring survival of RECQL4-deficient and normal fibroblasts after γ-irradiation. The RTS cells (AG05013 and AG17524) and normal fibroblasts (GM00323 and GM00969) were treated with 0, 1, 2 or 3 Gy of γ-irradiation and survival efficiency was estimated after 14 days by cell proliferation assays. The results showed that both the RTS cells are moderately more sensitive to γ-irradiation than respective normal cells (the RTS and normal fibroblasts were age and sex matched), and that survival decreases with increasing dose of γ-irradiation (Fig. 1). We also observed that the growth rate of both the RECQL4 deficient fibroblasts was approximately 1.5 to 2-fold slower than the normal fibroblasts (Supplementary Fig S1).
Fig. 1. RECQL4-deficient fibroblasts are sensitive to γ-irradiation compared to normal control fibroblasts.
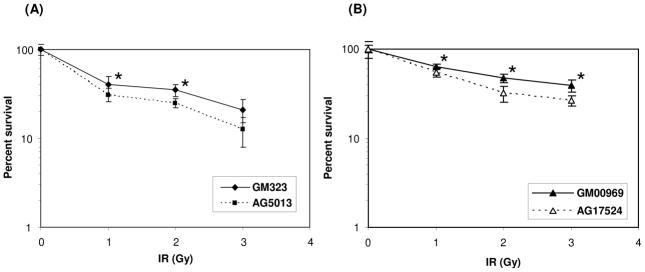
A, Normal (GM00323) and primary RTS fibroblasts (AG05013) cell lines were plated on a 60 mm petri dishes and treated with 0, 1, 2 and 3 Gy of γ-irradiation. Cells were allowed to grow for 14 days and fluorescent intensity was measured as described in Materials and Methods. B, Similar experiments were performed with normal fibroblasts (GM00969) and RTS cell line (AG17524). The experiments were performed in triplicate and the error bars represents the standard deviation (+/−). The degree of significance were calculated by Student’s t-Test method and the (*) represent the significant difference from the normal fibroblast with p value < 0.05.
RTS cells are defective in efficient repair of DNA DSBs
Sensitivity of RECQL4-deficient cells to γ-irradiation could be a result of inefficient processing of DSBs. To examine this possibility, two RECQL4-deficient (AG05013 and AG18371) and two normal cells (GM00323 and GM01864) were irradiated with 5 Gy of γ-irradiation and allowed to recover for 1, 3, 6 or 10 hours. Cells were then fixed and immunostained with γ-H2AX and 53BP1 antibodies and examined by confocal laser scanning microscopy. γ-H2AX and 53BP1 foci are widely used as markers for DSBs in the cell (Kinner et al., 2008; Schultz et al., 2000). RTS cells had a higher basal level of γ-H2AX foci than normal cells, suggesting inefficient repair of endogenous DNA DSBs perhaps due to problems at the replication forks (Supplementary Fig S2, lane 1). At early time points (i.e., 1 hour after irradiation), the number of 53BP1 foci in both the RECQL4 deficient cell lines were ~ 2-fold higher than in the control cells (Fig. 2, panels A,B, lane 2 and panels C,D). Furthermore, a significantly higher number of 53BP1 foci (2–3 fold) accumulated in γ-irradiated RTS cells than in normal fibroblasts (Fig. 2, panels A,B, lanes 3–5 and panels E,F) at late time points. The graph shows 53BP1 repair kinetics (percent 53BP1 foci with respect to time) and these focis disappeared more quickly in normal cells than in RTS cells suggesting inefficient repair of DSBs in RTS cells (Fig. 2, panels A,B, upper rows and panels E,F). Similar patterns of foci dispersal was also seen for γ-H2AX at the site of DSBs (Supplementary Fig S2, lanes 3–5). The slight differences in kinetics of γ-H2AX and 53BP1 foci formation and dispersal could be due to differences in recruitment and dissociation properties of γ-H2AX and 53BP1 at the site of DNA damage. These results suggest that RECQL4 deficient cells repair DSBs less efficiently than normal cells, which is consistent with reduced survival of RECQL4 deficient cells after exposure to γ-irradiation.
Fig. 2. The processing of DNA double strand breaks is delayed in RECQL4 defective primary fibroblasts.

After 5 Gy of γ-irradiation, the cells were allowed to recover under physiological conditions, fixed at indicated time points (0, 1, 3, 6 and 10 hrs) and immunostained with anti rabbit 53BP1 antibodies (as described in materials and methods). The images were captured with confocal microscope. Panel A and B show the persistence of more 53BP1 foci in RECQL4-deficient primary fibroblasts AG05013 and AG18371 (lower rows) compared to normal control fibroblasts GM00323 and GM01864 (upper rows), respectively at different time intervals as indicated. The first column represents the untreated cells (0 hour) as control. The total number of 53BP1 foci’s in both the RTS and normal control cell lines for each time point were counted and the average number of foci/ cell with respect to time are represented in panel C and D for two different cell lines, respectively. The repair kinetics of 53BP1 foci in both RTS and normal cell lines in terms of percentage of 53BP1 foci at each time point are represented in panel E and F for two different cell lines, respectively. In both panels, the total number of 53BP1 focis at 1 hr is taken as 100% foci for both normal and RTS cell lines.
Characterization of the laser microirradiation
Confocal laser scanning microscopy is a powerful technique for studying the spatiotemporal behaviors of DNA repair proteins in “real time” in living cells. This technique has been used to reveal novel functions of many DNA repair proteins including WRN and BLM (Mailand et al., 2007; Yano et al., 2008; Haince et al., 2008). Previous studies also show that WRN and BLM are recruited very early to laser-induced DSBs via their HRDC domain (Lan et al., 2005; Karmakar et al., 2006). Laser microirradiation produces a variety of DNA lesions, (DSBs, single-strand breaks (SSBs) and base modifications) in a spatially-restricted manner in nuclear DNA. The types of lesions formed after irradiation depends upon laser intensity and wavelength (Dinant et al., 2007; Lan et al., 2005). In this study, DSBs were induced with a 435 nm laser without cellular pre-treatment. We standardized the laser intensity so that SSBs could be distinguished from DSBs. At a laser intensity of <10%, only SSBs were produced, and XRCC1 (both GFP-tagged and endogenous), but not GFP-53BP1 or endogenous 53BP1 were recruited to the microirradiated site (Figure 3A, panels a and b, lanes 1 and 2). At a laser intensity of >10%, both SSBs and DSBs were produced, and both XRCC1 and 53BP1 were recruited to the microirradiated site (Fig. 3A, panels a and b, lanes 4 and 5). Live HeLa cells were also microirradiated using 8% or 18% laser intensity and co-immunostained with antibodies to XRCC1 and γ-H2AX (Fig. 3B). DSBs and γ-H2AX foci were generated by 18% but not by 8% laser intensity. Under these experimental conditions we then tested for the recruitment of GFP-tagged exogenous and endogenous RECQL4 protein to the site of DNA damage. The results showed that both endogenous and GFP-tagged RECQL4 were recruited to the DNA damage site at high laser intensity (Fig. 3A, compare lanes 3 and 6).
Fig. 3. The formation of DNA double strand breaks at the laser microirradiation site requires higher doses of laser irradiaiton.

A. Panel (a) shows the association of GFP tagged XRCC1, 53BP1 and RECQL4 in live HeLa cells at low (8%) and high (21%) doses of laser intensity. Transfected HeLa cells were microirradiated within the nucleus of the cell and images were captured for 2 min after photo bleaching. Panel (b) represents the association of endogenous XRCC1, 53BP1 and RECQL4 proteins at laser induced DNA damage site irradiated with low (8%) and high (21%) doses of laser intensity. Live HeLa cells were microirradiated and fixed after 5 min and immunostained with either XRCC1, 53BP1 or RECQL4 antibodies (see materials and methods). Arrow indicates the exact site of laser DNA damage. B. The live HeLa cells were microirradiated with the laser at 18% and 8% laser intensities. At 18% laser intensity recruitment of both endogenous γ-H2AX and XRCC1 was observed whereas at 8% laser intensity only XRCC1 is recruited and not the γ-H2AX. The arrow indicates the site of exact DNA damage. Asterisks (*) in the lower panel represents the DNA damage at 18% laser intensity.
RECQL4 accumulates at laser-induced DNA lesions
Kinetic studies were performed to examine the association and dissociation of RECQL4, WRN and BLM at laser-induced DNA damage in living cells. Similar to previous reports, in most cells, GFP-tagged RECQL4 was localized to the nucleus, but nucleo-cytoplasmic localization was observed in some cells [data not shown,(Burks et al., 2007; Yin et al., 2004)]. For this experiment, HeLa cells were transiently transfected with plasmids expressing GFP-RECQL4, GFP-WRN or GFP-BLM prior to laser-microirradiation, and cells were visualized at different time points after laser treatment. Previously, using a similar approach, WRN and BLM have been shown to be recruited to the DNA damage sites (Lan et al., 2005; Karmakar et al., 2006) and hence were used as a positive controls in this study (Fig. 4, panels A, B, C and D). The results showed that RECQL4 is recruited to DNA damage site within 5 seconds of microirradiation (Fig. 4, panels C and D).
Fig. 4. The recruitment kinetics of GFP tagged WRN, BLM and RECQL4 proteins at the site of laser induced DNA damage.

The HeLa cells were transfected with GFP tagged WRN, BLM or RECQL4 plasmids. After 24 hr post transfections, the cells were laser microirradiated within the nucleus with 435 nm laser light at 21% laser intensity. The images were captured for 60 seconds through FRAP channel using confocal microscope. The association of GFP-tagged WRN, BLM and RECQL4 proteins is shown at different time intervals after microirradiation in panel A, B, and C, respectively. Arrow represents the site of laser irradiation. The normalized intensity values of the kinetic of association of different proteins have been represented in panel D.
Endogenous γ-H2AX and RECQL4 colocalize after laser-induced DNA damage
Recruitment of endogenous γ-H2AX and RECQL4 to the microirradiation site was also examined in HeLa cells. Cells were fixed and immunostained with γ-H2AX and RECQL4 antibodies after 5 min of laser microirradiation and imaged. The images were analyzed using confocal microscopy and Volocity software. The results showed that endogenous γ-H2AX and RECQL4 colocalize within 5 minutes after laser treatment, suggesting that RECQL4 binds to DSBs (Fig. 5). A significant portion of the endogenous RECQL4 remains in the cytoplasm after irradiation, as reported previously (Yin et al., 2004; Werner et al., 2005).
Fig. 5. Endogenous γ-H2AX and RECQL4 colocalizes at the laser induced DNA damage site.

HeLa cells were laser microirradiated at 21% laser intensity followed by fixation after 5 min and immunostaining with anti rabbit γ-H2AX and anti goat RECQL4 antibodies. The untreated cell is shown in top left panel, γ-H2AX is shown in red (top middle), RECQL4 is depicted in green (top right) and the merge images of the two showing co-localization is shown in yellow (lower panels).
The NTS2 domain of RECQL4 is sufficient for recruitment to the DSB sites
RECQL4 possesses a characteristic central helicase domain which is a shared feature of RecQ helicases (Kitao et al., 1998). RECQL4 differs from WRN and BLM helicases in that it lacks the C-terminal RQC and HRDC domains. Two unique nuclear localization sequences are also present at the N-terminus of RECQL4 (Burks et al., 2007). To map the domain of RECQL4 responsible for its recruitment to the laser induced DSBs sites, we tested constructs containing either portions of the RECQL4 coding region, or the full length construct with portions deleted. The subcellular localization of each mutant was reported previously (Burks et al., 2007), and we confirmed the expression of each mutant by Western analyses (Supplementary Fig. S3). First, we tested three GFP-tagged constructs which contained portions of the RECQL4 coding region, namely the N-terminus (1-492), the N-terminus with central helicase domain (1-794) and the C-terminus (794-1208) (Figure 6). The results showed that the region spanning from aa 1-794 and aa 1-492 enabled recruitment to the DNA damaged site, whereas the C-terminal aa 794-1208 domain was not recruited, however this construct is cytoplasmic and thus may not have access to the DSB sites. This collective results suggest that at least one recruitment domain is present within the N-terminus of the RTS protein (Fig. 6, panels A and B, ii, iii, iv, v).
Fig. 6. Analysis of association of GFP tagged RECQL4 mutants at the site of laser DNA damage.

The different GFP-RECQL4 mutant constructs were transiently transfected in the HeLa cell and association kinetics were monitored at 21% laser intensity. Panel A, shows the schematics of different mutant and their association at DNA damage site (represented with + sign). Two Nuclear targeting sequences (NTS1 & NTS2) and the helicase domain are shown. Panel B show one representative images of each mutant association at the DNA damaged site.
To further map the domain within the N-terminus (1-492) of RECQL4, various mutants within the nuclear targeting sequence (NTS2, aa 363-492) were tested for their recruitment to the microirradiated site. It has been reported that the NTS2 domain of human RECQL4 includes a 130 amino acids segment within exons 5–8 which is necessary and sufficient for nuclear localization (Fig. 6, panel A,B, xi, xii) (Burks et al., 2007). Consistent with this observation, GFP-NTS2 (aa 363-492) was recruited to DNA damage in laser-treated cells, but NTS2-deleted RECQL4 (GFP-RECQL4 ΔNTS2) was not (Fig. 6, panel B, xi and xii). This confirms that NTS2 is sufficient for nuclear localization and for recruitment of RECQL4 to DNA damage. Further, the NTS2 domain was subdivided into NTS2A (aa 364-426) and NTS2B (aa 426-492). The NTS2A is encoded by parts of exons 5–7 of RECQL4, and NTS2A-deleted GFP-RECQL4 shows nuclear or nucleo-cytoplasmic localization pattern similar to wild type RECQL4 (Burks et al., 2007). Deletion of NTS2A domain (GFP-RECQL4ΔNTS2A), including a highly basic stretch of 22 amino acids named the “basic box” (GFP-RECQL4Δ Basic Box) does not affect the recruitment of RECQL4 to DNA damage (Fig. 6, panels A,B, vi, vii). In contrast, NTS2B deleted GFP-RECQL4 (GFP-RECQL4ΔNTS2B) show predominantly cytoplasmic localization (Burks et al., 2007). Furthermore, GFP-RECQL4ΔNTS2B and other mutants with deletions in the NTS2B domain (i.e., Δexon7 and ΔVLPLY, a highly conserved motif in all orthologues) are cytoplasmic and are not recruited to the site of microirradiation. This suggests their importance in the recruitment of full length RECQL4 to the site of the DNA damage. However, it is possible that these mutants were not recruited to the site of the microirradiation because of their predominant cytoplasmic localization. Thus, in conclusion, these results suggest that the NTS2 domain spanning from aa 363–492 contains at least one domain which is sufficient for the recruitment of RECQL4 to the DSBs. However, the possibility of other domains in the full length RECQL4 can not be ruled out and need further investigation.
GFP tagged-RECQL4 is recruited to DNA damage independent of functional WRN, BLM and ATM proteins
WRN and BLM interact physically and functionally, and BLM inhibits WRN exonuclease (von Kobbe et al., 2002). WRN and BLM also colocalize after cellular stress (von Kobbe et al., 2002). Because these results indicate potential interactions among RecQ helicases, the influence of WRN and BLM on dynamics of RECQL4 at the sites of DNA damage was investigated using cells lacking WRN or BLM. For this purpose, GFP-tagged RECQL4 was transiently expressed in SV40 transformed WRN (AG07066) or BLM (GM08505) and ATM (GM05849) deficient fibroblasts. The cells were microirradiated and analyzed by live cell imaging as described above. The results showed that RECQL4 is recruited efficiently to the microirradiated site in the absence of functional WRN, BLM or ATM proteins (Fig. 7).
Fig. 7. The recruitment of GFP tagged RECQL4 is independent of functional WRN, BLM and ATM proteins.

The association kinetics of transiently transfected GFP-RECQL4 protein at the site of laser induced DNA damage in SV40 transformed Normal fibroblasts (GM00637), BLM (GM08505), WRN (AG07066), and ATM (GM05849) mutant fibroblasts cells are shown in panel A, B, C and D, respectively. The normalized intensity value of the kinetic of association of RECQL4 in normal fibroblasts, BLM, WRN and ATM mutant cells have been represented in panel E.
The retention of GFP-tagged RECQL4 at the microirradiated site is short compared to WRN and BLM proteins
The efficiency of DNA repair depends on the dynamics of the DNA repair proteins at the site of DNA damage. WRN, BLM and RECQL4 are recruited early, but their rate of dissociation also affects the efficiency of the repair process. While there are many studies addressing the recruitment of DNA repair proteins to DNA damage, there is very little information about the disassociation dynamics of DNA repair proteins from such sites. Such studies require an environmental chamber, which we have utilized in these studies. Kinetics of protein dissociation from DNA damage site was examined in HeLa cells expressing GFP-tagged WRN, BLM or RECQL4. For this experiment, cells were microirradiated using 21% laser intensity, and placed in an environmental chamber, where they could be visualized by confocal microscopy under normal growth conditions throughout the experiment. The graph shows the dynamic association of the various RecQ helicases at the site of a lesion. Interestingly, RECQL4 arrives early and departs within 1 hr (Fig. 8. panels C and D). We also observed that the timing at which the WRN protein reaches its maximum intensity at the DNA damage site is different from BLM and RECQL4. WRN reaches maximal intensity at about one hr and then slowly departs. BLM reaches its maximal intensity at about 2hrs, but its disassociation is faster than for WRN. Collectively, we conclude that the initial recruitment is similar for the different RecQ helicases (Fig.4), but the maximal recruitment and rate of dissociation varies considerably for the different RecQ helicases. Thus, the different RecQ helicases have distinct roles in the response to DNA damage.
Fig. 8. Retention of RECQL4 at the DNA damaged site is shorter compared to WRN and BLM proteins.

The HeLa cells were transiently transfected with GFP tagged WRN, BLM and RECQL4 plasmids and after 24 hr post transfections, the cells were microirradiated at 21% laser intensity and the dissociation kinetics were monitored by live cell imaging. The cells were placed in an environmental chamber and the dissociation kinetics of GFP tagged WRN, BLM and RECQL4 at the site of laser induced DNA damage is shown in panel A, B and C, respectively. The time for image acquisition is shown for each panel. D. The percent maximum intensity values with respect to time of the dissociation of GFP tagged WRN, BLM and RECQL4 in HeLa cells have been represented below.
DISCUSSION
This report characterizes the role of RECQL4 in DNA DSB repair. Results presented here show that RECQL4-deficient cells are moderately more sensitive to γ-irradiation than normal fibroblasts (Fig. 1). The conflicting results in papers prior to identification of the role of RECQL4 in RTS may reflect that 30% of RTS patients carry mutations in a yet unidentified gene (Kitao et al., 1999; Wang et al., 2003). More recently, there have been divergent reports in the literature regarding the sensitivity of different primary RTS fibroblasts and murine fibroblasts towards ionizing radiation (Jin et al.,2008; Cabral et al., 2008, Hoki et al., 2003). Although WRN-deficient primary fibroblasts also demonstrate only modest sensitivity towards ionizing radiation (Yannone et al., 2001), WRN plays important roles in regulating HR- and NHEJ-dependent repair of DSBs. Therefore we propose that RECQL4 might also play important roles in DSB repair with regard to survival and efficient repair of damage.
We then examined γ-H2AX and 53BP1 foci formation and dispersal to show that RECQL4 deficient cells are indeed defective in efficient repair of DSBs. A comparison of the time course kinetics of foci dispersal in normal control fibroblast cells and RECQL4 deficient cells showed that there are approximately 2–3 fold more persistence of 53BP1 foci for >10 hours in RTS cells, suggesting that RECQL4-deficiency impairs efficient repair of DNA DSBs (Figure 2). Similarly, WRN deficient cells also have a higher basal level of γ-H2AX foci than control cells in experiments where WRN and control cells are age-matched. It has been proposed that an age-associated decrease in the efficiency of DSB repair causes genome instability in the context of both normal and premature aging (Sedelnikova et al., 2008). Also, in Xenopus the reduction in DSB induced γ-H2AX was significantly compromised in RECQL4 depleted extracts suggesting that RECQL4 functions in repair of DSB (Kumata et al., 2007).
This study used confocal laser scanning microscopy to image the kinetic behavior of RecQ helicases in response to laser-induced DNA damage in living cells. As previously shown for WRN and BLM protein (Lan et al., 2005; Karmakar et al., 2006), GFP-tagged RECQL4 is recruited rapidly to the site of laser-induced DNA damage (Fig. 4). The endogenous RECQL4 is also recruited to DSBs, where it colocalizes with γ-H2AX at the microirradiated site, but not to SSBs (Fig. 5). A previous study showed that a portion of RECQL4 foci are associated with regions of ssDNA at DSB sites in cells treated with etoposide and IR (Petkovic et al., 2005). A study using Xenopus oocyte extracts also suggested that RECQL4 accumulates in chromatin and promotes repair of DSBs (Kumata et al., 2007). These results suggest that RECQL4 associates with DSBs and promotes efficient processing and repair of DSBs.
This study shows that the NTS2 domain of RECQL4 is required and sufficient for recruitment of RECQL4 to the site of microirradiation (Fig. 6). RECQL4 is structurally unique and distinct from other RecQ helicases homologues, in that it does not possess the RQC and C-terminal HRDC domains, which are present in WRN and BLM. Both WRN and BLM are recruited to laser-induced DSBs via their HRDC domain (Lan et al., 2005; Karmakar et al., 2006). Karmakar et al. suggested that the HRDC domain plays a role in recruitment of BLM to DNA damage, because GFP-tagged RECQL1, which lacks an HRDC domain, is not rapidly recruited to DNA damage in irradiated cells (Karmakar et al., 2006). In this context, it is interesting that RECQL4, which also lacks the HRDC domain, is rapidly recruited to the site of microirradiation (this study). RECQL4, unlike other RecQ helicases, carries both nuclear targeting sequences at the N-terminus. Specifically, we show here that the NTS2 region of RECQL4 (aa 363–492) is sufficient for nuclear targeting and for recruitment of RECQL4 to DSB sites. This result is interesting as the N-terminus of RECQL4 is implicated as a critical region important for its essential roles in DNA replication (Sangrithi et al., 2005; Matsuno et al., 2006) and also is deleted through mis-splicing in the RECQL4 associated RAPADILINO syndrome. The N-terminus of the human RECQL4 protein also directly interacts with MCM10 and is important for assembly of pre-initiation complex (Xu et al., 2009). Our results also indicate that the chromatin binding domain resides within the N-terminus of RECQL4 which is also involved in various DNA metabolic pathways such as DNA replication and repair. However, additional studies are needed to determine whether the NTS2 region of RECQL4 mediates binding to DNA damage associated chromatin proteins or to DNA breaks themselves.
While there have been numerous studies examining the recruitment characteristics of DNA repair proteins, very little is known about the retention kinetics of RECQ helicase at DSB sites. In our studies we used an environmental chamber to maintain the cells under normal conditions over the long period of study. Interestingly, RECQL4 is retained at sites of DNA damage for only approximately one hour, while WRN and BLM are retained for longer periods of time (Fig. 8). Thus, RECQL4 might act at an early step in DSB repair, or it could act on a specialized transient DNA substrate, which is present at a low level in laser treated cells. These results also suggest that different RECQ helicases have distinct and specialized functions in the context of DSB repair which correlates with their specialized biochemical features and their responses to different genotoxic agents.
In cells DSB’s are repaired either by homologous recombination (HRR) or non homologous end-joining (NHEJ). NHEJ-mediated DSB repair is largely independent of terminal DNA sequence homology. Therefore, NHEJ is error-prone and produces deletions, insertions and translocations (Thompson & Schild, 2002). The HRR pathway repairs DSBs using homologous sequences on sister chromatids and to a lesser extent, on chromosome homologues (Johnson & Jasin, 2000; Liang et al., 1998). Previous studies have indicated that RECQL4 might be involved in both, HRR and NHEJ pathways. RECQL4 has been shown to form complex with Rad51 which is crucial for the repair of DSBs by the HRR pathway (Petkovik et al., 2005). In another study using Xenopus, RECQL4 has been shown to be loaded on to the chromatin in response to DSBs. Further, RECQL4 is loaded adjacent to Ku heterodimer binding site on damaged chromatin suggesting its possible involvement in NHEJ (Kumata et al., 2007). However, the mechanism of RECQL4 involvement in DSB repair is not yet understood. It is possible that RECQL4 participates in the repair process once it is loaded on to the chromatin at the site of DSB. In this complex RECQL4 can interact directly with various proteins involved in the HRR and NHEJ pathway at different stages of DSB repair and modulate their functions. Recent studies have also indicated that the RECQL4 might also be important in replication restart after repair (Xu et al., 2009). However, another likely possibility is that the function of RECQL4 may be indirect. Similar to WRN and BLM helicases, RECQL4 could function in resolving aberrant DNA structures formed during DNA replication and recombination repair processes and facilitate the loading of other repair factors at the site of the break. Therefore, the absence of RECQL4 in the cells would lead to genomic instability and could explain the heterogeneous premature aging phenotype and cancer susceptibility seen in RTS patients.
One of the limitations of this study is that most of the experiments are performed with overexpression of transfected proteins to study the cellular dynamics at the site of DNA damage. Nonetheless, this work strengthens the hypothesis that RECQL4 plays a significant role in the DNA damage response to DSBs. Additional studies are needed to further understand the molecular mechanisms that allow RECQL4 to promote genetic stability and facilitate an efficient response to DNA damage in human cells.
Experimental procedures
Cell lines and transfection
HeLa cells, SV40 transformed normal (GM00637), WRN (AG07066), BLM (GM08505), ATM (GM05849) fibroblasts cell lines, normal primary skin fibroblasts (GM00323, GM00969, GM01864) and RTS-deficient skin primary fibroblasts (AG05013, AG18371, AG17524) cell lines from Coriell Cell Repositories were used in the study. The RTS cell lines have confirmed RECQL4 mutations and do not show RECQL4 proteins in western analysis of the total cell extract (Kitao et al., 1999; Petkovic et al., 2005). The pair of RTS cells and normal fibroblasts cells used for survival assays and the foci formation assays are age and sex matched. The HeLa cells were cultured in DMEM media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37°C in a humidified atmosphere with 5% CO2. SV40 transformed normal, WRN, BLM and ATM fibroblasts were cultured in MEM media supplemented with 10% FBS. Normal primary skin fibroblasts cells and RTS-deficient primary fibroblasts were cultured in Amniomax II Complete media (Invitrogen, CA, USA). For live cell experiments the cells were plated in glass bottom Petri dishes (LabTec corp, USA) at 50% confluence 24 hours before the transfections. For transfection Fugene 6 was used as per manufacturer’s instructions (Roche, NJ, USA). For immunofluorescence studies, the cells were plated in 4 well chambered slides 24 hours before the treatments.
Cell proliferation assay
Cell proliferation was assessed using CyQuant (Molecular Probes, OR, USA). Both the normal cell line (GM00323 and GM00969) and the RTS cell line (AG05013 and AG17524) were plated in 60 mm petri dishes at a density of 1000 cells per plate. The cells were treated with 1, 2, 3 Gy of γ irradiation and then allowed to grow in Amniomax-II Complete media for 14 days at 37°C at 5% CO2. On the day of the analysis, the plates with adherent cells were washed once with 1X PBS and incubated with the CyQuant dye for 30 min in dark at 37 °C with 5% CO2. Fluorescence intensity, which is proportional to cell number, was measured on a Typhoon 8600 fluorescent scanner (Molecular Dynamics, CA, USA) with filters set at 480 nm excitation and 520 nm emission. Fluorescence data was analyzed and the number of colonies in each plate was counted by Image QuantTL software (Molecular Dynamics).
Analysis of γ-H2AX and 53BP1 foci
The γH2AX and 53BP1 foci were measured in two normal primary fibroblasts cell lines (GM00323 and GM01864) and two RTS cell lines (AG05013 and AG18371). Approximately, 25,000 primary fibroblasts cells were seeded on chambered slides, grown overnight and then treated with 5 Gy of γ-irradiation. Cells were allowed to recover for 1, 3, 6 or 10 hr, under normal growth conditions and then immediately fixed and stained (described in next section) with either mouse monoclonal γH2AX antibody (Upstate Biotechnoligy, 1:200) or rabbit polyclonal 53BP1 antibody (Abcam, 1:200) and Alexa goat anti-mouse 647 or donkey anti-rabbit 647 secondary antibody (Molecular Probes, OR, USA). Ten images representing about 15–20 cells per well were taken using a Nikon TE2000 spinning disk microscope with five laser imaging modules and a CCD camera (Hamamatsu). The data were analyzed using Volocity version 4.3.1 build 6 (Improvision).
Immunocytochemistry and antibodies
The cells were laser microirradiated and stained with antibodies against γH2AX, 53BP1, XRCC1, RECQL4. After microirradiation the cells were fixed with 3.7% formaldehyde in PBS for 10 min at room temperature. Then cells were washed 3 times with PBS, permeabilized with 0.2% Triton X 100 in PBS for 5 min at room temperature. The cells were washed with PBS and blocked in 5% fetal bovine serum overnight at 4 °C. The cells were incubated with different primary antibodies for 1 hr at 37 °C in a humidified chamber. Mouse monoclonal anti-phosphorylated H2AX (1:200, Upstate Biotechnology), goat polyclonal anti-RECQL4 (K16) (1:50, Santa Cruz Biotech), rabbit polyclonal anti-53BP1 (Abcam, 1:200) and rabbit polyclonal anti-XRCC1 (1:200, Santa Cruz Biotech) antibodies were used. Bound antibodies were visualized by incubating the cells with secondary antibodies, namely Alexa FITC (488) conjugated chicken anti-goat, Alexa 647-conjugated goat anti mouse and Alexa-conjugated 647 donkey anti rabbit. The images were captured with a confocal microscope (Nikon eclipse TE2000) and analyzed by volocity software (Improvision).
Western blotting
After separation in SDS-PAGE gel the proteins were transferred to PVDF membrane and the membrane was blocked with 5% fat free milk in 1X TBS-T for 1 hr at room temperature followed by incubation with primary rabbit anti GFP antibody (1: 5000, Novus Biologicals) overnight at 4 °C. The membranes were then washed thrice in 1X TBST, 5 min each, and then incubated in HRP conjugated secondary anti rabbit antibody (1:20,000, Sigma, USA) for 1 hr. After washing the membrane was developed by ECL plus kit following manufacturer’s protocol (GE Healthcare, USA).
Laser irradiation and confocal microscopy
We employed a Nikon Eclipse 2000E spinning disk confocal microscope with five laser imaging modules and a CCD camera (Hamamatsu, Tokyo, Japan). The set-up integrated a Stanford Research Systems (SRS) NL100 nitrogen laser by Micropoint ablation system (Photonics Instruments, St. Charles, IL). Site specific DNA damage was induced using the SRS NL100 nitrogen laser that was passed through a dye cell to emit at 435 nm wavelength. The power of the laser was attenuated through Improvison’s Volocity software 4.3.1 (Improvision/PerkinElmer, Coventry, England) in terms of percent intensity. Positions internal to the nuclei of either live HeLa cells or HeLa cells transfected with GFP tagged plasmids were targeted via a 40X oil objective lens. The laser intensities of 8% and 21% were used. Images were captured at various time points and analyzed using Volocity version 4.3.1 build 6 (Improvision). Experiments were performed using an environmental chamber attached to the microscope to maintain the normal atmosphere of the cells (i.e. 37°C and 5% CO2).
Fluorescence Recovery after Photobleaching (FRAP)
FRAP analysis was carried out with live cells transfected with GFP tagged WRN, BLM and RECQL4 plasmids. The same set up as the microirradiation was used to perform the photobleaching. Fluorescence recovery was monitored as described in the figure legends and the data for recovery was corrected for the background intensity and loss of total fluorescence. Each experiment was performed at least three times and the data presented here are mean intensity values obtained in a given experiments.
Supplementary Material
An equal number (1000) of normal and RTS fibroblasts were plated on a 60 mm petri dish and allowed to grow under normal conditions for 1, 3, 5, 7 and 9 days. The cells were harvested at indicated time points and counted using Z1 Coulter counter (Beckman Coulter, Inc., CA, USA). The graph shows the average number of cells plotted with respect to number of days. The experiments were performed in triplicate and the error bars represents the mean standard error (+/−).
After 5 Gy of γ-irradiation, the cells were allowed to recover and fixed at indicated time points (0, 1, 3, 6 and 10 hrs) and immunostained with anti mouse γ-H2AX antibodies (as described in materials and methods). The images were captured with confocal microscope. The persistence of γ-H2AX foci’s in RTS-deficient primary fibroblasts AG05013 (lower row) compared to normal control fibroblasts (GM00323, upper row), at different time intervals as indicated. The first column represents the untreated cells (0 hour) as control.
Various GFP-tagged RECQL4 deletion constructs were transiently transfected in the HeLa cells. After 24 hour post transfections the cells were lysed and the total cell extract were subjected to western blotting with rabbit polyclonal anti GFP antibody. Asterisks (*) shows the band corresponding to expressed proteins. (M) represent protein marker and the size of the protein markers are indicated on left.
Acknowledgments
We would like to thank Drs. Suhasini Avvaru and Venkateswarlu Popuri for critical reading of the manuscript. This work was supported by funds from the Intramural Research Program of the National Institute on Aging, NIH.
Footnotes
Author contributions: Conceived and designed the experiments: VB, DKS, PK. Performed the experiments: DKS, PK, MA, SS, AM, LB. Analyzed the data: VB, DKS, PK, DC, SP. Wrote the paper: DKS, VB.
References
- Anbari KK, Ierardi-Curto LA, Silber JS, Asada N, Spinner N, Zackai EH, Belasco J, Morrissette JD, Dormans JP. Two primary osteosarcomas in a patient with Rothmund-Thomson syndrome. Clinical Orthopaedics and Related Research. 2000;378:213–223. doi: 10.1097/00003086-200009000-00032. [DOI] [PubMed] [Google Scholar]
- Bohr VA. Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance. Trends Biochem Sci. 2008;33:609–620. doi: 10.1016/j.tibs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosh RM, Jr, Bohr VA. Human premature aging, DNA repair and RecQ helicases. Nucleic Acids Res. 2007;35:7527–7544. doi: 10.1093/nar/gkm1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burks LM, Yin JH, Plon SE. Nuclear import and retention domains in the amino terminus of RECQL4. Gene. 2007;391:26–38. doi: 10.1016/j.gene.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Cabral RE, Queille S, Bodemer C, de PY, Neto JB, Sarasin A, ya-Grosjean L. Identification of new RECQL4 mutations in Caucasian Rothmund-Thomson patients and analysis of sensitivity to a wide range of genotoxic agents. Mutat Res. 2008;643:41–47. doi: 10.1016/j.mrfmmm.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Capp C, Wu J, Hsieh TS. Drosophila RecQ4 has a 3′-5′DNA helicase activity that is essential for viability. J Biol Chem. 2009 doi: 10.1074/jbc.M109.008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der Kaloustian VM, McGill JJ, Vekemans M, Kopelman HR. Clonal lines of aneuploid cells in Rothmund-Thomson syndrome. Am J Med Genet. 1990;37:336–339. doi: 10.1002/ajmg.1320370308. [DOI] [PubMed] [Google Scholar]
- Dinant C, de Jager M, Essers J, van Cappellen WA, Kanaar R, Houtsmuller AB, Vermeulen W. Activation of multiple DNA repair pathways by subnuclear damage induction methods. Journal of Cell Science. 2007;120:2731–2740. doi: 10.1242/jcs.004523. [DOI] [PubMed] [Google Scholar]
- Durand F, Castorina P, Morant C, Delobel B, Barouk E, Modiano P. Rothmund-Thomson syndrome, trisomy 8 mosaicism and RECQ4 gene mutation. Annales de Dermatologie et de Venereologie. 2002;129:892–895. [PubMed] [Google Scholar]
- Fan W, Luo JY. RecQ4 Facilitates UV Light-induced DNA Damage Repair through Interaction with Nucleotide Excision Repair Factor Xeroderma Pigmentosum Group A (XPA) Journal of Biological Chemistry. 2008;283:29037–29044. doi: 10.1074/jbc.M801928200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haince JF, McDonald D, Rodrigue A, Dery U, Masson JY, Hendzel MJ, Poirier GG. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. Journal of Biological Chemistry. 2008;283:1197–1208. doi: 10.1074/jbc.M706734200. [DOI] [PubMed] [Google Scholar]
- Hickson ID. RecQ helicases: Caretakers of the genome. Nature Reviews Cancer. 2003;3:169–178. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- Hoki Y, Araki R, Fujimori A, Ohhata T, Koseki H, Fukumura R, Nakamura M, Takahashi H, Noda Y, Kito S, Abe M. Growth retardation and skin abnormalities of the Recql4-deficient mouse. Hum Mol Genet. 2003;12:2293–2299. doi: 10.1093/hmg/ddg254. [DOI] [PubMed] [Google Scholar]
- Im JS, Ki SH, Farina A, Jung DS, Hurwitz J, Lee JK. Assembly of the Cdc45-Mcm2–7-GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proc Natl Acad Sci U S A. 2009;106:15628–15632. doi: 10.1073/pnas.0908039106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Liu H, Zhang Y, Otta SK, Plon SE, Wang LL. Sensitivity of RECQL4-deficient fibroblasts from Rothmund-Thomson syndrome patients to genotoxic agents. Hum Genet. 2008;123:643–653. doi: 10.1007/s00439-008-0518-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RD, Jasin M. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. Embo Journal. 2000;19:3398–3407. doi: 10.1093/emboj/19.13.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmakar P, Seki M, Kanamori M, Hashiguchi K, Ohtsuki M, Murata E, Inoue E, Tada S, Lan L, Yasui A, Enomoto T. BLM is an early responder to DNA double-strand breaks. Biochemical and Biophysical Research Communications. 2006;348:62–69. doi: 10.1016/j.bbrc.2006.07.037. [DOI] [PubMed] [Google Scholar]
- Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678–5694. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitao S, Ohsugi I, Ichikawa K, Goto M, Furuichi Y, Shimamoto A. Cloning of two new human helicase genes of the RecQ family: biological significance of multiple species in higher eukaryotes. Genomics. 1998;54:443–452. doi: 10.1006/geno.1998.5595. [DOI] [PubMed] [Google Scholar]
- Kitao S, Shimamoto A, Goto M, Miller RW, Smithson WA, Lindor NM, Furuichi Y. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nature Genetics. 1999;22:82–84. doi: 10.1038/8788. [DOI] [PubMed] [Google Scholar]
- Kumata Y, Tada S, Yamanada Y, Tsuyama T, Kobayashi T, Dong YP, Ikegami K, Murofushi H, Seki M, Enomoto T. Possible involvement of RecQL4 in the repair of double-strand DNA breaks in Xenopus egg extracts. Biochimica et Biophysica Acta-Molecular Cell Research. 2007;177:556–564. doi: 10.1016/j.bbamcr.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Lan L, Nakajima S, Komatsu K, Nussenzweig A, Shimamoto A, Oshima J, Yasui A. Accumulation of Werner protein at DNA double-strand breaks in human cells. Journal of Cell Science. 2005;118:4153–4162. doi: 10.1242/jcs.02544. [DOI] [PubMed] [Google Scholar]
- Liang F, Han MG, Romanienko PJ, Jasin M. Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc Natl Acad Sci U S A. 1998;95:5172–5177. doi: 10.1073/pnas.95.9.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindor NM, Furuichi Y, Kitao S, Shimamoto A, Arndt C, Jalal S. Rothmund-Thomson syndrome due to RECQ4 helicase mutations: Report and clinical and molecular comparisons with Bloom syndrome and Werner syndrome. American Journal of Medical Genetics. 2000;90:223–228. doi: 10.1002/(sici)1096-8628(20000131)90:3<223::aid-ajmg7>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Macris MA, Krejci L, Bussen W, Shimamoto A, Sung P. Biochemical characterization of the RECQ4 protein, mutated in Rothmund-Thomson syndrome. DNA Repair. 2006;5:172–180. doi: 10.1016/j.dnarep.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- Matsuno K, Kumano M, Kubota Y, Hashimoto Y, Takisawa H. The N-terminal noncatalytic region of Xenopus RecQ4 is required for chromatin binding of DNA polymerase alpha in the initiation of DNA replication. Molecular and Cellular Biology. 2006;26:4843–4852. doi: 10.1128/MCB.02267-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orstavik KH, Mcfadden N, Hagelsteen J, Ormerod E, Vanderhagen CB. Instability of Lymphocyte Chromosomes in A Girl with Rothmund-Thomson Syndrome. Journal of Medical Genetics. 1994;31:570–572. doi: 10.1136/jmg.31.7.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkovic M, Dietschy T, Freire R, Jiao RJ, Stagljar I. The human Rothmund-Thomson syndrome gene product, RECQL4, localizes to distinct nuclear foci that coincide with proteins involved in the maintenance of genome stability. Journal of Cell Science. 2005;118:4261–4269. doi: 10.1242/jcs.02556. [DOI] [PubMed] [Google Scholar]
- Sangrithi MN, Bernal JA, Madine M, Philpott A, Lee J, Dunphy WG, Venkitaraman AR. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell. 2005;121:887–898. doi: 10.1016/j.cell.2005.05.015. [DOI] [PubMed] [Google Scholar]
- Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol. 2000;151:1381–1390. doi: 10.1083/jcb.151.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurman SH, Hedayati M, Wang Z, Singh DK, Speina E, Zhang Y, Becker K, Macris M, Sung P, Wilson DM, 3rd, Croteau DL, Bohr VA. Direct and indirect roles of RECQL4 in modulating base excision repair capacity. Hum Mol Genet. 2009;18:3470–3483. doi: 10.1093/hmg/ddp291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedelnikova OA, Horikawa I, Redon C, Nakamura A, Zimonjic DB, Popescu NC, Bonner WM. Delayed kinetics of DNA double-strand break processing in normal and pathological aging. Aging Cell. 2008;7:89–100. doi: 10.1111/j.1474-9726.2007.00354.x. [DOI] [PubMed] [Google Scholar]
- Siitonen HA, Kopra O, Kaariainen H, Haravuori H, Winter RM, Saamanen AM, Peltonen L, Kestila M. Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Human Molecular Genetics. 2003;12:2837–2844. doi: 10.1093/hmg/ddg306. [DOI] [PubMed] [Google Scholar]
- Stinco G, Governatori G, Mattighello P, Patrone P. Multiple cutaneous neoplasms in a patient with Rothmund-Thomson syndrome: Case report and published work review. Journal of Dermatology. 2008;35:154–161. doi: 10.1111/j.1346-8138.2008.00436.x. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Kohno T, Ishimi Y. DNA helicase activity in purified human RECQL4 protein. J Biochem. 2009;146:327–335. doi: 10.1093/jb/mvp074. [DOI] [PubMed] [Google Scholar]
- Thompson LH, Schild D. Recombinational DNA repair and human disease. Mutation Research-Fundamental and Molecular Mechanisms of Mutagenesis. 2002;509:49–78. doi: 10.1016/s0027-5107(02)00224-5. [DOI] [PubMed] [Google Scholar]
- Van Maldergem L, Siitonen HA, Jalkh N, Chouery E, De Roy M, Delague V, Muenke M, Jabs EW, Cai J, Wang LL, et al. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. Journal of Medical Genetics. 2006;43:148–152. doi: 10.1136/jmg.2005.031781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennos EM, Collins M, James WD. Rothmund-Thomson Syndrome - Review of the World Literature. Journal of the American Academy of Dermatology. 1992;27:750–762. doi: 10.1016/0190-9622(92)70249-f. [DOI] [PubMed] [Google Scholar]
- von Kobbe C, Karmakar P, Dawut L, Opresko P, Zeng XM, Brosh RM, Hickson ID, Bohr VA. Colocalization, physical, and functional interaction between Werner and Bloom syndrome proteins. Journal of Biological Chemistry. 2002;277:22035–22044. doi: 10.1074/jbc.M200914200. [DOI] [PubMed] [Google Scholar]
- Wang LL, Gannavarapu A, Kozinetz CA, Levy ML, Lewis RA, Chintagumpala MM, Ruiz-Maldonado R, Contreras-Ruiz J, Cunniff C, Erickson RP, et al. Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J Natl Cancer Inst. 2003;95:669–674. doi: 10.1093/jnci/95.9.669. [DOI] [PubMed] [Google Scholar]
- Werner SR, Murthy S, Winata T, Prahalad AK, Yang J, Hock JM. Recql4 is involved in the cellular response to oxidative stress in osteosarcoma cells. Journal of Bone and Mineral Research. 2005;20:54. [Google Scholar]
- Werner SR, Prahalad AK, Yang J, Hock JM. RECQL4-deficient cells are hypersensitive to oxidative stress/damage: Insights for osteosarcoma prevalence and heterogeneity in Rothmund-Thomson syndrome. Biochem Biophys Res Commun. 2006;345:403–409. doi: 10.1016/j.bbrc.2006.04.093. [DOI] [PubMed] [Google Scholar]
- Woo LL, Futami K, Shimamoto A, Furuichi Y, Frank KM. The Rothmund-Thomson gene product RECQL4 localizes to the nucleolus in response to oxidative stress. Experimental Cell Research. 2006;312:3443–3457. doi: 10.1016/j.yexcr.2006.07.023. [DOI] [PubMed] [Google Scholar]
- Xu X, Liu Y. Dual DNA unwinding activities of the Rothmund-Thomson syndrome protein, RECQ4. EMBO J. 2009;28:568–577. doi: 10.1038/emboj.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Rochette PJ, Feyissa EA, Su TV, Liu Y. MCM10 mediates RECQ4 association with MCM2–7 helicase complex during DNA replication. EMBO J. 2009;19:3005–3014. doi: 10.1038/emboj.2009.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yannone SM, Roy S, Chan DW, Murphy MB, Huang SR, Campisi J, Chen DJ. Werner syndrome protein is regulated and phosphorylated by DNA-dependent protein kinase. Journal of Biological Chemistry. 2001;276:38242–38248. doi: 10.1074/jbc.M101913200. [DOI] [PubMed] [Google Scholar]
- Yano K, Morotomi-Yano K, Wang SY, Uematsu N, Lee KJ, Asaithamby A, Weterings E, Chen DJ. Ku recruits XLF to DNA double-strand breaks. EMBO Rep. 2008;9:91–96. doi: 10.1038/sj.embor.7401137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JH, Kwon YT, Varshavsky A, Wang WD. RECQL4, mutated in the Rothmund-Thomson and RAPADILINO syndromes, interacts with ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Human Molecular Genetics. 2004;13:2421–2430. doi: 10.1093/hmg/ddh269. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
An equal number (1000) of normal and RTS fibroblasts were plated on a 60 mm petri dish and allowed to grow under normal conditions for 1, 3, 5, 7 and 9 days. The cells were harvested at indicated time points and counted using Z1 Coulter counter (Beckman Coulter, Inc., CA, USA). The graph shows the average number of cells plotted with respect to number of days. The experiments were performed in triplicate and the error bars represents the mean standard error (+/−).
After 5 Gy of γ-irradiation, the cells were allowed to recover and fixed at indicated time points (0, 1, 3, 6 and 10 hrs) and immunostained with anti mouse γ-H2AX antibodies (as described in materials and methods). The images were captured with confocal microscope. The persistence of γ-H2AX foci’s in RTS-deficient primary fibroblasts AG05013 (lower row) compared to normal control fibroblasts (GM00323, upper row), at different time intervals as indicated. The first column represents the untreated cells (0 hour) as control.
Various GFP-tagged RECQL4 deletion constructs were transiently transfected in the HeLa cells. After 24 hour post transfections the cells were lysed and the total cell extract were subjected to western blotting with rabbit polyclonal anti GFP antibody. Asterisks (*) shows the band corresponding to expressed proteins. (M) represent protein marker and the size of the protein markers are indicated on left.