

SUMMARY
Trypanosoma brucei, the causal agent for sleeping sickness, depends on ergosterol for growth. Herein, we describe the effects of a mechanism-based inhibitor, 26-fluorolanosterol (26FL), which converts in vivo to a fluorinated substrate of the sterol C24-methyltransferase essential for sterol methylation and function of ergosterol and missing from the human host. 26FL showed potent inhibition of ergosterol biosynthesis and growth of procyclic and bloodstream forms while it has no effect on cholesterol biosynthesis or growth of human epithelial kidney cells. During exposure of cloned TbSMT to 26-fluorocholesta-5,7,24-trienol, the enzyme is gradually killed as a consequence of the covalent binding of the intermediate C25 cation to the active site (kcat/kinact = 0.26 min-1/0.24 min-1; partition ratio of 1.08) whereas 26FL is non-productively bound. These results demonstrate that poisoning of ergosterol biosynthesis by a 26-fluorinated Δ24-sterol is a promising strategy for developing new treatment for trypanosomiasis.
INTRODUCTION
The parasitic protozoan, Trypanosoma brucei, the etiological agent for sleeping sickness, is naturally transmitted by the blood-sucking tsetse fly (Glossina) (Van Den Abbeele et al., 1999). Sleeping sickness is endemic in 36 African countries, with 300,000 new cases and approximately 10,000 deaths per year and > 60 million people at risk (WHO, 2013). Disease development of Human African Trypanosomiasis (HAT) is marked by an initial hemolymphatic stage which can progress to the central nervous system after the parasite crosses the blood brain barrier yielding a late meningoencephalitic stage that is deadly. Current chemotherapy options are stage-specific and limited to stage 1 drugs - pentamidine and suramin, and stage 2 drugs - melarsoprol and eflornithine and are compromised by adverse side reactions, diminishing efficacy in several geographical regions, and complexity of use (Barrett, 2010; Ferrins et al., 2013). Many of these drugs have been in use since the 1950’s. Taken together, there is a pressing need to develop chemotherapeutics with novel mechanisms of action for the treatment of life-threatening HAT infections.
Phylogenomic and chemical studies reveal distinct differences in the genetic and metabolite compositions of protozoan and animal sterol metabolomes which produce ergosterol and cholesterol, respectively (Fugi et al., 2014; Nes, 2011; Roberts et al., 2003). This diversity in sterol metabolism between T. brucei and its human host has presented the intriguing possibility that protozoan biochemistry could be selectively inhibited. Indeed, our recent discovery shows there is an essential requirement for hormonal levels of ergosterol to “spark” (signal) cell proliferation of T. brucei bloodstream forms (Figure 1A) which cannot be satisfied solely by cholesterol salvaged from the host (Haubrich et al., 2015). These studies, plus the demonstration that inhibitors shown to block enzymes in the post-squalene segment of ergosterol biosynthesis pathway prevent growth of many protozoan parasites without effect on cultured animal cells (Gigante et al., 2009; Gros et al., 2006; Gunatilleke et al., 2012; Lepesheva et al., 2007; Lepesheva and Waterman, 2011; Lorente et al., 2004; Lovieno et al., 2014; Porta et al., 2014; Rahman and Pascal., 1990; Zhou et al., 2007) confirms our idea. Consequently, there is an effort underway to probe ergosterol biosynthesis targets with the aim to identify new drugs to treat specific neglected tropical diseases considered ergosterol-dependent.
Figure 1.
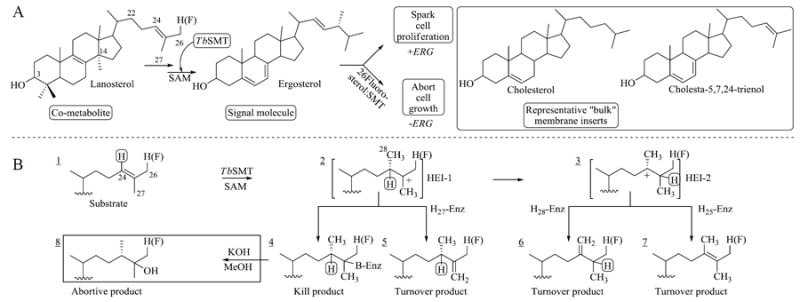
Overall biosynthetic pathway from lanosterol to ergosterol (ERG) showing functional aspects of different sterols (A) and C24-methylation reaction scheme showing conversion of Δ24-substrate to a kill or turnover methyl product (B). Formation of these alternative outcomes requires preliminary formation of a C24-methyl high energy intermediate (HEI) cation. 26-Fluorolanosterol, 26FL.
Most of the earlier research on lead development for new drugs resulted from the accidental observation of the biological effects of natural products or from screening organic compounds (Pink et al., 2005), whereas little attention has been directed toward druggable enzymes of ergosterol biosynthesis. In our search for effective ergosterol biosynthesis inhibitors to treat protozoan infections, we were guided by the seminal work on the repurposed drug - eflornithine (DL-α-difluoromethylornithine, DFMO), a fluorinated substrate mimic of L-ornithine decarboxylase (ODC) shown to control flux through the polyamine pathway, associated with cell growth and differentiation. Mechanism-based inactivation of this enzyme proceeds via pyridoxal phosphate-assisted decarboxylative elimination of fluoride to produce a fluorinated Michael acceptor that is captured by an active site cysteine residue (Andrews et al., 2014; Poulin et al., 1992). In addition, inhibition of this enzyme in Gambian sleeping sickness interferes with spermidine formation that, thereby produce parasite death (Heby et al., 2003). Importantly, a contributing factor to the treatment success of eflonithine is the significant differences in sensitivities of the parasite and animal ODC to the inhibitor.
Recently, the steroidal inhibitor 14α-methylenecyclopropyl Δ7-24(25)-dihydrolanosterol (MCP) fashioned to complex with T. brucei sterol 14α-demethylase (14-SDM) and prevent cell growth has emerged as an alternative to medical azoles to disrupt ergosterol homeostasis (Hargrove et al., 2012). The proposed mechanism of action of MCP to interfere with sterol 14-demethylation by irreversible binding to TbSDM differs from azoles which interact with the CYP51 enzyme as adduct to the heme iron as well as noncovalently with the substrate binding site (Hargrove et al., 2011). An entirely analogous strategy is well established in the area of aromatase inhibitors, where suicide substrates of the estrogen biosynthetic pathway enzymes have been shown to be potent drugs in breast cancer therapy (Bruegemeier et al., 2005). Azoles, however, suffer the disadvantage of being general P450 inhibitors, their efficacy depending on a greater affinity for the protozoan P45014SDM versus the mammalian P45014SDM, and some of them can bind concurrently to P450 enzymes and related steroid side chain metabolizing enzymes showing a lack of specificity (Lepesheva and Waterman, 2011). A more exact approach in the design of anti-parasite agents has been the development of phyla-specific steroidal inhibitors of sterol C24-methyltransferase (24-SMT) catalysis, such as, 25-azalanosterol. These high energy intermediate analogs can block ergosterol biosynthesis and halt cell growth by inhibiting specifically sterol methylation which does not occur in animals (Liu and Nes, 2009).
Given the key role of fluorinated small molecules as pharmacological agents in regulation of cholesterol biosynthesis and metabolism (Kauffman et al., 2000), and the demonstration that fluorinated cholesterols can replace cholesterol as a membrane insert and support growth of fungal sterol auxotrophs (Kauffman et al., 2000; Matsumori et al., 2008; Zundel et al., 1989), there has been remarkably little work reported on the influence of fluorinated compounds in ergosterol production and processing by protozoa. In our group, we reported fluoro substituents attached to the cycloartenol side chain at C24 or C26 that do not affect binding affinities to soybean 24-SMT (EC 2.1.1.143) as a consequence of the steric similarity between a fluorine and hydrogen atom (Patkar et al., 2013; Wang et al., 2008). On the other hand, the fluorine can exert a strong influence on the electronic environment that thereby affects intermediate cation formation and timing of the C24-methylation reaction. These effects promote partitioning toward covalent modifications over turnover catalyzed by 24-SMT (i.e., route 1→2→4 versus 5, 6 or 7, Figure 1B). This property of enzyme inactivation for ergosterol biosynthesis inhibitors could, unlike the ammonium-side chain containing sterols or medical azoles, disrupt ergosterol homeostasis without markedly affecting the sterol composition of treated-cells, the basis for a safe yet potent drug that could be used in combination therapy with existing therapeutics.
In this paper, we provide metabolite differences and biological effects together with mechanistic understanding for a novel class of fluorinated trypanocide evaluated using cultures of T. brucei and human epithelial kidney (HEK) cells and the cloned TbSMT. We were interested to identify a highly efficient substrate-mediated inhibitor of TbSMT (EC 2.1.1.41) that in its covalent inactivation affords a low partition ratio in which relatively few of the intermediate molecules formed are necessary to kill the enzyme while those that convert to methyl product leave the active site and become inert metabolites. The results reported here provide a new route to fluorinated functionality for “suicide substrate” poisoning of an essential enzyme in ergosterol biosynthesis. We also observed that 26FL is a steroidal prodrug that produces growth inhibition in the low micromolar range complementary to the previously described use of fluorinated substrate-based inhibitors that target the enzyme that controls flux through the polyamine pathway in T. brucei.
RESULTS
26-Fluorolanostero l(26-FL) Inhibits Growth of T. brucei Cells but not of HEK Cells
To assess the growth effect of a fluorosterol designed as a mechanism-based inhibitor of TbSMT, cell-based studies were performed with 26FL. The compound added at various concentrations to the medium of T. brucei procyclic (PCF) and bloodstream (BSF) forms and human epithelial kidney (HEK) cell cultures generated IC50 values of approximately 3 μM for PCF and 16 μM for BSF (Figure 2) while 26FL had no effect on HEK cell growth up to 100 μM (data not shown). The dose of 26FL causing 50% cell growth inhibition correlates to a moderate therapeutic index [ED50HEK/ED50bloodstream] of 6.2 with cells remaining viable even at the IC99 value of the drug (Haubrich et al., 2015).
Figure 2.
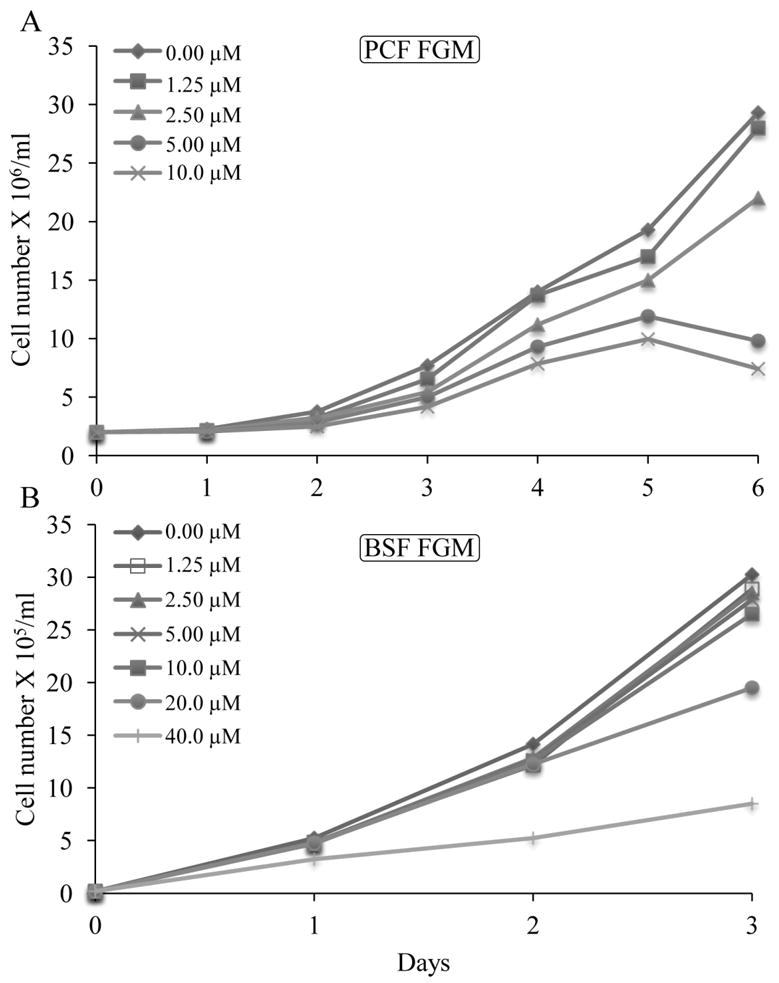
Growth response of Trypanosoma brucei procyclic forms (PCF) or bloodstream form (BSF) to increasing concentrations of 26-fluorolanosterol. Growth curves were performed in triplicate conducting three independent experiments described in the Experimental Procedures; error bars are not shown because, in most cases, they approximate the data symbols.
Exploitation of metabolic differences
To determine the underlying biochemistry responsible for the differences in growth response to 26FL, we considered the inhibitor to block the target enzyme, 24-SMT, in the ergosterol biosynthesis pathway. In agreement with our proposal, treated PCF showed a dose-dependent drop in ergosterol content that corresponds to zero-line growth inhibition equivalent to the inoculum. GC-MS analysis of neutral lipids of PCF treated cells at the IC50 of 26-FL show a loss of ergosterol at RRTc 1.11 (Figure 2, Panel A), otherwise the sterol profile is similar to the previously reported control (Lepesheva and Waterman, 2011). In our GC conditions, ergosterol and cholesta-5,7,24-trienol effectively co-elute, however, ergosterol can be detected in the mass spectrum at approximately 1-3 % the sterol mixture or at even lower concentrations at approximately .5% total sterol through selected ion monitoring (SIM) for appropriate ions (e.g., M+, 396 and M+- 33, 363 (-CH3- -H2O)) (Zhou et al., 2007) (Figure S1). At the IC50 of 26FL, trace amounts of three new fluorinated metabolites (<1% total sterol) were evident at the baseline of the chromatogram. When the drug is added at the IC90 value, the fluorinated compounds accumulate (~5% of total sterol) to greater levels in the sterol mixture than at IC50 values which allow for an interpretation of their mass spectra; two of them elute on the front and back-side of the peak corresponding to an ergosterol isomer- ergosta-5,7,25(27)-trienol (RRTc- 1.17, M+ 396) and a third compound elutes late in the chromatogram with a mobility typical of an “oxysterol”. Based on knowledge of chromatographic and mass spectral identification of the endogenous metabolites of the ergosterol biosynthesis pathway and oxysterols that contain a hydroxyl group in the side chain, coupled with the GC retention factor calculated for a 26-hydrogen to 26-fluorine moiety of 1.06 (Nes et al., 1988; Nes et al., 2012; Patkar et al., 2013; Xu et al., 1988), two new fluorinated steroidal “monols” were identified as 26-fluorocholesta-5,7,24-trienol (RRTc-1.16, M+ 400) and 26-fluoroergosta-8,25(27)-dienol (RRTc- 1.21, M+ 414, M+ - HF 396; M+- CH3 – HF, 381, M+ - H2O- HF- 2H; Figure 3B1 and 3B2, respectively). Mass spectra of the fluorinated sterol metabolites contain contaminant ions from ergosta-5,7,25(27)-trienol (Figure S1B). A third new compound eluting at RRTc 1.39 (M+ missing, M+- HF 414, M+ - H2O- HF, 378; Figure 3B-3) is tentatively identified as the steroidal “diol” 26-fluoro 3,25-dihydroxyergost-8-enol. A similar enzyme-generated diol with the cycloartenol nucleus was identified from incubation of 26-fluorocycloartenol with soybean 24-SMT (Patkar et al., 2013). The parent compound 26FL was also present in the fluorinated sterol mixture as a minor component in the GC tail corresponding to the diol at RRTc of 1.40 (M+ 444). The identification of an apparent abortive product of the C24-methylation reaction in the form of a “diol” (3C as *-3) is consistent with the intermediate cation intercepted by an active site nucleophile generating enzyme inactivation (Patkar et al., 2013). Thus, most of the 26FL accumulated by cells (< 95%) is converted to 26-fluorinated metabolites and the apparent inactivation of TbSMT by enzyme-activated 26-fluorozymosterol is likely reason for blocked zymosterol conversion to ergosta-8,25(27)-dienol and loss of ergosterol in cells.
Figure 3.
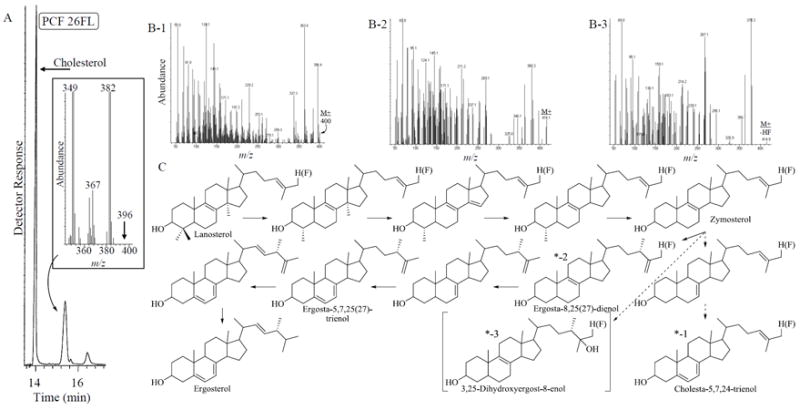
GC-MS analysis of neutral lipids from 26-fluorolanosterol-treated procyclic forms (at IC50 concentration of inhibitor): Panel A shows the total ion chromatogram of total sterols isolated from treated cells with inset showing the mass spectrum corresponding to the GC peak of cholesta-5,7,24-trienol (M+ 382) and ergosterol (M+ 396). Panels B1 and B2 represent the mass spectra from GC peaks on the front and back side of the peak corresponding to cholesta-5,7,24-trienol at the IC90 concentration of inhibitor. Panel C represent the GC peak eluting late at RRTc 1.39 at the IC90 concentration of the inhibitor. Panel C represents the uncommon ergosterol biosynthesis pathway in Trypanosoma brucei with structures having a star detected in the GC-MS analysis of 26-fluorolanosterol treatments.
Sterol analyses were conducted on neutral lipids of HEK cells cultured in the presence and absence of 26FL (at 100 μM). GC-MS analysis of the neutral lipids of control (Figure 4A) and treated-cells (Figure 4B) showed exclusively cholesterol or cholesterol accompanied by a set of 26-fluorosterol compounds that represent approximately 40% total cellular sterol; of the 26FL accumulated by cells, approximately half of it converts to inert products. Based on the 26H- to 26F - retention factor for standards bearing a 26-hydrogen atom (lanosterol RRTc, 1.31, M+ 426) and 26-fluorine atom (26-fluorolanosterol RRTc, 1.41, M+ 444; Figure 4D, star-3) and comparison of the RRTc and mass spectra (Figure 4C) of the 26-fluorosterol metabolites to the data established for intermediates in the cholesterol biosynthesis pathway (Nes, 2011), three metabolites detected in the treated cells are new (Figure 4C, compare with endogenous sterol mass spectra shown in Figure S2); 26-fluoro 4,4,-dimethyl cholesta-8,24-dienol (RRTc- 1.46, M+ 430; Figure 4D, star-4), 26-fluoro 4α-methyl cholesta-8,24-dienol (RRTc- 1.26, M+ 416; Figure 4D, star-2) and 26-fluoro cholesta-8,24-dienol co-eluting with cholesta-7,24-dienol (RRT-1.12, M+ 402 and M+ 384, respectively, Figure 4D, star- 1). Selective ion monitoring for molecular weights of compounds that could represent sterols with C24-reduced side chains (e.g., M+ 404 for 26-fluorocholesterol) failed to reveal any additional fluorosterol metabolites. It is worth mentioning as in the 26-fluorolanosterol-treated PCF incubation where ergosta-8,25(27)-dienol accumulates, in the 26-fluorolanosterol-treated HEK cells olefinic sterols with a double bond in the side chain Δ24(25)-position accumulate as well which suggest a general blockage of sterol reductase enzymes in these cultures. The accumulation of fluorinated sterols in HEK cells appears to have no apparent harmful effect on growth as expected (Zundel et al., 1989).
Figure 4.
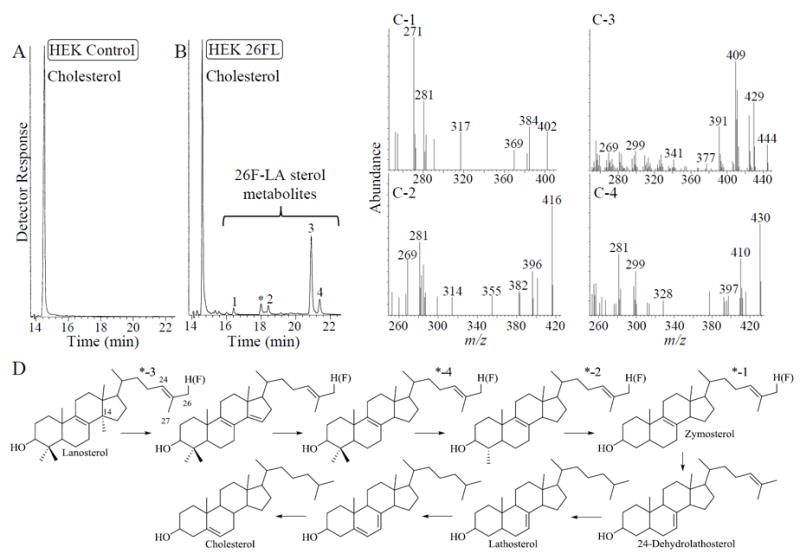
GC-MS analysis of neutral lipids from 26-fluorolanosterol-treated HEK cells. Panel A is total ion chromatogram of HEK control while Panel B is total ion chromatogram of 26-fluorolanosterol-treated cells. Panel C shows the mass spectra for the four 26-fluorosterols detected in the chromatogram and labeled in peaks 1 to 4; peak with star is a non-steroidal compound. Panel D shows the conical pathway for conversion of lanosterol to cholesterol with starred compounds corresponding to 26-fluorosterol metabolites shown in Panel B.
26-Fluorolanosterol is a Reversible Inhibitor of TbSMT
Inhibitor dissociation constants (Ki) for C24-methylation of 26-fluorolanosterol were based on initial reaction rate measurements with varied inhibitor concentrations (Nes et al., 2003; Nes et al., 2012). The fluorinated analog, 26FL, was incubated with cloned TbSMT under the standard reaction conditions used previously for the conversion of zymosterol to methyl product. As judged by GC-MS analysis, there is no product formation by TbSMT catalysis (Figure 5A) even after prolonged incubation or with different concentrations of protein up to 5 mg. This substrate, although not productively bound, exhibited a Ki value of 85 μM with competitive-type kinetics relative to zymosterol (Figure 5B) similar to the previously reported Ki value of lanosterol of 126 μM (Haubrich et al., 2015). Substrate protection is evident from the kinetics where addition of analog slows down the rate of enzyme inhibition while addition of substrate overcomes the inhibition by the analog. These results and the active conversion to metabolite following uptake by cells indicate that 26FL itself is not likely responsible for the growth inhibition.
Figure 5.
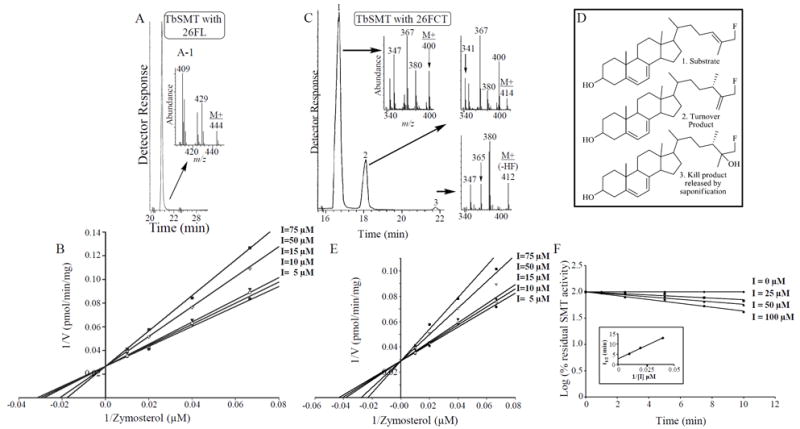
Enzyme-generated product distributions and kinetic analyses of 26-fluorolanosterol (26FL) and 26-fluorocholesta-5,7,24-trienol (26FCT) incubated with TbSMT. Panel A, Total ion chromatogram of 26-fluorolanosterol incubated with TbSMT and Panel B, Lineweaver-Burk plot of 26FL inhibition of TbSMT activity. Panel C, Capillary GC separation of methylated products synthesized from 26FCT by TbSMT with Panel D showing the corresponding structures. Panel E, Linweaver-Burk plot of 26FCT inhibition of TbSMT activity. Panel E, Time-dependent inactivation of TbSMT by 26FCT. Values plotted are the mean of duplicate determinations which varied by less than 5%.
Products of the TbSMT C24-Methylation with 26-Fluorocholesta-5,7,24-trienol
In order to determine whether the 26FL metabolite 26-fluorozymosterol could show suicide substrate properties toward TbSMT in a cell-free system we considered to prepare 26-fluorozymosterol as a substrate. However, due to a paucity of zymosterol for the synthetic work another analog was chosen to test with the enzyme. Because cholesta- 5,7,24-trienol shows similar competency to zymosterol in acceptance by TbSMT (Lepesheva and Waterman, 2011), we reasoned the fluorinated analog of cholesta-5,7,24- trienol could be prepared from commercially available ergosterol and tested as an alternative analog. 26-FCT paired with SAM was incubated with cloned TbSMT. GC-MS analysis (Figure 5C) showed that the early eluting compound at 18 minutes possesses a Δ5,7 system (ions at m/z 128, 143, 157, 158 and 159 similar to the substrate (Figure S3) and C24-methyl side chain (ions at m/z 414 (M+), 400, 380, 367, 349) typical of a methylated fluorosterol. The late eluting compound at approximately 21.9 minutes also contained a Δ5,7 system (ions at m/z 128, 143, 157, 158 and 159) and a C24-alkylated side chain with a hydroxyl group (ions at m/z 432 (= M+ missing), 412, 400, 365 and 347); these enzyme-generated products correspond to 26-fluoroergosta-5,7,25(27)-trienol and 26-fluoro 3,25-dihydroxyergosta-5,7-dienol, respectively (Figure 5D). The percent conversion of fluorinated analog to methyl product is approximately 25% while the ratio of C24- methyl turnover product to C24-methyl abortive product (Figure 1B) is approximately 8. The identification of a key abortive product (as the fluorinated diol), suggesting the ternary complex is arrested following formation of the first high energy intermediate, agrees with the proposed reaction steps of sterol C24-methylation catalyzed by TbSMT outlined in Figure 1B. It is noteworthy in the total ion current chromatogram in Figure 3A, that the retention factor for the 26-fluoro-25-hydroxy group (sterol 2 = RRTc, 1.29) versus 26-fluoro-Δ25(27) -group (sterol 3 = RRTc, 1.50) of 1.16 determined in the enzyme assay with 26-fluorocholesta-5,7,24-trienol matches the retention factor of 1.16 for the same side chain pair in 26-flouro- ergosta-8,25(27)-dienol (RRTc 1.21) and 26-fluoro 3,25-dihydroxyergost-8-enol (1.39) identified in the whole cells studies from incubation of 26-fluorolanosterol. The substrate analog evidently was capable to uncouple the reaction sequence catalyzed by TbSMT affording the production of the corresponding steroidal diol, the abortive product representative of the methyl intermediate bound to the enzyme.
Determination of Kinetic Parameters
To assess the relative rate of enzyme-catalyzed methylation of 26-FCT versus zymosterol, both substrates were examined in the linear range of enzymatic activity (5- 150 μM) with the same lysate preparation of recombinant enzyme. The fluoroanalog is methylated at approximately 9 % the rate of zymosterol conversion to ergosta-8,25(27)-dienol while yielding similar Km values of 25 ± 2 μM, comparable to our previous finding of a Km 47 μM for zymosterol and a Km of 25 μM for cholesta-5,7,24-trienol, respectively (Liu et al., 2011; Zhou et al., 2006). Using a concentration of 50 micrograms of native tetramer TbSMT approximated from the SDS-PAGE gel of the enzyme preparation reported previously (Zhou et al., 2006), the kcat values are estimated for 26-fluorocholesta-5,7,24-trienol of 0.26 min-1 compared to an estimated kcat for zymosterol of 2.88 min-1 which is the same value as before (Zhou et al., 2006). However, these turnover numbers are approximately an order of magnitude greater from our previously reported value of 0.6 min-1. In the latter case, the enzyme was purified to homogeneity using a series of chromatographic steps that involve anion exchange columns (Zhou et al., 2006). We tracked down the observed differences in catalytic competence and determined they arise from the allosteric capabilities of the enzyme operating in its native tetrameric form. Previously, we showed sigmoidal behavior of the pure enzyme toward SAM (S-adenosyl-L-methinione) (Nes et al., 1998). Now, we discovered that ATP bound to the 24-SMT in pre-Q column preparations is released from the active form during the purification procedure involving a Q-column chromatography step. Importantly, ATP at physiological concentrations of 400 μM (Rodriguez and Parks, 1981) is a positive allosteric effector of cloned yeast 24-SMT and TbSMT activity and restores completely the dramatic loss of enzyme activity following Q-column chromatography (Marshall, 2001; and unpublished results). When cAMP is added to fungal cultures, the effect on ATP levels is associated with an increase in ergosterol, presumably by activation of 24-SMT (Sardari et al., 2003).
The product outcome from incubation of 26FCT with TbSMT showed reaction with SAM and that the substrate analog binds specifically to the active site. Additionally, the kinetic behavior shows binding is almost as tight as that of the natural substrate. Finally, replacing the C26 hydrogen in the zymosterol system by fluorine retards the rate of C24- methylation, a reaction known to proceed through multiple carbonium high energy intermediates generates a single 26-fluoro abortive product. Thus, it is quite possible that the coupled methylation and deprotonation reactions catalyzed by TbSMT are discrete steps (Figure 1B) whereas the same reaction catalyzed by the yeast 24-SMT operates in a concerted manner to produce a single Δ24(28) - product (Nes et al., 1998).
Further evaluation of 26-fluorocholesta-5,7,24-trienol as an analog for TbSMT was based upon inhibition kinetics of 26FCT against varying concentrations of zymosterol and fixed amounts of SAM using an E. coli preparation of the cloned enzyme. The double-reciprocal plot of sterol methylation activity in the presence of several concentrations of the fluorinated compound is shown in Figure 5E. The data clearly indicate that the sterol is a competitive inhibitor of the sterol methylase with respect to zymosterol showing a Ki of 75 μM. These data and that of product distributions from incubation with 26FCT are consistent with the observation of accumulation of 26-fluoroergosta-8,25(27)-dienol in treated cells. Thus, the notion that 26-fluorozymosterol is a substrate mimic of zymosterol is confirmed by kinetic studies as well as by metabolic conversions catalyzed by TbSMT.
Inactivation of TbSMT
Stable metabolites of 26FL did not inhibit TbSMT through a reversible mechanism of inhibition. To assess the alternative irreversible mechanism of inhibition, we performed time-course experiments using the cloned enzyme which confirmed inactivation accompanying the methyl intermediate formation with half-maximal values for each occurring within 10 minutes (Figure 5, Panel F). TbSMT activity declined by a pseudo-first-order process with a half-life of 2.82 min-1. The rate of inactivation of TbSMT was saturable, with a maximum rate of inactivation, kinact of 0.24 min-1 ±0.01 min-1 and a Ki of 89 μM, similar to that reported from the steady-state Ki determinations reported in Figure 5, Panel E. These values compare favorably with the kinetic parameters for the normal substrate zymosterol. As expected, no inactivation occurred when the incubations were carried out with sterol only, consistent with absolute dependency of the co-substrates for the reaction to generate covalently bound product (Figure 1, Panel B).
DISCUSSION
Drug discovery for neglected tropical diseases falls into two broad categories of molecular target-based and phenotypic approaches (Gilbert, 2013). Significant problem with NTD drug resistance includes specificity and variant mechanisms of resistance (Barrett et al., 2011; Chitanga et al., 2011). These overarching problems need to be addressed in order for any single compound to treat trypanosomiais patients successfully. In the search for the next generation of anti-parasitic agents, work on trypanosome sequences coupled with biochemical investigations has pinpointed the sterol C24-methyltransferase in the ergosterol biosynthesis pathway as a potential drug target. The substrate specificity of this crucial catalyst reveals limited steroidal modifications that could be processed by the enzyme. Nonetheless, structure-activity analyses of a range of steroidal inhibitors incubated with 24-SMTs across kingdoms provide foundation for a tractable chemical start point. It has been demonstrated that substrate mimics compete with the natural acceptor of 24-SMT for a common catalytic site, and may occur with reversible inhibitors (azasterols) as enzyme-bound transition state analogs or with irreversible inhibitors (fluorinated olefins) as enzyme-bound abortive intermediates. Whichever mechanism applies to the steroidal inhibitor in vitro, it is clear that complete blockage of 24-SMT activity by these compounds can offer a proof-of-principle for such enzyme-targeted approaches. When suicide substrates compared to tight binding steroidal inhibitors of 24-SMT show relevant differences in their effects on ergosterol biosynthesis in treated cells, it could represent sequence-specific recognition in the formation of a covalent complex, the basis for phyla-specific inhibitors. Criteria for anti-parasite hits, leads and drug candidates in the design and testing of new enzyme inhibitors to block TbSMT in T. brucei have been met by our test analogs in this and previous studies- i.e., the compound is active with IC50 of ≤ 10 μM, active in vivo against parasites at a doses of ≤ 100 mg per kg and not overly toxic in animals at efficacious dose such that analogs can be obtained (Pink et al., 2005).
Preferential inhibition is gained in the suicide substrate approach to enzyme inactivation in ergosterol biosynthesis which exploits analog specificity of TbSMT. The inhibition constant Ki for a substrate analog describes its level of specificity and the reversible equilibrium between enzyme and inhibitor for the initial inhibitor binding step; the Ki dissociation constant of enzyme-inhibitor complex (EI), I is inhibitor concentration, S is substrate concentration and P the product concentration while kcat describes the catalytic turnover number and kinhib, expressed by the kinact constant, describes the time-dependent rate of enzyme inactivation (Walsh, 1984) (Scheme 1).
Scheme 1.
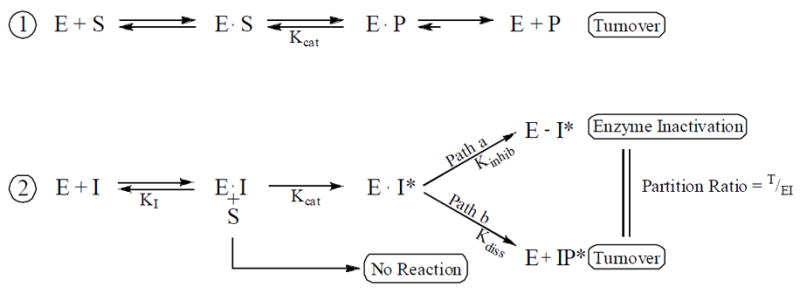
Kinetic schemes for the interaction of TbSMT with substrate analogs.
In 1- binding of enzyme (E) and Δ24-sterol substrate (S) followed by enzyme catalysis with formation of methyl product (P). In 2- inhibitor (I) binds to enzyme followed by activation via methylation (E▾I*) and this intermediate can partition along path b and dissociate from the active site (E + IP*) or enter into path a and irreversibly bind to the enzyme (E-I*).
Mechanistically for 26-flourinated analogs to act as an irreversible inhibitor the compounds should serve as substrates of the Michaelis complex and convert to a methylated intermediate cation (I*) similar to the processing of zymosterol by TbSMT. The resulting fluorinated intermediate has the ability to capture an apoprotein residue through competing processes that involve partitioning toward deprotonation and turnover (E + IP*) typical of the natural substrate (path b) or inactivation (E-I*) characteristic of the steroidal inhibitor (path a) as shown in Scheme 1. The functional inhibition of the enzyme is determined by the relative rates of each of these pathways and in particular the formation of the E-I* species.
One example of the in vitro potency of sterol C24-methyltransferase inhibitors is through comparison via the ratios of their Km/Ki values, thus the larger the ratio the greater the competitive inhibition. When tested against TbSMT, The 26-fluoro substituted analogs exhibited Km/Ki ratios for 26FL of 25/85 (= 0.29) and for 26FCT of 25/75 (= 0.33) (this study) compared to a Km/Ki ratio for 25-azalanosterol of 47/.039 (= 1205) reported previously (Zhou et al., 2006). While it is notable that the azasteroids are three orders of magnitude greater in effectiveness as inhibitors of cell-free TbSMT, they do not show a corresponding potency in terms of in vivo growth inhibition of cultured T. brucei cells. Thus 26FL, like 25-azalanosterol, inhibit PCF growth at approximately 1 μM.
Another way to assess altered catalytic capacity of TbSMT to fluorinated analog is through examination of both concentration-dependent and time-dependent inhibition kinetics. Here, the partition ratio is the key indicator of potential in vivo specificity and utility. The substrate partition ratio [kcat/kinact] for a TbSMT mechanism-based inhibitor is operationally defined as the number of inhibitor molecules that are methylated per unit time before one enzyme molecule is irreversibly inactivated. While the efficacy of these compounds can vary according to the chemical design, e.g., addition of a terminal methylene cyclopropane, sulfur, or fluorine substituent in the sterol side chain (Kanagasabai et al., 2004; Nes et al., 2012; Zhou et al., 2006), the lower the partition ratio, the less drug is needed to kill a given enzyme and potential toxic product release becomes null. A completely efficient suicide substrate that targets TbSMT would generate a partition ratio close to 1 and in so doing show desirable drug properties of specificity and low toxicity (Walsh, 1984). In studies of 26FCT tested with TbSMT, we determined the analog is a poor substrate for turnover; however, it irreversibly inactivated the enzyme in a saturable way implying that a Michaelis-complex forms, but partitions to favor the inactivation pathway. The partition ratio of 1.08 shows 26FCT to be one of the most highly efficient mechanism-based inhibitors thus far reported in the pharmaceutical literature.
Another critical factor in suicide substrate effectiveness for inhibition of ergosterol biosynthesis relates to their ability to be accumulated by cells and then out-compete the natural substrate for acceptance by the sterol methyltransferase enzyme. In previous work with suicide substrates designed to inhibit 24-SMT in pathogenic fungi, the compounds showed predicted in vitro activity but failed to be taken up by the fungi (Ganapathy et al., 2011). If these analogs are accumulated by the parasite, they must successfully bind to TbSMT at concentrations several times higher than the Km values of zymosterol which correlates to its endogenous steady-state concentration of 0.1% total sterol yielding 145 fg/cell. In the current study, 26FL-treated cells showed evidence for uptake and metabolism of the drug to include products of TbSMT catalysis. However, despite no significant change in the endogenous sterol profile except for a loss of ergosterol, the detection of 26-fluorosterol metabolites at the approximate level of 1% is consistent with 26-fluorozymosterol formed to a level that outcompetes the zymosterol available for TbSMT. Notably, as the exogenous amount of 26FL supplied to the cells was increased, providing for more endogenous 26-fluorozymosterol, the cell growth fell accordingly. In contrast to our observations with 25-azalanosterol-treated procyclic cells where cells at the drug IC90 generated a higher total endogenous sterol content than treated cells at IC50 or controls (thereby suggesting an up-regulation of ergosterol biosynthesis enzymes), for the 26FL-treated cells there was no significant change in the endogenous sterol composition as the concentration of drug increased from IC50 or IC90. Thus, suicide substrates of TbSMT may be considered the ultimate physiological inhibitor for ergosterol biosynthesis and as a potential therapeutic because it remains bound to the enzyme for the lifetime of the target in the cell and its effect specific on ergosterol formation.
SIGNIFICANCE
Trypanosomes have evolved to be ruthless parasites of their animal hosts. In this and our previous studies, we endeavored to develop a new strategy to treat trypanosomasis that involves the inhibition of crucial enzymes in the ergosterol biosynthesis pathway of T. brucei. This advance, particularly suited for specific combinations of mechanistically-distinct inhibitors of TbSMT and TbSDM, can be fashioned to kill rather than simply halt cell proliferation and in so doing overcome conventional resistance factors. Indeed, by incubating a medical azole with 25-azalanosterol paired at their IC50 values of 1-3 μM in the growth media of T. brucei showed synergy resulting in cell death (Haubrich et al., 2015).
This is the first time that fluorinated sterols have been tested as anti-parasitic agents. Our results show the promise of these compounds as prodrugs, specifically 26-fluorolanosterol, that can enter the ergosterol biosynthesis pathway, convert to a substrate analog of an indispensible enzyme and through catalysis inhibit sterol C24-methylation and cell growth. The partition ratio for 26FCT against TbSMT of 1.08 compares favorably for eflonithine against ODC of approximately 3.3 (Pegg et al., 1987), which suggest the basis of selective and potent antitrypanosomal action of fluorinated inhibitors could be the relatively slow turnover of TbSMT which in complex with activated analogs completely inactivates the catalyst, as reported for eflornithine against TbODC (Wang, 1991). This constitutive property of inhibitor-protein interactions may explain the minimal change in the course and equilibrium of metabolic sequences in treated T. brucei cells which receive the bulk amount of membrane sterol (cholesterol) from the host. Taking into account the above-described experiments and their results regarding 26- fluorinated sterols, indicate that mechanism-based inhibitors targeting the sterol methylation enzyme might be a promising and useful additional agent in the therapeutic armamentarium for the treatment of sleeping sickness and other protozoan infections.
EXPERIMENTAL PROCEDURES
Strain, culture procedures and cellular sterol analysis
A T. brucei strain 427 was grown at Meharry Medical College in SDM-79 medium (procyclic form) with 10% heat-inactivated FBS (Atlanta Biologicals), referred to as full-growth medium (FGM) at 27 °C. Blood-stream forms originating from cells grown in rats (Sprague Dawley male rats from Harlan Laboratories) fed the standard rat chow (PicLab Rodent diet) were cultured at 37 o0 in HMI-9 medium supplemented with 10% heat-inactivated FBS as previously described (Haubrich et al., 2015). Human epithelial kidney cells (HEK 293T) purchased from ATCC (Manassas, VA) and grown in RPMI medium supplemented with 20% heat-inactivated FBS. Both cultures were maintained in a humidified atmosphere containing 5% CO2. Cell counting was done with a Neubauer hemocytometer. The procyclic (PCF) and bloodstream forms (BSF) cultures were seeded at different densities of 2-3 × 106 \ cells/ml or 1 × 105 cells/ml, respectively, in tissue-culture flasks. Cell densities were maintained in the range of 0.5-1 × 107 cells/ml for the PCF and 0.1-1 × 106 cells/ml for the BSF by re-inoculation in fresh medium. Cells were harvested by centrifugation. HEK cell viability of HEK cells was monitored by the trypan blue dye exclusion method. To evaluate the growth inhibition effect, the cells were incubated with 26-fluorolanosterol as a 1% (v/v) solution in dimethyl sulfoxide in the concentration range 0-100 μM. Cells treated with 1% DMSO alone served as controls. The concentrations of 26FL causing 50% growth inhibition (ED50, 50% effective dose) was calculated from dose-response curves. The selectivity of the inhibitor for the parasite was estimated as ratio ED50 (HEK 293T) / ED50 BSF) (Hoet et al., 2004). Protozoan cells, collected by centrifugation, were saponified (10% KOH in methanol at reflux for 30 min) and the neutral lipid obtained by dilution with water and extraction into hexanes was analyzed by GC-MS.
Recovery and analysis of sterols from cultured cells
Procyclic and bloodstream forms and HEK cells were harvested at growth arrest in the presence or absence of 26- fluorolanosterol at the IC50 and IC90 concentrations of the compound for TB cells or 30 μM compound for HEK cells. Cell pellets were split with an internal standard of 5α-cholestane added to one of the cell pellets. Cells were saponified with 10% methanolic KOH and extracted with hexanes. The neutral lipids were analyzed by GC-MS against cholesterol as a chromatographic reference. Mass spectra data were compared to those obtained from authentic standards.
Enzyme assay and product analysis
The activity assays and analysis of enzyme-generated products were performed in analogous fashion to Zhou et al., 2007. A brief description of these protocols using the cloned TbSMT is described in the Supplemental Experimental Procedures.
Mechanism-based inactivation experiments
The soluble enzyme fraction containing TbSMT (100,000 g supernatant fraction, 75 μg, 600 μl) in the standard assay buffer was incubated at 35 °C for up to 10 min at 0 μM (control), 25, 50 and 100 μM 26- fluorocholesta-5,7,24-trienol at 2, 5, 8 and 10 min and the samples placed on dry ice ethanol-pre-cooled test tubes to prevent catalysis. The samples were then thawed on ice and the unbound inhibitor and SAM removed by repeated filtration using a Centricon (YM-10 membrane). Following treatment, the remaining enzyme activity was measured by comparison with the enzymatic activity of assays carried out under the standard assay conditions with saturated concentrations of zymosterol (100 μM) paired with 3H-SAM (100 μM). The logarithmic percentage of enzyme activity was plotted against incubation time of the enzyme-inhibitor mixture to determine the half-life (T½) of inactivation as previously described (Marshall, 2011; Nes et al., 2008). Chemical labeling of TbSMT by 26-fluorosterol was carried out using 3 mg/mL in standard lysate assay in overnight incubation. The abortive product covalently bound to the enzyme was released from the enzyme-sterol complex by saponification and the resulting neutral lipids composed of steroidal monols and diols (from ester formation in the active site) analyzed by GC-MS.
Compound Synthesis
The synthetic scheme (Figure S4) and detailed experimental procedures for the preparation and characterization of the 26-fluorosterols tested in this study are described in Supplemental Experimental Procedures.
Supplementary Material
Supplemental information includes a set of chromatographic data and mass spectra for representative standards and compounds isolated from treated cells, a description of chemical synthesis and characterization of 26-fluorolanosterol and 26-fluorocholesta- 5,7,24-trienol, a description of activity assays and product analysis using cloned TbSMT and can be found with this article online.
Highlights.
Target-specific fluorinated drug effect on Trypanosoma brucei PCF and BSF growth
26-Fluorinated Δ24-sterol is a mechanism-based poison of ergosterol biosynthesis
In vivo the Tbsterol C24-methyltransferase is inactivated by 26-fluorinated sterols
Chemical and kinetic evaluation of 26-fluorinated steroids as suicide substrates
Acknowledgments
This work was supported by grants from the National Institutes of Health (R21AI 119782) and National Science Foundation (MCB-0929212) to W.D.N. and from the National Institutes of Health (GM081146) to M.C. The involvement of Alicia Howard with the art in the graphical abstract is appreciated.
Footnotes
AUTHOR CONTRIBUTION
W.D.N. and M.C. designed the experiments; D.J.L., P.P., U.K.S., M.B.M., and B.A.H. performed the kinetic and growth experiments; M.B.M, and D.J.L. generated the standards and prepared the fluorinated compounds; W.D.N. and M.C. analyzed the data; W.D.N. wrote the manuscript; D.J.L., P.P., U.K.S., M.B.M., B.A.H., M.C. and W.D.N. edited the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrews KT, Fisher G, Skinner-Adams TS. Drug repurposing and human parasitic diseases. Intern J Parasitol Drugs Drug Resist. 2014;4:95–111. doi: 10.1016/j.ijpddr.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett MP. Potential new drugs for human African trypanosomiasis: some progress at last. Curr Opin Infect Dis. 2010;23:603–608. doi: 10.1097/QCO.0b013e32833f9fd0. [DOI] [PubMed] [Google Scholar]
- Barrett MP, Vincent IM, Burchmore RJ, Kazibwe AJ, Matovu E. Drug resistance in human African trypanosomiasis. Future Microbiol. 2011;6:1037–1047. doi: 10.2217/fmb.11.88. [DOI] [PubMed] [Google Scholar]
- Bruegemeier, Hackett JC, Diaz-Cruz ES. Aromatase inhibitors in the treatment of breast cancer. Endocrine Rev. 2005;26:331–345. doi: 10.1210/er.2004-0015. [DOI] [PubMed] [Google Scholar]
- Chitanga S, Marcotty T, Namangala B, Van den Bossche P, Abbeele JVD, Delespaux V. High prevelance of drug resistance in animal trypanosomes without a history of drug exposure. Plos Negl Trop Dis. 2011;5:e1454. doi: 10.1371/journal.pntd.0001454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrins L, Rahmani RS, Baell JB. Drug discovery and human African trypanosomiasis: a disease less neglected? Future Med Chem. 2013;5:1801–1841. doi: 10.4155/fmc.13.162. [DOI] [PubMed] [Google Scholar]
- Fugi MA, Gunasekera K, Ochsenreiter T, Guan X, Wenk MR, Maser P. Genome profiling of sterol synthesis shows convergent evolution in parasites and guides chemotherapeutic attack. J Lipid Res. 2014;55:929–938. doi: 10.1194/jlr.M048017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathy K, Kanagasabai R, Nguyen TTM, Nes WD. Purification, characterization, and inhibition of sterol C24-methyltransferase from Candida albicans. Arch Biochem Biophys. 2011;505:194–201. doi: 10.1016/j.abb.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Gigante F, Kaiser M, Brun R, Gilbert IH. SAR studies on azasterols as potential anti-trypanosomal and anti-leishmanial agents. Bioorgan & Med Chem. 2009;17:5950–5961. doi: 10.1016/j.bmc.2009.06.062. [DOI] [PubMed] [Google Scholar]
- Gilbert IH. Drug discovery for neglected diseases: Molecular target-based and phenotypic approaches. J Med Chem. 2013;56:7719–7726. doi: 10.1021/jm400362b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros L, Lorente SO, Jimenez CJ, Yardley V, Rattray L, Wharton H, Little S, Croft SL, Ruiz-Perez LM, Gonzlez-Pacanowska D, Gilbert IH. Evaluation of azasterols as anti-parasites. J Med Chem. 2006;49:6094–6103. doi: 10.1021/jm060290f. [DOI] [PubMed] [Google Scholar]
- Gunatilleke SS, Calvet CM, Johnston JB, Chen C-K, Erenburg G, Gut J, Engel JC, Ang KKH, Mulvaney J, Chen S, Arkin MR, McKerrow JH, Podust M. Diverse inhibitor types targeting the Trypanosoma cruzi CYP51. Plos Neg Trop Dis. 2012;6:e1736. doi: 10.1371/journal.pntd.0001736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargrove TY, Liu J, Nes WD, Waterman MR, Lepesheva GI. Substrate preferences and catalytic parameters determined by structural characteristics of sterol 14α-demethylase CYP51 from Leishmania infantum. J Biol Chem. 2011;286:26838–26848. doi: 10.1074/jbc.M111.237099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargrove TY, Wawrzak Z, Liu J, Waterman MR, Nes WD, Lepesheva GI. Structural complex of sterol 14a-demethylase (CYP51) with 14α-methylenecyclopropyl-Δ7-24,25-dihdrolanosterol. J Lipid Res. 2012;53:311–320. doi: 10.1194/jlr.M021865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubrich BA, Singha UK, Miller MB, Nes CR, Anyatonwu H, Lecordier L, Patkar P, Leaver DJ, Villalta F, Vanhollebeke B, Chaudhuri M, Nes WD. Discovery of an ergosterol-signaling factor that regulates Trypanosoma brucei growth. J Lipid Res. 2015;56:331–341. doi: 10.1194/jlr.M054643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heby O, Roberts SC, Ullman B. Polyamine biosynthetic enzymes as drug targets in parasitic protozoa. Biochem Soc. 2003;31:415–419. doi: 10.1042/bst0310415. [DOI] [PubMed] [Google Scholar]
- Hoet S, Opperdoes F, Brun R, Quentin-Leclercq J. Natural products active against African trypanosomes: a step towards new drugs. Nat Prod Rep. 2004;21:353–364. doi: 10.1039/b311021b. [DOI] [PubMed] [Google Scholar]
- Kanagasabai R, Zhou W, Liu J, Nguyen TTM, Veeramachaneni P, Nes WD. Disruption of ergosterol biosynthesis, growth and the morphological transition in Candida albicans by sterol methyltransferase inhibitors containing sulfur in the sterol side chain. Lipids. 2004;39:737–746. doi: 10.1007/s11745-004-1290-7. [DOI] [PubMed] [Google Scholar]
- Kauffman JM, Westerman PW, Carey MC. Fluorocholesterols, in contrast to hydroxycholesterols, exhibit interfacial properties similar to cholesterol. J Lipid Res. 2000;41:991–1003. [PubMed] [Google Scholar]
- Lepesheva GI, Ott RD, Hargrove TY, Kleshchenko YY, Schuster I, Nes WD, Hill GC, Villalta F, Waterman MR. Sterol 14α-demethylase as a potential target for antitrypanosomal therapy: Enzyme inhibition and parasite cell growth. Chem & Biol. 2007;14:1283–1293. doi: 10.1016/j.chembiol.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva GI, Waterman MR. Sterol 14α-demethylase (CYP51) as a therapeutic target for human trypanosomiasis and leishmaniasis. Curr Top Med Chem. 2011;11:2060–2071. doi: 10.2174/156802611796575902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Nes WD. Steroidal triterpenes: Design of substrate-based inhibitors of ergosterol and sitosterol synthesis. Molecules. 2009;14:4690–4706. doi: 10.3390/molecules14114690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Ganapathy K, Wywial E, Bujmicki JM, Nwogwugwu CA, Nes WD. Effect of substrate features and mutagenesis of active site tyrosine residues on the reaction course catalyzed by Trypanosma brucei sterol C24-methyltransferase. Biochem J. 2011;439:413–422. doi: 10.1042/BJ20110865. [DOI] [PubMed] [Google Scholar]
- Lorente SO, Rodrigues JCF, et al. Novel azasterols as potential agents for treatment of leishmaniasis and trypanosomiasis. Antimicrob Agents Chemotherap. 2004;48:2937–2950. doi: 10.1128/AAC.48.8.2937-2950.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovieno A, Miller D, Ledee DR, Alfonso EC. Cysticidal activity of antifungals against different genotypes of Acanthamoeba. Antimicrobiol Agents Chemotherp. 2014;58:5626–5628. doi: 10.1128/AAC.02635-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JA. Studies on the enzymology of sterol methyltransferase from Saccharomyces cerevisiae. Doctoral Dissertation, Texas Tech University. 2001:1–95. [Google Scholar]
- Matsumori N, Kasai Y, Oishi T, Murata M, Nomura K. Orientation of fluorinated cholesterol in lipid bilayers analyzed by 19F tensor calculation and solid-state NMR. J Amer Chem Soc. 2008;130:4757–4766. doi: 10.1021/ja077580l. [DOI] [PubMed] [Google Scholar]
- Nes WD, Xu S, Haddon WF. Evidence for similarities and differences in the biosynthesis of fungal sterols. Steroids. 1988;53:533–558. doi: 10.1016/0039-128x(89)90030-5. [DOI] [PubMed] [Google Scholar]
- Nes WD, McCourt BS, Zhou W-X, Ma J, Marshall JA, Peek L-A, Brennan M. Overexpression, purification, and stereochemical studies of the recombinant (S)-adenosyl-L-methionine: Δ24(25) -to Δ24(28)-sterol methyltransferase from Saccharomyces cerevisiae. Arch Biochem Biophys. 1998;353:297–311. doi: 10.1006/abbi.1998.0665. [DOI] [PubMed] [Google Scholar]
- Nes WD, Song Z, Dennis AL, Zhou W, Nam J, Miller M. Biosynthesis of phytosterols: Kinetic mechanism for the enzymatic C-Methylation of sterols. J Biol Chem. 2003;278:34505–34516. doi: 10.1074/jbc.M303359200. [DOI] [PubMed] [Google Scholar]
- Nes WD, Jayasimha P, Song Z. Yeast sterol C24-methyltransferase: Role of highly conserved tyrosine in catalytic competence studied by site-directed mutagenesis and thermodynamic analysis. Arch Biochem Biophys. 2008;477:316–323. doi: 10.1016/j.abb.2008.05.016. [DOI] [PubMed] [Google Scholar]
- Nes WD. Biosynthesis of cholesterol and other sterols. Chem Rev. 2011;111:6423–6451. doi: 10.1021/cr200021m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nes CR, Singha UK, Liu J, Villalta F, Waterman MR, Lepesheva GI, Chaudhuri M, Nes WD. Novel sterol metabolic network of Trypanosoma brucei procyclic and bloodstream forms. Biochem J. 2012;443:267–276. doi: 10.1042/BJ20111849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patkar P, Haubrich BA, Qi M, Nguyen TT, Thomas CD, Nes WD. C-24-methylation of 26-fluorocycloartenols by recombinant sterol C24-methyltransferase from soybean: evidence for channel switching and its phylogenetic implications. Biochem J. 2013;456:253–262. doi: 10.1042/BJ20121818. [DOI] [PubMed] [Google Scholar]
- Pegg AE, McGovern KA, Wiest L. Decarboxylation of α-difluoroethylornithnie by ornithine decarboxylase. Biochem J. 1987;241:305–307. doi: 10.1042/bj2410305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pink R, Hudson A, Mouries M-A, Bendig M. Opportunities and challenges in anti-parasitic drug discovery. Nature Rev Drug Discov. 2005;4:727–740. doi: 10.1038/nrd1824. [DOI] [PubMed] [Google Scholar]
- Porta EOJ, Carvalho PB, Avery MA, Tekwani BL, Labadie GR. Click chemistry decoration of amino sterols as promising strategy to develop new leishmanicidal drugs. Steroids. 2014;79:28–36. doi: 10.1016/j.steroids.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Poulin R, Lu L, Ackerman B, Bey P, Pegg AE. Mechanism of the irreversible inactivation of mouse ornithine decarboxylase by α-difluromethylornithine. J Biol Chem. 1992;267:150–158. [PubMed] [Google Scholar]
- Rahman MD, Pascal RA., jr Inhibitors of ergosterol biosynthesis and growth of the trypanosomatid protozoan Crithidia fasiculata. J Biol Chem. 1990;265:4989–4996. [PubMed] [Google Scholar]
- Roberts CW, Mcleod R, Rice DW, Ginger M, Chance ML, Goad LJ. Fatty and sterol metabolism: potential antimicrobial targets in apicomplexan and trypanosomatid parasitic protozoa. Mol Biochem Parasitol. 2003;126:129–142. doi: 10.1016/s0166-6851(02)00280-3. [DOI] [PubMed] [Google Scholar]
- Rodriguez RJ, Parks LW. Physiological response of Saccharomyces cerevisiae to 15-azasteroid-mediated growth inhibition. Antimicrob Agents Chemotherap. 1981;20:184–189. doi: 10.1128/aac.20.2.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardari S, Mori Y, Kurosawa T, Daneshtalab M. Can J Microbiol. 2003;49:344–349. doi: 10.1139/w03-045. [DOI] [PubMed] [Google Scholar]
- Trzaskos JM, Magolda RL, Favata MF, Fischer RT, Johnson PR, Chen HW, Ko SS, Leonard DA, Gaylor JL. Modulation of 3-hydroxy-3- methylglutaryl-CoA reductase by 15α-fluorolanst-7-en-3β-ol. J Biol Chem. 1993;268:22591–22599. [PubMed] [Google Scholar]
- Van Den Abbeele J, Claes Y, Van Bockstacle D, Le Ray D, Coosemans M. Trypanosoma brucei spp. Development in the tsetse fly: characterization of the post-mesocyclic stages in the foregut and proboscis. Parasitol. 1999;118:469–478. doi: 10.1017/s0031182099004217. [DOI] [PubMed] [Google Scholar]
- Walsh CT. Suicide substrates, mechanism-based enzyme inactivators: Recent Developments. Annu Rev Biochem. 1984;53:494–535. doi: 10.1146/annurev.bi.53.070184.002425. [DOI] [PubMed] [Google Scholar]
- Wang CC. A novel suicide inhibitor strategy for antiparasitic drug development. J Cell Biochem. 1991;45:49–53. doi: 10.1002/jcb.240450111. [DOI] [PubMed] [Google Scholar]
- Wang J, Liu J, Song Z, Nes WD. Sterol C24-methyltransferase: mechanistic studies of the C-methylation reaction with 24-fluorocycloartenol. Bioorg Med Chem Lett. 2008;18:232–235. doi: 10.1016/j.bmcl.2007.10.089. [DOI] [PubMed] [Google Scholar]
- World Health Organization, Trypanosomiasis, human African sleeping sickness. [July, 14 2013]; http://www.who.int/trypanosomiasis_african/en/index.html.
- Xu S, Norton RA, Crumley FG, Nes WD. Comparison of the chromatographic properties of sterols, select additional steroids and triterpenoids: Gravity-flow liquid chromatography, thin-layer chromatography, gas-liquid chromatography, and high performance liquid chromatography. J Chromatogr. 1988;452:377–398. doi: 10.1016/s0021-9673(01)81462-x. [DOI] [PubMed] [Google Scholar]
- Zhou W, Lepesheva GI, Waterman MR, Nes WD. Mechanistic analysis of a multiple product sterol methyltransferase implicated in ergosterol biosynthesis in Trypanosoma brucei. J Biol Chem. 2006;281:6290–6296. doi: 10.1074/jbc.M511749200. [DOI] [PubMed] [Google Scholar]
- Zhou W, Cross AM, Nes WD. Cholesterol import fails to prevent catalyst-based inhibition of ergosterol synthesis and cell proliferation of Trypanosoma brucei. J Lipid Res. 2007;48:665–673. doi: 10.1194/jlr.M600404-JLR200. [DOI] [PubMed] [Google Scholar]
- Zundel M, Nambiar KP, Boswell G, Bloch K. 6-Fluorocholesterol as a growth factor for the yeast mutant GL7. Biochemistry. 1989;28:5161–5164. doi: 10.1021/bi00438a037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental information includes a set of chromatographic data and mass spectra for representative standards and compounds isolated from treated cells, a description of chemical synthesis and characterization of 26-fluorolanosterol and 26-fluorocholesta- 5,7,24-trienol, a description of activity assays and product analysis using cloned TbSMT and can be found with this article online.