

Abstract
Clinical percutaneous delivery of synthetically engineered hydrogels remains limited due to challenges posed by crosslinking kinetics – too fast leads to delivery failure, too slow limits material retention. To overcome this challenge, we exploit supramolecular assembly to localize hydrogels at the injection site and introduce subsequent covalent crosslinking to control final material properties. Supramolecular gels were designed through the separate pendant modifications of hyaluronic acid (HA) by the guest-host pair cyclodextrin and adamantane, enabling shear-thinning injection and high target site retention (>98%). Secondary covalent crosslinking occurred via addition of thiols and Michael-acceptors (i.e., methacrylates, acrylates, vinyl sulfones) on HA and increased hydrogel moduli (E=25.0±4.5kPa) and stability (>3.5 fold in vivo at 28 days). Application of the dual-crosslinking hydrogel to a myocardial infarct model showed improved outcomes relative to untreated and supramolecular hydrogel alone controls, demonstrating its potential in a range of applications where the precise delivery of hydrogels with tunable properties is desired.
Keywords: hydrogel, injectable, supramolecular assembly, Michael-addition, hyaluronic acid
1. Introduction
Hydrogels are water-swollen polymeric networks that are used widely in biomedical applications as scaffolds for tissue reconstruction and regeneration or as delivery vehicles for cells, pharmaceuticals, or other cargo. Injectable hydrogels hold particular value in translational medicine as they may be implanted with minimally invasive methods.[1–5] To accomplish this, in vivo self-assembly either through physical or covalent mechanisms is often employed. Unfortunately, neither of these mechanisms is without issue for widespread use in biomedical applications.
Hydrogels assembled by physical mechanisms may be formed from numerous materials and interactions. These include biologically derived materials such as alginate, fibrin, gelatin, Matrigel or decellularized extracellular matrix;[2–4, 6, 7] however, these systems are limited in application, due to a high degree of batch-to-batch variability and minimal control over important material properties (e.g., mechanics and degradation). Thermoresponsive hydrogels, such as poly(N-isopropyl acrylamide), block copolymers, and polymer blends[8, 9] often display rapid sol-gel transition on injection to aid in localized retention.[10] However, these systems may not be suitable for percutaneous delivery (e.g., catheters) where materials are subject to necessary prolonged exposure at 37°C within the catheter, prior to injection. Under these conditions, the sol-gel transition may occur within the device and prevent desired delivery. Synthetic hydrogels including self-assembling peptides and α-cyclodextrin/PEG pseudopolyrotaxanes have also been extensively investigated, as discussed in a recent review.[1–3, 7] These systems are synthetically well defined and are stable toward external stimuli, such as temperature. However, they require potentially slow formation of higher order assemblies such as entanglements or microcrystalline domains in order to recover mechanical strength, compromising material retention at the target site. Moreover, extension of these systems to include end capped polyrotaxanes has led to highly elastic and even thermoresponsive systems.[11] Yet, extension of these systems to include in vivo formation has yet to be demonstrated.
As an alternative to these systems, supramolecular self-assembly based on the direct association of molecular components has recently emerged as a means of preparing hydrogels through specific, non-covalent crosslinks. Such assembly produce inherently dynamic networks that enable shear-thinning and self-healing properties, and thus injectability.[12] Both binary and ternary associating systems, such as those based on heterodimeric peptide/protein[13, 14] or host macrocycle interactions,[15, 16] have demonstrated shear-thinning abilities in conjunction with rapid network recovery. Similarly, cationically terminated linear or dendritic binders in conjunction with clay nanosheets have been used to form shear-thinning and self-healing nanocomposites through ionic interactions.[17] Such recovery characteristics may aid in retention at the injection site,[9, 18, 19] particularly for tissues that are under mechanical stress (e.g., nucleus pulpous) or those that undergo continual dynamic motion (e.g., cardiac tissue). Unfortunately, these materials are inherently limited in that they typically exhibit low mechanical strength[1, 12] and may exhibit rapid erosion dependent on the valence of crosslinking groups and their affinity.[1, 14, 20]
For more physically demanding applications, covalently crosslinked systems may be more appropriate. In addition to increased relative mechanical strength, hydrogels formed through covalent means display great versatility with the allowed inclusion of controlled network degradation and the introduction of biological cues such as cell adhesion and bioactive factor delivery. Indeed, numerous chemical mechanisms have been employed for in vivo crosslinking including redox-initiated[21] and externally triggered[22, 23] radical polymerizations, as well as various addition reactions including Schiff-base, Michael-addition, and Huisgen cycloaddition chemistries.[3] Michael-addition reactions remain prominent in the field, due in part to mild reaction conditions, bioorthogonal mechanisms, and readily tailorable reaction kinetics.[24] These properties are highly beneficial toward injectable applications where the hydrogel must form in vivo without cross-reactivity (e.g., with protein amine residues) or other biological consequence (e.g., exothermic necrosis). The ability to control crosslinking kinetics is also of utmost importance, as clinical procedures may require the hydrogel to remain in an injectable state for an hour or more, making rapid crosslinking reactions unsuitable. For ease of clinical application, the hydrogel must therefore undergo crosslinking with slow reaction kinetics to prevent premature crosslinking and delivery failure. While such slow reaction kinetics may allow delivery, it is realized that slow crosslinking results in undesirable loss of material from the injection site.[18, 25] Thus, there is basic design flaw with currently developed hydrogels for applications as an injectable material.
To address these inherent limitations of current injectable hydrogel systems, a generally applicable dual-crosslinking mechanism is developed herein. The hydrogels first undergo physical assembly ex vivo through supramolecular self-assembly mediated by guest (adamantine, Ad) and host (β-cyclodextrin, CD) pendant groups. This mechanism enables shear-thinning delivery with high retention at the target site. Following injection, a secondary covalent crosslinking occurs in situ via Michael-addition to stabilize the network. Crosslinking kinetics of this secondary network are clinically appropriate and controlled through both the Michael-acceptor reactivity and catalytic conditions. The novel combination of autonomous physical and covalent crosslinking demonstrated is a generally applicable method for retention and subsequent reinforcement of injectable materials amenable to clinical application and may serve as a platform for numerous ventures in bioengineering and regenerative medicine. To demonstrate this, the materials developed are utilized in a mechanical stabilization approach for treatment of myocardial infarctions.
2. Results and Discussion
2.1. Guest-host hydrogels for shear-thinning delivery and target site retention
β-Cyclodextrin is a macrocycle composed of 7 D-glucose units arranges in a toroidal fashion through 1,4-glucosidic bonds, enabling it to include a range of hydrophobic molecules in its interior cavity.[26] Of these potential guest molecules, adamantane is widely regarded as having one of the greatest affinities (Ka≈105 M−1)[12] due both to its hydrophobicity and complementary size. Together, the molecules form a guest-host pair known to interact in a one-to-one fashion upon mixing in aqueous conditions (Figure 1a). Hyaluronic acid (HA) is a linear polysaccharide found in native ECM and is well suited to applications in translational medicine, as it plays a role in embryogenesis, angiogenesis, cell migration and scar reduction.[27] Importantly, HA also provides carboxyl and hydroxyl functionalities, useful as reactive handles for ease of modular chemical modification. For assembly of guest-host (GH) hydrogels, HA was modified (Figure 1b) either by coupling of 1-adamantane acetic acid via esterification (guest, Ad-HA) or aminated β-cyclodextrin via amidation (host, CD-HA) as previously described.[15]
Figure 1.

Guest-host hydrogel formation, enabling shear-thinning injection and hydrogel retention. a) Interaction of adamantane (Ad, guest) and β-cyclodextrin (CD, host) in formation of a reversible guest-host (GH) complex crosslink, and corresponding synthesis (b) for specific guest (Ad-HA) and host (CD-HA) macromers. c) Schematic of supramolecular hydrogel formation utilizing guest-host complexation. d) Oscillatory time sweeps of individual macromers and hydrogel formed at 3.5wt%; storage modulus (G’, filled symbols) and loss modulus (G’’, empty symbols) at 1.0 Hz, 1.0% strain. e–f) Shear-thinning and recovery characterization, demonstrating shear yielding behavior at high strain (e) for GH hydrogel (3.5wt%) and corresponding recovery under repeated deformation (f) of 1.0 (low) and 100% strain (high, shaded) at 10Hz. g) Short axis MRI cross-section of an explanted porcine heart showing retention of injected hydrogels (left, injection sites indicated), with corresponding 3D reconstruction (right) of nine 300µL GH hydrogel injections (purple) in a porcine whole heart explant (red); imaged at 3T.
Guest-host complexation drives supramolecular assembly of a physically crosslinked hydrogel. Owing to the linear polymer architecture and pendant functionality of the guest and host groups, macromer components self-assemble through the binary complexes, which act cooperatively to result in a net avidity between polymer chains.[15, 28] These interactions were demonstrated by rheological measurements, showing that individual macromers were viscous solutions and mixing resulted in a hydrogel composed of non-covalent bonds (Figure 1c,d). Owing to the reversible nature of the guest-host complex, the networks formed are dynamic. Hydrogels thus displayed stress relaxation at low frequency, indicative of bond restructuring (Figure S1); the relaxation behavior of these networks was previously characterized.[15]
As a result of the dynamic bonding structure, hydrogels are capable of shear-induced flow and rapid recovery. In particular, GH hydrogels exhibited yielding behavior and transition to liquid-like flow under high strain (>35%, Figure 1e) and displayed characteristic shear-thinning behavior under continuous flow conditions (Figure S2). Owing to the high valence of polymer modifications (>40 groups per HA chain) and moderate association constant of the guest-host complex, near-instantaneous reassembly of the networks was exhibited with >85% recovery of G’ within 3s following shear-thinning (Figure 1f). As a direct result of these flow and recovery characteristics, GH hydrogels are easily injectable, self-healing, and readily re-form even upon injection into aqueous media (Movie S1, S2, and S3).
Finally, we sought to demonstrate the capacity of GH hydrogels for injection site retention. Towards this, magnetic resonance imaging (MRI) has been useful to image HA hydrogels, as T2 relaxation behavior of the multiple hydroxyl moieties enables ease of imaging both in vitro and in vivo.[22, 29] For this investigation, hydrogels were injected into explanted porcine myocardium and subjected to T2 weighted MRI, allowing visualization of material in situ. The pattern of nine injections was observed both in the tissue cross-section and corresponding 3D reconstruction (Figure 1g). Hydrogel injections were quantitatively evaluated at higher field strength and resolution to evaluate retained volumes and morphology over time, demonstrating GH hydrogels were initially well retained (>98% in all cases), and the hydrogel retention was independent of injection volume (50–300µL examined; Figure S3). Hydrogels were sustained within the tissue for greater than one week in vitro and exhibited minimal morphological changes over time with the exception of modest swelling (Figure S4 & Table S1). In summary, the guest-host hydrogel mechanism developed affords a supramolecular hydrogel through dynamic binary guest-host associations. The GH hydrogel is capable of shear-yielding and rapid mechanical recovery, thus enabling injectable delivery with high target site retention.
2.2 Dual-crosslinking hydrogels with controlled Michael-addition kinetics for injectable delivery
Michael-addition (MA) crosslinking (Figure 2a) occurs through the condensation of a nucleophile, such as a thiol, and an activated olefin. Common thiolated small molecule crosslinkers[30] were avoided in material design, opting rather for the modification of HA macromers to limit diffusion from the reaction site. Initial attempts were made to functionalize HA with thiols (HA-SH) via cystamine amidation; however, the product could not be isolated in soluble form following disulfide bond reduction and lyophilization. This is presumably a result of a decreased pKa of the thiol due to the regional chemical structure,[31] leading to accelarated generation of the thiolate anion and resulting interchain disulfide bond formation. HA was thus modified by esterification of HA with 3,3’-dithiodipropionic acid followed by reduction with DTT. The product was isolated by precipitation from EtOH and subsequent lyophilization from acidified water (HCl, pH 3.5). The degree of HA modification, determined by 1H-NMR, demonstrated complete reduction of the disulfide bond with no change in modification before and after reduction. Moreover, GPC analysis demonstrated that HA-SH was recovered without degradation of the polymer or formation of interchain disulfide bonds (Supplemental Methods).
Figure 2.
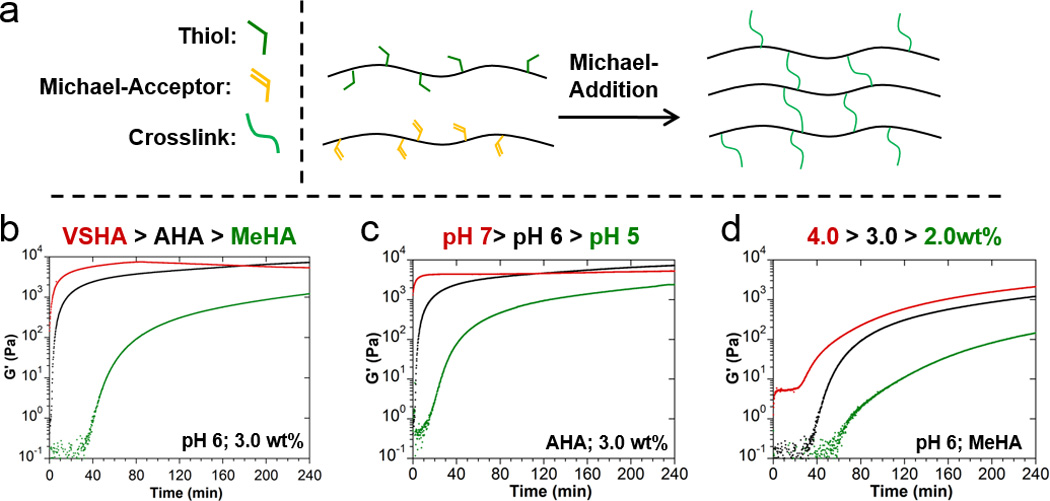
Michael-Addition hydrogel formation, tuning of crosslinking kinetics. a) Schematic of covalent crosslink formation via generalized Michael-addition reaction between a thiol-modified HA macromer and Michael-acceptor (VSHA: vinyl sulfone HA, AHA: acrylated HA, MeHA: methacrylated HA). b–d) Real-time rheological observation (storage modulus (G’), 1.0 Hz, 1.0% strain) of Michael-addition crosslinking with variations in the Michael-acceptor reactivity (b), pH (c), and macromer concentration (d).
Reactants and reaction conditions were directly investigated through real-time rheological observation to identify conditions with a range of gelation times from minutes to hours. As addition reaction kinetics are highly dependent on the structure and electrophilic activation of the Michael-acceptor, various Michael-acceptor modifications of HA were performed including the methacrylate (MeHA), acrylate (AHA), vinyl sulfone (VSHA), and maleimide (MaHA) derivatives. Reaction pH was also considered, as the thiolated anion (generated via deprotonation in basic conditions) is the reactive species.[24, 31] Gel times were observed to decrease with increasing reactivity of the Michael-acceptor (Figure 2b), pH (Figure 2c), and polymer concentration (Figure 2d). These trends were consistent across the entirety of the parameter space investigated (Figure S5). From these results, MeHA was determined to be a viable Michael-acceptor for clinical use, as it exhibited gel times greater than 45 minutes if the polymer concentration and pH were properly controlled. Based on these trials in conjunction with guest-host hydrogel behavior, we selected hydrogels of approximately 3.5wt% as optimal for pursuit of subsequent in vivo applications.
To allow combination of physical and covalent crosslinking mechanisms for dual-crosslinking (DC) hydrogels (Figure 3c), thiolated Ad-HA and methacrylated CD-HA were prepared by modular, sequential synthesis (Figure 3b). The degree of HA modification was maintained at approximately 20% for all groups, and sequential reactions were observed to be independent of prior modifications. Despite the secondary polymer modification for Michael-addition, GH hydrogels retained their native mechanical properties prior to liberation of the reactive mercapto group by reduction with DTT (Figure S6) and were qualitatively observed to have similar injectable properties. To demonstrate the necessity and efficacy of the dual-crosslinking approach toward retention, hydrogel injections were performed into explanted porcine myocardium followed by washing in PBS for 24 hours. MRI imaging (Figure 3d) showed that MA gels alone did not have sufficient initial mechanics for retention, even in a stationary tissue explant. Such a crosslinking approach is therefore insufficient for therapeutic use, despite its suitability for percutaneous injection. Conversely, the GH and DC gels were both well retained at the injection site. Rheological time sweeps show that upon initial formation, DC hydrogels have moduli similar to that of GH hydrogels, yet subsequent crosslinking via the desired Michael-addition resulted in a more rigid viscoelastic solid with increased shear modulus (Figure 3e). Following this covalent crosslinking step, cessation of flow and bulk relaxation behaviors was observed as evidenced by oscillatory frequency sweeps demonstrating the loss of bulk relaxation behavior at low frequency (Figure S7). Allowing crosslinking to proceed to completion, hydrogels with compressive moduli of 25.0±4.5kPa were obtained, whereas GH hydrogels did not have sufficient mechanics for unconfined compression testing. In sum, secondary modification of HA to allow Michael-addition was accomplished in a modular fashion, allowing in situ stabilization without compromising the injectable behavior of the GH gel.
Figure 3.

Dual-Crosslinking hydrogel formation, retention, and altered biophysical properties. a) Addition of thiol and methacrylate in formation of a covalent crosslink (a), and corresponding synthesis (b) of guest Michael-donor (Ad-HA-SH) and host Michael-acceptor (CD-MeHA) macromers. c) Schematic of dual-crosslinking (DC) hydrogel formation. d) MRI cross-section of explanted porcine myocardium injected with the Michael-addition (MA), guest-host (GH), and dual-crosslinking (DC) hydrogels; imaged at 9.4T (for interpretation in color as well as histological confirmation, the reader is referred to Figure S8 and S9). e) Oscillatory time sweeps (1.0 Hz, 1.0% strain) of GH and DC hydrogels immediately after mixing. f) Cumulative in vitro erosion profiles (mean±SD; n=3) for GH and DC hydrogels.
In addition to modification of the hydrogel stiffness, secondary covalent crosslinking is expected to alter hydrogel degradation. Physically crosslinked hydrogels, including the GH system, undergo mass loss dominated by surface erosion through stochastically governed network disassembly.[1, 14, 15] Rapid erosion and release of encapsulated cargo, often on the order of hours to days, is exhibited and known to be dependent on the affinity of the heterodimeric interactions and their valence.[1, 14, 20, 32] The GH hydrogel system demonstrated surprising stability toward erosion in vitro (65.6±1.9% remaining at day 28), likely owing to the high valence of functional groups. Mass loss, however, was relatively rapid as compared to the DC hydrogels, which exhibited a significant reductions in mass loss at all observed time points beyond day 3 (Figure 3f). For DC hydrogels, mass loss was still observed, likely resulting from macromer surface erosion preceded by the expected slow hydrolysis of thioether-ester bonds formed through Michael-addition.[33]
As established in vitro, the material systems described are intended to afford physically crosslinked hydrogels with injectable capacity and optional in vivo stabilization through covalent dual-crosslinking. However, in vitro degradation is not always a reliable predictor of in vivo behavior.[34] Our lab and others have shown that near-IR fluorescent labeling has great utility in tracking material degradation and release of biological factors in vivo.[35, 36] To examine the potential for crosslinking and degradation in vivo, hydrogels were injected subcutaneously in mice (Figure 4a). An innovative approach was used to provide fluorescence using a custom peptide sequence (GCKKG-Cy7.5). Standard solid phase peptide synthesis allowed preparation of the fluorescent peptide in high purity, where the amine terminus was capped by the free acid of Cyanine 7.5 during the synthesis. Despite harsh reaction conditions, absorbance and emission spectra demonstrated the expected fluorescence properties (λmax abs/em = 800/808 nm). Subsequent reaction with CD-MeHA under basic conditions enabled fascicle near-IR labeling of the macromer used to form the hydrogel. Upon injection, a visible gel formed under the tissue, and degradation was monitored over 4 weeks (Figure 4a,b). Over the time course, GH gels approached complete degradation, while DC gels remained as visible and palpable solids until the experiment endpoint (Figure S10). Quantification of fluorescence intensity (Figure 4c) again showed significant differences between groups for all time points beyond day 3. Results are in qualitative agreement with in vitro degradation studies and demonstrate the capacity of the material systems toward injectable delivery as well as the ability to implement secondary crosslinking in vivo. In sum, both in vitro and in vivo results clearly demonstrate the ability of the materials to form GH hydrogels that flow for injectable delivery, rapidly recover for retention at the target site, and which undergo optional secondary covalent crosslinking to modulate their biophysical properties.
Figure 4.

In vivo degradation behavior modulated by dual-crosslinking. a–c) Near-IR fluorescence imaging of guest-host (GH) and dual-crosslinking (DC) hydrogels after injection subcutaneously in the right flank of mice (a) and monitored over 28 days (b: fluorescent images, c: normalized signal intensity (mean±SEM; n=3)). d–f) Epicardial injection and retention in a rat infarct model. Injections were performed in the infarct border (d: schematic, e: visualization immediately after injection). f) Fluorescence imaging of hearts explanted at the terminal time point (top) and quantification of fluorescence signal intensity (bottom, (mean±SEM; n>5)).
2.3 Therapeutic potential: mechanical stabilization of myocardial infarct
To demonstrate the therapeutic potential of the material systems developed, they were investigated in the context of a preventative treatment for heart failure (HF) resultant from myocardial infarction (MI). Heart failure is a clinical condition in which the contractile functions of the heart have been impaired to a level where normal bodily functions are compromised.[37] In nearly 70% of cases, the condition has been attributed to maladaptive left ventricular (LV) remodeling following myocardial infarction (MI),[38] making this the greatest cause of cardiovascular related death.[39] Extensive work has shown these remodeling events are a result of initial expansion of the non-contractile and compliant infarct, altering normal stress distributions and precipitating progressive remodeling.[37, 40] DC hydrogels exhibit compressive moduli (25.0±4.5kPa) greater than that of native myocardium,[41] and comparable to that of materials shown to have efficacy in attenuating remodeling through mechanical stabilization of the infarct.[21, 29, 41, 42] A combination of experimental and computational approaches has recently demonstrated that this may be due to reductions in the local and global myofiber stresses, related to anisotropic stiffening of the tissue.[29] GH hydrogels are likewise investigated, as soft material injections have shown some efficacy in the treatment of MI,[43] though they may be limited in their ability to mechanically restrain the infarct to resist initial expansion. Due to the inability of MA hydrogels to be retained at the injection site in vitro, they were excluded from in vitro studies. We used a rodent model of MI to establish the efficacy of these material platforms in attenuating LV remodeling, as small animal models allow for relatively large sample size and thus reliable data analysis.[44]
Adult male Wistar rats underwent permanent ligation of the left anterior descending artery (LAD) to induce MI, and a series of injections (6–8; 75uL total per animal) was performed into the myocardium in the infract border zone region (Figure 4d). Importantly, both GH and DC hydrogels were observed to be well retained at the injection site despite the forces of myocardial contraction (Figure 4e) with continued retention at 1 day observed histologically (Figure S12). With respect to long-term retention, near-IR imaging of the explanted tissue (Figure 4f) shows diffuse fluorescence throughout the tissue for the GH hydrogel group, whereas DC hydrogels remain localized, even at 4 weeks post-MI, with a nearly 4-fold greater net retention. MI and sham groups showed similar, negligible background fluorescence indicating that the signal is not dependent on inflammation or remodeling of the myocardial tissue.
Histological, geometric, and functional outcomes were evaluated at 4 weeks post-MI to assess the efficacy of treatments, with complete analysis provided in Table S2. Treatment of MI by hydrogel injections resulted in desirable modulation of the tissue response. In particular, mechanical stabilization of the infarct is expected to protect the border zone from altered stress distributions and thereby reduce the infarct size. Histological evaluation revealed a trend toward reduction in the infarct size for DC treatment relative to GH, as quantified by fibrotic area (Figure 5a, Table S2). Importantly, a significant increase in border zone vascular density, including both capillary and arteriole densities, was observed for the DC relative to GH treatments (Figure S13). Histological analysis is in agreement with prior observations of border zone protection by infarct stabilization and the angiogenic potential of HA.[21, 36, 45] Progressive remodeling post-MI is also known to result in dilation of the left ventricle, negatively affecting its contractile function. Echocardiographic measurement of the LV inner diameter at end systole (LVIDs, Figure 5b) and end diastole (LVIDd) showed significantly attenuated LV dilation relative to MI control for both the GH and DC treatment groups. In sum, histological and geometric outcomes demonstrate attenuation of infarct expansion through protection of the border zone and resultant prevention of LV dilation.
Figure 5.

Histological, geometric, and functional outcomes after hydrogel injection. a) Histological cross-sections of myocardial infarction controls (MI) or after injection of either guest-host (GH) or dual-crosslinking (DC) hydrogels, demonstrating reduced scar formation (purple color in images) with hydrogel treatment at 28 days post-infarction. b–f) Geometric and functional outcomes of LV inner diameter at end systole (b), end-systolic pressure volume relationship (c) fractional shortening (d), ejection fraction (e), and cardiac output (f) for MI and sham controls, as well as after treatment with GH and DC hydrogels. Values represent mean±SEM, with sham values indicated by the shaded regions.
Continued contractile function of the LV is, however, the ultimate measure of treatment efficacy. The end-systolic pressure volume relationship (ESPVR) is a measure of contractility, valuable in hemodynamic analysis due to its independence from load and heart rate.[46] Determination of ESPVR (Figure S14) showed a trend toward increased contractility for both material treatments, but was significant only for DC hydrogels. Examination of functional outcomes, including fractional shortening (Figure 5d), ejection fraction (Figure 5e), and cardiac output (Figure 5f) likewise showed improved function over MI in both treatment groups, all with a consistent trend for stabilization by DC hydrogels to improve outcomes relative to GH. Moreover, the functional assessments of DC hydrogel treatments approached those of sham controls and were not statistically different for any measure performed (p>0.28), with the exception of ejection fraction, which was improved over MI but was statistically different than sham controls. Analysis of outcomes demonstrates the attenuation of adverse LV remodeling for both GH and DC hydrogels, with distinct histological and functional improvement afforded DC hydrogel treatment capable of mechanically stabilizing the infarct border zone.
3. Conclusion
In conclusion, the material system developed harnesses the beneficial features of both supramolecular assembly and addition reaction crosslinking in an orthogonal and complementary fashion to create a materials system with unique properties. Namely, the utilization of a physically crosslinked hydrogel enables initial retention at the injection site through shear-thinning delivery. This retention ability facilitates the employment of slow covalent crosslinking for stabilization in vivo, occurring on timescales that enable ease of clinical application. In recent years, there has been a significant drive toward percutaneous delivery of materials for both cosmetic and therapeutic purposes, with significant attention given to material retention as well as appropriate control of biophysical properties. Indeed, the material concept developed herein is a generalizable approach which addresses a fundamental challenge in percutaneous delivery: the ability to maintain hydrogel presence despite clinically required slow covalent crosslinking. The approach may be of great utility in many applications, including soft tissue reconstruction as well as toward therapeutic needs including nucleus pulpous replacement or treatment of MI.
4. Experimental Section
Hyaluronic Acid Macromer Modification
Guest and host macromers were prepared by methods previously described.[15] Briefly, adamantane modified hyaluronic acid (Ad-HA) was prepared by coupling 1-adamantane acetic acid to HA-TBA via BOC2O/DMAP mediated esterification. Cyclodextrin modified HA (CD-HA) was prepared by coupling β-CD-HDA to HA-TBA through amidation reaction with BOP. Thiolated hyaluronic acid (HA-SH) was prepared by analogous esterification of HA with 3,3’-dithiopropionic acid and subsequent reduction by DTT. Methacrylated HA (MeHA) was prepared by reaction of HA with methacrylic anhydride, similar to previous reports. Dual-crosslinking macromers were formed by sequential combination of these methods. Detailed syntheses of all compounds are provided in the Supplementary Methods.
Hydrogel Formation
For all studies, hydrogels were prepared from stock solutions of the individual macromers in PBS at the desired concentration. For hydrogel formation by guest-host assembly, the two component solutions were combined and mixed by manually stirring to ensure a homogenous hydrogel then centrifuged to remove entrapped air. Hydrogels were formulated such that adamantane and β-cyclodextrin were present in stoichiometric balance, and the concentration refers to the overall weight percent of combined macromers. Formation of hydrogels via the Michael-addition and dual-crosslinking mechanisms was similarly performed, with the pH of dissolution buffers adjusted to obtain the desired pH.
Mechanical Characterization
Rheological characterization was performed using an AR2000 stress-controlled rheometer (TA Instruments) fitted with a 20 mm diameter cone and plate geometry, 59 min 42 s cone angle, and 27 µm gap. Rheological properties were examined at 37 °C by oscillatory frequency sweeps (0.01–100 Hz; 1% strain), time sweeps (1.0; 1% strain), strain sweeps (10 Hz; 0.05–250% strain), and continuous flow experiments (linearly ramped: 0–0.5 s−1 and returned). For shear recovery experiments, shear-thinning was performed at 100% strain with recovery at 1.0% strain, each at 10 Hz. Compressive mechanics were performed on samples (n=3) following crosslinking at pH 8.0 overnight at 37 °C with a dynamic mechanical analyzer (DMA, Q800 TA Instruments) at a strain rate of 10%/min. Moduli were calculated at a strain of 10–20%.
MRI Imaging and Analysis
Cardiac explants were imaged (conditions and pulse sequences detailed in Supplemental Methods) using either a 3T or 9.4T MRI Scanner (Siemens). Images were converted into NIFTI format using ImageJ software. Reconstruction was carried out in ITK-SNAP by automated segmentation with manual edge correction, allowing three-dimensional reconstruction and quantification of retained hydrogel volumes.
In Vitro Degradation
Acrylamide molds were used to contain the hydrogels within a 5 mm diameter 6 mm high depression. Hydrogels were prepared as described and 30 µL of the desired hydrogel loaded into the depression. Hydrogels were covered with 1 mL PBS and allowed to erode at 37°C. The buffer was replaced twice weekly. Following 28 days, hydrogels were degraded in 1mg/mL hyaluronidase to allow determination of remaining hydrogel content necessary for data normalization. Macromer release was quantified by uronic acid assay.
In Vivo Degradation
To allow for degradation to be directly observed in vivo, CD-MeHA was labeled by the near-IR dye Cy7.5 through preparation of the peptide GCKKG-Cy7.5 and subsequent Michael-addition of the peptide to form the derivatized HA macromer (detailed in Supplementary Methods). After injection (25µL), degradation was assessed biweekly over 4 weeks (Pearl Impulse, LI-COR). The fluorescence signal (λex/λem=785/820 nm) was integrated over identically sized regions of interest centered over the injection site for all animals. Values were baseline corrected to images of mice prior to injection (n=11), and normalized to the peak intensity for the material group.
Myocardial Infarct Model
MI was induced in adult male Wistar rats using an established and highly reproducible model. Briefly, under anesthesia and with mechanical ventilation, a left thoracotomy was performed to expose the heart. Suture ligation of the left anterior descending (LAD) coronary artery afforded an anterolateral infarct comprising ~20% of the left ventricular wall, which was visually confirmed by color change resulting from myocardial ischemia.[36] The animals were randomized into 3 groups and received 6–8 separate intramyocardial injections (75 µL per animal) of saline (n=22), GH hydrogel (n=8), or DC hydrogel (n=8) into the border zone of the infarct. Sham procedures underwent saline injection without ligation. Animals were recovered and subject to endpoint analysis at 28 days post-infarct. Detailed methods are provided in Supplementary Methods.
Statistical Analysis
All data is reported as means ± standard error (SEM) or standard deviation (SD), for in vitro data. For degradation studies, comparison between groups was performed by Students t-test with two-tailed criteria and significance determined at p<0.05. For the infarct model, statistical significance was determined by one-way ANOVA with post hoc testing to compare between groups. Bonferroni correction was used to account for multiple comparisons, with α=0.05.
Supplementary Material
Acknowledgements
The authors thank Drs. W. Gramlich, C. Highley, and B. Purcell for helpful discussion concerning material synthesis and experimental design, L. Wang for assistance with rheological data collection, and R. Rai, J. Patel, and G. Hung for assistance with implementation and analysis of the animal models. This work was made possible by financial support from the National Institutes of Health (R01 HL111090, R01 HL089315) and a Predoctoral Fellowship (C.B.R) and Established Investigator Award (J.A.B.) from the American Heart Association.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Christopher B. Rodell, Department of Bioengineering, University of Pennsylvania, Philadelphia, Pennsylvania, 19104, United States
John W. MacArthur, Division of Cardiovascular Surgery, Department of Surgery, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, 19104, United States
Shauna M. Dorsey, Department of Bioengineering, University of Pennsylvania, Philadelphia, Pennsylvania, 19104, United States
Ryan J. Wade, Department of Bioengineering, University of Pennsylvania, Philadelphia, Pennsylvania, 19104, United States Department of Materials Science, University of Pennsylvania, Philadelphia, Pennsylvania, 19104, United States.
Leo L. Wang, Department of Bioengineering, University of Pennsylvania, Philadelphia, Pennsylvania, 19104, United States
Y. Joseph Woo, Department of Cardio-Thoracic Surgery, Stanford University School of Medicine, Stanford, California, 94305, United States.
Jason A. Burdick, Email: burdick2@seas.upenn.edu, Department of Bioengineering, University of Pennsylvania, Philadelphia, Pennsylvania, 19104, United States.
References
- 1.Guvendiren M, Lu HD, Burdick JA. Soft Matter. 2012;8:260. [Google Scholar]
- 2.Kretlow JD, Klouda L, Mikos AG. Adv. Drug Delivery Rev. 2007;59:263. doi: 10.1016/j.addr.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 3.Li YL, Rodrigues J, Tomas H. Chem. Soc. Rev. 2012;41:2193. doi: 10.1039/c1cs15203c. [DOI] [PubMed] [Google Scholar]
- 4.Yu L, Ding JD. Chem. Soc. Rev. 2008;37:1473. doi: 10.1039/b713009k. [DOI] [PubMed] [Google Scholar]
- 5.Ko DY, Shinde UP, Yeon B, Jeong B. Prog. Polym. Sci. 2013;38:672. [Google Scholar]
- 6.Badylak SF, Freytes DO, Gilbert TW. Acta Biomaterialia. 2009;5:1. doi: 10.1016/j.actbio.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 7.Yan CQ, Pochan DJ. Chem. Soc. Rev. 2010;39:3528. doi: 10.1039/b919449p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klouda L, Mikos AG. Eur. J. Pharm. Biopharm. 2008;68:34. doi: 10.1016/j.ejpb.2007.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gupta D, Tator CH, Shoichet MS. Biomaterials. 2006;27:2370. doi: 10.1016/j.biomaterials.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 10.Moon HJ, Ko DY, Park MH, Joo MK, Jeong B. Chem. Soc. Rev. 2012;41:4860. doi: 10.1039/c2cs35078e. [DOI] [PubMed] [Google Scholar]
- 11.Bin Imran A, Esaki K, Gotoh H, Seki T, Ito K, Sakai Y, Takeoka Y. Nat Commun. 2014;5 [Google Scholar]
- 12.Appel EA, del Barrio J, Loh XJ, Scherman OA. Chem. Soc. Rev. 2012;41:6195. doi: 10.1039/c2cs35264h. [DOI] [PubMed] [Google Scholar]
- 13.Foo C, Lee JS, Mulyasasmita W, Parisi-Amon A, Heilshorn SC. Proc. Natl. Acad. Sci. U. S. A. 2009;106:22067. doi: 10.1073/pnas.0904851106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu HD, Charati MB, Kim IL, Burdick JA. Biomaterials. 2012;33:2145. doi: 10.1016/j.biomaterials.2011.11.076. [DOI] [PubMed] [Google Scholar]
- 15.Rodell CB, Kaminski AL, Burdick JA. Biomacromolecules. 2013;14:4125. doi: 10.1021/bm401280z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Appel EA, Loh XJ, Jones ST, Biedermann F, Dreiss CA, Scherman OA. J. Am. Chem. Soc. 2012;134:11767. doi: 10.1021/ja3044568. [DOI] [PubMed] [Google Scholar]
- 17.Wang Q, Mynar JL, Yoshida M, Lee E, Lee M, Okuro K, Kinbara K, Aida T. Nature. 2010;463:339. doi: 10.1038/nature08693. [DOI] [PubMed] [Google Scholar]; Tamesue S, Ohtani M, Yamada K, Ishida Y, Spruell JM, Lynd NA, Hawker CJ, Aida T. J. Am. Chem. Soc. 2013;135:15650. doi: 10.1021/ja408547g. [DOI] [PubMed] [Google Scholar]
- 18.Lee F, Chung JE, Kurisawa M. J. Controlled Release. 2009;134:186. doi: 10.1016/j.jconrel.2008.11.028. [DOI] [PubMed] [Google Scholar]
- 19.Park KM, Yang JA, Jung H, Yeom J, Park JS, Park K-H, Hoffman AS, Hahn SK, Kim K. ACS Nano. 2012;6:2960. doi: 10.1021/nn204123p. [DOI] [PubMed] [Google Scholar]; Parisi-Amon A, Mulyasasmita W, Chung C, Heilshorn SC. Adv. Healthcare Mater. 2013;2:428. doi: 10.1002/adhm.201200293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu HD, Soranno DE, Rodell CB, Kim IL, Burdick JA. Adv. Healthcare Mater. 2013;2:1028. doi: 10.1002/adhm.201200343. [DOI] [PubMed] [Google Scholar]
- 21.Tous E, Ifkovits JL, Koomalsingh KJ, Shuto T, Soeda T, Kondo N, Gorman JH, Gorman RC, Burdick JA. Biomacromolecules. 2011;12:4127. doi: 10.1021/bm201198x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hillel AT, Unterman S, Nahas Z, Reid B, Coburn JM, Axelman J, Chae JJ, Guo Q, Trow R, Thomas A, Hou Z, Lichtsteiner S, Sutton D, Matheson C, Walker P, David N, Mori S, Taube JM, Elisseeff JH. Sci. Transl. Med. 2011;3:93ra67. doi: 10.1126/scitranslmed.3002331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gramlich WM, Holloway JL, Rai R, Burdick JA. Nanotechnology. 2014;25 doi: 10.1088/0957-4484/25/1/014004. [DOI] [PubMed] [Google Scholar]
- 24.Mather BD, Viswanathan K, Miller KM, Long TE. Prog. Polym. Sci. 2006;31:487. [Google Scholar]
- 25.Martens TP, Godier AFG, Parks JJ, Wan LQ, Koeckert MS, Eng GM, Hudson BI, Sherman W, Vunjak-Novakovic G. Cell Transplant. 2009;18:297. doi: 10.3727/096368909788534915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szejtli J. Chem. Rev. 1998;98:1743. doi: 10.1021/cr970022c. [DOI] [PubMed] [Google Scholar]
- 27.Burdick JA, Prestwich GD. Adv. Mater. 2011;23:H41. doi: 10.1002/adma.201003963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Semenov A, Charlot A, Auzely-Velty R, Rinaudo M. Rheol. Acta. 2007;46:541. [Google Scholar]
- 29.Kichula ET, Wang H, Dorsey SM, Szczesny SE, Elliott DM, Burdick JA, Wenk JF. Ann Biomed Eng. 2014;7:1546. doi: 10.1007/s10439-013-0937-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoyle CE, Bowman CN. Angewandte Chemie-International Edition. 2010;49:1540. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 31.Lutolf MP, Tirelli N, Cerritelli S, Cavalli L, Hubbell JA. Bioconjugate Chem. 2001;12:1051. doi: 10.1021/bc015519e. [DOI] [PubMed] [Google Scholar]
- 32.van de Manakker F, Braeckmans K, el Morabit N, De Smedt SC, van Nostrum CF, Hennink WE. Adv. Funct. Mater. 2009;19:2992. [Google Scholar]
- 33.Schoenmakers RG, van de Wetering P, Elbert DL, Hubbell JA. J. Controlled Release. 2004;95:291. doi: 10.1016/j.jconrel.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 34.Bruggeman JP, de Bruin BJ, Bettinger CJ, Langer R. Biomaterials. 2008;29:4726. doi: 10.1016/j.biomaterials.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]; Grayson ACR, Voskerician G, Lynn A, Anderson JM, Cima MJ, Langer R. J. Biomater. Sci., Polym. Ed. 2004;15:1281. doi: 10.1163/1568562041959991. [DOI] [PubMed] [Google Scholar]
- 35.Soranno DE, Lu HD, Weber HM, Rai R, Burdick JA. J Biomed Mater Res A. 2014;102:2173. doi: 10.1002/jbm.a.34902. [DOI] [PMC free article] [PubMed] [Google Scholar]; Artzi N, Oliva N, Puron C, Shitreet S, Artzi S, Ramos AB, Groothuis A, Sahagian G, Edelman ER. Nat. Mater. 2011;10:704. doi: 10.1038/nmat3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacArthur JW, Purcell BP, Shudo Y, Cohen JE, Fairman A, Trubelja A, Patel J, Hsiao P, Yang E, Lloyd K, Hiesinger W, Atluri P, Burdick JA, Woo YJ. Circulation. 2013;128:S79. doi: 10.1161/CIRCULATIONAHA.112.000343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jessup M, Brozena S. N. Engl. J. Med. 2003;348:2007. doi: 10.1056/NEJMra021498. [DOI] [PubMed] [Google Scholar]
- 38.Gheorghiade M, Bonow RO. Circulation. 1998;97:282. doi: 10.1161/01.cir.97.3.282. [DOI] [PubMed] [Google Scholar]
- 39.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. C. Amer Heart Assoc Stat, S. Stroke Stat, Circulation. 2012;125:E2. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holmes JW, Borg TK, Covell JW. Annu. Rev. Biomed. Eng. 2005;7:223. doi: 10.1146/annurev.bioeng.7.060804.100453. [DOI] [PubMed] [Google Scholar]; Gorman RC, Jackson BM, Burdick JA, Gorman JH. J Cardiovasc Transl. 2011;4:73. doi: 10.1007/s12265-010-9244-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ifkovits JL, Tous E, Minakawa M, Morita M, Robb JD, Koomalsingh KJ, Gorman JH, Gorman RC, Burdick JA. Proc. Natl. Acad. Sci. U. S. A. 2010;107:11507. doi: 10.1073/pnas.1004097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wall ST, Walker JC, Healy KE, Ratcliffe MB, Guccione JM. Circulation. 2006;114:2627. doi: 10.1161/CIRCULATIONAHA.106.657270. [DOI] [PubMed] [Google Scholar]
- 43.Davis ME, Motion JPM, Narmoneva DA, Takahashi T, Hakuno D, Kamm RD, Zhang SG, Lee RT. Circulation. 2005;111:442. doi: 10.1161/01.CIR.0000153847.47301.80. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hsieh PCH, MacGillivray C, Gannon J, Cruz FU, Lee RT. Circulation. 2006;114:637. doi: 10.1161/CIRCULATIONAHA.106.639831. [DOI] [PubMed] [Google Scholar]; Lin Y-D, Luo C-Y, Hu Y-N, Yeh M-L, Hsueh Y-C, Chang M-Y, Tsai D-C, Wang J-N, Tang M-J, Wei EIH, Springer ML, Hsieh PCH. Sci. Transl. Med. 2012;4 doi: 10.1126/scitranslmed.3003841. [DOI] [PubMed] [Google Scholar]; Singelyn JM, Sundaramurthy P, Johnson TD, Schup-Magoffin PJ, Hu DP, Faulk DM, Wang J, Mayle KM, Bartels K, Salvatore M, Kinsey AM, DeMaria AN, Dib N, Christman KL. J Am Coll Cardiol. 2012;59:751. doi: 10.1016/j.jacc.2011.10.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patten RD, Hall-Porter MR. Circulation-Heart Failure. 2009;2:138. doi: 10.1161/CIRCHEARTFAILURE.108.839761. [DOI] [PubMed] [Google Scholar]
- 45.Yoon SJ, Fang YH, Lim CH, Kim BS, Son HS, Park Y, Sun K. J Biomed Mater Res B. 2009;91B:163. doi: 10.1002/jbm.b.31386. [DOI] [PubMed] [Google Scholar]
- 46.Kass DA, Beyar R, Lankford E, Heard M, Maughan WL, Sagawa K. Circulation. 1989;79:167. doi: 10.1161/01.cir.79.1.167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.