

Abstract
Background
Mucopolysaccharidoses (MPS) are common lysosomal storage disorders causing typically progressive skeletal and ocular abnormalities.
Objectives
To describe the clinic features, metabolic profile and a unique mutation in a domestic shorthair (DSH) kitten with MPS VII.
Animals
Affected kitten and 80 healthy cats.
Methods
Serum lysosomal enzyme activities and urinary glycosaminoglycan (GAG) accumulation were assessed. Exons of the β‐glucuronidase gene (GUSB) were sequenced from genomic DNA and genotyping was conducted.
Results
A 3‐month‐old DSH cat was presented for stunted growth, paresis, facial dysmorphia, multiple skeletal deformities, and corneal opacities. Evaluation of blood smears disclosed metachromatic granules in leukocytes and a urinary mucopolysaccharide spot test was positive. The proband had no GUSB activity but normal or increased activities for other lysosomal enzymes. Sequencing of the GUSB gene from the proband and comparison to the sequence of 2 healthy cats and the published feline genome sequence demonstrated 2 unique single base transitions (c.1421T>G and c.1424C>T) in exon 9, altering 2 adjacent codons (p.Ser475Ala and p.Arg476Trp). These amino acid changes are in a highly conserved domain of the GUSB protein and nontolerable to maintain function. Moreover, the p.Arg476Trp mutation previously has been identified in human patients. None of the other clinically healthy cats had these mutations.
Conclusions and Clinic Importance
The diagnostic approach to MPS disorders is delineated. This is only the second mutation known to cause MPS VII in cats. Similarly, 2 different mutations have been described in MPS VII dogs, thereby showing the molecular heterogeneity of MPS VII in companion animals.
Keywords: Lysosomal storage disease, Ocular disease, Skeletal deformities, β‐glucuronidase
Abbreviations
- 4MU
4‐methylumbelliferyl
- Ala, A
alanine
- Arg, R
arginine
- bp
base pair
- diH2O
deionized water
- DNA
deoxyribonucleic acid
- dNTP
deoxynucleotide
- DSH
domestic shorthair
- EDTA
ethylenediamine tetra‐acetic acid
- GAGs
glycosaminoglycans
- gDNA
genomic DNA
- GUSB
β‐glucuronidase
- HS
hot‐start
- KOD
kodakaraenis
- MPS
mucopolysaccharidosis
- OMIA
online Mendelian inheritance in animals
- PCR
polymerase chain reaction
- RFLP
restriction fragment length polymorphism
- Ser, S
serine
- SIFT
sorting intolerant from tolerant
- SNP
single nucleotide polymorphism
- TIM
triosephosphate isomerase
- Trp, W
tryptophan
A variety of lysosomal storage disorders including mucopolysaccharidoses (MPS), sphingolipidoses, and glycoproteinoses have been discovered and are well characterized in several animal species, and dogs, cats, and mice serve as excellent disease models of these rare inborn errors of metabolism in humans. Although most lysosomal storage diseases are associated with neurologic signs, MPS (except type III) disorders mostly cause progressive skeletal and ocular abnormalities with secondary neurologic signs. In MPS, degradation of the glycosaminoglycans (GAGs) chondroitin, dermatan, or heparan sulfate is hampered because of deficient activity of a single lysosomal acid hydrolase. Isolated cases of domestic shorthair (DSH) cats with MPS I, VI, and VII have been reported, and MPS VI seems to be somewhat common in Siamese cats.1
Mucopolysaccharidosis type VII (MPS VII), also known as Sly syndrome in humans (Online Mendelian Inheritance in Man 253220), is a rare autosomal recessive trait caused by more than 50 different mutations in the β‐glucuronidase gene (GUSB).2, 3, 4 In addition to the well‐known human disorder, MPS VII also has been identified in several animal species including mice5, 6, 7 and dogs (Online Mendelian inheritance in animals [OMIA] 000667‐9615).8, 9, 10, 11 In dogs, the same missense mutation in GUSB (p.Arg166His) was found in mixed breed9 and German shepherd dogs,10 whereas another mutation (p.Pro281Leu) was discovered recently in Brazilian Terriers.11 Mucopolysaccharidosis VII also was reported in 3 domestic shorthair (DSH) cats (OMIA 000667‐9685). The first case was reported from Switzerland,12 and another 2 cases were identified in the United States (Michigan and California).13, 14 However, only the cat from Michigan has been studied at the molecular genetic level.13 Here, we describe the clinicopathologic features of MPS VII caused by a new GUSB mutation in a DSH kitten.
Materials and Methods
Animals
Aside from the samples from the proband and related cats, EDTA‐preserved blood, serum, buccal swabs, and urine samples from other domestic shorthair (DSH) cats were available for these studies at the Metabolic Genetics Screening Laboratory at the School of Veterinary Medicine, University of Pennsylvania. Diagnostic studies are offered as part of a diagnostic evaluation for suspected inborn errors of metabolism in animals and were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania. Samples were shipped on ice overnight after collection and urine and serum samples were stored frozen until further analysis.
Urinary Metabolic Analyses
Urinary GAG concentrations were analyzed semiquantitatively (toluidine blue mucopolysaccharide spot test)15 and quantitatively (normalized by the sample's creatinine concentration using a GAG Isolation & Concentration Kit and Blyscan GAG Assay Kit).1
Serum Lysosomal Enzyme Activities
Five lysosomal enzyme activities were determined in serum by fluorescence assay using 4‐methylumbelliferyl (4MU) substrates following previously established protocols in our laboratory.13 The storage diseases and corresponding enzymes were investigated based upon prior reports in cats and included (1) Mucopolysaccharidoses: α‐L‐iduronidase (IDUA, EC 3.2.1.76 for MPS I), N‐acetylgalactosamine‐4‐sulphatase (4S, EC 3.1.6.12 for MPS VI), β‐D‐glucuronidase (GUSB, EC 3.2.1.31 for MPS VII), and (2) Glycoproteinoses: α‐mannosidase (MANB, EC 3.2.1.24 for α‐mannosidosis) and α‐L‐fucosidase (FUCA, E 3.2.1.51 for α‐fucosidosis).
Molecular Genetic Studies
Genomic DNA (gDNA) was extracted from peripheral blood leukocytes and buccal swabs using DNA extraction kits,2 and concentrations were measured.3
A total of 12 exons including the 5′untranslated region (UTR), exon‐intron junctions and the 3′UTR of the feline GUSB gene were examined using the Hot‐start (HS) Polymerase Chain Reaction (PCR) Kit4 and Sanger sequencing techniques. A total of 11 PCR primer pairs were designed5 and synthesized6 (Table 1). The PCR reaction mixture contained 100 ng (2 μl) of gDNA as a template, 25 μL of 2X PCR buffer, 10 μL of 10 mM deoxynucleotides (dNTPs, 2.5 mM each), 0.75 μL of each forward and reverse primers (20 μM), 1 μL of Kodakaraenis (KOD) HS DNA polymerase (1 U/μL), and sterile deionized water (diH2O) up to a total volume of 50 μL. The PCR protocol included an initial HS at 94°C for 2 minutes, 35 cycles of denaturing at 94°C for 10 seconds, annealing at 60–62°C for 30 seconds, and extending at 68°C for 30–45 seconds (times varied based on the length of the amplicons). A negative control, comprised of diH2O instead of gDNA, was used to detect possible contamination. The PCR products were separated by 1% agarose gel electrophoresis, extracted using a QIAquick Gel Extraction Kit,7 sequenced at the Gene Sequencing Core Facility of the School of Medicine at the University of Pennsylvania and analyzed using Lasergene software.8
Table 1.
PCR primers and conditions used in sequencing feline GUSB gene
| Exon | Sequence (5′‐ 3′) | Tm (°C) | Extension (Second) | Amplicon (bp) |
|---|---|---|---|---|
| 1 | Forward: CATCTACCTGTATTTCTGCGGATACAA | 60 | 30 | 507 |
| Reverse: CCGTTTCCTCCCTCGCAGTC | ||||
| 2 | Forward: TTCTTCCTGCAGGCCAGGTG | 60 | 30 | 386 |
| Reverse: TGGGAACCACACACCCCAAG | ||||
| 3 | Forward: CGCCCTGACTTGCTGTTCCT | 60 | 30 | 322 |
| Reverse: CCAGGGCCTCACGTCTGCTA | ||||
| 4 | Forward: GCAGGGTGGCCCTAGCAGAC | 62 | 30 | 421 |
| Reverse: GCCCGTGCCAGTTCAGACAG | ||||
| 5 | Forward: CACTGCAGGTTGGGCTCCAG | 62 | 30 | 442 |
| Reverse: GCCTCCCTACCAAGCGGTGA | ||||
| 6–8 | Forward: GCCAGCACGGATGCCTCACA | 62 | 45 | 949 |
| Reverse: GCCTGCCCCAGAGCCAACCT | ||||
| 9 | Forward: TCGGGAGGGAGGAAGGGTTT | 60 | 30 | 302 |
| Reverse: CTCCTCCCAGCAGCCTGTGTT | ||||
| 10 | Forward: TTTTCTGCAGCCCCCTGTTG | 60 | 30 | 368 |
| Reverse: GACGGGTGGGAGAGGAGCAC | ||||
| 11 | Forward: TGGTCACGTCACATGGATCT | 60 | 30 | 366 |
| Reverse: TAGAGCTTTCCCCCTCGTTT | ||||
| 12a | Forward: CCCGTTTGGCCCATCAGAAG | 60 | 45 | 1,016 |
| Reverse: GGAGCAAGACAGGTGGCATCA | ||||
| 12b | Forward: TCCTTTTGGATGATGCCACCTG | 60 | 45 | 1,090 |
| Reverse: GCTCCCCCTGAATGCTCCAC |
Sources: NCBI‐Genbank (Gene ID: 493879, http://www.ncbi.nlm.nih.gov, Ensembl (Felis_catus_6.2, ENSFCAG00000006928), http://www.ensembl.org and UCSC Genome Bioinformatics (ICGSC Felis_catus_6.2/felCat5), http://genome.ucsc.edu.
Genotyping methods for mutant and normal (wild‐type) alleles were developed9 and performed with 80 gDNA samples from EDTA blood or buccal swabs of clinically healthy DSH cats including a littermate of the proband. A gDNA mixture of the affected proband and a clinically healthy cat at a 1 : 1 ratio (both genotypes were confirmed by GUSB gene sequencing) was used as a control to detect the heterozygous state. The DNA fragment around the putative mutations was amplified by real‐time PCR using a 15 μL reaction mixture containing 7.5 μL of 2X TaqMan® Universal PCR master mix (no AmpErase® Uracil N‐Glycosylase), 0.375 μL of 40X assay mix consisting of unlabeled PCR primers (forward: ACGCTGATTGCTCACACCAA; reverse: TCATAGTTGGAGTTGGTCACAAAGG), and fluorescently labeled TaqMan® 3′‐Minor groove binder probes (for c. 1421T>G transition, VIC® dye‐labeled AGCCTTGGATCCCTCC for normal, and FAM™ dye‐labeled AGCCTTGGATCCCGCC for mutant; for c.1424C>T substitution, VIC® dye‐labeled CTTGGATCCCTCCCGG for normal, and FAM™ dye‐labeled CTTGGATCCCTCCTGG for the mutation), 6.125 μL of diH2O and 1 μL of template gDNA. The cycling conditions utilized universal thermal cycling parameters: initial heating step at 95°C for 10 minutes, 40 cycles of 15 seconds at 92°C for denaturing and 60 seconds at 60°C for annealing/extending, and 10 minutes at 72°C for final extension.
To further determine the impact of the amino acid substitutions on the GUSB protein structure and function, the Sorting Intolerant from Tolerant (SIFT) algorithm software was applied to predict the effect of the amino acid change.10 The scores range from 0.00 (unacceptable) to 1 (normal); a score of <0.05 is deleterious.
Results
Clinic Case Report
A 3‐month old, female kitten from a farm in Trenton, Ontario, Canada was noted to have weakness in the rear legs. No prior and family history was available but an apparently healthy littermate was adopted by the same owner at the same time. On physic examination, the diseased kitten appeared stunted for its age, had a rounded head and pectus excavatum, and had hind limb paralysis. The kitten was able to pull itself forward with its forelimbs. No obvious pain was identified on palpation, and motor functions appeared to be delayed. The clinical signs progressed over the next 3 months to tetraparesis and ocular opacities developed (Fig 1A–C). The proband's skeletal radiographs disclosed pectus excavatum, kyphosis, shortened and fused vertebral bodies, dysostosis multiplex, hypoplastic epiphyses, thickened diaphyses, irregularly fragmented endplates, and increased intervertebral distances (Fig 1D). Based on the progressive clinical features, a systemic congenital orthopedic or metabolic disorder was suspected. A blood smear stained with Wright–Giemsa disclosed many metachromatic granules (also referred to as Alder‐Reilly bodies) within lysosomes of neutrophils and lymphocytes, representing GAGs. This finding was suggestive of MPS rather than other lysosomal storage disorders (Fig 1E).
Figure 1.
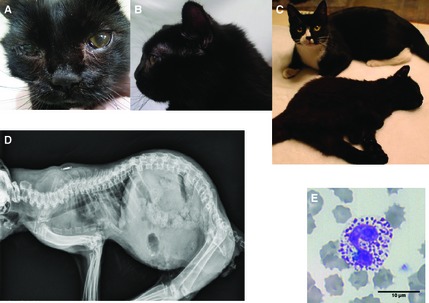
Clinical features of a 6‐month‐old DSH cat with MPS VII. (A) Facial dysmorphia and corneal opacities. (B) Flattened nose, large rounded head, and short neck. (C) Tetraparesis, and short stature compared with the healthy littermate (back). (D) Skeletal radiograph showed pectus excavatum, kyphosis, fused and shortened vertebral bodies, increased intervertebral distances, dysostosis multiplex, hypoplastic epiphyses, and thickened diaphysis. (E) Blood smear stained with Wright–Giemsa solution showing a neutrophil with many coarse metachromatic granules, which represent cellular GAGs accumulation.
Biochemical Analysis
The proband's urinary excretion of GAGs was evaluated by 2 methods. The semiquantitative toluidine blue mucopolysaccharide spot test was strongly positive, and quantitative analysis identified a large amount of urinary GAGs (Fig 2). These results of urinary GAGs analysis further indicated that MPS was likely.
Figure 2.
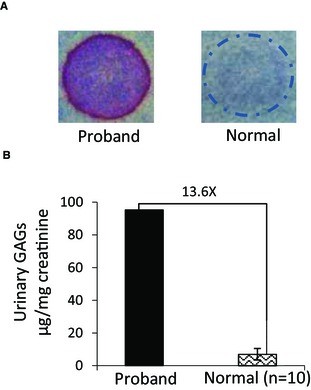
Urinary GAGs analysis from the proband with MPS VII and clinically healthy DSH cats. (A) Mucopolysaccharide spot test. Proband: strongly positive result; a clinically healthy cat: negative result. (B) Quantitative assay. The proband's urinary GAGs concentration (95.2 μg/mg creatinine) compared with the clinically healthy, age‐matched controls (mean ± SD: 7.0 ± 3.3 μg/mg creatinine, n = 10); a 13.6X increase is shown.
Assessment of lysosomal enzymes (Table 2) identified a severe deficiency of serum GUSB activity, whereas the other 2 enzymes (IDUA and 4S) involved in GAG catabolism were increased when compared with healthy controls. The other 2 enzyme activities tested were normal. The lysosomal enzyme activity profile led to a final diagnosis of MPS VII.
Table 2.
Serum lysosomal enzyme activities in a cat with MPS VII and normal controls
| Enzyme | Diseasea | Cat with MPS VII | Normal cats Mean ± SD (n = 10) | |
|---|---|---|---|---|
| Activityb | %c | |||
| Mucopolysaccharidoses | ||||
| α‐L‐iduronidase | MPS I | 31.9 | 215.5 | 14.8 ± 6.7 |
| N‐acetylgalactosamine‐4‐sulphatase | MPS VI | 17.4 | 175.8 | 9.9 ± 3.6 |
| β‐D‐glucuronidase | MPS VII | 0.0 | 0.0 | 269.5 ± 32.5 |
| Glycoproteinoses | ||||
| α‐mannosidase | α‐Mannosidosis | 3015 | 100.4 | 3002 ± 779 |
| α‐L‐fucosidase | α‐Fucosidosis | 98.7 | 97.5 | 101.27 ± 34.0 |
Disease corresponds to the specific abnormal function of the enzyme tested.
Specific activity: nM 4 MU/h/mL serum.
Percentage activity of healthy controls (based on the mean of 10 healthy cats).
Molecular Genetic Study
In cats, the GUSB gene is located on chromosome E3, and has 12 exons coding for 651 amino acids. Comparison of the proband's GUSB sequence to the published genome sequence of cats (NCBI‐Genbank, Gene ID: 493879 and Ensembl: ENSFCAG00000006928) and results from 2 clinically healthy cats sequenced simultaneously identified homozygosity for 2 single nucleotide substitutions in exon 9: a thymine (T) base located at cDNA position 1421 was changed to guanine (G) (c.1421T>G), and just 3 bases away, a cytosine (C) base was changed to thymine (T) (c.1424C>T). These 2 base changes resulted in 2 adjacent amino acid substitutions with a serine changed to alanine (p.Ser475Ala) and an arginine replaced by a tryptophan (p.Arg476Trp; Fig 3). Moreover, this cat did not have the missense mutation previously reported in another DSH cat (c.1074G>A, p.Glu351Lys in exon 7).13 In addition, 6 single nucleotide polymorphisms (SNPs) were observed in the 3′UTR region of the proband's GUSB gene: c.2046C>T, c.2645T>C, c.2880A>C, c.2934C>T, c.3261A>C and c.3307A>C. These variants also were observed in 2 healthy cats and therefore were not considered responsible for the GUSB deficiency.
Figure 3.
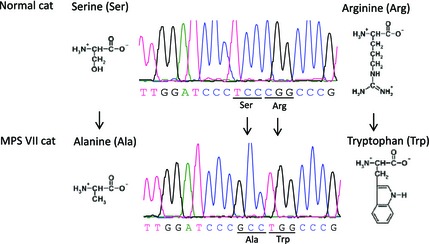
The results of sequencing analysis on GUSB gene of a cat with MPS VII. The results reveal two missense point mutations in exon 9 of the GUSB gene, (c.1421T>G, c.1424C>T), which cause two adjacent and detrimental amino acid substitutions (p.Ser475Ala and p.Arg476Trp).
Multiple alignments of the GUSB amino acid sequence performed across multiple eukaryotic species indicated that the p.Ser475Ala and p.Arg476Trp substitutions reside in a highly conserved region of the third domain of the GUSB enzyme (Table 3). The SIFT analysis indicated that neither the p.Ser475Ala (SIFT score = 0.03) nor the p.Arg476Trp (SIFT score = 0.00) were tolerable. Thus, both amino acid changes were considered highly deleterious to the structure and function of the GUSB protein.
Table 3.
Comparison of multiple eukaryotic species' GUSB amino acid sequence alignments around the substitution sites. The two amino acid substitutions, p.S475A (p.Ser475Ala) and p.R476W (p.Arg476Trp) occurred in a highly conserved region of the GUSB protein, and were predicted to be pathogenic, and likely disrupt the structure of the GUSB enzyme. (Source: http://www.ncbi.nlm.nih.gov/protein)
| Eukaryotic specie | GUSB amino acid sequence |
|---|---|
| Proband | …466IAHTKALDPAWPVTFVTNSNY486… |
| Felis catus | IAHTKALDPSRPVTFVTNSNY |
| Homo sapiens | IAHTKSLDPSRPVTFVSNSNY |
| Canis familiaris | IAHTKALDPSRPVTFVTNSNY |
| Pan troglodytes | IAHTKALDPSRPVTFVSNSNY |
| Bos taurus | IAHTKALDPSRPVTFVTNTNY |
| Gallus gallus | IAHTKALDPSRPVTFVSDTNY |
| Danio rerio | ISHTKSLDSSRPVTFITDSNY |
Genotyping the c.1421T>G and c.1424C>T mutations with 2 real‐time PCR assays readily discriminated the mutant and normal alleles and also detected the heterozygotes made by an admixture of a sample from a healthy cat and the affected cat (Fig 4). Screening of an additional 80 DSH cats, including a littermate, 2 nearby cats and 77 cats from various parts of the United States showed only the normal haplotype. Parents were not available for study.
Figure 4.
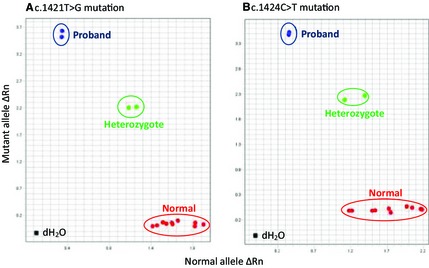
Allelic discrimination genotyping results of fluorescence real‐time PCR for detecting 2 GUSB mutations utilizing gDNA from the proband, clinically healthy DSH cats, and a mixture of both. End point fluorescence intensities (Rn values) for the normal allele are expressed on the X‐ axis and for the mutant allele on the Y‐axis. (A) c.1421T>G mutation fluorogenic probe. (B) c.1424C>T mutation fluorogenic probe. Both allelic discrimination plots clearly show the 3 genotypes for the 2 mutations. The proband has mutant (duplicate assay) genotypes (blue dots). The 10 clinically normal DSH cats, including a littermate of the proband, have normal alleles (red dots). The mixed gDNA sample from the proband and a healthy cat reflects the heterozygote states (duplicate assay, green dots). dH 2O indicates a negative template DNA control.
Discussion
This study describes the diagnostic evaluation of a kitten with multisystemic skeletal malformations, ocular opacities, positive urinary mucopolysaccharide spot test and metachromatic leukocyte inclusions suggestive of MPS. Serum enzyme and molecular genetic studies identified a complete deficiency of GUSB enzyme activity caused by 2 adjacent amino acid changes in the GUSB gene. Although 1 of the mutations (p.Arg476Trp) has been reported in human patients with MPS VII, our study represents the second report of a disease‐causing mutation in GUSB in cats. Although clinical features can be suggestive of a storage disorder, other congenital skeletal malformations and congenital hypothyroidism16 also should be considered as differential diagnoses.
The accumulation of GAGs in tissues, leukocytes and urine are among the most important and simple biochemical markers to suggest MPS. Urinary GAG concentration can readily be quantified by simple kit methods and chondroitin, dermatan, and heparan sulfates can be identified by electrophoresis.17 Although urinary GAG concentration can be moderately increased in growing animals, the 13.6‐fold increase in the proband's urine over that of age‐matched normal cats was indicative of MPS. Additional evaluation of GAGs may have further suggested a specific type of MPS (chondroitin and dermatan sulfates with MPS VII), but was not done in this cat.
Because the 11 known MPS disorders share many clinical features such as early age of onset, dwarfism, facial dysmorphia, bone and joint abnormalities, organomegaly and neurological signs with variable degree of severity, it was necessary to determine which specific lysosomal enzyme was deficient.18 Enzyme activities measured in serum, as done in this kitten, and leukocytes are standard methods, but recently we also showed that because of the extraordinary stability of most lysosomal enzymes, dried blood spot samples can be used to measure these enzyme activities and document a GUSB deficiency.19 The undetectable activity of GUSB in the proband's serum led to the final diagnosis of MPS VII. As previously reported for MPS VII and other MPS disorders, the other enzyme activities in the same pathway are increased likely because of the compensated transcription factor EB regulation mechanism.20 Indeed, in the proband's serum, activities of other MPS enzymes that degrade GAGs (IDUA and 4S) were markedly increased, whereas enzymes involved in glycoprotein catabolism (MANB and FUCA) had activities in the reference range.
It was not surprising that the kitten from Canada in this report did not have the point missense mutation causing substitution of a highly conserved amino acid (p.Glu351Lys) reported previously in a DSH cat from Michigan despite showing very similar clinical signs and biochemical features of MPS VII.13 Both likely represent independent new mutation events because of subsequent breeding of related cats heterozygous for the mutant alleles to produce affected animals (homozygous for the mutation).
The structure of the GUSB monomeric enzyme is well conserved among species (80% similarity to human GUSB at the amino acid level [UCSC Genome Bioinformatics, ICGSC Felis_catus_6.2/felCat5, http://genome.ucsc.edu]) and is composed of 3 functional domains, including a jelly roll barrel (amino acid residues 22–223, exons 1–4), an immunoglobulin constant region (amino acid residues 224–342, exons 5–6), and a triosephosphate isomerase (TIM) barrel domain with its catalytic site (amino acid residues 343–632, exons 7–12).21 In the affected cat, the identified single base changes (c.1421T>G and c.1424C>T) in exon 9 of GUSB result in p.Ser475Ala and p.Arg476Trp substitutions of the GUSB protein; this area resides in the middle of the TIM barrel domain which is highly conserved among mammals.
To further determine the effect of the p.Ser475Ala and p.Arg476Trp amino acid substitutions (in which polar amino acids were replaced by nonpolar amino acids) on the GUSB protein structure, a SIFT analysis was performed. Based upon the SIFT analysis results, it was predicted that either amino acid substitution would be highly deleterious to the GUSB protein's structure and function. Furthermore, the p.Arg476Trp missense mutation previously was reported in human patients and caused a severe MPS VII disorder.22 It is unknown how much the adjacent p.Ser475Ala missense mutation worsened the deleterious effect of the p.Arg476Trp substitution.
We established species‐specific allelic discrimination based real‐time PCR methods for both the c.1421T>G and c.1424C>T variants which readily differentiated the mutant alleles exclusively found in the proband from the normal alleles in all other cats and identified the heterozygote state (Fig 4). The proband was from Trenton Ontario, Canada and no other cats except 1 littermate and 2 healthy cats from that farm were available for study and were found to have the normal haplotype. Although this is a simple screening test, the mutant haplotype will likely only be found in the region from which the cat originated, except when cats travel with people to other areas. Such a situation occurred with this kitten, when the family moved to Iowa. Thus, as in the case of MPS VII in Michigan, general screening for these 2 mutations is not recommended. Rather, animals with clinical signs suspicious of MPS should be tested utilizing blood smear examination, the urine mucopolysaccharide spot test, and enzyme activity testing of serum or dried blood spot screening tests.
Currently, MPS VII and other MPS disorders lack an efficient clinical treatment despite intense experimental investigations offering promising results with novel gene transfers, such as bone marrow transplantation for MPS VII in mice23 and dogs,24 enzyme replacement treatment for MPS VII in mice,25 and gene treatment for MPS VII in dogs.26, 27, 28 These treatments have demonstrated a great capacity to prevent many of the clinical features but once clinical signs occur, MPS VII is not reversible. Symptomatic treatment, pain alleviation, and physiotherapy can be offered before euthanasia is elected.
In conclusion, we describe the clinical, biochemical and molecular findings in a DSH cat with MPS VII. We identified a pair of adjacent GUSB mutations that reflects only the second molecular characterization of MPS VII in cats.
Acknowledgments
Grant support: This study was supported in part by NIH OD 010939.
Conflict of Interest Declararion: Urs Giger and Shurnevia Strickland are affiliated with the not‐for‐profit PennGen Laboratories at the University of Pennsylvania which offers MPS testing.
Off‐label Antimicrobial Declaration: Authors declare no off‐label use of antimicrobials.
Footnotes
GAGs Isolation & Concentration Kit and the Blyscan GAGs Assay Kit, Biocolor Ltd., Carrickfergus, Antrim, UK.
QIAamp DNA Blood Mini Kit, QIAGEN, Valencia, CA
NanoDrop 2000, UV‐Vis Spectrophotometer, Thermo Scientific, Wilmington, DE
Novagen‐EMD Company, Gibbstown, NJ
DNAStar, Inc., Madison, WI
Integrated DNA Technologies, Inc., Coralville, IA
QIAquick Gel Extraction Kit, QIAGEN, Valencia, CA
Lasergene software, DNAStar, Inc., Madison, WI
ABI 7500 instrument and “Custom TaqMan® SNP Genotyping Assay”, Applied Biosystems, Life Technologies, Grand Island, NY
J. Craig Venter Institute, Rockville, MD, http://sift.jcvi.org
References
- 1. Haskins ME, Giger U. Chapter 24: Lysosomal storage diseases In: Kaneko JJ, Harvey JW, Bruss ML, eds. Clinical Biochemistry of Domestic Animals, 6th ed San Diego, CA: Academic Press; 2008:731–750. [Google Scholar]
- 2. Oshima A, Kyle JW, Miller RD, et al. Cloning, sequencing, and expression of cDNA for human beta‐glucuronidase. Proc Natl Acad Sci USA 1987;84:685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sly WS, Vogler C, Grubb JH, et al. Active site mutant transgene confers tolerance to human beta‐glucuronidase without affecting the phenotype of MPS VII mice. Proc Natl Acad Sci USA 2001;98:2205–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tomatsu S, Montano AM, Dung VC, et al. Mutations and polymorphisms in GUSB gene in mucopolysaccharidosis VII (Sly syndrome). Hum Mutat 2009;30:511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Birkenmeier EH, Davisson MT, Beamer WG, et al. Murine mucopolysaccharidosis type VII. Characterization of a mouse with beta‐glucuronidase deficiency. J Clin Invest 1989;83:1258–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vogler C, Birkenmeier EH, Sly WS, et al. A murine model of mucopolysaccharidosis VII. Gross and microscopic findings in beta‐glucuronidase‐deficient mice. Am J Pathol 1990;136:207–217. [PMC free article] [PubMed] [Google Scholar]
- 7. Vogler C, Levy B, Galvin N, et al. Early onset of lysosomal storage disease in a murine model of mucopolysaccharidosis type VII: undegraded substrate accumulates in many tissues in the fetus and very young MPS VII mouse. Pediatr Dev Pathol 2005;8:453–462. [DOI] [PubMed] [Google Scholar]
- 8. Haskins ME, Desnick RJ, DiFerrante N, et al. Beta‐glucuronidase deficiency in a dog: A model of human mucopolysaccharidosis VII. Pediatr Res 1984;18:980–984. [DOI] [PubMed] [Google Scholar]
- 9. Ray J, Bouvet A, DeSanto C, et al. Cloning of the canine beta‐glucuronidase cDNA, mutation identification in canine MPS VII, and retroviral vector‐mediated correction of MPS VII cells. Genomics 1998;48:248–253. [DOI] [PubMed] [Google Scholar]
- 10. Silverstein DDC, Carmichael KP, Wang P, et al. Mucopolysaccharidosis type VII in a German Shepherd Dog. J Am Vet Med Assoc 2004;224:553–557, 532–533. [DOI] [PubMed] [Google Scholar]
- 11. Hytönen MK, Arumilli M, Lappalainen AK, et al. A novel GUSB mutation in Brazilian terriers with severe skeletal abnormalities defines the disease as mucopolysaccharidosis VII. PLoS ONE 2012;7:e40281. doi:10.1371/journal.pone.0040281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gitzelmann R, Bosshard NU, Superti‐Furga A, et al. Feline mucopolysaccharidosis VII due to beta‐glucuronidase deficiency. Vet Pathol 1994;31:435–443. [DOI] [PubMed] [Google Scholar]
- 13. Fyfe JC, Kurzhals RL, Lassaline ME, et al. Molecular basis of feline beta‐glucuronidase deficiency: An animal model of mucopolysaccharidosis VII. Genomics 1999;58:121–128. [DOI] [PubMed] [Google Scholar]
- 14. Schultheiss PC, Gardner SA, Owens JM, et al. Mucopolysaccharidosis VII in a cat. Vet Pathol 2000;37:502–505. [DOI] [PubMed] [Google Scholar]
- 15. Berry HK, Spinanger J. A paper spot test useful in study of Hurler's syndrome. J Lab Clin Med 1960;55:136–138. [PubMed] [Google Scholar]
- 16. Traas AM, Abbott BL, French A, et al. Congenital thyroid hypoplasia and seizures in 2 littermate kittens. J Vet Intern Med 2008;22:1427–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wegrowski Y, Maquart FX. Chapter 17: Cellulose acetate electrophoresis of GAGs Methods in Molecular Biology In: Iozzo RV, ed. Proteoglycan Protocols. Methods in Molecular Biology™ Volume 171. Totowa, NJ: Humana Press; 2001:175–179. [DOI] [PubMed] [Google Scholar]
- 18. Yu C, Sun Q, Zhou H. Enzymatic screening and diagnosis of lysosomal storage diseases. N Am J Med Sci 2013;6:186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sewell AC, Haskins ME, Giger U. Dried blood spots for the enzymatic diagnosis of lysosomal storage diseases in dogs and cats. Vet Clin Pathol 2012;41:548–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sardiello M, Palmieri M, di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science 2009;325:473–477. [DOI] [PubMed] [Google Scholar]
- 21. Hassan MI, Waheed A, Grubb JH, et al. High resolution crystal structure of human β‐glucuronidase reveals structural basis of lysosome targeting. PLoS ONE 2013;8:e79687. doi:10.1371/journal.pone.0079687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vervoort R, Islam MR, Sly WS, et al. Molecular analysis of patients with beta‐glucuronidase deficiency presenting as hydrops fetalis or as early mucopolysaccharidosis VII. Am J Hum Genet 1996;58:457–471. [PMC free article] [PubMed] [Google Scholar]
- 23. Sands MS, Barker JE, Vogler C, et al. Treatment of murine mucopolysaccharidosis type VII by syngeneic bone marrow transplantation in neonates. Lab Invest 1993;68:676–686. [PubMed] [Google Scholar]
- 24. Sammarco C, Weil M, Just C, et al. Effects of bone marrow transplantation on the cardiovascular abnormalities in canine mucopolysaccharidosis VII. Bone Marrow Transplant 2000;25:1289–1297. [DOI] [PubMed] [Google Scholar]
- 25. Sands MS, Vogler C, Kyle JW, et al. Enzyme replacement therapy for murine mucopolysaccharidosis type VII. J Clin Invest 1994;93:2324–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ponder KP, Melniczek JR, Xu L, et al. Therapeutic neonatal hepatic gene therapy in mucopolysaccharidosis VII dogs. Proc Natl Acad Sci USA 2002;99:13102–13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sleeper MM, Wang P, O'Donnell P, et al. Comparison of vector type and timing of gene therapy to correct cardiovascular manifestations of mucopolysaccharidosis VII. Circulation 2010;122:A15413. [Google Scholar]
- 28. Smith LJ, Martin JT, O'Donnell P, et al. Effect of neonatal gene therapy on lumbar spine disease in mucopolysaccharidosis VII dogs. Mol Genet Metab 2012;107:145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]