

Abstract
Stimulus responsive release of Circulating Tumor Cells (CTCs), with high recovery rates from their capture platform, is highly desirable for off-chip analyses. Here, we present a temperature responsive polymer coating method to achieve both release as well as culture of viable CTCs captured from patient blood samples.
The Circulating Tumor Cell (CTC) is an important biomarker in cancer management. Enumeration of CTCs has been proven of prognostic value in multiple cancer types including breast, prostate and colorectal cancer1–3. Recent studies, however, have revealed the limited clinical relevance of CTC enumeration alone in interventional trials4. Additionally, single CTC transcriptomic studies have revealed the wide heterogeneity of CTCs5, further indicating the need for in depth molecular and functional characterization of CTCs. Indeed, there have been increased efforts to implement molecular and genomic characterization of CTCs for informing clinical trial design, treatment selection, and ultimately, precision cancer management6. To fulfil such goals, a technology that not only allows for efficient CTC capture but also versatile to accommodate various downstream characterization of CTC both on-chip and off-chip is highly desirable.
We have previously reported a very successful size-based strategy for CTC isolation7, and its variation employing slot-shaped pores for viable CTC capture and analysis8. These Parylene-C based microfilters not only capture and retain CTCs with high efficiency but also allow in situ analyses, which require extensive manipulation of CTCs. However, the strong retention of CTCs hinders those experiments that require that the captured cells be transferred onto other platforms. For example, microfluidic platforms, such as the Fluidigm™ single cell analysis system5 or the DEPArray™ system9 require suspended cells as input, and novel ex vivo culture methods require CTCs to be cultured on specialized surfaces, such as ultra-low attachment plates10. All of these platforms, which have great promise as a companion chemo-sensitivity test, are incompatible with many of the capture platforms, including our microfilters. Thus, a facile route to viably recover CTCs captured from the blood of cancer patients is highly desirable.
Several platforms that allow for viable CTC capture and release have been reported based on immobilized antibodies conjugated to a cleavable linker11, Poly (N-iso-propylacrylamide) (PIPAAm)12, or electroactive films13. However, these systems are affinity-based and can be potentially biased by the target antigen. For example, the most commonly targeted antigen for CTC capture, EpCAM, can be absent within certain CTC populations possessing mesenchymal phenotypes14. In contrast, label free, size-based isolation and release of CTCs can provide a method to study the CTC population in a potentially more comprehensive manner, such as the centrifuge-on-a-chip method reported by Mach et al.15 Here, we demonstrated that by combining our filtration-based platform with the PIPAAm coating method, we could achieve an antigen-agnostic, efficient method to effectively capture and isolate viable CTCs from blood.
PIPAAm is a polymer that undergoes a reversible lower critical solution temperature (LCST) phase transition at a solution temperature of 32°C16. Traditionally, this property of PIPAAm has been widely explored for tissue engineering applications. Typically, cells are cultured on PIPAAm coated surfaces at 37°C when PIPAAm is hydrophobic. The cells can then be detached as a sheet when the culture temperature is shifted to below 32°C, where the PIPAAm coated surface becomes hydrated17, 18.
In our formulation, epithelial cancer cells are bound to the parylene C membrane by non-specific electrostatic interactions instead of extracellular matrix (ECM) mediated adhesion. This necessitated a modification of the conventional PIPAAm based release strategy. The epithelial cell capture is performed at room temperature (below 32°C) and cell release is enabled by placing the filter in our culture media maintained at 37°C. At this temperature, the PIPAAm polymer layer becomes hydrophobic, thereby releasing the electrostatically bound cells.
To coat PIPAAm onto the slot filter, we first dissolved PIPAAm in butanol at 10% (w/v) concentration. The PIPAAm solution was then spin coated onto the slot filter at a top speed of 6000 rpm for one minute19. The coated filter was air-dried and stored at room temperature before use (Fig. 1A,B). We compared the pore sizes before and after coating by phase-contrast microscopy (Supplementary Fig. 1) and as anticipated, found a 7% decrease in pore length and 15% decrease in pore width after PIPAAm coating (Supplementary Fig. 1). The effect of this pore size decrease was enhancement in capture efficiency (Table 1), along with decrease in enrichment factor, with more erythrocytes and leukocytes seen post capture (Supplementary Fig. 2A). This is consistent with our previous report on slot filters with a 5μm pore width8. However, this decrease in enrichment factor does not hamper the functionality of the PIPAAm coated slot filter for viable CTC capture and release, since the additional erythrocytes and leukocytes captured can be easily removed by a gentle wash of fresh media at day 1 (Supplementary Fig. 2B). This did not affect cell viability, proliferation or metabolic rate post capture (Fig 2, 4, Supplementary Fig. 4). To perform CTC capture, the coated filter was cut into 6 mm by 6 mm squares and fit into a filtration cassette (Top acrylic piece: H 4 mm, L 30mm, W 18 mm, bottom acrylic piece: H 8mm, L 30mm, W 18 mm) (Fig. 1) Methodologically, CTCs are captured onto PIPAAm coated slot filters at room temperature with PIPAAm coating in its hydrophilic state. Post capture, a mild reverse flow was applied to release cells trapped in the pores and then the filter was incubated in the release medium at 37°C to induce the phase transition. The cells bound to filter are then released (Fig. 1).
Fig. 1.
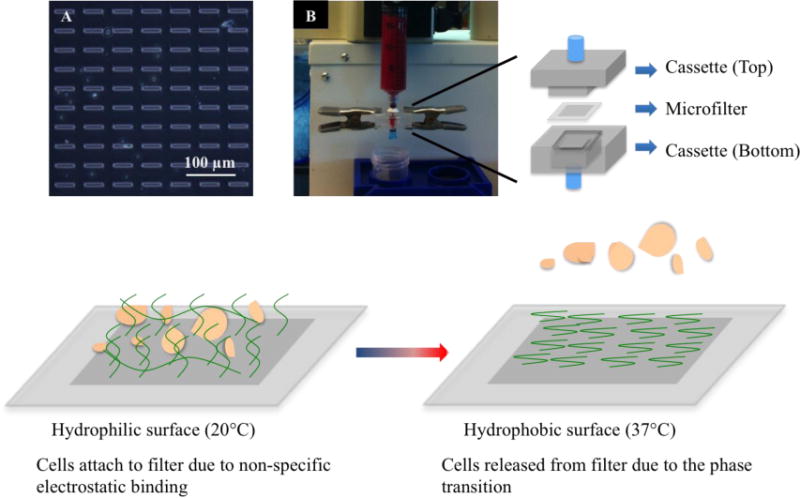
PIPAAm coated slot filters to capture and release circulating tumor cells from blood. (Top Panel) (A) Microscopic view (400× magnification) of the slot filter post PIPAAm coating; (B) Filtration set-up with syringe pump to capture CTCs from blood using PIPAAm coated slot filters, the PIPAAm coated slot filter is sandwiched between the top cassette and the bottom cassette. (Bottom Panel) Scheme of using temperature change to release captured CTCs from PIPAAm coated slot filters.
Table 1.
Capture, Release and Retrieval efficiency of PIPAAm coated slot filters. Capture efficiency is calculated by dividing cell numbers captured on filter before release by cell numbers spiked into blood. Release efficiency is calculated by dividing cell numbers released from the filter by cell numbers captured on the filter before release. Retrieval Efficiency is calculated by dividing cell numbers released from filter by cell numbers spiked into blood.
| Cell Line | Filter | Capture Efficiency | Release Efficiency | Retrieval Efficiency |
|---|---|---|---|---|
| SkBr-3 | Non-coated | 89% +− 3% | 7% +− 1% | 6% +− 1% |
| PIPAAm-coated | 94% +− 9% | 82% +− 5% | 77% +− 5% | |
| LMTS-GFP | PIPAAm-coated | 87% +− 10% | 79% +− 14% | 69% +− 12% |
Fig. 2.
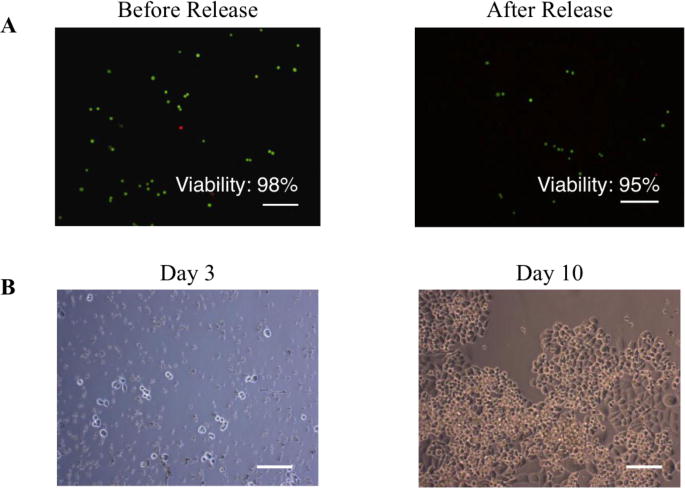
SK-Br-3 breast cancer cells captured and released from blood remained viable and expanded in culture rapidly. (A) SKBr-3 cells released from the filter was tested with a Live-Dead Assay. 95% (540 out of 567 cells counted) of the cells remained viable (Green) following PIPAAm-filter release. Dead cells are labelled in red. (B) Cells released from PIPAAm coated slot filter remained viable and expanded in culture. Scale Bar: 100 μm
Fig.4.
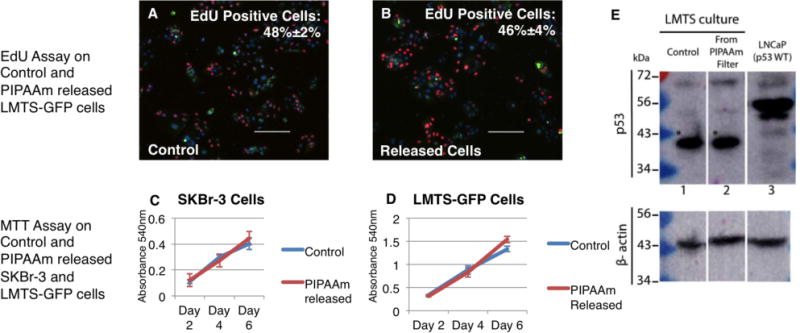
Measurement of proliferation rates and metabolism rates of tumor cells before and after PIPAAm coated slot filter capture and release Proliferation rate of the LMTS-GFP cells was measured using an EdU assay. All cells were labeled with Hoechst (Blue) and express GFP (Green), and newly proliferated cells were labeled with EdU (Red). Comparable proliferation rates were observed between LMTS-GFP cells plated as controls (A) and spiked in LMTS-GFP cells retrieved from blood by PIPAAm coated slot filters (B). Metabolism rate and growth curves of SKBr-3 cells and LMTS-GFP cells were measured using an MTT Assay. No significant differences in metabolism rate and proliferation rate were seen between released cells from PIPAAm coated slot filter and control cells for either (C) SKBr-3 cells or (D) the LMTS-GFP CRC line. We also probed the captured and released LMTS-GFP cells for mutated p53 protein as seen in control cells. (E) Extracts from parental control (lane 1) and LMTS-GFP cells cultured following release from the PIPAAm filter (lane 2) were probed by immunoblot for the p53 tumor suppressor protein. The LMTS line harbors an E298* truncation mutation with a predicted molecular weight of approximately 43 kDa (asterisk). LNCaP cells (Lane 3) were run as a control for wild type p53. β-actin was used as a loading control as seen on bottom panel of E.
To test the PIPAAm coated slot filters, we performed detailed analyses of tumor cell capture and release efficiency using ~1,000 SKBr-3 cells labeled by Carboxyfluorescein Succinimidyl Ester (CFSE) spiked into 7.5 mL of healthy donor’s blood as a model system (details in Supplementary Method). Healthy donors’ blood samples in this study were obtained under a protocol approved by the University of Miami (Miami, FL, USA) IRB (20150020) following an informed consent. Results from 3 replicates are shown in Supplementary Table 1. The coating method did not hamper the capture efficiency of the filter itself. Overall, we achieved capture, release and retrieval efficiency averages of 94%±9%, 82%±5% and 77%±5% respectively (Table 1, summarized from data in Supplementary Table 1, 2). The release and retrieval efficiency was increased as compared with those of non-coated filter (7%±1% release efficiency and 6%±1% retrieval efficiency) (Table 1, Supplementary Table 3).
To test the viability of the cells released from the filter, we spiked ~1,000 SK-Br-3 cells into 7.5 mL of healthy donor’s blood, captured and released them from PIPAAm coated slot filter using the method described above. A Live-Dead® assay (Life Technologies, Grand Island, NY) was performed to evaluate the cell viability before spike into blood and after release. The pre-spike viability was 98% (592 out of 602 cells counted) and the viability of cells captured and released from blood was 95% (540 out 567 cells counted) (Fig. 2A). We also cultured, in parallel experiment, the cells released from blood McCoy’s 5A culture medium (Gibco, Life Technologies, Grand Island, NY). Images were taken at day 3 and day 10. As shown in Fig. 2B, cells released from the filter remained viable and expanded rapidly in culture establishing their viability post cell capture from blood and release from the filter.
The ultimate application of this technology is to capture patient-derived CTCs from blood and release them with minimal impact on their viability. The released CTCs can then be subject to optimal culture surfaces/conditions, which would have been impossible on cells retained on filter. To demonstrate the feasibility of this application, we used conditionally reprogrammed cells (CRCs)20 as a model system. The CRC approach enables the establishment of continuous primary cell cultures from all epithelial tissues and could potentially be applied for CTCs culture establishment. Using the CRC method, a tumor cell line was established from a non-small cell Lung carcinoma that had Metastasized To the Spine and termed LMTS. This line was stably labeled with green fluorescent protein (GFP) and termed LMTS-GFP. Although Parylene C can be favorable for culturing certain immortalized cell line21, 22, it was not the optimal surface to culture LMTS-GFP cells (Fig. 3A). Thus, in order to retrieve LMTS-GFP cells from blood and expand them in culture, we tested the PIPAAm coated slot filter for viable cell release, post capture. First, we tested the capture, release and retrieval efficiency of LMTS-GFP cells on the PIPAAm coated filters. ~1,000 LMTS-GFP cells were spiked into 7.5 mL of healthy donor’s blood and the capture and release experiment was performed in triplicate. As indicated in Table 1, efficiencies of capture, release and retrieval produced averages values of 87%±10%, 79%±14% and 69%±12%, respectively. With the confirmative results showing high efficiency retrieval of LMTS-GFP cells from blood, we then tested the culture of these cells retrieved from blood. We spiked ~1,000 LMTS-GFP cells into 3 mL of healthy donor’s blood. The blood was then diluted to 6 mL volume using PBS. Post capture, the released cells (Fig. 3C) were then cultured in a 48 well plate (Greiner Bio-One, Monroe, NC) in irradiated J2 conditioned F+Y media prepared as described20, 23. As shown in Fig. 3D, released cells remained viable at Day 5 and expanded in culture.
Fig.3.
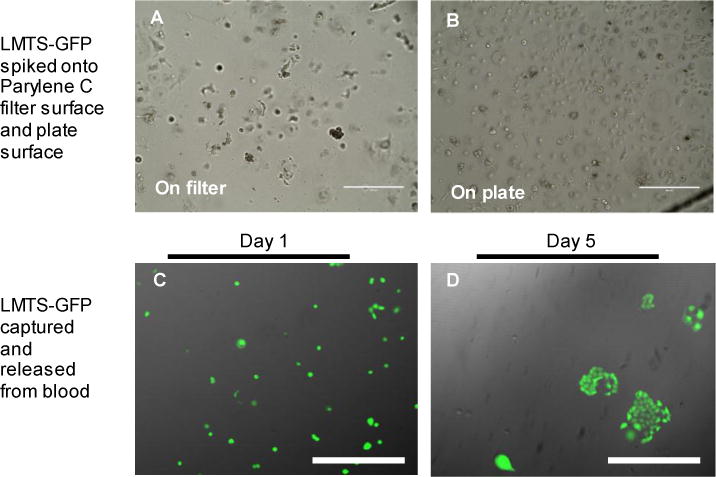
Capture and release of LMTS-GFP CRCs using PIPAAm coated slot filters. (Top Panel) Comparison of LMTS-GFP cells cultured on Parylene C surfaces (A) and on culture plates (B). Parylene C surfaces do not support the growth of LMTS-GFP CRCs. Scale Bar 400 μm. (Bottom Panel) LMTS-GFP cells were spiked into healthy donor’s blood and retrieved by the PIPAAm coated slot filter. Images were taken under phase contrast microscopy and the FITC channel and merged for display. Images were taken at day 1 (C) and day 5 (D). Scale Bar: 100 μm.
In order to confirm that the process of retrieving tumor cells from blood does not alter the cells’ proliferation rate, metabolism and biochemical properties, we analyzed the tumor cells retrieved from blood by MTT and EdU assays as shown Fig 4. For the EdU assay, the modified thymidine analog EdU (Life Technologies, Grand Island, NY) was integrated into newly synthesized DNA and then labeled with Alexa Fluor 594® dye. Cells were incubated with EdU for 2 hours and then fixed for labeling. We then enumerated the total number of cells (Hoechst+ cells) and newly proliferated cells (Hoeschst+, EdU+ cells) and calculated the percentage of EdU+ cells. For SKBr-3 cells, EdU+ cells constituted 29%±6% of the control cell population and 32%±8% of the PIPAAm released cells population, with no significant difference between groups (p value=0.44) (Supplementary Fig.4). For LMTS-GFP cells, EdU+ cells constituted 48%±2% of the control cell population (Fig.4A) and 46%±4% of the PIPAAm released cells population (Fig.4B), with no significant difference between groups (p value=0.38). To test the growth curve and metabolism rate of the PIPAAm released cells, we have also performed MTT assay (Life Technologies, Grand Island, NY) on released SKBr-3 cells and LMTS-GFP cells. As seen in Fig. 4 C and D, the growth curves and metabolism rates remained the same for both SKBr-3 and LMTS-GFP cells pre- and post- filter capture and release. To verify that the biochemical markers of LMTS-GFP cells were not altered, we probed for the p53 E298* truncation mutation harbored by LMTS-GFP cell line in cells we spiked into blood and retrieved using PIPAAm coated slot filters. As seen in Fig. 4E, LMTS-GFP cells retrieved from blood by PIPAAm coated slot filters contain the same truncated p53 tumor suppressor protein as seen in control LMTS cells.
To test the applicability of our strategy for human patient samples, we tested this technology with 4 metastatic breast cancer patient samples. Briefly, we collected 15 mL of peripheral blood by venipuncture from each patient under a protocol approved by the University of Miami (Miami, FL, USA) IRB (20130312) following an informed consent. Blood samples were split evenly into 2 tubes of 7.5 mL blood for each patient. One tube of blood was processed using our round-pore microfilter, which has been demonstrated previously to capture greater number of CTCs as compared with CellSearch® in the same cohort of patients24. The other tube was processed using the PIPAAm coated slot filter. Post-release, the release medium was spun onto a slide for CTC enumeration. Both the microfilter and the slide with released cells were then subjected to immunofluorescence staining for pan-cytokeratin and CD45, markers for CTCs identification. As data shown in Supplementary Fig. 5, the CTC enumeration using PIPAAm coated slot filter or round-pore filter in parallel samples were comparable. Thus, with the PIPAAm coating, we showed the ability to recover CTCs captured from human patient samples with a high retrieval rate.
Conclusions
Circulating Tumor Cells (CTCs) have been established as an important biomarker in cancer and they may play promising roles in cancer management. Molecular and cellular analyses of CTCs can reveal valuable information for cancer prognosis and may help drive precision medicine. Several platforms commonly used in CTC capture and analyses have limitations in allowing for molecular and cellular analyses of CTCs since the cells captured are either fixed or immobilized. Further, affinity-based systems, which allow for viable CTCs capture and release, can potentially be biased by the choice of target antigen. In contrast, by combining our antigen-agnostic filtration based platform with the versatile PIPAAm coating method, we can achieve capture and release of viable CTCs from blood at a high efficiency. This will lay the foundation for the in depth characterization of viable CTCs, including single cell phenotypic and genomic analysis as well as ex vivo CTCs culture. Based on our proof-of-principle demonstration with human patient samples, future work is under way to employ this technology for clinical applications.
Supplementary Material
Acknowledgments
This work was supported by NIH R01 5R01CA141077-04, R21 5R21CA182050-02, R33 1R21CA123027-01/03 (Cote), 1UC4DK104208-01 (Agarwal) and P30 CA051008-21 (Weiner) grants and DoD W81XWH-09-1-0050, TR000102-04 GHUCCTS-PCSP, and W81XWH-13-1-0327 (Albanese) grants. We thank all the patients who have donated blood samples to support this work. We thank Drs. Guiseppe Giaconne, Ritesh Parajuli, and Marc E. Lippman for their assistance in clinical sample acquirement, and Drs. Carmen Gomez, Ralf Landgraf, Stephan Züchner, Toumy Guettouche, Diana Lopez for their insightful discussions. Zheng Ao thanks partial support and assistance from the Sheila and David Fuente Graduate Program in Cancer Biology, Sylvester Comprehensive Cancer Center.
Notes and references
- 1.Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW, Hayes DF. The New England journal of medicine. 2004;351:781–791. doi: 10.1056/NEJMoa040766. [DOI] [PubMed] [Google Scholar]
- 2.de Bono JS, Scher HI, Montgomery RB, Parker C, Miller MC, Tissing H, Doyle GV, Terstappen LW, Pienta KJ, Raghavan D. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:6302–6309. doi: 10.1158/1078-0432.CCR-08-0872. [DOI] [PubMed] [Google Scholar]
- 3.Cohen SJ, Punt CJ, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY, Picus J, Morse MA, Mitchell E, Miller MC, Doyle GV, Tissing H, Terstappen LW, Meropol NJ. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 2009;20:1223–1229. doi: 10.1093/annonc/mdn786. [DOI] [PubMed] [Google Scholar]
- 4.Smerage JB, Barlow WE, Hortobagyi GN, Winer EP, Leyland-Jones B, Srkalovic G, Tejwani S, Schott AF, O’Rourke MA, Lew DL, Doyle GV, Gralow JR, Livingston RB, Hayes DF. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2014;32:3483–3489. doi: 10.1200/JCO.2014.56.2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Powell AA, Talasaz AH, Zhang H, Coram MA, Reddy A, Deng G, Telli ML, Advani RH, Carlson RW, Mollick JA, Sheth S, Kurian AW, Ford JM, Stockdale FE, Quake SR, Pease RF, Mindrinos MN, Bhanot G, Dairkee SH, Davis RW, Jeffrey SS. PloS one. 2012;7:e33788. doi: 10.1371/journal.pone.0033788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alix-Panabières C, Pantel K. Nature Reviews Cancer. 2014;14:623–631. doi: 10.1038/nrc3820. [DOI] [PubMed] [Google Scholar]
- 7.Zheng S, Lin H, Liu JQ, Balic M, Datar R, Cote RJ, Tai YC. Journal of chromatography A. 2007;1162:154–161. doi: 10.1016/j.chroma.2007.05.064. [DOI] [PubMed] [Google Scholar]
- 8.Xu T, Lu B, Tai YC, Goldkorn A. Cancer research. 2010;70:6420–6426. doi: 10.1158/0008-5472.CAN-10-0686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peeters DJ, De Laere B, Van den Eynden GG, Van Laere SJ, Rothe F, Ignatiadis M, Sieuwerts AM, Lambrechts D, Rutten A, van Dam PA, Pauwels P, Peeters M, Vermeulen PB, Dirix LY. British journal of cancer. 2013;108:1358–1367. doi: 10.1038/bjc.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, Desai R, Zhu H, Comaills V, Zheng Z, Wittner BS, Stojanov P, Brachtel E, Sgroi D, Kapur R, Shioda T, Ting DT, Ramaswamy S, Getz G, Iafrate AJ, Benes C, Toner M, Maheswaran S, Haber DA. Science. 2014;345:216–220. doi: 10.1126/science.1253533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng Y, Zhang Y, Sun S, Wang Z, Wang M, Yu B, Czajkowsky DM, Liu B, Li Y, Wei W, Shi Q. Scientific reports. 2014;4:7499. doi: 10.1038/srep07499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hou S, Zhao H, Zhao L, Shen Q, Wei KS, Suh DY, Nakao A, Garcia MA, Song M, Lee T, Xiong B, Luo SC, Tseng HR, Yu HH. Advanced materials. 2013;25:1547–1551. doi: 10.1002/adma.201203185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiao Y, Zhou H, Xuan N, Cheng M, Rao Y, Luo Y, Wang B, Tang R. ACS applied materials & interfaces. 2014 doi: 10.1021/am505072z. [DOI] [PubMed] [Google Scholar]
- 14.Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, Concannon KF, Donaldson MC, Sequist LV, Brachtel E, Sgroi D, Baselga J, Ramaswamy S, Toner M, Haber DA, Maheswaran S. Science. 2013;339:580–584. doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mach AJ, Kim JH, Arshi A, Hur SC, Di Carlo D. Lab on a chip. 2011;11:2827–2834. doi: 10.1039/c1lc20330d. [DOI] [PubMed] [Google Scholar]
- 16.Okano T, Bae YH, Jacobs H, Kim SW. Journal of Controlled Release. 1990;11:255–265. [Google Scholar]
- 17.Yamada N, Okano T, Sakai H, Karikusa F, Sawasaki Y, Sakurai Y. Die Makromolekulare Chemie, Rapid Communications. 1990;11:571–576. [Google Scholar]
- 18.Kumashiro Y, Yamato M, Okano T. Ann Biomed Eng. 2010;38:1977–1988. doi: 10.1007/s10439-010-0035-1. [DOI] [PubMed] [Google Scholar]
- 19.Agarwal A, Goss JA, Cho A, McCain ML, Parker KK. Lab on a chip. 2013;13:3599–3608. doi: 10.1039/c3lc50350j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, Timofeeva OA, Nealon C, Dakic A, Simic V, Haddad BR, Rhim JS, Dritschilo A, Riegel A, McBride A, Schlegel R. The American journal of pathology. 2012;180:599–607. doi: 10.1016/j.ajpath.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang TY, Yadav VG, De Leo S, Mohedas A, Rajalingam B, Chen CL, Selvarasah S, Dokmeci MR, Khademhosseini A. Langmuir. 2007;23:11718–11725. doi: 10.1021/la7017049. [DOI] [PubMed] [Google Scholar]
- 22.M. Kaminska, W. Okroj W Fau – Szymanski, W. Szymanski W Fau – Jakubowski, P. Jakubowski W Fau – Komorowski, A. Komorowski P Fau – Nosal, H. Nosal A Fau – Szymanowski, M. Szymanowski H Fau – Gazicki-Lipman, H. Gazicki-Lipman M Fau – Jerczynska, Z. Jerczynska H Fau – Pawlowska, B. Pawlowska Z Fau – Walkowiak and B. Walkowiak
- 23.Palechor-Ceron N, Suprynowicz FA, Upadhyay G, Dakic A, Minas T, Simic V, Johnson M, Albanese C, Schlegel R, Liu X. The American journal of pathology. 2013;183:1862–1870. doi: 10.1016/j.ajpath.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin HK, Zheng S, Williams AJ, Balic M, Groshen S, Scher HI, Fleisher M, Stadler W, Datar RH, Tai YC, Cote RJ. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:5011–5018. doi: 10.1158/1078-0432.CCR-10-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.