

Abstract
MenE is an o-succinylbenzoyl-CoA (OSB-CoA) synthetase in the bacterial menaquinone biosynthesis pathway and is a promising target for the development of novel antibacterial agents. The enzyme catalyzes CoA ligation via an acyl-adenylate intermediate, and we have previously reported tight-binding inhibitors of MenE based on stable acyl-sulfonyladenosine analogues of this intermediate, including OSB-AMS (1) which has an IC50 value of ≤ 25 nM for the Escherichia coli MenE. Herein, we show that OSB-AMS reduces menaquinone levels in S. aureus, consistent with its proposed mechanism of action, despite the observation that the antibacterial activity of OSB-AMS is ~1000-fold lower than the IC50 for enzyme inhibition. To inform the synthesis of MenE inhibitors with improved antibacterial activity, we have undertaken a structure–activity relationship (SAR) study stimulated by the knowledge that OSB-AMS can adopt two isomeric forms in which the OSB side chain exists either as an open-chain keto acid or a cyclic lactol. These studies revealed that negatively charged analogues of the keto-acid form bind, while neutral analogues do not, consistent with the hypothesis that the negatively-charged keto-acid form of OSB-AMS is the active isomer. X-ray crystallography and site-directed mutagenesis confirm the importance of a conserved arginine for binding the OSB carboxylate. Although most lactol isomers tested were inactive, a novel difluoroindanediol inhibitor (11) with improved antibacterial activity was discovered, providing a pathway toward the development of optimized MenE inhibitors in the future.
Introduction
Novel antibiotics are urgently needed to treat infections caused by drug-resistant strains of pathogens such as multi- and extensively drug-resistant Mycobacterium tuberculosis and methicillin-resistant Staphylococcus aureus (MRSA)[1–4]. Approximately one-third of the world's population is infected with active or latent M. tuberculosis and the mortality rate is growing rapidly in both developed and developing countries due to drug-resistant strains[5–7]. Furthermore, community-acquired MRSA is the cause of more than 7 million hospitalizations from skin and soft tissue infections annually in the US alone[8, 9]. Treatments of these infections are estimated to cost $10–20 billion annually in the US[10].
Lipoquinones such as ubiquinone (CoQ), phylloquinone, and menaquinone are biological quinones that play pivotal roles in all living organisms[11–16]. CoQ is the primary redox-active electron carrier in the electron transport chain in humans and most aerobically growing Gram-negative bacteria[17, 18], while phylloquinone plays this role in photosynthetic organisms[19]. Menaquinone is the sole electron transport chain quinone in Gram-positive bacteria, Mycobacteria spp., and all anaerobically growing bacteria regardless of Gram-variability[20–22]. Although menaquinone also serves as a co-factor to many vitamin K-dependent enzymes in humans such as γ-glutamyl carboxylases[18, 23], de novo biosynthesis of menaquinone only occurs in certain prokaryotes while humans obtain menaquinone almost exclusively through diet (ingestion of phylloquinone[24]) or from gut flora. Thus, enzymes in the menaquinone biosynthetic pathway are attractive therapeutic targets for treating diseases caused by menaquinone-producing pathogens.
Due to the unique and essential role of menaquinone in bacterial growth, virulence, and survival, inhibitors of menaquinone biosynthesis have been developed for use as chemical tools to interrogate pathway essentiality and as a prelude to novel therapeutic discovery. Organisms such as M. tuberculosis and S. aureus synthesize menaquinone from chorismate via a pathway that includes at least nine distinct enzymes (Scheme 1)[16, 25–27]. Inhibitors of MenD[28, 29], MenC[30], MenE[31], MenB[32, 33], and MenA[34–37] have been reported. These pharmacological studies validate genetic approaches that have demonstrated the essentiality of men genes in bacteria including M. tuberculosis[38, 39] and Bacillus subtilis[25, 40], and suggest that inhibitors of menaquinone biosynthesis may lead to therapeutics that target both replicating as well as latent populations of M. tuberculosis[34, 35, 41, 42].
Scheme 1. Classical de novo menaquinone biosynthesis pathway.

This pathway consists of at least nine enzymes that catalyze the formation of menaquinone from chorismate[12, 13, 16]. The fifth enzyme, MenE, is an acyl-CoA ligase which ligates CoA to OSB via an OSB-AMP intermediate. (OSB = o-succinylbenzoate; AMP = adenosine monophosphate; DHNA-CoA = 1,4-dihydroxy-2-napthoyl-CoA)
Our own efforts have focused on MenE, the o-succinylbenzoyl-CoA synthetase that belongs to the ANL (acyl-CoA synthetase, nonribosomal peptide synthetase adenylation domain, luciferase)[43] family and catalyzes the ligation of CoA to o-succinylbenzoate (OSB) via an ordered Bi Uni Uni Bi Ping-Pong mechanism in the presence of a divalent cation and ATP (Scheme 1)[31, 44]. In the first half-reaction, MenE catalyzes the reaction of OSB and ATP to form a tightly-bound OSB-AMP intermediate coupled with release of inorganic pyrophosphate (PPi). In the second half-reaction, CoA attacks the mixed anhydride intermediate to form OSB-CoA with release of AMP[31, 44–47]. Genetic knockout and genome-wide transposon bioinformatic experiments have demonstrated that MenE is essential for the survival, growth, and virulence of Mycobacteria spp., Bacillus spp., Lactococcus lactis, and other pathogenic and nonpathogenic bacteria[38, 40, 48–50]. MenE homologs are also proposed to be essential in photosynthetic organisms such as Synechocystis spp. and Arabidopsis[51, 52].
We have previously described the development and characterization of MenE inhibitors based on acyl-AMS (5'-O-(N-acylsulfamoyl)adenosine) analogues known to inhibit adenylate-forming enzymes[44, 47]. We reported that OSB-AMS (1) is a tight-binding inhibitor of the S. aureus, M. tuberculosis, and E. coli MenE enzymes, with low-nM Kiapp values with respect to OSB[44]. We noted that a free carboxylate on the OSB side chain is required for potent MenE inhibition, and reported that OSB-AMS exists in equilibrium between an open chain keto-acid and a closed lactol form (Figure 1). To guide future modifications of the scaffold, we have now evaluated which form of the inhibitor is the active pharmacophore using analogues of OSB-AMS that mimic either the open keto-acid or closed lactol isomers in enzyme inhibition assays, binding assays, and X-ray crystallographic studies. We have also analyzed the mechanism of inhibition of these OSB-AMS analogues, and evaluated their antimicrobial activities against pathogenic bacteria.
Figure 1. OSB-AMS (1) analogs.

Open keto-acid and closed lactol isomers of OSB-AMS (1) are in equilibrium at physiological pH. We synthesized analogues (3–11) of both conformations to test the active pharmacophore. Compounds 3–8 are keto-acid analogues which favor the open-ring conformation and compounds 9–11 are lactol analogues which favor the close-ring structure. Compound 2 is a meta-carboxylate analogue to test the importance of the ortho position of the carboxylate. Synthesis of these analogues can be found in the supplemental section.
Materials and Methods
Sequence Alignment
To determine the sequence homology between the MenE enzymes from different organisms, the amino acid sequences of MenE from E. coli (K-12, ecMenE), S. aureus (RN4220, saMenE), and M. tuberculosis (Erdman, mtMenE) were aligned using INRA Multalin[53] and Alignment Annotator[54]. Overall, ecMenE shares 29% and 30% sequence identity with saMenE and mtMenE respectively, while mtMenE shares 26% sequence identity with saMenE. The same procedures were performed to align the acyl-CoA synthetase superfamily.
Site-directed mutagenesis
High fidelity Phusion polymerase (NEB) was used for standard PCR amplification and mutagenesis. Based on the nucleotide sequence of ecMenE (from K-12), the following primers were designed, synthesized, and used for the mutation of ecMenE:
|
| ||
| Mutation | Forward 5'–3' | Reverse 5'–3' |
|
| ||
| R195K | GGAATTATGTGGAAGTGGTTATACGC | GCGTTAAACCACTTCCACATAATTCC |
|
| ||
| R195Q | GGAATTATGTGGCAGTGGTTATACGC | GCGTATAACCACTGCCACATAATTC |
|
| ||
Circular Dichroism
CD experiments were performed using a Chirascan CD spectrometer. MenE was diluted to 10 μM in 350 μL 20 mM NaHPO4 buffer pH 7.4 containing 150 mM NaCl and 1 mM MgCl2. Far-UV wavelength (190–260 nm) spectra were collected in triplicate using a 1 mm cuvette in 1 nm increments. Data were collected using Chirascan software and reprocessed using MATLAB. The mean residue ellipticity (deg cm2 dmol−1 residue−1) was calculated using the following equation:
where the path length is in mm, protein concentration is in M, and n is the number of peptide bonds.
Enzyme assays
Enzyme inhibition studies were performed in 20 mM NaHPO4 buffer pH 7.4 containing 150 mM NaCl and 1 mM MgCl2 using a MenE-MenB coupled assay in which MenE is rate limiting[44, 47]. IC50 values were determined in reaction mixtures containing OSB (60 μM), ATP (240 μM), CoA (240 μM), mtMenB (2.5 μM) and varying inhibitor concentrations (5–250 μM). Reactions were initiated by addition of ecMenE (25 nM) and the production of DHNA-CoA was monitored at 392 nm (ε392 4000 M−1 cm−1). To determine kcat and KM, OSB was varied from 10–250 μM, while Kiapp was determined by varying both inhibitor and OSB from 5–250 μM and 60–350 μM, respectively.
X-ray crystallography
Co-crystallization trials used the ecMenE R195K mutant (~15 mg/mL) and ~200 μM OSB-AMS and initial crystallization conditions were obtained by sitting-drop vapor diffusion with Hampton Research HR2-110 buffer A6. The optimized growth condition was found to be 0.2 M MgCl2 (6·H2O), 0.1 M Tris HCl pH 8.5, 18–22% w/v PEG 4,000, with hanging-drop vapor diffusion at 298.15 K. Diffraction data were collected at 100 K at the X6A beamline at Brookhaven National Laboratories using an ADSC Q270 CCD detector. Data were indexed, integrated, and scaled using HKL2000[55], and the structure was solved by molecular replacement using MolRep[56]. The search model consisted of the large N-terminal domain of saMenE (PDB: 3IPL) and the small C-terminal domain of 4-chlorobenzoate:CoA synthetase (PDB: 1T5D). The model was refined through successive rounds of manual model building using COOT[57] and restrained refinement using REFMAC5[58]. The ligand was added directly to the difference Fourier map only after the refinement converged. Ligand restraints were generated using the PRODRG server[59]. The data collection and refinement statistics are provided in Table S1 and Table S2, respectively.
Isothermal titration calorimetry
Isothermal titration calorimetry was performed at 22 °C using a MicroCal VP-ITC instrument. A 1 mM solution of OSB-AMS (1) (dissolved in 20 mM NaHPO4 buffer pH 7.4 containing 150 mM NaCl and 1 mM MgCl2 at 22 °C) was titrated in 4 μL increments into the 1.8 mL cell containing a 25 μM solution of ecMenE (wild-type, R195K, or R195Q) in the same buffer. The data were fit to a single binding site model with the Origin software package.
Fluorescence binding assays
ITC experiments with difluoroindanediol 11 did not show a measurable change in enthalpy, and thus ITC was unable to quantify the binding of this compound to the enzyme. Instead, to determine the Kd for 11, we used a direct binding assay in which the change in the intrinsic tryptophan fluorescence of ecMenE was monitored (see Figure S1 for the binding isotherm). A solution of 50 μM 11 was titrated into 300 nM ecMenE in 20 mM NaHPO4 buffer pH 7.4 containing 150 mM NaCl and 1 mM MgCl2 at 25 °C. The solution was stirred continuously and fluorescence measurements were made with a Quanta Master fluorimeter using excitation and emission wavelengths of 280 and 332 nm, respectively. Slit widths were optimized to 4 and 2 nm for excitation and emission, respectively. Data were corrected for the inner filter effect and then fit to the following equation using MATLAB:
Antibacterial activity
Minimum inhibitory concentrations (MIC) were determined using visual growth inspection of cells grown in transparent 96-well plates. E. coli, B. subtilis (ATCC 6051), S. aureus (ATCC BAA-1762), and M. tuberculosis (H37Rv) were grown to mid-log phase (OD600 of 0.6–0.8) in LB, Miller Hinton, synthetic broth, or 7H9 with 0.5% Glycerol, 0.05% Tween, and 10%OADC media at 37 °C in an orbital shaker. A final inoculum concentration of 1–2 × 106 cells per well was treated with inhibitor at final concentrations ranging from 0.5 – 500 μg/mL. The MIC was defined as the minimum concentration at which a well showed no obvious growth by visual inspection (MIC-99). Growth rescue studies were performed by supplementing minimal media (synthetic broth) with 10 μM menaquinone-4 (MK4) and following the same procedure.
Cytotoxicity
To obtain insight into the potential cytotoxicity of our MenE inhibitors, we evaluated the in vitro cytotoxicity using Vero monkey kidney cells. Briefly, 105 cells/well were aliquoted into 96-well culture plates in serum rich media. The cells were incubated for 24–36 h at 37 °C in 5% CO2. The media was then aspirated and replaced with 200 μL of serum-free fresh media. Cells were incubated for 5 h at 37 °C in 5% CO2, after which compounds dissolved in serum-free cell media were added, giving a concentration range of 0.97 – 250 μg/mL. The cells were incubated for 24 – 36 h at 37 °C in 5% CO2. Cell death was assessed by incubating 20 μL of cell suspension from each well with 20 μL Trypan Blue for 5 min. The ratio of viable/dead cells was determined using a hemocytometer where stained cells were scored as dead and non-stained cells were scored as viable. The cytotoxic concentration was defined as the minimum inhibitor concentration which gave ~90% cell death.
Quantification of menaquinone levels in S. aureus
The effect of MenE inhibitors on menaquinone levels in S. aureus was determined as follows. Cultures of S. aureus ATCC BAA-1762 (5 mL synthetic broth medium with 10% glucose) were incubated overnight in a 37 °C shaker in the presence or absence of OSB-AMS below its MIC (15.6 μg/mL). The Blight and Dyer (1959) lipid extraction protocol was used to isolate the menaquinone-containing fraction from the cells[60]. Briefly, 0.75 mL of 1:2 (v/v) CHCl3:MeOH was added to 0.2 mL of culture. The mixture was vortexed thoroughly, and 0.25 mL of CHCl3 was added followed by further vortexing after which 0.25 mL of H2O was added. The mixture was then vortexed and centrifuged at 500 × g for 5 minutes at rt. The bottom phase was recovered, transferred to a glass vial and 200 μL was analyzed by APCI LC-MS/MS in positive ion mode using a Thermo TSQ Quantum Access (Thermo-Fisher) Triple Quadrupole Mass Spectrometer. Menaquinone levels were quantified using standard curves based on menaquinone-4 (MK4) or menaquinone-9 (MK9) (Sigma). Samples were introduced into the mass spectrometer by flow injection at 100 μL/min with 2:1 MeOH/CHCl3 as the solvent. Multiple Reaction Monitoring (MRM) was performed at 30 eV and MK4, MK5 and MK6 were quantified using the standard curve for MK4 whereas MK7, MK8, and MK9 were quantified using MK9. Ubiquinone-4 (CoQ4) was used as an internal standard and experiments were performed in triplicate.
Results and Discussion
Synthesis of OSB-AMS Analogues
OSB-AMS has a carboxylate group ortho to the succinyl substituent that can undergo cyclization through attack of the carboxylate on the succinyl ketone to generate a lactol isomer (Figure 1). To explore the importance of this isomeric equilibrium for enzyme inhibition and to investigate SAR toward the development of optimized MenE inhibitors, we synthesized one analogue in which the carboxylate and succinyl substituents were meta to one another (MSB-AMS, 2) as well as a series of keto-acid (3–8) and lactol analogues (9–11) of OSB-AMS (Figure 1, Scheme S1).
Synthesis of MSB-AMS proceeded by a Suzuki coupling of previously reported vinyl bromide 12[44, 47, 61] and aryl boronic acid 13, followed by ozonolysis and deprotection of the t-butyl ester to give the acyl side chain 15. The acyl side chain was then coupled to protected AMS scaffold 16, prepared as previously described[44, 47, 61], followed by global deprotection to give MSB-AMS (2). OSB-AMS analogues 3–9 were synthesized analogously using this general method of initial synthesis of the acyl side chain, subsequent coupling to the protected AMS scaffold 16, and global deprotection.
Alternative synthetic strategies (Scheme S1f, g) were necessary to access the lactam (10) and difluoroindanediol (11) analogues due to reactivity associated with their individual structures. Briefly, the lactam analogue 10 was synthesized from TBS-protected MeOSB-AMS analogue 19 (Scheme S1f), accessed via previously reported synthetic steps[44, 47, 61]. This intermediate was treated with anhydrous ammonia followed by global deprotection to yield lactam 10. Difluoroindanediol 11 was synthesized by alkylation of difluoroindandione 20 followed by deprotection to give the acyl side chain 22. This acyl side chain was then coupled to the protected AMS scaffold 16 to give intermediate 23 before subsequent reduction, and global deprotection to afford difluoroindanediol 11.
Inhibition of MenE
We next determined IC50 values for inhibition of E. coli MenE (ecMenE) by compounds 1–11 using a MenE-MenB coupled assay (Table 1). MSB-AMS (2), as well as the oxazole (5), nitro (6), boronic acid (7), and trifluoroethanol (8) analogues of the keto acid form, and the lactone (9) and lactam (10) analogues of the lactol form, showed no inhibition of ecMenE up to a concentration of 100 μM. In contrast, the tetrazole analogue 3 inhibited ecMenE with an IC50 of 2.2 ± 0.4 μM, while the squarate analogue 4 showed more potent inhibition with an IC50 of 0.17 ± 0.05 μM. Interestingly, the difluoroindanediol analogue 11 was also an effective inhibitor with an IC50 of 1.5 ± 0.1 μM and a Kd of 120 ± 23 nM was determined using a fluorescence perturbation binding assay (Figure S1). These results indicate that analogues of both the open and closed isomers are able to inhibit MenE. Crucially, analogues 3 and 4 have acidic protons on the ortho substituent (tetrazole 3 pKa = 3.4, squaric acid 4 pKa = 1.3) such that these groups carry a negative charge at neutral pH like the natural OSB substrate. These data suggest that enzyme inhibition requires a negative charge in the inhibitor close to the position normally occupied by the OSB carboxylate group. Difluoroindanediol 11 has a pKa of 11.5 and may also be deprotonated when bound to the enzyme. Furthermore, we believe that the net neutral charge of the nitro analogue 6 substantially decreases its ability to hydrogen bond or form ionic interactions despite being a respectable carboxylate bioisostere when interacting with a metal cofactor[62, 63]. Finally, we speculate that the boronic acid analogue (7) (pKa > 13) forms a boronate complex with the ortho-ketone on the succinate chain, raising the pKa of the boronic acid such that the inhibitor is uncharged at neutral pH.
Table 1.
In vitro enzyme inhibition of OSB-AMS analogues using ecMenE
| Inhibitor1 | IC50 (μM)2 | pKa3 | |
|---|---|---|---|
| 1 (OSB-AMS) |

|
0.025 ± 0.005 | 4 |
| 2 (MSB-AMS) |

|
> 100 | 4 |
| 3 (tetrazole) |

|
2.2 ± 0.4 | 3.4 |
| 4 (squarate) |

|
0.17 ± 0.05 | 1.3 |
| 5 (oxazole) |

|
> 100 | NA |
| 6 (nitro) |

|
> 100 | NA |
| 7 (boronic acid) |

|
> 100 | > 14 |
| 8 (trifluoroethanol) |

|
> 100 | 11.5 |
| 9 (lactone) |

|
> 100 | NA |
| 10 (lactam) |

|
> 100 | NA |
| 11 (difluoroindanediol) |

|
1.5 ± 0.1 | 11.5 |
MSB-AMS 2 is an isomeric meta- substituted analogue. Analogues 3–8 are mimics of the keto-acid isomer of OSB-AMS while analogues 9–11 are mimics of the lactol isomer.
IC50 values for inhibition of E. coli MenE (ecMenE). All IC50 measurements were performed in triplicate.
pKa of the OSB analogue. NA, not applicable.
Role of R195 in OSB-CoA formation and inhibition of MenE
We previously used computational docking to build a model of OSB-AMS (1) bound to saMenE[44]. Using this approach, we identified R222 within 4.7 Å of the OSB carboxylate in the active site of saMenE. Sequence alignment reveals that R222 is conserved in other MenE homologs and corresponds to R90 in mtMenE and R195 in ecMenE (Figure 2). To explore the role of this conserved residue, we replaced R195 in ecMenE with Lys or Gln by site-directed mutagenesis. Circular dichroism spectra of these mutants showed no significant alteration in the secondary structure (Figure S2). We then analyzed the change in catalytic efficiency (kcat/KM) of the mutant enzymes compared to wild-type ecMenE (Table 2) using the MenE-MenB coupled assay[64]. These studies revealed that the kcat/KM decreased by 15-fold for R195K MenE compared to wild-type MenE, while the R195Q mutant had no detectable activity up to an OSB concentration of 240 μM. Further analysis demonstrated that the effect of the R195K mutation on activity was primarily a result of a 16-fold increase in KM for OSB while kcat for product formation was unchanged (Table 2).
Figure 2. Sequence alignment of MenE homologs from pathogenic bacteria.

E. coli (K-12), S. aureus (MRSA, Rosenbach), M. tuberculosis (H37Rv) were aligned using INRA hierarchical clustering[53] and Alignment Annotator[54]. Blue arrow indicates the conserved arginine identified in the active site of saMenE (R222) by docking OSB-AMS into the crystal structure of apo saMenE[44]. The homologous residue in ecMenE is R195 and R90 in mtMenE. Red arrows indicate relevant residues interacting with OSB-AMS seen in the ecMenE crystal structure and conserved in saMenE and mtMenE.
Table 2.
Catalytic parameters and ITC data for the interaction of OSB and OSB-AMS with wt and mutant ecMenE
| ecMenE | (μM)1 | kcat (min−1)1 | kcat/KM (μM−1min−1)1 | (nM)2 | ΔH (kcal/mol)2 | ΔG (kcal/mol)2 | ΔΔG (kcal/mol)2 |
|---|---|---|---|---|---|---|---|
| wt | 1 ± 0.02 | 46 ± 0.12 | 46 ± 0.023 | 44 ± 11 | −2.0 ± 0.1 | −10.0 | |
| R195K | 16 ± 1.4 | 47 ± 0.33 | 3 ± 0.2 | 394 ± 36 | −2.5 ± 0.20 | −8.8 | 1.2 |
| R195Q | Not Active | 4500 ± 112 | −3.1 ± 0.10 | −7.3 | 2.7 |
Kinetic parameters were obtained using the MenE-MenB coupled assay[12, 64]. Measurements were performed in 20 mM NaHPO4 buffer pH 7.4 containing 150 mM NaCl and 1 mM MgCl2 at 25°C. kcat and KM values are reported for wild-type and R195K MenE. Replacement of R195 with lysine resulted in a 15-fold decrease in kcat/KM. No activity could be detected for the R195Q mutant even at an OSB concentration of 240 μM. All measurements were performed in triplicate.
Binding of OSB-AMS to ecMenE determined by ITC. A 1 mM solution of inhibitor (dissolved in 20 mM NaHPO4 buffer pH 7.4 containing 150 mM NaCl and 1 mM MgCl2 at 25°C) was titrated in 4 μL increments into the 1.8 mL cell containing 25 μM solution of ecMenE in the same buffer. The data were fit to a single binding site model. Measurements were made in triplicate. Replacement of R195 with Lys and Gln results in a ~ 10 and 100-fold increase in Kd of the inhibitor for the enzyme, respectively.
To investigate the role of R195 in enzyme inhibition, we evaluated the binding of OSB-AMS to ecMenE R195K and R195Q by isothermal calorimetry and demonstrated that replacement of R195 with Lys or Gln decreased binding affinity of the inhibitor to ecMenE by 9- or 102-fold, respectively (Table 2, Figure S3).
X-ray crystal structure of the ecMenE (R195K)·OSB-AMS complex
To underpin our efforts to develop potent MenE inhibitors and extend the modeling studies with saMenE, we set out to obtain the X-ray structure of MenE in complex with OSB-AMS (1). These efforts were most successful with the R195K mutant of ecMenE resulting in a 2.4 Å resolution structure of R195K ecMenE co-crystallized with OSB-AMS (PDB: 5C5H). The structure was solved by molecular replacement using the structures of saMenE and 4-chlorobenzoate:CoA ligase (CBAL) from Alcaligenes sp. AL3007 (PDB: 3IPL and 1T5D, ~29% sequence identity) as search models. Data collection and refinement statistics are given in Table S1 and Table S2, respectively.
MenE is a member of the adenylate-forming enzyme superfamily in which ATP is used to activate a carboxylate for subsequent attack of a nucleophile. One of the best characterized members of this family is CBAL, which has been extensively studied by Gulick, Dunaway-Mariano, and colleagues[65–67]. Both MenE and CBAL are comprised of a larger N-terminal domain and a smaller C-terminal domain, and structures of CBAL in complex with an adenylate intermediate as well as CoA thioester product analogue reveal that ligand binding causes the two domains to move relative to each other as the reaction proceeds. Domain alternation reconfigures the active site from a conformation that catalyzes acyl-adenylate formation to one that facilitates CoA binding and thioester formation[68–70].
In Figure 3, we show the structure of the OSB-AMS:ecMenE·complex overlayed with that of apo saMenE (PDB: 3IPL). These structures differ in the relative orientations of domains 1 and 2. However both structures are representative of the adenylate-bound conformation observed for CBAL (PDB: 3CW8), in which G408 in region A8 (399-GRVDDMIISG-408) is removed from the active site whereas K492 in region A10 (486-PKNALNK-492) is located in the active site[65–67]. The corresponding residues in ecMenE (saMenE) are G358 (G402) and K437 (K483) (Figure S4), and in Figure 3 it can be seen that G358/G402 are located away from the MenE binding site, whereas K437 is close to the bound OSB-AMS in ecMenE. Note that K483 is disordered in the structure of saMenE.
Figure 3. Overlaid structures of OSB-AMS: R195K ecMenE and apo saMenE.

Structure overlay of the OSB-AMS:ecMenE complex with apo saMenE (3IPL.pdb). These structures differ in the relative orientation of large domain 1 (silver) and small domain 2 (beige for E. coli and cyan for S. aureus), but represent the adenylate-bound conformation in which G358/G402 (yellow) in the A8 core motif is removed from the active site whereas the K437/K483 (red) is located in the active site. G358 and K437 are residues from E. coli MenE. G402 and K483 are residues from S. aureus. K483 is disordered in the S. aureus structure.
OSB-AMS binding site and keto-acid isomeric form of the inhibitor
Residues that interact with OSB-AMS (1) are highlighted in Figure 4 and Figure S5 and include T142, H186, S188, K195 (R195), S222, T272, D336, R350, and K437, which are all conserved in E. coli, S. aureus, and M. tuberculosis MenE (Figure 2). Residues T142 (Motif 1, A3, P-loop), T272 (Motif II, A5), D336 (Motif III, A7), R350 (A8, hinge) and K437 (A10) are components of the conserved sequence motifs in the adenylate-forming enzyme superfamily (Figure S4) and are, thus, involved in the general chemical reaction that leads to acyl-adenylate formation. Residues S188, K195 (R195), S222 and T277 are clustered around the OSB portion of OSB-AMS and likely confer substrate specificity upon MenE.
Figure 4. X-ray structure of OSB-AMS: R195K ecMenE showing interactions with OSB-AMS (A).
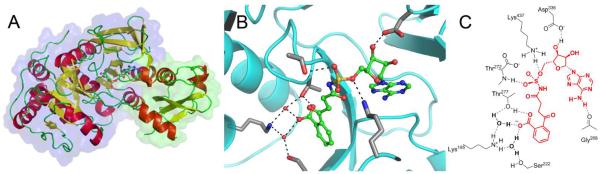
The overall structure of ecMenE:OSB-AMS is shown with the larger N-terminal (domain I) and the smaller C-terminal domain (domain II) highlighted by transparent surface representations in blue and green, respectively. The ligand is provided in ball-and-stick representation. (B) The structure of the bound ligand, OSB-AMS, is shown in the active site. The ligand with green carbon atoms in ball-and-stick representation and side chains that hydrogen bond with the ligand are shown with grey carbon atoms. (C) A schematic of OSB-AMS in the ecMenE active site. The ecMenE sidechains interacting with OSB-AMS are shown in black, the ligand is shown in red, and hydrogen bonding interactions are illustrated with dashed lines.
The electron density of the OSB-AMS ligand is well-defined and consistent with the keto-acid isomer rather than the lactol isomer (Figure S6). In addition, the OSB carboxylate interacts with K195 via a water-mediated ionic bridge comprised of two conserved water molecules (Figure 4). We posit that R195 in wild-type EcMenE also participates in this water mediated interaction, although a direct interaction with the OSB carboxylate cannot be ruled out. In either case, the X-ray structure is consistent with our model of OSB-AMS bound to saMenE[44] as well as the site-directed mutagenesis studies reported above. In particular, the experimentally observed change in binding free energy (ΔΔG) of OSB-AMS to ecMenE is consistent with the removal of one (R195K) or two (R195Q) water-mediated hydrogen bond interactions with the ligand, suggesting that the R195 guanadinium group in wild-type ecMenE makes two interactions with the OSB carboxylate moiety. These studies further support the notion that the OSB substrate binds to MenE as its open-chain keto acid isomer.
Antibacterial activity and cytotoxicity
To assess the antibacterial activity of the biochemically-active MenE inhibitors OSB-AMS (1), tetrazole 3, squarate 4, and difluoroindanediol 11, we determined their ability to inhibit growth of B. subtilis, MRSA, M. tuberculosis and E. coli using a 96-well plate inhibitor screening assay (Table 3). E. coli was included as a control since this Gram-negative organism does not produce menaquinone under aerobic conditions[71] and, as expected, none of the compounds inhibited growth of E. coli up to 500 μg/mL concentration. Meanwhile, OSB-AMS (1) had MIC values of 62.5, 31.25, and 125 μg/mL against B. subtilis, MRSA, and M. tuberculosis, respectively. Notably, while tetrazole 3 and squarate 4 showed little or no antibacterial activity (MIC ≥ 250 μg/mL), difluoroindanediol 11 had MIC values of 31.25, 15.6, and 15.6 μg/mL against B. subtilis, MRSA and M. tuberculosis, respectively, 2- to 8-fold more potent than OSB-AMS. One possibility is that the negatively charged sidechain of OSB-AMS (1), tetrazole 3, and squarate 4 limits their bacterial permeability and, hence, their antimicrobial activities, despite their potent biochemical activities (Table 1). In contrast, difluoroindanediol 11 has a sidechain pKa of 11.5 (presumed to correspond to the secondary alcohol) and is neutral at physiological pH, which may afford improved cell permeability and, hence, increased antibacterial activity, despite its 60-fold less potent biochemical inhibition of ecMenE compared to OSB-AMS. We note that the acyl sulfamate is expected to be negatively charged in all cases (pKa ~ 1.5–2.0).
Table 3.
Antimicrobial and cytotoxic activity of lead OSB-AMS analogues.
| Inhibitor | MIC (μg/mL)1 | MIC + MK4 (μg/mL)2 | Cytotoxicity (μg/mL)3 | ||||
|---|---|---|---|---|---|---|---|
| E. coli | B. subtilis | MRSA | M. tuberculosis | MRSA | M. tuberculosis | Vero | |
| 1 | > 250 | 62.5 | 31.25 | 125 | > 250 | > 250 | 125 |
| 3 | > 250 | 250 | > 500 | ND | > 500 | ND | ND |
| 4 | > 250 | 250 | > 500 | ND | > 500 | ND | ND |
| 11 | > 250 | 31.25 | 15.6 | 15.6 | > 250 | > 250 | > 250 |
MIC values were obtained against E. coli (K-12), B. subtilis (ATCC 6057), methicillin-resistant S. aureus (ATCC BAA-1762), and M. tuberculosis (H37Rv). Inoculum levels for each MIC measurement ranged from 1×106 to 2 ×106 cells/mL. All MICs were performed in technical and experimental triplicate. ND, not determined.
MICs performed with exogenous 10 μg/mL MK4 added to the synthetic growth media.
Cytotoxicity values were obtained against Vero (monkey kidney epithelial) cells. Measurements were performed in technical and experimental triplicate.
Finally, the antibacterial activity of the four MenE inhibitors was assessed in the presence of menaquinone-4 (MK4, 10 μM). All bacteria that were sensitive to the MenE inhibitors were rescued by supplementation with MK4, supporting the target specificity and mechanism of action of the inhibitors (Table 3). The cytotoxicity of OSB-AMS and difluoroindanediol 11 in Vero cells was 125 and >250 μg/mL, respectively (Table 3). Thus, difluoroindanediol 11 exhibits useful selectivity for antibacterial activity over mammalian cell cytotoxicity.
Effect of OSB-AMS on menaquinone levels in S. aureus
To provide direct insight into the mode of action of the MenE inhibitors, we analyzed the effect of OSB-AMS on menaquinone levels in S. aureus by LC-MS/MS (Figure 5). S. aureus produces a series of menaquinones that differ in the number of isoprene units that comprise the side chain. In our analysis, menaquinone-8 (MK8) was the major species with significant quantities of MK7 and MK9. Treatment of S. aureus with OSB-AMS resulted in a ~3–5 fold decrease in the levels of all menaquinones, consistent with the antibacterial activity of this compound resulting from a direct effect on menaquinone biosynthesis.
Figure 5. Effect of OSB-AMS on menaquinone levels in MRSA.

Menaquinone levels were quantified by LC-MS/MS using standard curves generated with MK4 and MK9 (see experimental methods section for more details). A distribution of MKs are present in untreated MRSA with MK8 the most abundant, consistent with other reports[72]. Treatment with 15.6 μM OSB-AMS (half-MIC) results in a ~60% decrease in MK levels consistent with MenE inhibition. Error bar shown as experimental triplicates.
Discussion
Menaquinone is a lipid-soluble, redox-active cofactor in the bacterial respiratory chain of many pathogens including S. aureus and M. tuberculosis. Enzymes involved in menaquinone biosynthesis are attractive targets for antibacterial drug discovery, and we have previously reported rationally-designed inhibitors of MenE, an ATP-dependent ligase that catalyzes the formation of OSB-CoA in the pathway. Our acyl-sulfonyladenosine inhibitors are stable, non-hydrolyzable analogues of the acyl-adenylate intermediate that is formed during the MenE catalyzed reaction, and the parent inhibitor OSB-AMS (1) (Figure 1) is a nanomolar inhibitor of the MenE enzymes from E. coli, S. aureus, and M. tuberculosis[44]. However, OSB-AMS has relatively weak antibacterial activity, possibly due to issues with cell penetration/efflux, and consequently we have undertaken a series of studies herein to explore the mode of action of OSB-AMS and to generate SAR for the design of novel MenE inhibitors with improved antibacterial activity.
As expected, OSB-AMS had no effect on E. coli grown aerobically (MIC > 500 μg/mL), as this organism only utilizes menaquinone under anaerobic conditions. However, OSB-AMS also has only moderate antibacterial activity against menaquinone-dependent bacteria, with MIC values of 30–125 μg/mL against S. aureus, M. tuberculosis and B. subtilis, in contrast to its potent biochemical inhibition of MenE. Despite this, there is evidence that OSB-AMS does directly affect menaquinone biosynthesis in these organisms: sub-MIC concentrations of OSB-AMS reduce the level of menaquinone in S. aureus, and the antibacterial activity of OSB-AMS can be complemented by the addition of MK4 to the media. We speculated that the disconnect between enzyme inhibition and antibacterial activity might be a consequence of poor cell penetration and/or efflux, and thus we synthesized analogues of OSB-AMS to explore further the relationship between MenE biochemical inhibition and cellular antimicrobial activity. Because OSB-AMS can exist in two isomeric forms (Figure 1), a specific goal of our studies was to determine the bioactive isomer of our lead compound.
Previous modeling studies using the apo structure of saMenE identified a putative interaction between a conserved arginine (R222 in saMenE, R195 in ecMenE) and the OSB carboxylate. Site-directed mutagenesis supports the importance of R195 for both substrate ligation and MenE inhibition: replacement of R195 in ecMenE with Lys resulted in a 16-fold increase in Km for OSB, while an R195Q mutant had no detectable catalytic activity. In addition, the affinity of OSB-AMS for ecMenE decreased 9- and 102-fold for the R195K and R195Q ecMenE mutants, respectively, supporting a direct interaction between K195 and the OSB carboxylate. Further weight to this conclusion can now be provided by the structure of OSB-AMS bound to the R195K ecMenE mutant reported here, which is the first liganded structure of a MenE enzyme. Our structure reveals a water-mediated interaction between K195 and the OSB-AMS carboxylate, which we posit is also present in enzyme–inhibitor and enzyme–substrate complexes involving wild-type MenE. Finally, the importance of the ortho-carboxylate group was also evaluated by synthesizing the meta-substituted congener MSB-AMS (2). This analogue did not affect MenE activity up to a concentration of 250 μM.
In addition to substantiating the importance of R195, the X-ray structure of the ecMenE(R195K)·OSB-AMS complex also provides insight into the active isomeric form of the inhibitor: electron density of the ligand is consistent with the keto-acid isomer of OSB-AMS (Figure S6). To further explore the importance of the keto-acid versus lactol isomers of OSB-AMS for enzyme inhibition, we synthesized a series of OSB-AMS analogues that mimic either of the two isomeric species. OSB-AMS has an IC50 value of 25 nM for ecMenE, and we discovered analogues of both the keto-acid and lactol isomers that inhibit the enzyme with IC50 values in the 0.2–2 μM range. Given the key interaction between the OSB carboxylate and R(K)195 in the active site of MenE revealed by structural studies and site-directed mutagenesis, we speculate that the presence of a negatively charged substituent in tetrazole 3 and squarate 4 is likely critical for inhibition of MenE. In addition, although the pKa of difluoroindanediol 11 is 11.5, it is possible that one of the hydroxyl groups could be deprotonated when bound in the MenE active site and interact with R(K)195. However, the corresponding open-chain trifluoroethanol analogue 8 also has a side chain pKa of 11.5 but does not inhibit MenE, suggesting an alternative scenario in which difluoroindanediol 11 may adopt a distinct binding mode compared to OSB-AMS. The difluoroindanediol lactol analogue 11 is particularly interesting as it exhibits promising antibacterial activity (MIC 15–30 μg/mL) despite inhibiting ecMenE 60-fold less potently than OSB-AMS. This may result from improved cell penetration as the difluoroindanediol is expected to be neutral at physiological pH. Taken together, our studies provide critical mechanistic understanding that will drive future efforts to develop MenE inhibitors with improved enzyme inhibition and cellular activity.
Supplementary Material
Acknowledgements
We thank Dr. Stephen Walker for assistance with the BSL2 and cell culture studies, Dr. Robert Rieger from the Proteomics Center at Stony Brook University for MK quantification and analysis, Dr. Huilin Li (BNL/SBU) for assistance with protein crystallization, and Dr. George Sukenick, Rong Wang, Dr. Hui Liu, Hui Fang, and Dr. Sylvi Rusli (MSKCC) for expert NMR and mass spectrometry support. X-ray data collection was performed at beamline X6A of the National Synchrotron Light Source in Brookhaven National Laboratories and we thank Vivian Stojanoff, Jean Jakoncic, and Edwin Lazo (BNL) for assistance with synchrotron data collection.
Funding: This work was supported by the NIH (R01 GM100477 to DST, R01 GM102864 to PJT, T32 GM073546–Gross to CEE, P41 GM103403–Ealick to KR, and CCSG P30 CA008748–Thompson). The National Synchrotron Light Source and is supported by NIH agreement GM-00080 (supplement to a PSI program) and DOE contract DE-AC02-98CH10886.
Footnotes
Supporting Information This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Silver LL. Challenges of antibacterial discovery. Clin Microbiol Rev. 2011;24(1):71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simmons KJ, Chopra I, Fishwick CW. Structure-based discovery of antibacterial drugs. Nat Rev Microbiol. 2010;8(7):501–510. doi: 10.1038/nrmicro2349. [DOI] [PubMed] [Google Scholar]
- 3.Paul S, et al. New and emerging pathogens. Part 5. Those crazy cocci: more virulent and resistant to antibiotics than ever. MLO Med Lab Obs. 1996;28(6):40–42. 44. 46 passim. [PubMed] [Google Scholar]
- 4.Fischbach MA, Walsh CT. Antibiotics for emerging pathogens. Science. 2009;325(5944):1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harper C. Tuberculosis, a neglected opportunity? Nat Med. 2007;13(3):309–12. doi: 10.1038/nm0307-309. [DOI] [PubMed] [Google Scholar]
- 6.Nathan C. Drug-resistant tuberculosis: a new shot on goal. Nat Med. 2014;20(2):121–123. doi: 10.1038/nm.3470. [DOI] [PubMed] [Google Scholar]
- 7.Keener AB. Oldie but goodie: Repurposing penicillin for tuberculosis. Nat Med. 2014;20(9):976–978. doi: 10.1038/nm0914-976. [DOI] [PubMed] [Google Scholar]
- 8.McKenna M. Vaccine development: Man vs MRSA. Nature. 2012;482(7383):23–25. doi: 10.1038/482023a. [DOI] [PubMed] [Google Scholar]
- 9.Hersh AL, et al. National trends in ambulatory visits and antibiotic prescribing for skin and soft-tissue infections. Arch Intern Med. 2008;168(14):1585–1591. doi: 10.1001/archinte.168.14.1585. [DOI] [PubMed] [Google Scholar]
- 10.Zimlichman E, et al. Health care-associated infections: a meta-analysis of costs and financial impact on the US health care system. JAMA Intern Med. 2013;173(22):2039–2046. doi: 10.1001/jamainternmed.2013.9763. [DOI] [PubMed] [Google Scholar]
- 11.Pandya KP, King HK. Ubiquinone and menaquinone in bacteria: a comparative study of some bacterial respiratory systems. Arch Biochem Biophys. 1966;114(1):154–157. doi: 10.1016/0003-9861(66)90316-x. [DOI] [PubMed] [Google Scholar]
- 12.Bentley R, Meganathan R. Biosynthesis of vitamin K (menaquinone) in bacteria. Microbiol Rev. 1982;46(3):241–280. doi: 10.1128/mr.46.3.241-280.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meganathan R, Kwon O. Biosynthesis of Menaquinone (Vitamin K) and Ubiquinone (Coenzyme Q) Ecosal Plus. 2009;3 doi: 10.1128/ecosalplus.3.6.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furt F, et al. A bimodular oxidoreductase mediates the specific reduction of phylloquinone (vitamin K(1)) in chloroplasts. Plant J. 2010;64(1):38–46. doi: 10.1111/j.1365-313X.2010.04305.x. [DOI] [PubMed] [Google Scholar]
- 15.Martius C. The metabolic relationships between the different K vitamins and the synthesis of the ubiquinones. Am J Clin Nutr. 1961;9((4)Pt 2):97–103. doi: 10.1093/ajcn/9.4.97. [DOI] [PubMed] [Google Scholar]
- 16.Meganathan R. Biosynthesis of menaquinone (vitamin K2) and ubiquinone (coenzyme Q): a perspective on enzymatic mechanisms. Vitam Horm. 2001;61:173–218. doi: 10.1016/s0083-6729(01)61006-9. [DOI] [PubMed] [Google Scholar]
- 17.Wallace BJ, Young IG. Role of quinones in electron transport to oxygen and nitrate in Escherichia coli. Studies with a ubiA- menA- double quinone mutant. Biochim Biophys Acta. 1977;461(1):84–100. doi: 10.1016/0005-2728(77)90071-8. [DOI] [PubMed] [Google Scholar]
- 18.Dowd P, et al. Vitamin K and energy transduction: a base strength amplification mechanism. Science. 1995;269(5231):1684–1691. doi: 10.1126/science.7569894. [DOI] [PubMed] [Google Scholar]
- 19.Schultz G, Soll J. Biosynthesis of alpha-tocopherol (vitamin E), phylloquinone (2-methyl-3-phytylnaphtoquinone, vitamin K1) and other prenylquinones in plants. On the problem of inability of the biosynthesis in animals--a survey (author's transl) Dtsch Tierarztl Wochenschr. 1980;87(11):410–412. [PubMed] [Google Scholar]
- 20.Collins MD, Goodfellow M, Minnikin DE. Isoprenoid quinones in the classification of coryneform and related bacteria. J Gen Microbiol. 1979;110(1):127–136. doi: 10.1099/00221287-110-1-127. [DOI] [PubMed] [Google Scholar]
- 21.Nahaie MR, et al. Polar lipid and isoprenoid quinone composition in the classification of Staphylococcus. J Gen Microbiol. 1984;130(9):2427–2437. doi: 10.1099/00221287-130-9-2427. [DOI] [PubMed] [Google Scholar]
- 22.Hiratsuka T, et al. An alternative menaquinone biosynthetic pathway operating in microorganisms. Science. 2008;321(5896):1670–1673. doi: 10.1126/science.1160446. [DOI] [PubMed] [Google Scholar]
- 23.Shearer MJ, Newman P. Recent trends in the metabolism and cell biology of vitamin K with special reference to vitamin K cycling and MK-4 biosynthesis. J Lipid Res. 2014;55(3):345–362. doi: 10.1194/jlr.R045559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakagawa K, et al. Identification of UBIAD1 as a novel human menaquinone-4 biosynthetic enzyme. Nature. 2010;468(7320):117–121. doi: 10.1038/nature09464. [DOI] [PubMed] [Google Scholar]
- 25.Taber HW, Dellers EA, Lombardo LR. Menaquinone biosynthesis in Bacillus subtilis: isolation of men mutants and evidence for clustering of men genes. J Bacteriol. 1981;145(1):321–327. doi: 10.1128/jb.145.1.321-327.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burt VT, Pennock JF. Biosynthetic studies on the origin of the napthoquinone nucleus of menaquinone-4 (vitamin K) in the shore crab Carcinus maenas. Biochem Soc Trans. 1977;5(6):1714–1716. doi: 10.1042/bst0051714. [DOI] [PubMed] [Google Scholar]
- 27.Truglio JJ, et al. Crystal structure of Mycobacterium tuberculosis MenB, a key enzyme in vitamin K2 biosynthesis. J Biol Chem. 2003;278(43):42352–42360. doi: 10.1074/jbc.M307399200. [DOI] [PubMed] [Google Scholar]
- 28.Fang M, et al. Succinylphosphonate esters are competitive inhibitors of MenD that show active-site discrimination between homologous alpha-ketoglutarate-decarboxylating enzymes. Biochemistry. 2010;49(12):2672–2679. doi: 10.1021/bi901432d. [DOI] [PubMed] [Google Scholar]
- 29.Fang M, et al. Using substrate analogues to probe the kinetic mechanism and active site of Escherichia coli MenD. Biochemistry. 2011;50(40):8712–8721. doi: 10.1021/bi201202n. [DOI] [PubMed] [Google Scholar]
- 30.Pulaganti M, et al. Molecular modeling and docking studies of O-succinylbenzoate synthase of M. tuberculosis--a potential target for antituberculosis drug design. Appl Biochem Biotechnol. 2014;172(3):1407–1432. doi: 10.1007/s12010-013-0569-4. [DOI] [PubMed] [Google Scholar]
- 31.Tian Y, et al. Bacillus anthracis o-succinylbenzoyl-CoA synthetase: reaction kinetics and a novel inhibitor mimicking its reaction intermediate. Biochemistry. 2008;47(47):12434–12447. doi: 10.1021/bi801311d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li X, et al. CoA Adducts of 4-Oxo-4-Phenylbut-2-enoates: Inhibitors of MenB from the M. tuberculosis Menaquinone Biosynthesis Pathway. ACS Med Chem Lett. 2011;2(11):818–823. doi: 10.1021/ml200141e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X, et al. Synthesis and SAR studies of 1,4-benzoxazine MenB inhibitors: novel antibacterial agents against Mycobacterium tuberculosis. Bioorg Med Chem Lett. 2010;20(21):6306–6309. doi: 10.1016/j.bmcl.2010.08.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurosu M, Crick DC. MenA is a promising drug target for developing novel lead molecules to combat Mycobacterium tuberculosis. Med Chem. 2009;5(2):197–207. doi: 10.2174/157340609787582882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dhiman RK, et al. Menaquinone synthesis is critical for maintaining mycobacterial viability during exponential growth and recovery from non-replicating persistence. Mol Microbiol. 2009;72(1):85–97. doi: 10.1111/j.1365-2958.2009.06625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurosu M, Begari E. Vitamin K2 in electron transport system: are enzymes involved in vitamin K2 biosynthesis promising drug targets? Molecules. 2010;15(3):1531–1553. doi: 10.3390/molecules15031531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li K, et al. Multitarget drug discovery for tuberculosis and other infectious diseases. J Med Chem. 2014;57(7):3126–3139. doi: 10.1021/jm500131s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48(1):77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 39.Zhang YJ, et al. Global assessment of genomic regions required for growth in Mycobacterium tuberculosis. PLoS Pathog. 2012;8(9):e1002946. doi: 10.1371/journal.ppat.1002946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kobayashi K, et al. Essential Bacillus subtilis genes. Proc Natl Acad Sci U S A. 2003;100(8):4678–4683. doi: 10.1073/pnas.0730515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Debnath J, et al. Discovery of selective menaquinone biosynthesis inhibitors against Mycobacterium tuberculosis. J Med Chem. 2012;55(8):3739–3755. doi: 10.1021/jm201608g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gengenbacher M, et al. Nutrient-starved, non-replicating Mycobacterium tuberculosis requires respiration, ATP synthase and isocitrate lyase for maintenance of ATP homeostasis and viability. Microbiology. 2010;156(Pt 1):81–87. doi: 10.1099/mic.0.033084-0. [DOI] [PubMed] [Google Scholar]
- 43.Gulick AM. Conformational dynamics in the Acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem Biol. 2009;4(10):811–827. doi: 10.1021/cb900156h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu X, et al. Stable analogues of OSB-AMP: potent inhibitors of MenE, the o-succinylbenzoate-CoA synthetase from bacterial menaquinone biosynthesis. Chembiochem. 2012;13(1):129–136. doi: 10.1002/cbic.201100585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heide L, Arendt S, Leistner E. Enzymatic synthesis, characterization, and metabolism of the coenzyme A ester of o-succinylbenzoic acid, an intermediate in menaquinone (vitamin K2) biosynthesis. J Biol Chem. 1982;257(13):7396–7400. [PubMed] [Google Scholar]
- 46.Sharma V, Hudspeth ME, Meganathan R. Menaquinone (vitamin K2) biosynthesis: localization and characterization of the menE gene from Escherichia coli. Gene. 1996;168(1):43–48. doi: 10.1016/0378-1119(95)00721-0. [DOI] [PubMed] [Google Scholar]
- 47.Lu X, et al. Mechanism-based inhibitors of MenE, an acyl-CoA synthetase involved in bacterial menaquinone biosynthesis. Bioorg Med Chem Lett. 2008;18(22):5963–5966. doi: 10.1016/j.bmcl.2008.07.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akerley BJ, et al. A genome-scale analysis for identification of genes required for growth or survival of Haemophilus influenzae. Proc Natl Acad Sci U S A. 2002;99(2):966–971. doi: 10.1073/pnas.012602299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abicht HK, et al. Non-enzymic copper reduction by menaquinone enhances copper toxicity in Lactococcus lactis IL1403. Microbiology. 2013;159(Pt 6):1190–1197. doi: 10.1099/mic.0.066928-0. [DOI] [PubMed] [Google Scholar]
- 50.Chen WH, et al. OGEE: an online gene essentiality database. Nucleic Acids Res. 2012;40(Database issue):D901–906. doi: 10.1093/nar/gkr986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wade Johnson T, et al. The menD and menE homologs code for 2-succinyl-6-hydroxyl-2,4-cyclohexadiene-1-carboxylate synthase and O-succinylbenzoic acid-CoA synthase in the phylloquinone biosynthetic pathway of Synechocystis sp. PCC 6803. Biochim Biophys Acta. 2003;1557(1–3):67–76. doi: 10.1016/s0005-2728(02)00396-1. [DOI] [PubMed] [Google Scholar]
- 52.Kim HU, et al. The AAE14 gene encodes the Arabidopsis o-succinylbenzoyl-CoA ligase that is essential for phylloquinone synthesis and photosystem-I function. Plant J. 2008;54(2):272–283. doi: 10.1111/j.1365-313X.2008.03416.x. [DOI] [PubMed] [Google Scholar]
- 53.Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988;16(22):10881–10890. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gille C, et al. Alignment-Annotator web server: rendering and annotating sequence alignments. Nucleic Acids Res. 2014;42(Web Server issue):W3–6. doi: 10.1093/nar/gku400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 56.Vagin A, Teplyakov A. An approach to multi-copy search in molecular replacement. Acta Crystallogr D Biol Crystallogr. 2000;56(Pt 12):1622–1624. doi: 10.1107/s0907444900013780. [DOI] [PubMed] [Google Scholar]
- 57.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 58.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 59.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 8):1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 60.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37(8):911–917.. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 61.Cisar JS, et al. Exploiting ligand conformation in selective inhibition of non-ribosomal peptide synthetase amino acid adenylation with designed macrocyclic small molecules. J Am Chem Soc. 2007;129(25):7752–7753. doi: 10.1021/ja0721521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zakharian TY, Di Costanzo L, Christianson DW. (S)-2-amino-6-nitrohexanoic acid binds to human arginase I through multiple nitro-metal coordination interactions in the binuclear manganese cluster. J Am Chem Soc. 2008;130(51):17254–17255. doi: 10.1021/ja807702q. [DOI] [PubMed] [Google Scholar]
- 63.Kelly TR, Kim MH. Relative Binding-Affinity of Carboxylate and Its Isosteres - Nitro, Phosphate, Phosphonate, Sulfonate, and Delta-Lactone. Journal of the American Chemical Society. 1994;116(16):7072–7080. [Google Scholar]
- 64.Meganathan R, Bentley R. Menaquinone (vitamin K2) biosynthesis: conversion of o-succinylbenzoic acid to 1,4-dihydroxy-2-naphthoic acid by Mycobacterium phlei enzymes. J Bacteriol. 1979;140(1):92–98. doi: 10.1128/jb.140.1.92-98.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu R, et al. Mechanism of 4-chlorobenzoate:coenzyme a ligase catalysis. Biochemistry. 2008;47(31):8026–8039. doi: 10.1021/bi800698m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reger AS, et al. Structural characterization of a 140 degrees domain movement in the two-step reaction catalyzed by 4-chlorobenzoate:CoA ligase. Biochemistry. 2008;47(31):8016–8025. doi: 10.1021/bi800696y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu R, et al. The mechanism of domain alternation in the acyl-adenylate forming ligase superfamily member 4-chlorobenzoate: coenzyme A ligase. Biochemistry. 2009;48(19):4115–4125. doi: 10.1021/bi9002327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Branchini BR, et al. Bioluminescence is produced from a trapped firefly luciferase conformation predicted by the domain alternation mechanism. J Am Chem Soc. 2011;133(29):11088–11091. doi: 10.1021/ja2041496. [DOI] [PubMed] [Google Scholar]
- 69.Sundlov JA, et al. Crystal structure of firefly luciferase in a second catalytic conformation supports a domain alternation mechanism. Biochemistry. 2012;51(33):6493–6495. doi: 10.1021/bi300934s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bandarian V, et al. Domain alternation switches B(12)-dependent methionine synthase to the activation conformation. Nat Struct Biol. 2002;9(1):53–56. doi: 10.1038/nsb738. [DOI] [PubMed] [Google Scholar]
- 71.Newton NA, Cox GB, Gibson F. The function of menaquinone (vitamin K 2) in Escherichia coli K-12. Biochim Biophys Acta. 1971;244(1):155–166. doi: 10.1016/0304-4165(71)90132-2. [DOI] [PubMed] [Google Scholar]
- 72.Wakeman CA, et al. Menaquinone biosynthesis potentiates haem toxicity in Staphylococcus aureus. Mol Microbiol. 2012;86(6):1376–1392. doi: 10.1111/mmi.12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.