

Abstract
Selective serotonin reuptake inhibitors (SSRIs) are widely used antidepressants, but the mechanisms by which they influence behavior are only partially resolved. Adult hippocampal neurogenesis is necessary for some of the responses to SSRIs, but it is unknown whether the mature dentate gyrus granule cells (mature DG GCs) also contribute. We deleted Serotonin 1A receptor (5HT1AR; a receptor required for the SSRI response) specifically from DG GCs and found that the effects of the SSRI fluoxetine on behavior and the Hypothalamic-Pituitary-Adrenal (HPA) axis were abolished. By contrast, mice lacking 5HT1ARs only in young adult born granule cells (abGCs) showed normal fluoxetine responses. Importantly, 5HT1AR deficient mice engineered to express functional 5HT1ARs only in DG GCs responded to fluoxetine, indicating that 5HT1ARs in DG GCs are sufficient to mediate an antidepressant response. Taken together, these data indicate that both mature DG GCs and young abGCs must be engaged for an antidepressant response.
Keywords: 5HT1AR, Htr1a, Dentate Gyrus, Hippocampus, Depression, Anxiety, Antidepressant, Granule Cells, Adult Neurogenesis, Serotonin, SSRI, Fluoxetine, HPA Axis, Mature Granule Cells, Adult Born Granule Cells
Elucidation of the neurobiological basis of depression and anxiety and identification of improved treatments for patients are two of the foremost challenges in modern psychiatry. Mood disorders impact 7% of the world’s population and severe forms of depression affect 2–5% of the US population1. In addition, anxiety and depression have a high comorbidity with co-occurrence rates up to 60% in patients2. Imaging and post-mortem studies implicate several areas including prefrontal and cingulate cortices, hippocampus, amygdala, and thalamus in mood disorders3,4. Together, these brain regions operate a series of highly interacting circuits that likely mediate the progression of depression and the antidepressant response4. Identification of mechanisms within these brain regions should lead to improved therapies.
In the hippocampus, chronic treatment with selective serotonin reuptake inhibitors (SSRIs), the most widely used class of antidepressants, increases the dendritic spine density of pyramidal neurons in CA subfields and stimulates multiple stages of adult neurogenesis in the dentate gyrus (DG)5,6. Chronic, but not acute, SSRI treatment results in increased proliferation of dividing neural precursor cells, as well as faster maturation and integration of young adult born granule cells (abGCs) into the DG6,7. The young abGCs are highly active for a few weeks after functional integration into the neuronal network8,9, but eventually become functionally indistinguishable from the mature developmentally born granule cells when they are approximately 8 weeks old9,10. Focal radiological or genetic strategies to ablate or impair the neurogenic niche result in loss of some antidepressant-mediated behaviors, demonstrating that young abGCs are required for some antidepressant effects7,11,12. A few lines of evidence indicate that mature DG GCs (a population consisting of both developmentally born and adult born neurons that are older than 8 weeks) may also play a role in mediating mood and the antidepressant response. Stress induces profound changes in the DG. The hippocampus is vulnerable to various hormones induced by stress such as glucocorticoids, and rat adrenalectomy results in the death of most DG GCs13. Furthermore, infusion of peptides, such as brain-derived neurotrophic factor (BDNF), vascular endothelial growth factor (VEGF), and Activin, into the DG yield an antidepressant-like response14,15. In addition, optogenetic manipulations of ventral DG GCs demonstrate a role in anxiety-related behaviors16. Importantly, humans suffering from major depressive disorder have fewer DG GCs than controls17. A recent study also found that DG volume significantly decreases as the number of depressive episodes increases18. These data suggest that delineation of whether mature DG GCs contribute to depression and the antidepressant response is necessary. A better understanding of how SSRIs modulate neuronal circuitry in vivo to confer behavioral changes is crucial for the development of novel, more effective, and faster acting antidepressants. Moreover, approaches that target specific serotonin receptors or downstream pathways, rather than generally elevating serotonin (as SSRIs do), may also lead to improved treatment strategies.
Human genetic and imaging studies demonstrate that differences in Serotonin 1A receptor (5HT1AR) levels or regulation are associated with depression, anxiety, and the response to antidepressants19,20. A C(−1019)G polymorphism in the promoter region of the 5HT1AR gene associates with mood-related variables, including depression and the response to antidepressant treatment19,21. Germline 5HT1AR-deficient mice do not show behavioral or neurogenic responses to fluoxetine7. In addition, chronic treatment with the 5HT1AR agonist 8-hydroxy-2-(di-n-propylamino) tetralin (8-OH-DPAT) results in increased neurogenesis and decreased anxiety7. Taken together, these results demonstrate that 5HT1ARs are a major target of elevated serotonin and are required for the beneficial effects of antidepressant treatment. Therefore, tissue specific deletions of 5HT1AR populations will both determine the subset of 5HT1ARs and help identify the circuitry that mediates the antidepressant response. In the ventral DG, 5HT1ARs are highly expressed in mature DG GCs22. It is unknown whether 5HT1ARs are expressed in neural progenitors or young abGCs in the DG.
In this study, we sought to examine the independent roles of both mature DG GCs and young abGCs in the antidepressant response by deleting 5HT1AR from both populations through the usage of tissue-specific promoters.
RESULTS
Creation of floxed 5HT1AR mice
In order to study tissue specific 5HT1AR-deficiencies, we created mice with loxP sites flanking the single exon and the 3′ untranslated region containing the polyadenylation signal of the 5HT1AR gene (Figure 1a). We engineered the mice so that upon Cre-mediated excision of the 5HT1AR exon and the 3′ untranslated region, a yellow fluorescent protein (YPet) is expressed under control of the 5HT1AR promoter. Initial experiments, including assessment of the behavioral and neurogenic response to fluoxetine and labeling of 5HT1ARs with the radioactive ligand I-125 MPPI, demonstrated that mice homozygous for the floxed 5HT1AR allele (fl1A) were indistinguishable from wild-type (WT) littermates (Supplementary Figure 1 and data not shown). Therefore, homozygous fl1A mice are referred to as “Control” mice throughout the study.
Figure 1.
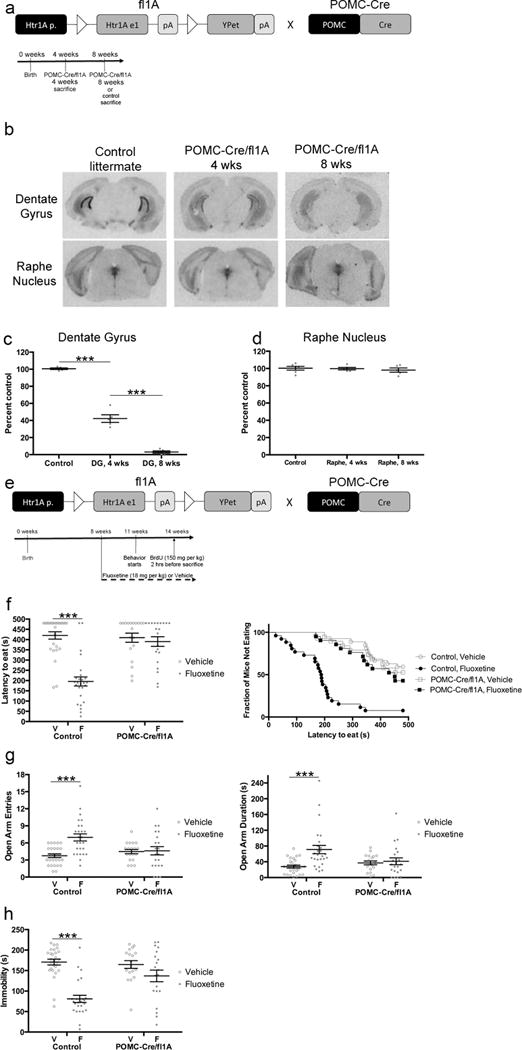
5HT1ARs in DG GCs are required for the behavioral effects of fluoxetine. a) Floxed 1A mice were crossed with POMC-Cre mice. White triangles indicate loxP sites. Htr1A p.: 5HT1AR promoter; Htr1A e1: 5HT1AR exon; pA: polyadenylation signal. Timeline is for panels b–c. POMC-Cre/fl1A mice were sacrificed at 4 or 8 weeks and compared to control littermates sacrificed at 8 weeks. n=5 per group. b) I-125 MPPI labeling. Sections are from ventral dentate gyrus or dorsal raphe nucleus. c) I-125 MPPI quantification in DG. One-Way ANOVA: F(2,12)=324.2, p<.0001. *** indicates p<.0001 (Tukey’s). n=5 per group. d) I-125 MPPI quantification in raphe nucleus. One-Way ANOVA: F(2,13)=.280, p=.7600. n=5–6 per group. e) Timeline for panels f–h. Control or POMC-Cre/fl1A mice were administered fluoxetine (18 mg/kg/day) or vehicle starting at 8 weeks of age. Behavior started three weeks after initiation of fluoxetine. n=19–27 per group. f) NSF results. Both bar graphs (left) and survival curves (right) indicating latency to eat are shown. *** indicates p<.0001 (Kaplan-Meier Survival Analysis with Bonferroni correction and Mantel-Cox p-values). g) EPM results. Open arm entries (left) and open arm duration (right) are shown. Both open arm entries (F(1,89)=8.120, p=.0054) and open arm duration (F(1,89)=6.435, p=.0129) were analyzed by Two-Way ANOVA. In the left panel, *** indicates p<.0001 (Tukey’s). In the right panel, *** indicates p=.0003 (Tukey’s). h) FST results. Immobility duration (F(1,86)=9.769, p=.0024) was analyzed by Two-Way ANOVA. *** indicates p<.0001 (Tukey’s). Mean lines and error bars throughout the figure represent mean ± SEM. V: Vehicle. F: Fluoxetine.
5HT1ARs on DG GCs are necessary for the behavioral effects of fluoxetine
We first investigated the functional roles of 5HT1ARs in all DG GCs. To this end, we crossed the floxed 5HT1AR mice with POMC-Cre mice16,23, where Cre is highly and selectively expressed in all GCs of the DG and in the arcuate nucleus of the hypothalamus (Figure 1a). I-125 MPPI autoradiography demonstrated a near complete deletion of 5HT1ARs in the DG (>90%) when bigenic POMC-Cre/fl1A mice were 8 weeks old (Figure 1b–c). This deletion was specific to the DG, as 5HT1AR levels were unchanged throughout the rest of the brain, including in the Raphe nucleus (Figure 1b and d).
We next assessed the behavioral response to antidepressants by treating Control and POMC-Cre/fl1A mice with either vehicle or fluoxetine (18mg/kg/day for 21 days) (Figure 1e). First, we tested the mice in Novelty Suppressed Feeding (NSF), which involves a 24-hour food deprivation and then placement into a large, brightly lit, and novel arena containing food in the center. Chronic, but not acute, antidepressant administration decreases the latency for mice to enter the center of the anxiogenic arena and bite the food pellet7,12. As expected, chronic fluoxetine treatment decreased the latency for Control mice to feed (Figure 1f; p<.0001; all statistics available in Supplementary Table 1). However, fluoxetine was ineffective in the POMC-Cre/fl1A mice (Figure 1f; p=.5071). At baseline there were no effects of genotype. There were no differences between groups in the percentage of weight lost during the deprivation or the amount of food consumed in the home cage (Supplementary Figure 2). Therefore, these data indicate that POMC-Cre/fl1A mice do not respond to fluoxetine in the NSF.
We also tested the behavior of this cohort of mice in the Elevated Plus Maze (EPM) (Figure 1g). In the EPM, chronic fluoxetine increases the number of open arm entries and the duration of time spent in the open arms12. Two-Way ANOVAs revealed significant genotype/treatment interactions for open arm entries (F(1,89)=8.120, p=.0054) and duration (F(1,89)=6.435, p=.0129). As expected, fluoxetine significantly increased open arm entries and duration in Control mice (p<.0001 for entries, p=.0003 for duration). However, fluoxetine was ineffective in POMC-Cre/fl1A mice (p=.9980 for entries, p=.9859 for duration). At baseline there were no significant effects of genotype. Thus, similar to NSF, fluoxetine was ineffective in mice lacking 5HT1ARs in all GCs.
We also assessed the behavior of these mice in the Forced Swim Test (FST) (Figure 1h), which is commonly used to assess antidepressant-like effects12. Acute or chronic treatment with antidepressants decrease the time spent immobile in the FST. We found a significant genotype/treatment interaction in the FST (F(1,86)=9.769, p=.0024). Interestingly, fluoxetine decreased immobility in Control mice (p<.0001) but did not have any significant effects in POMC-Cre/fl1A mice (p=.2624). At baseline there were no significant effects of genotype. Taken together these data suggest that 5HT1ARs in DG GCs are critical for the behavioral response to SSRIs.
In addition to DG GCs, the POMC promoter also drives Cre expression in the arcuate nucleus of the hypothalamus. We did not detect any YPet expression in the arcuate nucleus of the hypothalamus (data not shown), and in situ hybridizations indicated that there were very low levels of 5HT1AR expression in the arcuate nucleus (Supplementary Figure 3). However, to confirm that the behavioral effects observed in the POMC-Cre/fl1A mice were due to loss of 5HT1ARs from DG GCs, we performed a very different manipulation to delete 5HT1ARs specifically from DG GCs. To this end, we bilaterally injected AAV8-CamKII-Cre or Control (AAV8-CamKII-GFP) virus into the DG of 4-week-old fl1A mice, and then 4 weeks later treated these mice with Vehicle or Fluoxetine (Figure 2a). We confirmed that the AAV8-CamKII-Cre virus mediated 5HT1AR deletion in the DG using I-125 MPPI (>80% deletion) and found that the nearby region CA1 was unaffected (Figure 2b–d). Importantly, we found that fluoxetine was ineffective in the NSF, EPM, and FST in fl1A mice injected with the AAV8-CamKII-Cre virus (Figure 2e–g; NSF: Control (AAV8-CamKII-GFP) Vehicle vs Fluoxetine: p=.0004, AAV8-CamKII-Cre Vehicle vs Fluoxetine: p=.3345; EPM Open Arm Entries: Two-Way ANOVA: F(1,53)=13.95, p=.0005; EPM Open Arm Duration: Two-Way ANOVA: F(1,53)=13.00, p=.0007; FST: Two-Way ANOVA: F(1,54)=5.385, p=.0241). Therefore, two very different manipulations both indicated that 5HT1ARs in DG GCs are necessary for the behavioral response to SSRIs.
Figure 2.
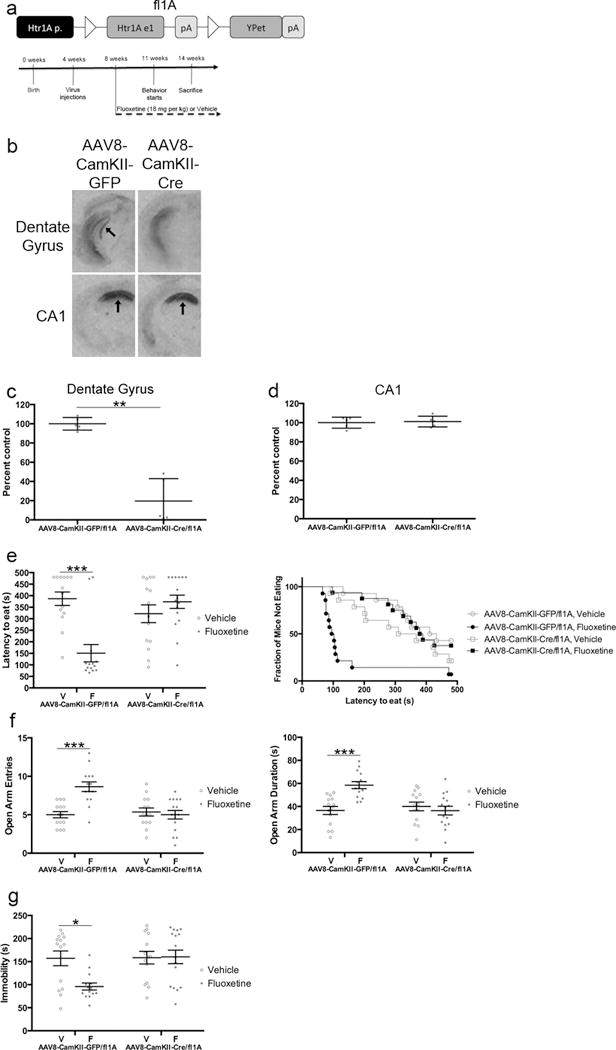
Viral-mediated deletion shows that 5HT1ARs in DG GCs are required for the behavioral effects of fluoxetine. a) Floxed 1A mice were injected with either AAV8-CamKII-Cre or AAV8-CamKII-GFP virus. Timeline is for panels e–g. fl1A mice were injected with virus at 4 weeks of age and were then administered fluoxetine (18 mg/kg/day) or vehicle starting at 8 weeks of age. Behavior started three weeks after initiation of fluoxetine. n=14–15 per group for behavior. b) I-125 MPPI labeling. Sections are from ventral dentate gyrus or dorsal CA1. In top left panel, arrow indicates dentate gyrus. In bottom panels, arrows indicate CA1. c) I-125 MPPI quantification in DG. ** indicates p=.0010 (Student’s t-test with Welch’s correction). n=5 per group. d) I-125 MPPI quantification in CA1. p=.7593 (Student’s t-test). n=5 per group. e) NSF results. Both bar graphs (left) and survival curves (right) indicating latency to eat are shown. *** indicates p=.0004 (Kaplan-Meier Survival Analysis with Bonferroni correction and Mantel-Cox p-values). f) EPM results. Open arm entries (left) and open arm duration (right) are shown. Both open arm entries (F(1,53)=13.95, p=.0005) and open arm duration (F(1,53)=13.00, p=.0007) were analyzed by Two-Way ANOVA. *** indicates p<.0001 for open arm entries and p=.0004 for open arm duration (Tukey’s). g) FST results. Immobility duration (F(1,54)=5.385, p=.0241) was analyzed by Two-Way ANOVA. * indicates p=.0142 (Tukey’s). Mean lines and error bars throughout the figure represent mean ± SEM. V: Vehicle. F: Fluoxetine.
5HT1ARs on young abGCs are not required for the effects of fluoxetine
The POMC-Cre/fl1A mice lacked 5HT1ARs in all DG GCs (both mature DG GCs and young abGCs). Therefore, we next examined the functional role of 5HT1ARs specifically in the young abGCs by crossing the fl1A mice with Nestin-CreERT2 mice24–26. In this line, 57.5±3.3% of total surviving young abGCs exhibit Cre-mediated recombination24,25. To this end, 8-week-old Nestin-CreER/fl1A (Figure 3a) or Control (Supplementary Figure 4) mice were pretreated either with tamoxifen (Tamoxifen 8 wks) to induce Cre-mediated recombination or with vehicle (Vehicle). The YPet expression pattern in the Nestin-CreER/fl1A mice indicated that the 5HT1AR promoter is not active (and thus 5HT1AR is not expressed) until young abGCs are 3–4 weeks old (Supplementary Figure 5). Therefore, we tried two different treatment protocols. First, chronic fluoxetine (18 mg/kg/day; Fluoxetine 8 wks) or vehicle (Vehicle) administration commenced in 8-week-old mice, concurrent with the tamoxifen or vehicle pretreatment (Figure 3a left timeline). The timing of this group was driven by the rationale that it takes several weeks for SSRIs to stably increase 5-HT levels in the hippocampus27, and that the increase in 5-HT levels would therefore align with expression of the 5HT1AR receptors. Second, fluoxetine (18 mg/kg/day; Fluoxetine 11 wks) administration commenced in 11-week-old mice (3 weeks after pretreatment), around the onset of 5HT1AR expression in young abGCs (Figure 3a right timeline). The timing of this second group was driven by the rationale that 5HT1ARs should be expressed when fluoxetine treatment begins. All of these groups of mice were then subjected to NSF, EPM, and FST12. Figure 3 shows the results from the Nestin-CreER/fl1A mice and Supplementary Figure 4 shows the results from the Control mice.
Figure 3.
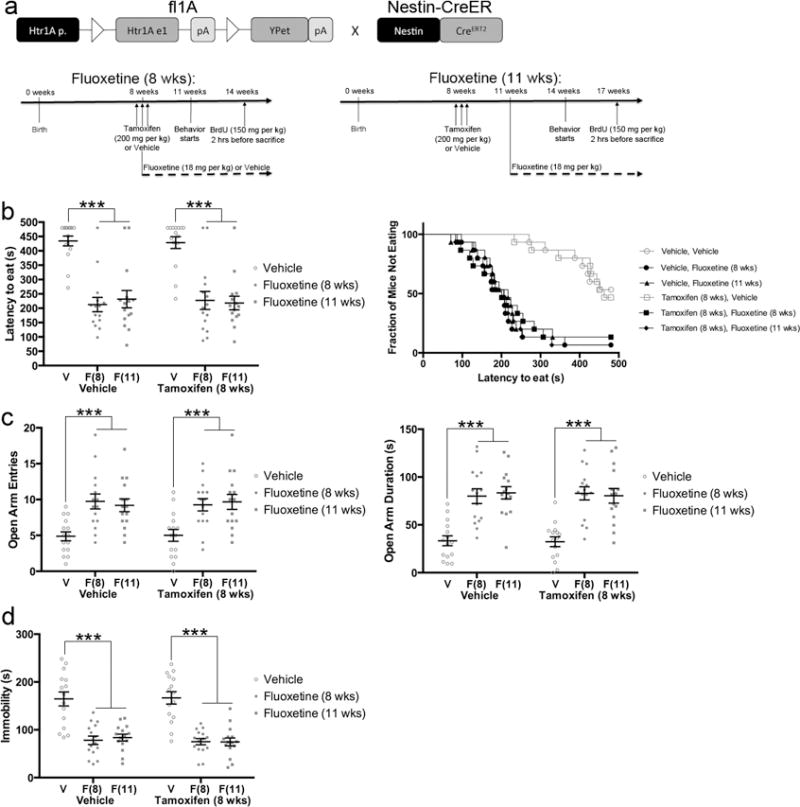
5HT1ARs in young abGCs are not required for the behavioral response to antidepressants. a) Floxed 1A mice were crossed with Nestin-CreERT2 mice. Timelines are for panels b–d. Nestin-CreER/fl1A mice were pretreated with 200mg/kg tamoxifen or vehicle (three days, twice per day). Daily fluoxetine (18 mg/kg) or vehicle treatment began either when the mice were 8 weeks old (left, concurrent with the tamoxifen) or when they were 11 weeks old (right). Behavior commenced three weeks after initiation of fluoxetine treatment. n=15 per group. b) NSF results. Both bar graphs (left) and survival curves (right) indicating latency to eat are shown. *** indicates p<.0001 or p=.0002 (only for Vehicle Vehicle vs Vehicle Fluoxetine (11 wks)) (Kaplan-Meier Survival Analysis with Bonferroni correction and Mantel-Cox p-values). c) EPM results. Open arm entries (left) and open arm duration (right) are shown. *** indicates p<.0001 (Two-Way ANOVA; treatment effect only). d) FST results. *** indicates p<.0001 (Two-Way ANOVA; treatment effect only). Mean lines and error bars throughout the figure represent mean ± SEM. V: Vehicle. F(8): Fluoxetine (8 wks). F(11): Fluoxetine (11 wks).
In NSF, EPM, and FST, we found that all groups of mice that were treated with fluoxetine showed a clear and significant decrease in latency to eat, increase in open arm entries and duration, and decrease in immobility, respectively (Figure 3b–d and Supplementary Figure 4). In all measures there were significant effects of treatment, but there were no significant effects of pretreatment (tamoxifen). Fluoxetine was effective when treatment commenced concurrent with tamoxifen or vehicle pretreatment and also when treatment commenced slightly before 5HT1AR expression in young abGCs (Figure 3b–d). In the NSF, there were no differences between groups in the percentage of weight lost during the deprivation or the amount of food consumed in the home cage (Supplementary Figure 2). For EPM and FST there were no significant genotype/treatment interactions. These data indicate that 5HT1ARs in young abGCs do not contribute to the behavioral response to SSRIs. This is strikingly different than ablation of young abGCs, which results in a partial loss of antidepressant effects7,12, and may be related to the fact that 5HT1ARs are expressed late in the differentiation process of young abGCs. Taken together with the results from the POMC-Cre/fl1A mice, these results strongly suggest that 5HT1ARs in mature DG GCs are required for the behavioral effects of fluoxetine across a battery of anxiety- and depression-related tests and also that mature DG GCs play an essential role in mounting an antidepressant response.
5HT1ARs on DG GCs are sufficient to mediate an antidepressant response
Since the behavioral data in the Nestin-CreER/fl1A, POMC-Cre/fl1A, and virus-injected mice demonstrate the necessity of 5HT1ARs in mature DG GCs, we also wanted to determine if these receptors are sufficient to mediate an antidepressant response. These experiments could help determine whether DG GC 5HT1ARs are a potential target for more specific treatments. To this end, we used transgenic mice that express 5HT1ARs in the DG under control of the Nrip2 promoter (Figure 4a)28. As previously reported, when these transgenic mice are crossed with mice that are germline deficient for 5HT1AR (termed 1A KO)29, then the resulting mice (termed DG-1A+) only express 5HT1ARs at high levels in the DG and at low levels in the central nucleus of the amygdala (CeA)28. Slice recordings found that while DG GCs in these mice show a robust response to serotonin, the CeA neurons do not28. Therefore, functional 5HT1ARs are expressed specifically in DG GCs in the DG-1A+ mice. I-125 MPPI experiments confirmed that in DG-1A+ mice 5HT1ARs were expressed in the DG and not in other brain regions such as the Raphe Nucleus (Figure 4b, Supplementary Figure 6)28.
Figure 4.
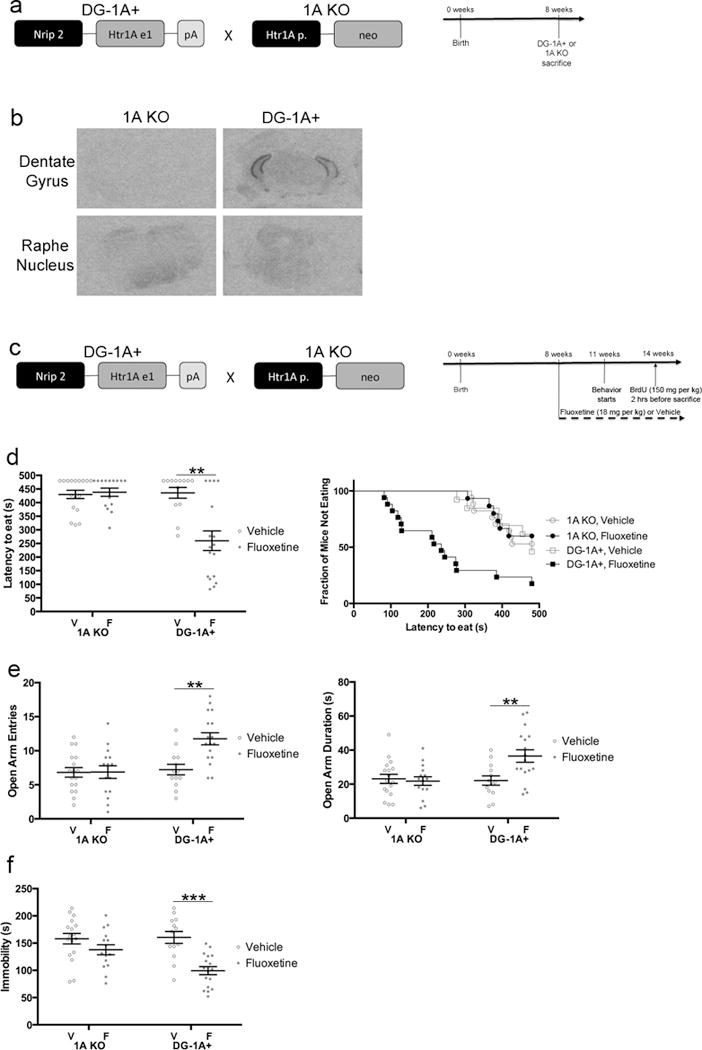
5HT1ARs in DG GCs are sufficient for the behavioral effects of fluoxetine. a) DG-1A+ mice were generated and crossed with 5HT1AR deficient mice (1A KO). Timeline is for panel b. 1A KO and DG-1A+ mice were sacrificed at 8 weeks. n=5 per group. b) I-125 MPPI labeling. Sections are from ventral dentate gyrus or dorsal raphe nucleus. c) Timeline for panels d–f. DG-1A+ or 1A KO mice were administered fluoxetine (18 mg/kg/day) or vehicle starting at 8 weeks of age. Behavior started three weeks after initiation of fluoxetine. n=13–17 per group. d) NSF results. Both bar graphs (left) and survival curves (right) indicating latency to eat are shown. ** indicates p=.0079 (Kaplan-Meier Survival Analysis with Bonferroni correction and Mantel-Cox p-values). e) EPM results. Open arm entries (left) and open arm duration (right) are shown. Both open arm entries (F(1,58)=7.204, p=.0095) and open arm duration (F(1,58)=6.773, p=.0117) were analyzed by Two-Way ANOVA. ** indicates p=.0022 in left panel and p=.0086 in right panel (Tukey’s). f) FST results. Immobility duration (F(1,58)=4.848, p=.0317) was analyzed by Two-Way ANOVA. *** indicates p=.0001 (Tukey’s). Mean lines and error bars throughout the figure represent mean ± SEM. V: Vehicle. F: Fluoxetine.
As expected, fluoxetine was ineffective in the NSF, EPM, and FST in 1A KO mice (Figure 4c–f, Supplementary Figure 7)7. By contrast, DG-1A+ mice treated with fluoxetine showed a significant decrease in latency to eat in the NSF, increase in open arm entries and duration in the EPM, and decrease in immobility in the FST (Figure 4c–f; NSF: 1A KO Vehicle vs Fluoxetine: p=.7412, DG-1A+ Vehicle vs Fluoxetine: p=.0079; EPM Open Arm Entries: Two-Way ANOVA: F(1,58)=7.204, p=.0095; EPM Open Arm Duration: Two-Way ANOVA: F(1,58)=6.773, p=.0117; FST: Two-Way ANOVA: F(1,58)=4.848, p=.0317). There were no significant effects of genotype (between 1A KO and DG-1A+ mice) at baseline. Taken together, these results strongly suggest that DG 5HT1ARs are sufficient for mediating the behavioral effects of fluoxetine across several anxiety- and depression-related tests and further demonstrate that DG GCs play a critical role in the antidepressant response.
5HT1ARs on DG GCs regulate fluoxetine-induced neurogenesis
The above results demonstrate that DG granule cell 5HT1ARs are critical for mediating the behavioral response to SSRIs. We therefore wanted to examine whether mature DG GCs also mediate other effects of antidepressants, such as increased adult hippocampal neurogenesis6,7. Data from germline 5HT1AR-deficient mice demonstrate that 5HT1ARs are also required for the effects of fluoxetine on neurogenesis (Supplementary Figure 7)7. Thus, we assessed several neurogenesis measures in all groups of mice that were behaviorally tested (Figure 5).
Figure 5.
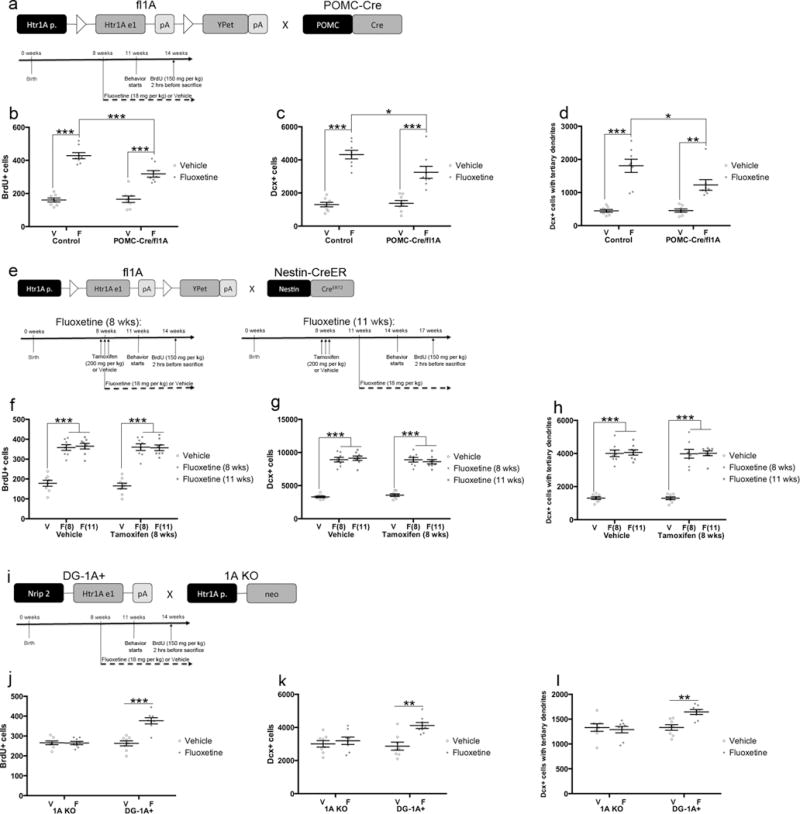
5HT1ARs in DG GCs are necessary and sufficient for the neurogenic effects of fluoxetine. a) Mice used for panels b–d were randomly chosen from behavioral cohort in Figure 1. n=8 per group. b) Proliferation results. The number of BrdU-positive cells was analyzed by Two-Way ANOVA (F(1,28)=10.66, p=.0029). *** indicates p<.0001 for Control Vehicle vs Fluoxetine and POMC-Cre/fl1A Vehicle vs Fluoxetine and p=.0007 for Control Fluoxetine vs POMC-Cre/fl1A Fluoxetine (Tukey’s). c) The number of young abGCs. The number of Dcx-positive cells was analyzed by Two-Way ANOVA (F(1,28)=5.292, p=.0291). *** indicates p<.0001, and * indicates p=.0247 for Control Fluoxetine vs POMC-Cre/fl1A Fluoxetine (Tukey’s). d) The number of young abGCs with tertiary dendrites. The number of Dcx-positive cells with tertiary dendrites was analyzed by Two-Way ANOVA (F(1,28)=4.954, p=.0343). *** indicates p<.0001, ** indicates p=.0015, and * indicates p=.0212 (Tukey’s). e) Mice used for panels f–h were randomly chosen from behavioral cohort in Figure 3. n=8 per group. f) Proliferation results. *** indicates p<.0001 (Two-Way ANOVA; treatment effect only) g) The number of young abGCs. *** indicates p<.0001 (Two-Way ANOVA; treatment effect only). h) The number of young abGCs with tertiary dendrites. *** indicates p<.0001 (Two-Way ANOVA; treatment effect only). i) Mice used for panels f–h were randomly chosen from behavioral cohort in Figure 4. n=8 per group. j) Proliferation results. The number of BrdU-positive cells was analyzed by Two-Way ANOVA (F(1,28)=23.7, p<.0001). *** indicates p<.0001 (Tukey’s). k) The number of young abGCs. The number of Dcx-positive cells was analyzed by Two-Way ANOVA (F(1,28)=6.311, p=.0180). ** indicates p=.0014 (Tukey’s). l) The number of young abGCs with tertiary dendrites. The number of Dcx-positive cells with tertiary dendrites was analyzed by Two-Way ANOVA (F(1,28)=8.031, p=.0084). ** indicates p=.0075 (Tukey’s). Mean lines and error bars throughout the figure represent mean ± SEM. V: Vehicle. F: Fluoxetine. F(8): Fluoxetine (8 wks). F(11): Fluoxetine (11 wks).
We first assessed brain sections from POMC-Cre/fl1A and Control mice to determine whether 5HT1ARs in all DG GCs are necessary for mediating the effects of fluoxetine on neurogenesis (Figure 5a–d). We found significant genotype/treatment interactions for the number of proliferating cells as measured by 5-bromo-2′-deoxyuridine (BrdU) incorporation (Figure 5b; F(1,28)=10.66, p=.0029), the number of young abGCs as measured by the number of Doublecortin (Dcx)-expressing cells (Figure 5c; F(1,28)=5.292, p=.0291), and maturation of young abGCs as measured by the number of Dcx-positive cells with tertiary dendrites (Figure 5d; F(1,28)=4.954, p=.0343). Importantly, the effects of fluoxetine were attenuated in POMC-Cre/fl1A mice relative to Control mice for all measures (Control Vehicle vs. Fluoxetine: p<.0001 for BrdU, Dcx, and Dcx with tertiary dendrites; POMC-Cre/fl1A Vehicle vs Fluoxetine: p<.0001 for BrdU, p<.0001 for Dcx, and p=.0015 for Dcx with tertiary dendrites; Control Fluoxetine vs POMC-Cre/fl1A Fluoxetine: p=.0007 for BrdU, p=.0247 for Dcx, and p=.0212 for Dcx with tertiary dendrites). There were no genotype effects at baseline for any measures. Taken together, these results suggest that DG 5HT1ARs are necessary for mediating the complete effects of fluoxetine on adult hippocampal neurogenesis.
We next assessed Nestin-CreER/fl1A and Control mice in order to determine the importance of young abGC 5HT1ARs in mediating the effects of fluoxetine on neurogenesis (Figure 5e–h and Supplementary Figure 4). In both Nestin-CreER/fl1A and Control mice, we found that fluoxetine significantly increased the number of proliferating cells, the number of young abGCs, and maturation of young abGCs (Figure 5f–h and Supplementary Figure 4). There were no significant effects of pretreatment (tamoxifen) or pretreatment/treatment interactions for any measures. Furthermore, fluoxetine was effective in all measures when treatment commenced concurrent with tamoxifen or vehicle pretreatment and when treatment commenced slightly before 5HT1AR expression in young neurons. Thus, the 5HT1AR-mediated effects of fluoxetine on adult neurogenesis do not require 5HT1AR expression in young abGCs and therefore most likely occur in a non-cell autonomous fashion. These data, in combination with the data from the POMC-Cre/fl1A mice, demonstrate that mature DG GCs are important regulators of the neurogenic response to antidepressants.
Finally, we assessed the effects of fluoxetine on adult hippocampal neurogenesis in 1A KO and DG-1A+ mice in order to determine whether DG granule cell 5HT1ARs are sufficient to mediate the neurogenic response to antidepressants (Figure 5i–l). We found significant genotype/treatment interactions for the number of proliferating cells (Figure 5j; F(1,28)=23.7, p<.0001), the number of young neurons (Figure 5k; F(1,28)=6.311, p=.0180), and the maturation of young neurons (Figure 5l; F(1,28)=8.031, p=.0084). As expected, 1A KO mice did not show a response to chronic fluoxetine treatment in any measures7. By contrast, DG-1A+ mice treated with fluoxetine had an increased number of proliferating cells (Figure 5j; p<.0001), an increased number of young neurons (Figure 5k; p=.0014), and increased maturation of young neurons (Figure 5l; p=.0075). There were no genotype effects at baseline for any measures. Taken together, these data demonstrate that DG granule cell 5HT1ARs are sufficient for mediating the neurogenic response to fluoxetine and also further demonstrate that mature DG GCs are critical mediators of the antidepressant response.
5HT1ARs on DG GCs regulate the neuroendocrine response to fluoxetine
To define the mechanism underlying why mature DG GCs are necessary for the antidepressant response, we next assessed the effects of 5HT1AR deletions on the Hypothalamic-Pituitary-Adrenal (HPA) axis response to stress. The major neuroendocrine response to stress is activation of the HPA axis, which results in production of corticosterone in the adrenal glands30. Alterations in the HPA axis can lead to depressive illness in humans and behavioral phenotypes in anxiety- and depression-related behavioral tasks in rodents12,31. Therefore, we assessed the responses of the HPA axis to chronic fluoxetine when mice were in their home cage and then again one week later, right after the same cohorts of mice were exposed to the Elevated Plus Maze (Figure 6).
Figure 6.
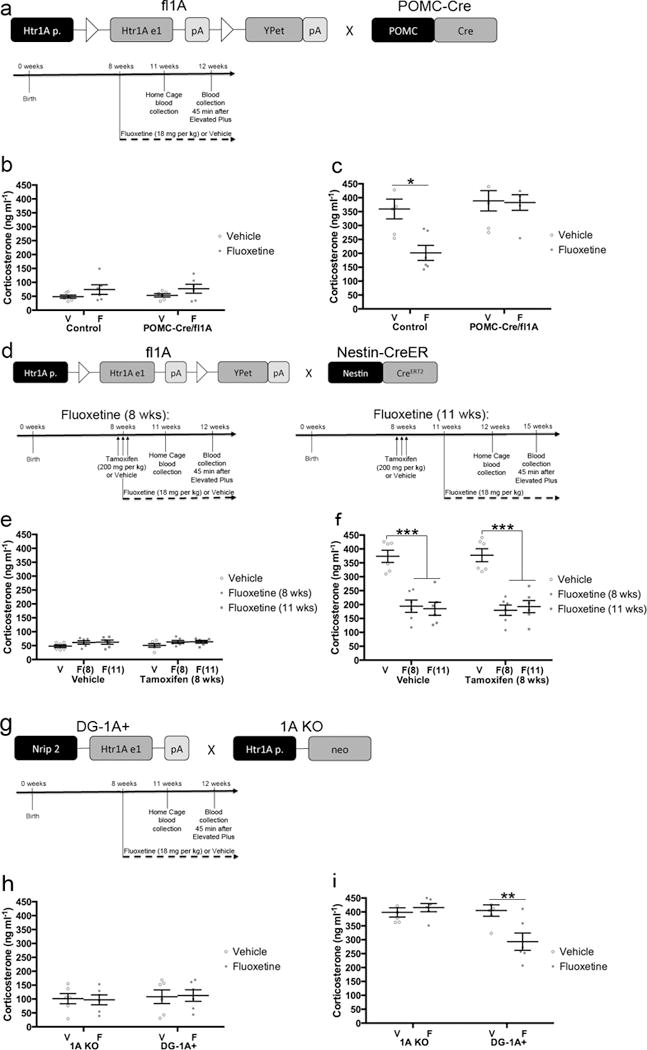
5HT1ARs in DG GCs are required for the neuroendocrine response to fluoxetine. a) Timeline for panels b–c. Control or POMC-Cre/fl1A mice were administered daily fluoxetine (18 mg/kg) or vehicle starting at 8 weeks of age. Blood was collected from mice in their home cage three weeks after the start of fluoxetine treatment and then again from the same mice one week later 45 minutes after EPM exposure. n=6 per group. b) Home cage plasma corticosterone levels. There were no differences (Two-Way ANOVA). c) Plasma corticosterone levels after EPM exposure. Corticosterone levels were analyzed by Two-Way ANOVA (F(1,20)=5.585, p=.0284). * indicates p=.0117 (Tukey’s). d) Timeline for panels e–f. Nestin-CreER/fl1A mice were pretreated with 200mg/kg tamoxifen or vehicle (three days, twice per day). Daily fluoxetine (18 mg/kg) or vehicle treatment began either when the mice were 8 weeks old (left, concurrent with the tamoxifen) or when they were 11 weeks old (right). Blood was collected from mice in their home cage three weeks after the start of fluoxetine treatment and then again from the same mice one week later 45 minutes after EPM exposure. n=6 per group. e) Home cage plasma corticosterone levels. There were no differences (Two-Way ANOVA). f) Plasma corticosterone levels after EPM exposure. *** indicates p<.0001 (Two-Way ANOVA; treatment effect only). Bars and error bars throughout the figure represent mean ± SEM. g) Timeline for panels h–i. 1A KO or DG-1A+ mice were administred daily fluoxetine (18 mg/kg) or vehicle starting at 8 weeks of age. Blood was collected from mice in their home cage three weeks after the start of fluoxetine treatment and then again from the same mice one week later 45 minutes after EPM exposure. n=6 per group. h) Home cage plasma corticosterone levels. There were no differences (Two-Way ANOVA). i) Plasma corticosterone levels after EPM exposure. Corticosterone levels were analyzed by Two-Way ANOVA (F(1,20)=8.878, p=.0074). ** indicates p=.0079 (Tukey’s). Mean lines and error bars throughout the figure represent mean ± SEM. V: Vehicle. F: Fluoxetine. F(8): Fluoxetine (8 wks). F(11): Fluoxetine (11 wks).
We began by examining the effects of chronic fluoxetine on the HPA axis in a new cohort of POMC-Cre/fl1A and Control mice (Figure 6a–c). When blood was collected from mice in their home cage, there were no significant differences in plasma corticosterone levels (Figure 6b). However, 45 minutes after mice were exposed to the Elevated Plus Maze, we found a significant genotype/treatment interaction for plasma corticosterone levels (Figure 6c; F(1,20)=5.585, p=.0284). In control mice, fluoxetine significantly attenuated the elevation in plasma corticosterone levels (p=.0117). However, fluoxetine did not alter plasma corticosterone levels in POMC-Cre/fl1A mice (p=.9991). There were no baseline differences between genotypes after exposure to the Elevated Plus Maze. These data indicate that DG 5HT1ARs are necessary for mediating the effects of fluoxetine on the HPA axis.
We next assessed the effects of chronic fluoxetine on the HPA axis in a new cohort of Nestin-CreER/fl1A mice (Figure 6d–f). There were no significant differences in plasma corticosterone levels when mice were in their home cage (Figure 6e), but exposure to the Elevated Plus Maze dramatically increased plasma corticosterone levels. Fluoxetine significantly attenuated this elevation in plasma corticosterone levels in all treated groups (Figure 6f). There was not a significant effect of pretreatment (tamoxifen) or a significant pretreatment/treatment interaction for plasma corticosterone levels. Furthermore, fluoxetine effectively attenuated the elevation in plasma corticosterone levels when treatment commenced concurrent with tamoxifen or vehicle pretreatment and when treatment commenced slightly before 5HT1AR expression in young neurons. Thus, mice lacking 5HT1ARs in young abGCs show a normal HPA axis response to stress and fluoxetine treatment. Taken together with the data from the POMC-Cre/fl1A mice, these data suggest that deletion of 5HT1ARs from mature DG GCs prevents the ability of fluoxetine to attenuate the stress response. This finding suggests that a critical role of mature DG GCs during the antidepressant response is to modulate HPA axis function.
To further assess the role of DG 5HT1ARs in the mediating the effects of fluoxetine on the HPA axis, we next assessed plasma corticosterone levels in a new cohort of 1A KO and DG-1A+ mice (Figure 6g–i). There were no significant differences in plasma corticosterone levels when mice were in their home cage (Figure 6h), but after exposure to the Elevated Plus Maze we found a significant genotype/treatment interaction for plasma corticosterone levels (Figure 6i; F(1,20)=8.878, p=.0074). In 1A KO mice, fluoxetine did not affect the EPM-induced elevation in plasma corticosterone levels (p=.9427). By contrast, fluoxetine significantly attenuated the elevation in plasma corticosterone levels in DG-1A+ mice (p=.0079). Taken together, these data demonstrate that 5HT1ARs in DG GCs are sufficient for mediating a HPA axis response to fluoxetine and also further demonstrate that mature DG GCs are critical mediators of the antidepressant response.
5HT1ARs on DG GCs regulate fluoxetine-induced growth factor expression
To determine how loss of 5HT1ARs from DG GCs might impact adult hippocampal neurogenesis, we next determined RNA expression levels of the growth factors BDNF and VEGF in the DG of Control and POMC-Cre/fl1A mice treated with Vehicle or Fluoxetine (Figure 7a–c). BDNF and VEGF expression levels are increased by fluoxetine treatment, and these increases are necessary for the effects of fluoxetine on behavior and adult hippocampal neurogenesis14,15,32–34. Furthermore, a selective 5HT1AR agonist increases while a selective 5HT1AR antagonist decreases VEGF levels in the hippocampus32. Two-Way ANOVAs revealed significant genotype/treatment interactions for BDNF (F(1,8)=10.68, p=.0114) and VEGF (F(1,8)=6.749, p=.0317) RNA expression levels. As expected, chronic fluoxetine treatment increased RNA expression levels of BDNF (p=.0004) and VEGF (p=.0003) in the DG of Control mice (Figure 7b–c). By contrast, the fluoxetine-induced increase in RNA expression levels of BDNF and VEGF were abolished or attenuated in POMC-Cre/fl1A mice (Figure 7b–c; BDNF: POMC-Cre/fl1A Vehicle vs Fluoxetine: p=.1038; VEGF: POMC-Cre/fl1A Vehicle vs Fluoxetine: p=.0210, Control Fluoxetine vs POMC-Cre/fl1A Fluoxetine: p=.0266). These data indicate that DG 5HT1ARs are necessary for mediating the fluoxetine-induced increase in BDNF and VEGF RNA expression levels.
Figure 7.
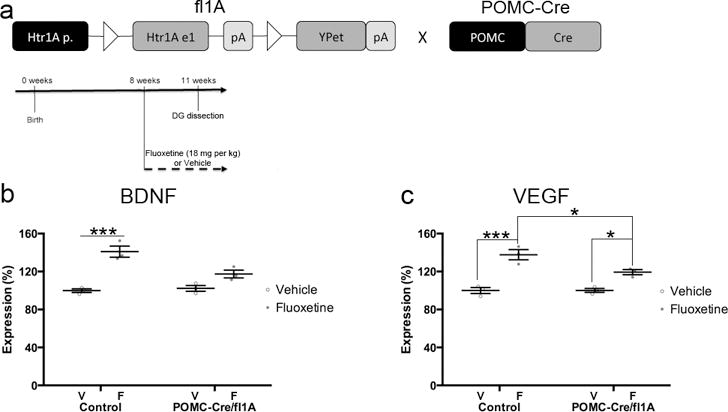
Fluoxetine-induced increases in BDNF and VEGF in the DG are attenuated in mice lacking 5HT1ARs in DG GCs. a) Timeline for panels b–c. Control or POMC-Cre/fl1A mice were administered daily fluoxetine (18 mg/kg) or vehicle starting at 8 weeks of age. DG was dissected and RNA was prepared three weeks after the start of fluoxetine treatment. n=3 per group. b) DG BDNF RNA expression levels were analyzed by Two-Way ANOVA (F(1,8)=10.68, p=.0114). *** indicates p=.0004 (Tukey’s). c) DG VEGF RNA expression levels were analyzed by Two-Way ANOVA (F(1,8)=6.749, p=.0317). *** indicates p=.0003, * indicates p=.0210 for POMC-Cre/fl1A Vehicle vs Fluoxetine, and p=.0266 for Control Fluoxetine vs POMC-Cre/fl1A Fluoxetine (Tukey’s). Mean lines and error bars throughout the figure represent mean ± SEM. V: Vehicle. F: Fluoxetine.
DISCUSSION
5HT1ARs on mature DG GCs mediate several effects of fluoxetine
This study demonstrates that mature DG GC 5HT1ARs are necessary and sufficient for the effects of fluoxetine on behavior, neurogenesis, and the neuroendocrine system. Importantly, serum fluoxetine levels were similar across all genotypes and pretreatments tested (Supplementary Figure 8). SSRIs block serotonin reuptake and increase serotonin levels throughout the brain. While anxiety behavior is influenced by 5HT1ARs, these effects are largely mediated during discrete developmental windows by the autoreceptor population of 5HT1ARs that are present on the serotonergic projections of the Raphe Nucleus27,29,35,36. Mice that are germline deficient in 5HT1ARs do not show a behavioral or neurogenic response to chronic fluoxetine treatment, but these mice lack both autoreceptors and heteroreceptors throughout life7,29. The POMC-Cre/fl1A and AAV8-CamKII-Cre/fl1A mice had a specific deletion of DG GC 5HT1A heteroreceptors limited to adulthood and the DG-1A+ mice only expressed functional 5HT1ARs in DG GCs. The data from these lines demonstrate that 5HT1ARs expressed in the DG are necessary and sufficient for the behavioral, neurogenic, and neuroendocrine effects of fluoxetine. Furthermore, because deleting 5HT1ARs from young abGCs did not affect the fluoxetine response, 5HT1ARs in mature DG GCs are critical for the effects of fluoxetine on behavior, neurogenesis, and the neuroendocrine system.
There are 14 distinct serotonin receptors, so the neuromodulatory effects of SSRI-enhanced serotonin levels could be mediated by several different subtypes37. 5HT1AR and 5HT4R are the most prominently expressed serotonin receptor subtypes in the DG22. Interestingly, 5HT4R agonists display antidepressant-like activities38,39 and fluoxetine is ineffective in several behavioral tasks in germline deficient 5HT4R mice40. 5HT1ARs inhibit cAMP signaling through coupling to Gi/o while 5HT4Rs stimulate cAMP formation through coupling to Gs37. Since these two receptors have opposing effects on intracellular signaling, the effects of antidepressants on mature DG GCs may require a balance that is only achieved when both receptors are present. Increased signaling through either 5HT4R (as seen here in the case of 5HT1AR deficiency) or through 5HT1AR (in the case of 5HT4R deficiency) may yield improper fluoxetine-mediated modulation of mature DG GCs that have consequences for the hippocampal circuitry. Future studies should further address this hypothesis by investigating mice with specific deletion of 5HT4Rs in the DG. In addition, the behavioral and neurogenic effects of fluoxetine in DG-1A+ mice were not as prominent as what was seen in a distinct cohort of WT mice (Supplementary Figure 7). Therefore, we cannot rule out that 5HT1ARs in other brain regions may also be important for the antidepressant response.
Mature DG GCs are critical mediators of the antidepressant response
One potential mechanism of how the mature DG GCs may modulate young abGCs is through highly regulated secretion of growth factors such as BDNF and VEGF14,15,32–34,41. Acute pharmacological manipulations of 5HT1ARs influence VEGF expression levels in the DG32. Interestingly, we found that fluoxetine-induced increases in BDNF and VEGF expression levels in the DG are attenuated in mice lacking 5HT1ARs in DG GCs. Although there is a precedent for 5HT1AR-mediated regulation of VEGF in the DG32, these results are still somewhat surprising given that 5HT1AR is an inhibitory receptor and that both BDNF and VEGF are induced by activity. Therefore, it is possible that DG 5HT1ARs mediate the effects of chronic fluoxetine on BDNF and VEGF expression levels via distinct 5HT1AR-related signaling cascades or through an indirect mechanism. Future work is necessary to further delineate these possibilities.
Complete ablation of young abGCs by methods such as focal irradiation consistently leads to alterations in the antidepressant response in only one behavioral test (NSF)7,12. By contrast, deletion of 5HT1ARs from all DG GCs leads to alterations in the antidepressant response in several behavioral tests (NSF, EPM, and FST). Therefore, complete ablation of young abGCs has much milder effects than modulation of all DG GCs. Enhancing adult neurogenesis through genetic methods does not result in an antidepressant-like response, indicating that increasing the number of young abGCs is not sufficient for mediating a change in mood-related behavior, at least in baseline conditions25. Our data suggests that the mature population of GCs must also be engaged for the antidepressant response, and that the young abGCs may work in concert with the mature population of GCs (Supplementary Figure 9).
We propose that during a response to fluoxetine treatment 5HT1ARs on mature DG GCs respond to the increase in serotonin levels by activating signaling cascades that result in the secretion of growth factors such as BDNF and VEGF that enhance adult neurogenesis (Supplementary Figure 9)14,15. In turn, the young abGCs, which are more plastic than the mature DG GCs, may modulate the activity of the mature DG GCs by acting on the local microcircuit (Supplementary Figure 9)42–44. The resulting combined activity of the both the mature DG GCs and the young abGCs is then required for the antidepressant response.
Antidepressants regulate the HPA axis through mature and young abGCs
While it remains unknown how the DG mediates the behavioral effects of antidepressants, a candidate mechanism can be found in the downstream circuitry. While stress profoundly regulates the DG, the hippocampus also provides negative feedback regulation to the HPA axis. Ventral hippocampal outputs to the ventral subiculum influence the HPA axis through GABAergic projections of areas such as the bed nucleus of the stria terminalis (BNST)45. Our data shows that deletion of 5HT1ARs from DG GCs blocks the effects of fluoxetine on the HPA axis. Interestingly, other studies have found that manipulations of adult neurogenesis result in altered plasma levels of corticosterone46,47. In addition, unpredictable chronic mild stress in mice reduces hippocampal neurogenesis and dampens the relationship between the hippocampus and the HPA axis48. This relationship can be restored by treatment with fluoxetine in a neurogenesis-dependent manner48. Therefore, our data and previous studies suggest that antidepressants regulate the HPA axis through modulation of both mature DG GCs and young abGCs. The output of the DG into this circuit is defined by the interaction between these two populations of cells. Since fluoxetine regulates both mature DG GCs and young abGCs, the DG provides an entry point for antidepressants to modulate a mood-related circuit and ultimately behavior.
Ventral dentate gyrus as a target for novel therapies
The hippocampus shows highly distinct afferent and efferent connectivity along its dorsoventral axis. The dorsal hippocampus connects with associational cortical regions important for cognitive functions while the ventral hippocampus connects with regions that mediate emotional affect such as the prefrontal cortex, nucleus accumbens, hypothalamus, and BNST49. Lesions of the dorsal hippocampus affect spatial memory while lesions of the ventral pole affect anxiety16,49. We found that 5HT1ARs in mature DG GCs are necessary and sufficient for the behavioral effects of antidepressants. Given that the vast majority of 5HT1ARs in the DG are expressed in the ventral pole22, 5HT1ARs are positioned to exhibit a specific influence on the limbic system and the HPA axis and thereby regulate mood- and anxiety-related behavior. Clinical trials with drugs that target 5HT1ARs, such as pindolol, have yielded disappointing results50. However, genetic methods that manipulate specific populations of 5HT1ARs are clarifying that distinct populations of 5HT1ARs play extremely different functional roles27,35. This is especially evident when comparing autoreceptors with heteroreceptors. Therefore, specific manipulation of 5HT1ARs or downstream intracellular effectors in the ventral DG may provide a more precise method for controlling mood and anxiety circuitry than drugs that generally increase serotonin throughout the brain or that target all populations of 5HT1ARs. Future work is necessary to delineate these potential targets for novel antidepressants. Our data also indicates that mature DG GCs are critical mediators of the antidepressant response. Interestingly, optogenetic elevation of activity in ventral DG GCs has anxiolytic effects16. Thus, we propose that pharmacological or electrical (such as deep brain stimulation) manipulations of the ventral DG are also novel potential treatments for mood and anxiety disorders.
ONLINE METHODS
Mice
Creation of Floxed 5HT1AR mice
The targeting vector for the fl1A mice consisted of a 5′ homology arm (-4993 to 4009, numbers correspond to the translation initiation site of 5HT1AR) with loxP, FRT, PGK-EM7-Neo minigene, FRT, loxP, YPet cDNA (YPet is a modified yellow fluorescent protein51), SV40 polyadenylation signal, 3′ homology arm (4010 to 5569, 1.6 kb), and diphteria toxin A subunit (DTA). The first loxP site was inserted just upstream of the coding region (1 to 1266) and the second loxP site was inserted after the Htr1a polyadenylation signal (3848 to 3853). The linearized targeting vector was electroporated into ES cells derived from the 129S6 strain (line CSL3) and G418 resistant clones were selected. Southern blots were then performed to verify the homologous recombination. To this end, genemic DNA was digested with BamHI and a 32P-labeled probe (7040–7879) was used. This probe detected a 13 kb band for the wild-type allele and 9 kb band for the knock-in allele. The recombinant ES cells were then injected into blastocysts from the C57BL/6 strain and chimeric mice were obtained. Germline transmitted mice were then crossed with ROSA-Flpe mice52 to remove the PGK-EM7-Neo minigene through FLP-FRT recombination. The offspring of these crosses were established as the fl1A mice. Nestin-CreER, POMC-Cre, 1A KO, and DG-1A+ mice were previously described7,16,23–25,28,29.
Husbandry
Mice were housed in groups of three to five per cage and had ad libitum access to food and water. Mice were maintained on a 12:12 light/dark schedule; all testing was conducted during the light period. Mouse protocols were approved by the Institutional Animal Care and Use Committee of Columbia University and the Research Foundation for Mental Hygiene, Inc. and were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Mice. Care was taken to minimize the number of mice used and their suffering.
Experimental Mice
Only male mice were used throughout the manuscript. All cohorts entailed littermates from several breeding cages. Mice of different genotypes, pretreatments, and treatments were all housed in the same cages.
Receptor Autoradiography
Quantitative measurements of receptor autoradiography were performed as previously described35. Briefly, mice were killed by cervical dislocation and decapitation. Extracted brains were frozen immediately on crushed dry ice (−75°C) and maintained at −80°C until sectioning. Brains were cryosectioned at a thickness of 18 μm and sections were thaw-mounted on Superfrost slides (Fisher Scientific). Sections were maintained at −80°C until processing. Mounted sections were processed for 4-(2′-methoxyphenyl)-1-[2′-(n-2″-pyridinyl)-p-[125I]iodobenzamido]ethylpiperazine (125I-MPPI) autoradiography and receptor levels were quantified as described27.
Drugs
Tamoxifen Administration
Eight-week-old Control or Nestin-CreER/fl1A mice were administered either Vehicle or Tamoxifen pretreatment as indicated in the figure timelines. Tamoxifen (Sigma) was suspended in a 50% honey/50% water mixture and was administered by oral gavage. 200 mg/kg of tamoxifen were administered twice per day to the mice when they were 54, 55, and 56 days old (a total of six administrations). Vehicle pretreated mice received a 50% honey/50% water mixture that did not contain tamoxifen.
Fluoxetine Administration
Fluoxetine (18 mg/kg/day in deionized water) or vehicle (deionized water) was delivered by oral gavage for three weeks prior to behavior testing. The oral gavage of fluoxetine or vehicle continued throughout the behavior testing and to experimental endpoints. On the days when mice were subjected to behavioral testing, fluoxetine or vehicle administrations were conducted after the mice completed the testing in order to avoid any acute effects.
Behavioral Testing
The behavioral testing was conducted in the following order: Elevated Plus Maze, Novelty Suppressed Feeding, and then Forced Swim Test. Mice were given three days between exposures to different behavioral tests. All behavioral testing was performed between 10am and 3pm.
Elevated Plus Maze
Elevated Plus Maze was performed as previously described12. Briefly, mice were placed into the central area facing one closed arm and allowed to explore the maze for 5 min. Testing took place in bright ambient light conditions (800–900 lux). The maze was cleaned with disinfecting wipes and paper towels between each run. Data were scored using TopScan software (CleverSys, Reston, VA).
Novelty Suppressed Feeding
NSF was performed as described12,53. The testing apparatus consisted of a plastic box (50×50×20 cm), the floor of which was covered with approximately 2 cm of wooden bedding. 24 hours prior to behavioral testing, all food was removed from the home cage. At the time of testing, a single pellet of food was placed on a white paper platform in the center of the box. A mouse was placed in a corner of the box, and a stopwatch was immediately started. The latency to eat (defined as the mouse sitting on its haunches and biting the pellet with the use of forepaws) was timed. Mice were in the testing arena for a total of 8 mins. Immediately after the testing period, the mice were transferred to their home cages, and the amount of food consumed by the mouse in the subsequent 5 mins was measured. Each mouse was weighed before food deprivation and before testing to assess the percentage of body weight loss.
Forced Swim Test
Forced Swim Test was performed as previously described12. Briefly, mice were placed into clear plastic buckets 20 cm in diameter and 23 cm deep, filled two-thirds of the way up with 26°C water and were videotaped. Mice were in the forced swim buckets for 6 mins, but only the last 4 mins were scored. Scoring was automated using Videotrack software (ViewPoint, France).
Virus injections
Mice were anesthetized with sodium pentobarbital (diluted 1/10 from stock of 50mg/mL and injected at a volume of 10ml/kg) and positioned in a stereotaxic frame (Stoelting) over a heated pad. Using a stereomicroscope (Leica), the head was shaved and disinfected. An incision was made in the scalp, exposing the skull. Craniotomies were opened bilaterally with a dental drill at −2.0 mm, +/−1.4 mm from the bregma line and midline respectively, and a stainless steel 33 gauge blunt needle attached to a 10μl syringe (World Precision Instruments, Inc. Sarasota, Fl) was inserted to 2.1mm depth from the top of the brain at injection craniotomy, corresponding to the dorsal dentate gyrus. For ventral dentate gyrus targeting, additional craniotomies were made at −3.5 mm, +/−2.8 mm from the bregma line and midline respectively, and injections were done at a depth of 3.6mm from the skull at bregma. All mice received bilateral dorsal and ventral dentate gyrus injections (a total of 4 injections per mouse; 0.2 μl over 2 min for dorsal and 0.3μl over 3 min for ventral) of either AAV8-CamKII-mCherry-Cre or AAV8-CamKII-eGFP (both obtained from UNC Vector core). The needle was left in situ for an additional 5 min to aid diffusion from the needle tip and prevent backflow. The needle was then slowly retracted and the scalp incision closed with Vetbond 3M. Each animal was monitored and received carprofen (s.c. 5 mg/kg) for pain management for 3 days. The mice were house for four weeks postoperatively prior to be beginning of drug administration.
Immunohistochemistry
Perfusions and sectioning
Mice were anesthetized between 12–2pm with ketamine and xylazine (100 mg/ml ketamine; 20 mg/ml xylazine) and were then perfused transcardially (cold saline for 2 min, followed by 4% cold paraformaldehyde in saline at 4°C). The brains were then dissected and cryoprotected in 30% sucrose with 0.1% NaN3 and stored at 4°C. Serial sections (35 μM) were cut through the entire hippocampus on a cryostat and stored in PBS with 0.1% NaN3.
BrdU administration and immunostaining
Mice were administered BrdU (150 mg/kg, i.p. dissolved in saline), 2 hrs before sacrifice in order to assess cell proliferation. For the immunohistochemistry experiments, the sections were washed three times for 10 mins in PBS, mounted onto slides, dried, and then exposed to Citrate Buffer (10 mM Citric Acid, pH 6.0 at 95°C) for 2 hrs. After a brief 1 min wash in PBS, the slides were then incubated overnight at room temperature with the primary antibody (Anti-BrdU rat monoclonal, Serotec, 1:100). The next day, the slides were washed two times for 5 mins with PBS, and were then incubated for 1 hr with the secondary antibody (Cy3-conjugated goat anti-rat, Molecular Probes, 1:200). The slides were then washed three times for 10 min in PBS and coverslipped.
Dcx immunostaining
For doublecortin staining, sections were rinsed in PBS, treated with 1% H2O2 in 1:1 PBS and methanol for 15 min to quench endogenous peroxidase activity (and to enhance dendritic staining), incubated in 10% normal donkey serum and 0.3% Triton X-100 for 30 min, and then incubated overnight at 4°C in primary antibody for doublecortin (goat;1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA). The next day, the sections were exposed to biotinylated donkey anti-goat (1:500) seconday antibody (Jackson ImmunoResearch, PA, USA) in PBS for 2 hrs at room temperature. The immunostaining was then developed using an avidin-biotin complex (Vector, CA, USA) and a DAB kit, and the sections were then mounted onto slides and coverslipped.
Cell counting
Cells were counted on a Zeiss (Oberkochen, Germany) Axioplan-2 upright microscope12,54. One out of every six sections through the hippocampus (12 total sections) was counted for each mouse.
Blood Collection and Corticosterone Level Measurements
Blood was collected by submandibular venipuncture55. The blood was collected into Eppendorf tubes that contained 10 μl of 0.5 M EDTA to prevent coagulation. The blood-EDTA mixture was then mixed by inversion and was placed on ice for no more than 15 min. The mixture was then centrifuged for 5 min at 1800 RMP. Plasma was collected and stored at -80° C. Blood was collected from mice in their home cage and then again from the same mice one week later, 45 minutes after exposure to the Elevated Plus Maze. All blood collection occurred between 12–2pm in order to minimize any potential effects of the diurnal corticosterone variation. The plasma corticosterone levels were then assessed using an enzyme immunoassay (ELISA) kit (Arbor Assays). The manufacturer’s instructions for the ELISA kit were followed.
qPCR
Dentate gyrus was dissected and then RNA was extracted using a RNA/DNA Purification kit (Norgen Biotek Corp). Conversion of total RNA into first strand cDNA was then accomplished by using Superscript III enzyme (Invitrogen, CA, USA). Quantitative PCR (qPCR) was carried out in 6.5ul reactions using Taqman Fast Advanced Mastermix and Taqman probes for BDNF, VEGF, and the housekeeping gene Rn18s (Life Technologies) on a StepOne Plus Real-Time PCR System (Applied Biosystems). Triplicate cycle thresholds (Ct values) were obtained for each dentate gyrus and averaged. The values for Rn18s were then used to normalize the expression values of BDNF and VEGF with the delta Ct method. After converting delta Ct values to percentage of the control group (the mean of Control + Vehicle was assigned a value of 100%), then the mean and SEM of each group was calculated.
In situ hybridizations
In situ hybridizations were performed as described22. The probe for 5HT1AR was also previously described22 (bases 2114-4089 in NM_008308). The probe for POMC corresponded to bases 35-945 in NM_008895. Briefly, after a paraformaldehyde treatment, a series of washes, and an acetylation step, prehybridization was carried out for 5 h at room temperature in hybridization buffer, consisting of 50 per cent formamide (Roche), 5xSSC (saline sodium citrate buffer), 5xDenhardts (Sigma), 0.25 mg ml−1 yeast tRNA (Ambion), and 0.4 mg ml−1 Salmon Sperm DNA (Stratagene). Sections were then incubated in hybridization buffer containing digoxigenin (DIG)-labelled cRNA probe at 60°C overnight. After another series of washes and a blocking step, the sections were incubated with alkaline phosphatase-conjugated anti-DIG antibody (1:5000 dilution; Roche) for 90 min at room temperature. After another series of washes to remove unbound antibody, the sections were incubated with freshly prepared nitroblue tetrazolium chloride/5-bromo-4-chloro-3-indolylphosphate p-toluidine salt (NBT/BCIP) color substrate (Roche) for up to 16 h at room temperature, after which the reaction was stopped by immersion into PBS. After ISH staining, the sections were counterstained with Nuclear Fast Red (Vectastain).
Statistics
All statistics are presented in Supplementary Table 1. Two-Way ANOVA assessing pretreatment × treatment (for Nestin-CreER/fl1A mice), genotype × treatment (for Control, POMC-Cre/fl1A, 1A KO, and DG-1A+ mice), or virus × treatment (for AAV8 injected mice) were used to assess Elevated Plus Maze, Forced Swim Test, Neurogenesis, and Corticosterone levels as indicated in the figures. A p-value of <.05 was considered significant. If significant interactions were found, then Tukey’s Method was used as the post hoc test to compare between individual groups. Since Novelty Suppressed Feeding does not show a regular distribution of data points, Kaplan-Meier Survival Analysis was used to assess the latency to eat results53. Multiple comparisons were subjected to a Bonferroni correction to determine significance. Student’s t-test and One-Way ANOVA were also used to assess I-125 MPPI results and serum fluoxetine levels when appropriate.
Supplementary Material
Acknowledgments
The authors thank Khaing Win and David Tora for technical support and Mazen Kheirbek, and David Leonardo for helpful discussions. This work was supported by NIMH R37MH068542 (RH), NIMH R01MH083862 (RH), HDRF MPPN8883 (RH), NYSTEM C029157 (RH), NIMH K01MH098188 (BAS), BBRF NARSAD Young Investigator 19658 (BAS), a Charles H. Revson fellowship (BAS), a German Research Foundation (DFG) postdoctoral fellowship (CA), NIMH T32MH015144 (ZRD), NIMH R01MH01844 (AD), funds from EMBL (CTG), and an EC Marie Curie Fellowship (NM). RH receives compensation as a consultant for Lundbeck and Roche.
Footnotes
AUTHOR CONTRIBUTIONS
BAS conceived and performed experiments, analyzed results, and wrote the manuscript. CA contributed to virus injections experiments. AH and MRL performed mouse husbandry and contributed to most experiments. AP, ZRD, and LJD contributed to experiments and analysis. TT, NM, and CTG performed mouse husbandry for 1A KO and DG-1A+ mice and experiments in Supplementary Figure 6. AD performed mouse husbandry for Nestin-CreER mice and contributed to analysis. KFT made the fl1A mice and performed the experiments in Supplementary Figure 3. Most experiments were performed in the lab of RH, who also conceived experiments and contributed to the analysis and writing of the manuscript.
References
- 1.Murray CJ, Lopez AD. Evidence-based health policy–lessons from the Global Burden of Disease Study. Science. 1996;274:740–743. doi: 10.1126/science.274.5288.740. [DOI] [PubMed] [Google Scholar]
- 2.Gorman JM. Comorbid depression and anxiety spectrum disorders. Depression and anxiety. 1996;4:160–168. doi: 10.1002/(SICI)1520-6394(1996)4:4<160::AID-DA2>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 3.Drevets WC. Neuroimaging and neuropathological studies of depression: implications for the cognitive-emotional features of mood disorders. Curr Opin Neurobiol. 2001;11:240–249. doi: 10.1016/s0959-4388(00)00203-8. [DOI] [PubMed] [Google Scholar]
- 4.Nestler EJ, et al. Neurobiology of depression. Neuron. 2002;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- 5.Hajszan T, MacLusky NJ, Leranth C. Short-term treatment with the antidepressant fluoxetine triggers pyramidal dendritic spine synapse formation in rat hippocampus. Eur J Neurosci. 2005;21:1299–1303. doi: 10.1111/j.1460-9568.2005.03968.x. [DOI] [PubMed] [Google Scholar]
- 6.Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20:9104–9110. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santarelli L, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301:805–809. doi: 10.1126/science.1083328. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt-Hieber C, Jonas P, Bischofberger J. Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature. 2004;429:184–187. doi: 10.1038/nature02553. [DOI] [PubMed] [Google Scholar]
- 9.Ge S, Yang CH, Hsu KS, Ming GL, Song H. A critical period for enhanced synaptic plasticity in newly generated neurons of the adult brain. Neuron. 2007;54:559–566. doi: 10.1016/j.neuron.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laplagne DA, et al. Functional convergence of neurons generated in the developing and adult hippocampus. PLoS Biol. 2006;4:e409. doi: 10.1371/journal.pbio.0040409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y, et al. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron. 2008;59:399–412. doi: 10.1016/j.neuron.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.David DJ, et al. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron. 2009;62:479–493. doi: 10.1016/j.neuron.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sloviter RS, et al. Selective loss of hippocampal granule cells in the mature rat brain after adrenalectomy. Science. 1989;243:535–538. doi: 10.1126/science.2911756. [DOI] [PubMed] [Google Scholar]
- 14.Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. 2002;22:3251–3261. doi: 10.1523/JNEUROSCI.22-08-03251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warner-Schmidt JL, Duman RS. VEGF is an essential mediator of the neurogenic and behavioral actions of antidepressants. Proc Natl Acad Sci USA. 2007;104:4647–4652. doi: 10.1073/pnas.0610282104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kheirbek MA, et al. Differential Control of Learning and Anxiety along the Dorsoventral Axis of the Dentate Gyrus. Neuron. 2013;77:955–968. doi: 10.1016/j.neuron.2012.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boldrini M, et al. Hippocampal granule neuron number and dentate gyrus volume in antidepressant-treated and untreated major depression. Neuropsychopharmacology. 2013;38:1068–1077. doi: 10.1038/npp.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Treadway MT, et al. Illness progression, recent stress, and morphometry of hippocampal subfields and medial prefrontal cortex in major depression. Biol Psychiatry. 2015;77:285–294. doi: 10.1016/j.biopsych.2014.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le François B, Czesak M, Steubl D, Albert PR. Transcriptional regulation at a HTR1A polymorphism associated with mental illness. Neuropharmacology. 2008;55:977–985. doi: 10.1016/j.neuropharm.2008.06.046. [DOI] [PubMed] [Google Scholar]
- 20.Strobel A, et al. Allelic variation in 5-HT1A receptor expression is associated with anxiety- and depression-related personality traits. Journal of neural transmission (Vienna, Austria : 1996) 2003;110:1445–1453. doi: 10.1007/s00702-003-0072-0. [DOI] [PubMed] [Google Scholar]
- 21.Fakra E, et al. Effects of HTR1A C(-1019)G on amygdala reactivity and trait anxiety. Arch Gen Psychiatry. 2009;66:33–40. doi: 10.1001/archpsyc.66.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanaka KF, Samuels BA, Hen R. Serotonin receptor expression along the dorsal-ventral axis of mouse hippocampus. Philos Trans R Soc Lond, B, Biol Sci. 2012;367:2395–2401. doi: 10.1098/rstb.2012.0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McHugh TJ, et al. Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science. 2007;317:94–99. doi: 10.1126/science.1140263. [DOI] [PubMed] [Google Scholar]
- 24.Dranovsky A, et al. Experience dictates stem cell fate in the adult hippocampus. Neuron. 2011;70:908–923. doi: 10.1016/j.neuron.2011.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sahay A, et al. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature. 2011 doi: 10.1038/nature09817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kheirbek MA, Tannenholz L, Hen R. NR2B-Dependent Plasticity of Adult-Born Granule Cells is Necessary for Context Discrimination. J Neurosci. 2012;32:8696–8702. doi: 10.1523/JNEUROSCI.1692-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richardson-Jones JW, et al. 5-HT(1A) Autoreceptor Levels Determine Vulnerability to Stress and Response to Antidepressants. Neuron. 2010;65:40–52. doi: 10.1016/j.neuron.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsetsenis T, Ma XH, Lo Iacono L, Beck SG, Gross C. Suppression of conditioning to ambiguous cues by pharmacogenetic inhibition of the dentate gyrus. Nat Neurosci. 2007;10:896–902. doi: 10.1038/nn1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramboz S, et al. Serotonin receptor 1A knockout: an animal model of anxiety-related disorder. Proc Natl Acad Sci USA. 1998;95:14476–14481. doi: 10.1073/pnas.95.24.14476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci. 2009;10:397–409. doi: 10.1038/nrn2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87:873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- 32.Greene J, Banasr M, Lee B, Warner-Schmidt J, Duman RS. Vascular endothelial growth factor signaling is required for the behavioral actions of antidepressant treatment: pharmacological and cellular characterization. Neuropsychopharmacology. 2009;34:2459–2468. doi: 10.1038/npp.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monteggia LM, et al. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci USA. 2004;101:10827–10832. doi: 10.1073/pnas.0402141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richardson-Jones JW, et al. Serotonin-1A autoreceptors are necessary and sufficient for the normal formation of circuits underlying innate anxiety. J Neurosci. 2011;31:6008–6018. doi: 10.1523/JNEUROSCI.5836-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gross C, et al. Serotonin1A receptor acts during development to establish normal anxiety-like behaviour in the adult. Nature. 2002;416:396–400. doi: 10.1038/416396a. [DOI] [PubMed] [Google Scholar]
- 37.Hannon J, Hoyer D. Molecular biology of 5-HT receptors. Behavioural Brain Research. 2008;195:198–213. doi: 10.1016/j.bbr.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 38.Lucas G, et al. Serotonin(4) (5-HT(4)) receptor agonists are putative antidepressants with a rapid onset of action. Neuron. 2007;55:712–725. doi: 10.1016/j.neuron.2007.07.041. [DOI] [PubMed] [Google Scholar]
- 39.Mendez-David I, et al. Rapid Anxiolytic Effects of a 5-HT Receptor Agonist Are Mediated by a Neurogenesis-Independent Mechanism. Neuropsychopharmacology. 2013 doi: 10.1038/npp.2013.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kobayashi K, Ikeda Y, Suzuki H. Behavioral destabilization induced by the selective serotonin reuptake inhibitor fluoxetine. Mol Brain. 2011;4:12. doi: 10.1186/1756-6606-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waterhouse EG, et al. BDNF promotes differentiation and maturation of adult-born neurons through GABAergic transmission. J Neurosci. 2012;32:14318–14330. doi: 10.1523/JNEUROSCI.0709-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lacefield CO, Itskov V, Reardon T, Hen R, Gordon JA. Effects of adult-generated granule cells on coordinated network activity in the dentate gyrus. Hippocampus. 2010 doi: 10.1002/hipo.20860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burghardt NS, Park EH, Hen R, Fenton AA. Adult-born hippocampal neurons promote cognitive flexibility in mice. Hippocampus. 2012 doi: 10.1002/hipo.22013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ikrar T, et al. Adult neurogenesis modifies excitability of the dentate gyrus. Frontiers in neural circuits. 2013;7:204. doi: 10.3389/fncir.2013.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jankord R, Herman JP. Limbic regulation of hypothalamo-pituitary-adrenocortical function during acute and chronic stress. Ann N Y Acad Sci. 2008;1148:64–73. doi: 10.1196/annals.1410.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schloesser RJ, Manji HK, Martinowich K. Suppression of adult neurogenesis leads to an increased hypothalamo-pituitary-adrenal axis response. Neuroreport. 2009;20:553–557. doi: 10.1097/WNR.0b013e3283293e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature. 2011;476:458–461. doi: 10.1038/nature10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Surget A, et al. Antidepressants recruit new neurons to improve stress response regulation. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fanselow MS, Dong HW. Are the Dorsal and Ventral Hippocampus Functionally Distinct Structures? Neuron. 2010;65:7–19. doi: 10.1016/j.neuron.2009.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McAskill R, Mir S, Taylor D. Pindolol augmentation of antidepressant therapy. The British journal of psychiatry : the journal of mental science. 1998;173:203–208. doi: 10.1192/bjp.173.3.203. [DOI] [PubMed] [Google Scholar]
- 51.Nguyen AW, Daugherty PS. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat Biotechnol. 2005;23:355–360. doi: 10.1038/nbt1066. [DOI] [PubMed] [Google Scholar]
- 52.Farley FW, Soriano P, Steffen LS, Dymecki SM. Widespread recombinase expression using FLPeR (flipper) mice. Genesis. 2000;28:106–110. [PubMed] [Google Scholar]
- 53.Samuels BA, Hen R. Novelty-Suppressed Feeding in the Mouse. In: Gould TD, editor. Mood and Anxiety Related Phenotypes in Mice: Characterization Using Behavioral Tests. II. Humana Press; Totowa, NJ, USA: 2011. pp. 107–121. [Google Scholar]
- 54.Wang J-W, David DJ, Monckton JE, Battaglia F, Hen R. Chronic fluoxetine stimulates maturation and synaptic plasticity of adult-born hippocampal granule cells. J Neurosci. 2008;28:1374–1384. doi: 10.1523/JNEUROSCI.3632-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Golde WT, Gollobin P, Rodriguez LL. A rapid, simple, and humane method for submandibular bleeding of mice using a lancet. Lab animal. 2005;34:39–43. doi: 10.1038/laban1005-39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.