

Abstract
Background
Heterogeneous bleeding phenotypes are observed in haemophilia A patients with the same mutation in the F8 gene. Specific mutations in the A2 domain of factor VIII are associated with mild haemophilia and a higher risk of inhibitor development. Double mutations in mild haemophilia A are rarely reported. In this study, we investigated the in vitro function of factor VIII, performing different specific and global coagulation assays, observed clinical characteristics and assessed the possible predictive diagnostic value of the differences.
Materials and methods
The clinical features of haemophiliacs with a mild phenotype were reviewed. Blood samples were obtained and analysed for mutations and coagulation assays: activated partial thromboplastin time, one-stage and chromogenic factor VIII activity, factor VIII antigen and rotational thromboelastometry.
Results
We report on a cohort of 22 patients with double Glu113Asp, Arg593Cys mutations. All our patients have a quantitative defect of factor VIII and preserved similar functional activity. Factor VIII activities measured by the one-stage or chromogenic method were not discrepant, although the chromogenic assay resulted in 20% lower factor VIII activities. Waveform analysis showed a lower maximum value of the second derivative curve (Max2) of APTT with curve shape alternation, while thromboelastometry (INTEM) showed low sensitivity in comparison to results in a normal population.
Discussion
In genotyping, the coexistence of a second mutation should never be excluded, especially in cases of discordant clinical presentation. Waveform analysis correlates better with factor VIII activity than thromboelastometry and the Max2 parameter could provide additional information in managing haemophilia patients. The utility of specific factor activity and global haemostatic assays in general practice still needs to be investigated.
Keywords: mild haemophilia A, double mutation, FVIII assay, waveform analysis, thromboelastometry
Introduction
Haemophilia A is a recessively inherited, X-linked bleeding disorder. In the mild form, the coagulant activity of factor VIII (FVIII:C) in patients’ plasma is reduced to 5–40%, being 30% on average1. The FVIII:C usually correlates with the clinical severity of haemophilia A; however, it is known that patients with the same FVIII:C can have different clinical bleeding phenotypes2,3. Patients with a mild form of the disease usually suffer from bleeding after trauma or surgery and rarely experience spontaneous haemarthroses4. Haemophilia is initially suspected when the activated partial thromboplastin time (APTT) is prolonged. The FVIII:C can be measured by a one- or two-stage clotting test or by the simplified chromogenic method. It is now clear that the one-stage clotting test or chromogenic method can result in discrepant results for FVIII:C, thus affecting the classification of haemophilia severity5,6.
Low procoagulant activity is caused by a deficiency or dysfunction of the FVIII protein due to mutations in different domains7. About 410 of more than 2,000 identified F8 mutations are single base substitutions and cause mild haemophilia A (HAMSTeR/HADB, http://www.factorviii-db.org/, last accessed on 22/06/2014). Almost 50% of missense mutations are clustered in the A2 domain8. These mutations can cause defects in protein folding and intracellular trafficking, defects in interactions with von Willebrand factor, thrombin activation, stability of FVIII, phospholipid binding and interactions with activated factor IX9,10. The amount of factor VIII protein (FVIII:Ag) in plasma does not always correlate with the protein’s activity. About 5% of patients with haemophilia A have a normal to slightly reduced level of dysfunctional FVIII in plasma as measured by the immunological method (type II defect)11.
The coexistence of multiple mutations in the same gene can have synergistic, neutral or opposing effects on the clinical phenotype12. There are 13 published cases of patients with double mutations in the F8 mutation database (HAMSTeRS/HADB, http://www.factorviii-db.org/, last accessed on 23/04/2014), four of them described as having a mild phenotype. Kentsis et al. reported that a double mutation can cause a more severe phenotype13, while the presence of an additional mutation should be considered in a discrepant phenotype.
Some mutations contribute to a higher risk of inhibitor development but the global incidence of this complication remains low in mild haemophilia: 2.7–13%9. Such mutations are located mainly within exons encoding C2 domains of the light chain of FVIII. The Arg593Cys mutation in the A2 domain has been identified as causing mild haemophilia with a higher risk of inhibitors14–16. Treatment modality (continuous infusion of the FVIII concentrate in association with surgical procedures), age at first exposure to exogenous FVIII, race and family history, and additional immunological markers involved in antibody production are all risk factors which have been associated with a higher risk of inhibitor development17–20.
We present a group of patients from Slovenia with the phenotype of mild haemophilia A and Arg593Cys, Glu113Asp mutations, either as a single or a double mutation. All cases had a prolonged APTT and FVIII activity, measured by a one-stage test, between 5% and 50%. In this cohort, we investigated possible discrepancies in coagulation results as determined by different FVIII:C assays and examined the value of APTT waveform analysis and rotational thromboelastometry (ROTEM®, ROTEM delta, Pentapharm GmbH, Munich, Germany). Possible synergistic effects of a double mutation on tests and clinical characteristics were explored and the potential predictive diagnostic value of tests was assessed.
Material and methods
Patients
In the Slovenian national registry, 48% of all 189 patients with haemophilia A have a mild phenotype. All of these patients (adults and children) were invited to participate in the study. Blood samples were obtained from the patients for mutation analysis and coagulation assays. In addition, samples from 20 healthy male adult subjects were obtained as a control group for the coagulation studies and 160 healthy unrelated DNA donors from Slovenia were used as healthy controls for the genetic studies. Blood was collected with the patients’ consent, using EDTA tubes for DNA extraction or 0.11 M trisodium citrate tubes for coagulation assays. Whole blood was analysed immediately (thromboelastometry) or processed to obtain plasma, which was stored in aliquots at −70 °C.
To investigate clinical features, we reviewed spontaneous joint, soft tissue and mucosal haemorrhagic events over a 6-year period (from 2007 to 2014). Factor VIII consumption was recorded for each patient (see Table I). At the time of study inclusion, seven patients were younger than 18 years. Forty-four percent of patients had A blood type and 39% had O blood type. In three cases, blood group was not determined during the patients’ life time. Fifteen of the 18 patients had no spontaneous bleeding episodes or needed replacement FVIII therapy only during trauma and/or invasive procedures. Ten patients, including the patient with a single mutation, did not receive any therapy during the observation period. Spontaneous bleeding episodes in three patients (17%) were treated with FVIII replacement. One patient developed inhibitors after a surgical procedure and continuous FVIII replacement therapy. He had 11 spontaneous soft tissue bleeds and four episodes of haemarthrosis in 2 years. Details of this patient have already been reported21.
Table I.
Haemorrhagic events and FVIII consumption in patients with Glu113Asp, Arg593Cys and the patient with Arg593Cys over the 6-year period.
| Patient/family | Age | Blood group | Observ. period (years) | FVIII:C (%) | Spontaneous bleeding | Other bleeds | Treatment | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| 1st | Chr | Joints | Soft tissue | Mucosae | FVIII (IU/kg) | Other | |||||
| 1/1 | 13 | A | 6 | 11.9 | 9.8 | - | - | - | Trauma, GI (ulcer) | 140 | BT |
| 2/2 | 12 | A | 6 | 8.3 | 7.6 | - | - | - | - | 0 | 0 |
| 3/2 | 2.5 | A | 2.5 | 6.5 | 4.2 | - | - | - | - | 0 | 0 |
| 4/3 | 44 | A | 6 | 8.2 | 6.7 | - | - | - | 2 traumas, tooth extraction | 279 | 0 |
| 5/3 | 38 | O | 6 | 5.8 | 3.1 | - | - | - | Tonsillectomy | 1,190 | Resuturing |
| 6/4 | 18 | ND | 6 | 8.9 | 6.9 | - | - | - | Trauma | 0 | 0 |
| 7/4 | 12 | ND | 6 | 9.1 | 7.1 | - | - | - | Trauma | 0 | 0 |
| 8/5 | 31 | O | 6 | 7.1 | 5.1 | - | - | - | - | 0 | 0 |
| 9/6 | 34 | O | 7 | 6.2 | 5.2 | 1 | - | - | Tooth extraction | 218 | 0 |
| 10/6 | 31 | O | 6 | 6.2 | 5.1 | - | - | - | - | 0 | 0 |
| 11/7 | 20 | ND | 6 | 7.6 | 7.1 | - | - | - | - | 0 | 0 |
| 12/7 | 51 | A | 6 | 7.3 | 6.5 | - | - | 1 | 3 traumas | 431 | 0 |
| 13/8# | 18 | A | 6 | 6.6 | 5.7 | 4 | 11 | - | 6 traumas, appendectomy, radiosynovect. | 1,229 | NS 624 mg, AF DDAVP$ |
| 14/9 | 5 | A | 5 | 7.3 | 4.6 | - | - | - | 5 traumas, urol. surgery, haemato-tympanon | 1,067 | DDAVP 2x |
| 15/10 | 54 | O | 6 | 8.1 | 7.9 | - | - | - | Tooth extraction | 272 | 0 |
| 16/11 | 36 | O | 6 | 8.2 | 8.2 | - | - | - | - | 0 | 0 |
| 17/12 | 34 | A | 6 | 12.2 | 10.5 | - | - | - | - | 0 | 0 |
| 18/13† | 48 | O | 6 | 14.1 | 13.5 | - | - | - | - | 0 | 0 |
Observ.: observation; FVIII:C: factor VIII coagulant activity; IU: International Unit; GI: gastrointestinal; BT: blood transfusion; 1st: one-stage assay; Chr: chromogenic assay; ND: not determined.
Development of inhibitors;
from 4/2008-2/2010 treatment with NovoSeven (NS) and antifibrinolytic (AF), from then on with DDAVP (negative inhibitors);
patient with a single mutation.
Mutation studies
Molecular studies were performed on DNA extracted from peripheral blood leucocytes as previously described22. To address whether identified mutations (screened against HAMSTeRS and HGMD [http://www.factorviii-db.org/ and http://www.hgmd.cf.ac.uk, last accessed on 23/04/2014]) could be polymorphisms, 160 healthy, unrelated DNA donors were used as healthy controls. The amino acid numbering matches the previously published system and is based upon the first amino acid of the mature protein being -19.
To detect the possible presence of both mutations in healthy individuals, we performed a high-resolution melting analysis of the polymerase chain reaction amplicons as previously described23. The high-resolution melting analysis was performed for the coding region and exon-intron boundaries of exons 4 and 12 of the F8 gene.
The F8 haplotypes of all selected patients were determined in order to investigate whether this group of patients shares a common ancestor. The primers to amplify within or adjacent to the F8 locus (DXS1073, DXS1108, DXS9897, F8Int22) were chosen as previously described24. Products were resolved with a 3500 Genetic Analyzer (Applied Biosystems, Norwalk, CT, USA) and analysed using GeneMapper software (Applied Biosystems).
Coagulation assays
APTT was determined by an ACL TOP 500 analyser (Instrumentation Laboratory [IL], Lexington, MA, USA) using the APTT-SP reagent (IL). The clot-reaction curve was measured kinetically at 671 nm. Waveform analysis involves calculation of the first derivative (velocity) and the second derivative (acceleration) of the clotting reaction by the ACL TOP software. The absolute maximum value of the second derivative (Max2) indicates the point of maximum inflection of the first derivative curve and represents the point of maximum coagulation acceleration25.
The one-stage FVIII:C (FVIII:C-1st) assay was performed using the ACL TOP 500 analyser by standard APTT methods with the APTT-SP reagent (IL) and FVIII-deficient plasma (IL).
The chromogenic assay for FVIII (FVIII:C-Chr) was performed using the Electachrome Factor VIII kit (IL) according to the manufacturer’s instructions for the microplate method, with the exception that the incubation time of the first step was extended. The prolongation from 120 to 420 sec was decided in order to make the assay more sensitive. Activated FVIII is inactivated more quickly than normally during a longer incubation time, allowing for a better detection of a mild discrepant FVIII phenotype26.
FVIII:Ag levels were determined with the Asserachrom VIII:Ag enzyme immunoassay kit (Diagnostica Stago, Asnieres, France), according to the package insert, by a manual method (Multiskan EX, Thermo Labsystem, Vantaa, Finland).
Rotation thromboelastometry was performed on ROTEM® using the INTEM predefined test. Processes of intrinsic activation involving thrombin generation, clot formation (fibrin polymerisation) and lyses are described with the following parameters: clotting time (CT), the period from the start of analysis until the start of clot formation; clot formation time (CFT), the period until an amplitude of 20 mm is reached; maximum clot firmness (MCF), the maximum amplitude of the curve; and α-angle, the angle of tangent to the curve at 2 mm amplitude, which represents the rate of clot polymerisation27.
Statistical analysis
All statistical calculations were performed and figures created with GraphPad Prism 6.01 for Windows (GraphPad Software, La Jolla, CA, USA). A paired t-test was used to compare methods and an unpaired t-test was used to compare patients vs controls in order to detect statistically significant differences (p<0.05).
Results
There are 90 patients with mild haemophilia A in the Slovenian Haemophilia Registry. We identified 22 patients from 12 unrelated families sharing the same double F8 mutations: the first in exon 4, p.Glu132Asp (c.396A>C, legacy numbering Glu113Asp), and the second in exon 12, p.Arg612Cys (c.1834C>T, legacy numbering Arg593Cys). One patient has a single Arg593Cys mutation. The high-resolution melting analysis clearly distinguished exon 4 and exon 12 F8 mutations in patients when compared to 160 normal controls. Altered melting curves were easily identified for both mutations, showing that they are not present in the general population (data not shown). We also determined four F8 microsatellite haplotypes. All patients with a double mutation had the same three alleles and one different allele, indicating that all probands with this mutation share a common ancestor.
Eighteen patients agreed to participate in additional coagulation studies. The clinical characteristics of the studied group are presented in Table I. All patients with a double mutation have prolonged APTT, ranging from 43 to 59 sec (reference range: 23–36 sec) and low FVIII:C-1st, ranging from 5.8 to 11.9% (mean, 8.0%). The patient with a single mutation has an average FVIII:C-1st of 13.1% (number of samples=3), which is also the highest factor level in our group of patients (Table II).
Table II.
Mean values and standard deviation (in brackets) of APTT, Max2, FVIII activities and antigen in double mutation and single mutation patients.
| N. of patients | APTT (s) | Max2 (dA/dt2) | FVIII:C-1st (%) | FVIII:C-Chr (%) | FVIII:Ag (%) | |
|---|---|---|---|---|---|---|
| Arg593Cys, Glu113Asp | 17 | 47 (4)* | 249 (43)* | 8.0 (1.8)* | 6.5 (1.9)* | 10.4 (1.9) |
| Arg593Cys | 1$ | 42 | 375 | 13.1 | 13.5 | 13.0 |
| Normal controls | 20 | 29 (2.8) | 635 (140) | 122.0 (39.0) | 113.9 (31.0) | 63–199# |
APTT: activated partial thromboplastin time; Max2: maximum value of the second derivative curve; FVIII:C-1st: one-stage clotting assay for factor VIII; FVIII:C-Chr: chromogenic assay for FVIII; FVIII:Ag: factor VIII protein.
Three independent samples from a single patient;
p<0.0001 (patients vs normal controls);
manufacturer’s expected values.
The APTT of the patients with a single mutation was shorter (42 sec) than the average APTT of the patients with double mutations (47 sec). The first and second derivative algorithm of the primary APTT curve locates the clotting time simultaneously with the light absorbance change during measurement. The point of Max2 represents the clotting time. The average Max2 in controls was 635 dA/dt2, the patient with a single mutation had a Max2 value of 375 dA/dt2 and the average Max2 value of the patients with double mutations was 249 dA/dt2 (Figure 1). When we compared the second derivative curves from controls and haemophiliacs, there were differences not only in the position and height of the peaks but also in the shape of the curves (Figure 2). All patients with haemophilia A had a smaller subsequent peak in the second derivative curve, meaning that the APTT reaction was accelerated twice in the process of fibrin formation. We found a weak correlation between FVIII:C-1st and Max2 (r=0.676), whereas no correlation was found between FVIII:C-1st and APTT.
Figure 1.
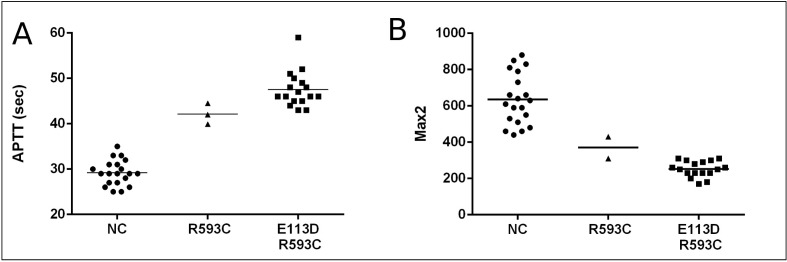
The (A) APTT and (B) Max2 in normal controls, a patients with a single mutation (R593C) and patients with double mutations (E113D, R593C).
The lines represent the means for each group.
APTT: activated partial thromboplastin time; Max2: maximum value of the second derivative curve.
Figure 2.
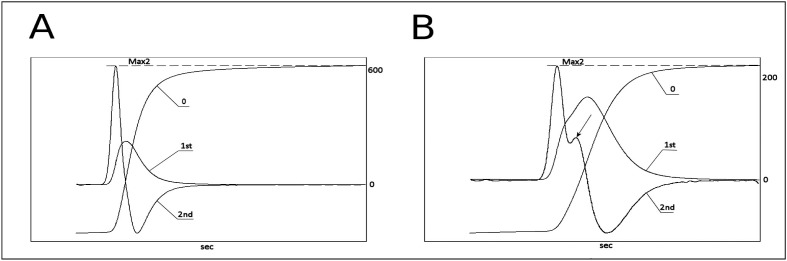
Typical APTT reaction curves of (A) normal subjects and (B) patients with mild haemophilia A with a double mutation.
0: APTT, 1st: first derivative curve, 2nd: second derivative curve. The Max2 value (dA/dt2) is marked with the dashed line; the arrow points to the subsequent peak of the second derivative curve.
Note: the scales on the right axes are not of the same magnitude.
APTT: activated partial thromboplastin time; Max2: maximum value of the second derivative curve.
The FVIII chromogenic assay was performed in order to assess the potential discrepancy between methods for determining FVIII:C. The FVIII:C-1st/FVIII:C-Chr activity ratio was between 1.0 and 1.9, meaning that the results were not discrepant (ratio<2.0) for any of the patients. The mean measured one-stage FVIII:C was approximately 20% higher than the measured chromogenic activity (8.0 vs 6.5%, p<0.0001). In three of the 17 double mutation patients, the FVIII:C-Chr activity was measured at less than 5% and these patients might be classified as having moderate haemophilia A. The patient with a single mutation had the highest FVIII activity (13.1 and 13.5%) regardless of the method used. Between-run coefficients of variation were 5% for the chromogenic assay and 5.5% for the one-stage method, according to manufacturer’s specifications.
We performed an immunoassay to determine whether the patients had a quantitative or qualitative defect of protein. The level of FVIII:Ag was proportional to FVIII activity in all 18 patients with either of the two mutations (Table II). Figure 3 shows results for FVIII activities and the antigen for our cohort of patients. Neither FVIII:C or FVIII:Ag correlated with the blood group in the 15 patients whose ABO phenotype was determined.
Figure 3.

FVIII one-stage (FVIII:C-1st), chromogenic (FVIII:C-Chr) activity and antigen (FVIII:Ag) for double (●) and single mutation (□) patients.
The INTEM test performed with ROTEM® represents a complement of the APTT coagulation test. The test is stated to be sensitive to acquired or congenital intrinsic factor deficiencies. However, only seven patients with either mutation had a CT above the manufacturer’s reference interval (100–240 sec), while the others had CT values in the normal range albeit above the normal mean value. Five patients had a low α-angle. The CFT and MCF showed even less or no deviation from reference ranges. All INTEM parameters of the patient with a single mutation were within the normal range (Figure 4). We found no correlation of INTEM with the bleeding phenotype in our group of patients.
Figure 4.

ROTEM® parameters in double mutation patients (○) and a single mutation patient (□).
The rectangles represent the normal ranges for parameters. CT: clotting time; CFT: clot formation time; MCF: maximum clot firmness.
Discussion
The HAMSTeRS/HADB database includes four double mutations described as causing a mild haemophilia A phenotype, three being double missense mutations and one occurring in combination with a polymorphism. In our study, we report for the first time 22 patients in the Slovenian population with a double mutation (Glu113Asp, Arg593Cys), all with phenotypically mild haemophilia A with FVIII:C-1st from 5.8–11.9% and a proportionally reduced antigen.
Both mutations have already been described and individually produce a mild phenotype28. The Glu113Asp mutation lies in the A1 domain in a Ca2+ binding region. The substitution of Glu with the smaller Asp residue in position 113 conserves a charge but weakens the binding of metal ions29. It is suggested that suboptimal occupancy of the metal binding site may therefore decrease the cofactor activity. The missense mutation Arg593Cys is localised in the A2 domain. The loss of ion bonds and formation of a disulphide bridge due to the change into cysteine cause an abnormal conformation and destabilise the FVIII molecule10,30. The Factor VIII Variant Database28 reported data for FVIII:C for both mutations. The average FVIII:C-1st for Glu113Asp and Arg593Cys single mutation patients were 9.7% and 15% respectively. Our double mutation patients’ average FVIII:C-1st activity was 8%. This value is lower than the majority of reported ones. All our patients were categorised as having type I or a quantitative defect while preserving some functional activity. These findings are consistent with those of Roelse et al.31, who reported that the Arg593Cys mutation causes intracellular accumulation and degradation of FVIII due to suboptimal folding of protein in transfected cells, and in contrast with some reports from the Factor VIII Variant Database describing a much higher antigen level of FVIII.
The unique finding of the double mutation prompted us to investigate whether these patients had a shared descent. Eighteen selected patients belong to 12 different families. Their haplotype profile indicates that all probands for the double mutation share a common ancestor. As we expected, the haplotype of the patient with a single mutation has a distinct pattern of allele sizes. A similar founder effect was found in the New Zealand and Dutch populations in a group of patients with the Arg593Cys mutation16,32. Our analysis also showed that this double mutation is not present in the healthy population in Slovenia.
It is still debatable which method for determining FVIII activity reflects patients’ phenotype better33. There was not a marked discrepancy in FVIII:C-1st/FVIII:C-Chr results in our patients; however, the results of the one-stage assay were, on average, 20% higher than the results obtained with the chromogenic method. Three of our patients had FVIII:C-Chr levels lower than 5%. None of them had a spontaneous bleeding episode in the observed period. They were treated with FVIII replacement therapy when having to undergo surgical procedures. A FVIII activity lower than 5% is concordant with some previous reports describing a moderate phenotype for the Arg593Cys mutation28. The overall bleeding tendency in our patients was not higher than that in the group with mild haemophilia A in general. Although there were seven patients with O blood type in the study, their FVIII activity was not lower than that in patients with A blood type, as we could have expected. The chromogenic FVIII method was chosen over the classical two-stage method, which is laborious and non-standardised with lower precision. The chromogenic method is accurate in the lower range and can be easily modified or/and automated. It is not influenced by the clotting specific reagent or the presence of lupus anticoagulants. It has largely replaced the two-stage assay and is now the reference method for determining the potency of FVIII concentrates34.
We demonstrated a lower Max2 magnitude and an additional small peak in the second derivative APTT curve in double and single mutation patients as compared to normal controls. Despite a very low thrombin concentration in the test, monitoring the subtle changes in light transmittance following the activation and re-calcification of plasma in our group of patients suggests that the normal sigmoid waveform pattern of the process of fibrin formation is altered. It is known that in haemophilia A patients the initiation phase of the tenase complex is delayed, the thrombin-dependent mechanism of clot stability is affected and the rate of fibrinopeptide formation is altered35. Similar findings support the sensitivity of waveform analysis in patients with haemostatic disorders, such as disseminated intravascular coagulation, sepsis, and high-titre haemophilia inhibitors36–39, and its contribution to diagnostics and evaluation of haemostasis. The sensitivity of APTT reagents to factor deficiency varies40. All our patients had FVIII activity levels below 30%, while the APTT-SP reagent sensitivity for FVIII deficiency is 40% according to the manufacturer. APTT-SP is suitable as a screening reagent and shows the additional potential to predict factor deficiency from waveform analysis in the routine phase of laboratory diagnostics. Its clinical utility in different forms of haemophilia and in optimising replacement therapy still needs to be investigated.
The modified version of thromboelastography, ROTEM®, is a whole blood method based on the activation of cellular and enzymatic components of coagulation. The method has been used in various clinical states as a perioperative screening technique for hyper- or hypocoagulability and (in the last few years) for assessing the bleeding tendency in patients with bleeding disorders41. Pathological results with INTEM are often obtained in patients with haemophilia or inhibitors42. The values of INTEM parameters (CT, CFT, MCF and the α-angle) in our group of patients with double or single mutations did not reliably differ from those in healthy individuals43. The most sensitive parameter was CT, which was prolonged in 38% of all observed patients in our study. These results are in agreement with previously reported data44 indicating that the sensitivity of ROTEM is poor when used in patients with mild haemophilia. The increased variability in INTEM parameters seen in our patients may be a reflection of a wide difference in factor FVIII:C-1st (range, 5.8% to 11%) compared to that in studies with patients with severe or moderate haemophilia, in which the thromboelastogram phenotype characterised the FVIII activity better45. Thromboelastometry and thromboelastography are not yet standardised methods, although great progress has been made in the past few years. In our study, we minimised errors in the pre-analytical phase, following the recommendations of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis group46.
Coagulation test results showed that the presence of two mutations has a slightly synergistic effect on FVIII activity. The patient with a single mutation had the highest FVIII activity, determined with the chromogenic and one-stage tests. The difference between the value in this patient and those with two mutations was evident, although not huge. This finding was supported by the difference in the Max2 parameter of the APTT waveform analysis, where the single mutation patient had higher Max2 values than any of the patients with a double mutation (375 versus 249 dA/dt2, respectively). Suboptimal binding of the bivalent ion in patients with an E113D mutation may additionally change the kinetics of fibrin formation in the APTT test and influence FVIII activity29.
The Arg593Cys mutation is a well-described mutation which has been found to be related to a higher risk of inhibitor formation9,30, especially in the context of intensive, perioperative FVIII use16. Eight of our patients were given FVIII replacement therapy in the observation period because of spontaneous bleeds, trauma or invasive procedures. Only one of them, a patient aged 12 years, developed inhibitors after a surgical procedure21. This patient had some of the previously described risk factors for inhibitor development: a new Cys residue introduced in the mutant protein structure30 and long-term perioperative FVIII replacement therapy16.
Conclusions
Double mutations are not common in patients with haemophilia A. The fact that many discordant clinical presentations are observed raises concern regarding the current practice in F8 mutation determination. In genotyping, the existence of a second mutation should always be excluded. The additional inhibitor risk mutation in our patients is of great importance for their clinical management. The coexistence of the second mutation in the same allele permits the genotyping of only one mutation for the purpose of prenatal screening. The presence of two mutations in our patients did not affect the clinical phenotype, despite a slightly lower FVIII:C. The utility of specific factor activity assays and global haemostatic assays is still under investigation. Haemophilia centres should use one-stage and chromogenic (or two-stage) assays in the diagnosis of possible mild haemophilia A with discrepancy. The ROTEM® global assay appears to be insensitive in mild haemophilia A, at least using our analytical conditions. Waveform analysis, on the other hand, correlates with FVIII activity better and could be a sensitive additional parameter in the diagnosis and management of haemophilia patients. However, the variability of these methods limits their generalised use in clinical practice.
Acknowledgements
We would like to thank Ms Rajka Bavčer and Ms Eva Đalić for technical assistance. The study was approved by the National Medical Ethics Committee of the Republic of Slovenia (approval n. 60/40/11).
Footnotes
Authorship contributions
JJ, ATB and MD designed the study, ATB, MD and JK performed the research, ATB wrote the paper, MD and JJ revised the paper, IPZ and MBD acquired clinical data and revised the paper.
The Authors declare no conflict of interest.
References
- 1.Franchini M, Favaloro EJ, Lippi G. Mild hemophilia A. J Thromb Haemost. 2010;8:421–32. doi: 10.1111/j.1538-7836.2009.03717.x. [DOI] [PubMed] [Google Scholar]
- 2.Santagostino E, Mancuso ME, Tripodi A, et al. Severe hemophilia with mild bleeding phenotype: molecular characterization and global coagulation profile. J Thromb Haemost. 2010;8:737–43. doi: 10.1111/j.1538-7836.2010.03767.x. [DOI] [PubMed] [Google Scholar]
- 3.Van den Berg HM, De Groot PH, Fischer K. Phenotypic heterogeneity in severe hemophilia. J Thromb Haemost. 2007;5(Suppl 1):151–6. doi: 10.1111/j.1538-7836.2007.02503.x. [DOI] [PubMed] [Google Scholar]
- 4.White GC, 2nd, Rosendaal F, Aledort LM, et al. Definitions in hemophilia. Recommendation of the scientific subcommittee on factor VIII and factor IX of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 2001;85:560. [PubMed] [Google Scholar]
- 5.Cid AR, Calabuig M, Cortina V, et al. One-stage and chromogenic FVIII:C assay discrepancy in mild haemophilia A and the relationship with the mutation and bleeding phenotype. Haemophilia. 2008;14:1049–54. doi: 10.1111/j.1365-2516.2008.01781.x. [DOI] [PubMed] [Google Scholar]
- 6.Bowyer AE, Van Veen JJ, Goodeve AC, et al. Specific and global coagulation assays in the diagnosis of discrepant mild hemophilia A. Haematologica. 2013;98:1980–7. doi: 10.3324/haematol.2013.088088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fang H, Wang L, Wang H. The protein structure and effect of factor VIII. Thromb Res. 2007;119:1–13. doi: 10.1016/j.thromres.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 8.McGinniss MJ, Kazazian HH, Jr, Hoyer LW, et al. Spectrum of mutations in CRM-positive and CRM-reduced hemophilia A. Genomics. 1993;15:392–8. doi: 10.1006/geno.1993.1073. [DOI] [PubMed] [Google Scholar]
- 9.D’Oiron R, Pipe SW, Jacquemin M. Mild/moderate haemophilia A: new insights into molecular mechanisms and inhibitor development. Haemophilia. 2008;14(Suppl 3):138–46. doi: 10.1111/j.1365-2516.2008.01730.x. [DOI] [PubMed] [Google Scholar]
- 10.Markoff A, Gerke V, Bogdanova N. Combined homology modelling and evolutionary significance evaluation of missense mutations in blood clotting factor VIII to highlight aspects of structure and function. Haemophilia. 2009;15:932–41. doi: 10.1111/j.1365-2516.2009.02009.x. [DOI] [PubMed] [Google Scholar]
- 11.Amano K, Sarkar R, Pemberton S, et al. The molecular basis for cross-reacting material-positive hemophilia A due to missense mutations within the A2-domain of factor VIII. Blood. 1998;91:538–48. [PubMed] [Google Scholar]
- 12.Chen J-M, Férec C, Cooper DN. Closely spaced multiple mutations as potential signatures of transient hypermutability in human genes. Hum Mutat. 2009;30:1435–48. doi: 10.1002/humu.21088. [DOI] [PubMed] [Google Scholar]
- 13.Kentsis A, Anewalt R, Ganguly A, et al. Discordant haemophilia A in male siblings due to a de novo mutation on a familial missense mutant allele. Haemophilia. 2009;15:971–2. doi: 10.1111/j.1365-2516.2009.02035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fijnvandraat K, Turenhout EA, van den Brink EN, et al. The missense mutation Arg593 --> Cys is related to antibody formation in a patient with mild hemophilia A. Blood. 1997;89:4371–7. [PubMed] [Google Scholar]
- 15.Oldenburg J, El-Maarri O, Schwaab R. Inhibitor development in correlation to factor VIII genotypes. Haemophilia. 2002;8(Suppl 2):23–9. doi: 10.1046/j.1351-8216.2001.00134.x. [DOI] [PubMed] [Google Scholar]
- 16.Eckhardt CL, Menke LA, van Ommen CH, et al. Intensive peri-operative use of factor VIII and the Arg593-->Cys mutation are risk factors for inhibitor development in mild/moderate hemophilia A. J Thromb Haemost. 2009;7:930–7. doi: 10.1111/j.1538-7836.2009.03357.x. [DOI] [PubMed] [Google Scholar]
- 17.Oldenburg J, Pavlova A. Genetic risk factors for inhibitors to factors VIII and IX. Haemophilia. 2006;12(Suppl 6):15–22. doi: 10.1111/j.1365-2516.2006.01361.x. [DOI] [PubMed] [Google Scholar]
- 18.Mauser-Bunschoten EP, Den Uijl IE, Schutgens RE, et al. Risk of inhibitor development in mild haemophilia A increases with age. Haemophilia. 2012;18:263–7. doi: 10.1111/j.1365-2516.2011.02629.x. [DOI] [PubMed] [Google Scholar]
- 19.Astermark J, Berntorp E, White GC, et al. The Malmö International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patients. Haemophilia. 2001;7:267–72. doi: 10.1046/j.1365-2516.2001.00510.x. [DOI] [PubMed] [Google Scholar]
- 20.Shepherd AJ, Skelton S, Sansom CE, et al. A large-scale computational study of inhibitor risk in non-severe haemophilia A. Br J Haematol. 2015;168:413–20. doi: 10.1111/bjh.13131. [DOI] [PubMed] [Google Scholar]
- 21.Dolničar MB, Rajić V, Kitanovski L, Debeljak M. Rituximab for the treatment of high titre inhibitors in mild haemophilia A. Blood Transfus. 2014;12(Suppl 1):s345–7. doi: 10.2450/2013.0229-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Debeljak M, Kitanovski L, Trampuš Bakija A, Benedik Dolničar M. Spectrum of F8 gene mutations in haemophilia A patients from Slovenia. Haemophilia. 2012;18:e420–3. doi: 10.1111/hae.12003. [DOI] [PubMed] [Google Scholar]
- 23.Kovač J, Macedoni Lukšič M, Trebušak Podkrajšek K, et al. Rare single nucleotide polymorphisms in the regulatory regions of the superoxide dismutase genes in autism spectrum disorder. Autism Res. 2014;7:138–44. doi: 10.1002/aur.1345. [DOI] [PubMed] [Google Scholar]
- 24.Harraway JR, Smith MP, George PM. A highly informative, multiplexed assay for the indirect detection of hemophilia A using five-linked microsatellites. J Thromb Haemost. 2006;4:587–90. doi: 10.1111/j.1538-7836.2006.01790.x. [DOI] [PubMed] [Google Scholar]
- 25.Nair PS, Shetty S, Ghosh K. Double mutations in haemophilia: muddling strangers or indifferent partners in crime? Haemophilia. 2010;16:970–1. doi: 10.1111/j.1365-2516.2010.02326.x. [DOI] [PubMed] [Google Scholar]
- 26.Rodgers SE, Duncan EM, Sobieraj-Teague M, Lloyd JV. Evaluation of three automated chromogenic FVIII kits for the diagnosis of mild discrepant haemophilia A. Int J Lab Hematol. 2009;31:180–8. doi: 10.1111/j.1751-553X.2007.01021.x. [DOI] [PubMed] [Google Scholar]
- 27.Theusinger OM, Nürnberg J, Asmis LM, et al. Rotation thromboelastometry (ROTEM) stability and reproducibility over time. Eur J Cardio-Thorac Surg. 2010;37:677–83. doi: 10.1016/j.ejcts.2009.07.038. [DOI] [PubMed] [Google Scholar]
- 28.Rallapalli P, Kemball-Cook G, Tuddenham EG, et al. Factor FVIII Variant Database. 2014. Manuscript in preparation. [Google Scholar]
- 29.Wakabayashi H, Su Y-C, Ahmad SS, et al. A Glu113Ala mutation within a factor VIII Ca2+-binding site enhances cofactor interactions in factor Xase. Biochemistry. 2005;44:10298–304. doi: 10.1021/bi050638t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hay CR. Factor VIII inhibitors in mild and moderate-severity haemophilia A. Haemophilia. 1998;4:558–63. doi: 10.1046/j.1365-2516.1998.440558.x. [DOI] [PubMed] [Google Scholar]
- 31.Roelse JC, De Laaf RT, Timmermans SM, et al. Intracellular accumulation of factor VIII induced by missense mutations Arg593-->Cys and Asn618-->Ser explains cross-reacting material-reduced haemophilia A. Br J Haematol. 2000;108:241–6. doi: 10.1046/j.1365-2141.2000.01834.x. [DOI] [PubMed] [Google Scholar]
- 32.Laurie AD, Sheen CR, Hanrahan V, et al. The molecular aetiology of haemophilia A in a New Zealand patient group. Haemophilia. 2007;13:420–7. doi: 10.1111/j.1365-2516.2007.01487.x. [DOI] [PubMed] [Google Scholar]
- 33.Makris M, Peyvandi F. Assaying FVIII activity: one method is not enough, and never was. Haemophilia. 2014;20:301–3. doi: 10.1111/hae.12446. [DOI] [PubMed] [Google Scholar]
- 34.Verbruggen B, Meijer P, Novákova I, Van Heerde W. Diagnosis of factor VIII deficiency. Haemophilia. 2008;14(Suppl 3):76–82. doi: 10.1111/j.1365-2516.2008.01715.x. [DOI] [PubMed] [Google Scholar]
- 35.Wolberg AS, Campbell RA. Thrombin generation, fibrin clot formation and hemostasis. Transfus Apher Sci. 2008;38:15–23. doi: 10.1016/j.transci.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toh CH, Ticknor LO, Downey C, et al. Early identification of sepsis and mortality risks through simple, rapid clot-waveform analysis. Implications of lipoprotein-complexed C reactive protein formation. Intensive Care Med. 2003;29:55–61. doi: 10.1007/s00134-002-1557-2. [DOI] [PubMed] [Google Scholar]
- 37.Smith EY, Charles LA, Van Cott EM. Biphasic activated partial thromboplastin time waveform and adverse events in non-intensive care unit patients. Am J Clin Pathol. 2004;121:138–41. doi: 10.1309/W4F7-892W-JE6Y-1W7Y. [DOI] [PubMed] [Google Scholar]
- 38.Kasuda S, Tanaka I, Shima M, et al. Effectiveness of factor VIII infusions in haemophilia A patients with high responding inhibitors. Haemophilia. 2004;10:341–6. doi: 10.1111/j.1365-2516.2004.00907.x. [DOI] [PubMed] [Google Scholar]
- 39.Shima M, Matsumoto T, Ogiwara K. New assays for monitoring haemophilia treatment. Haemophilia. 2008;14(Suppl 3):83–92. doi: 10.1111/j.1365-2516.2008.01737.x. [DOI] [PubMed] [Google Scholar]
- 40.Bowyer A, Kitchen S, Makris M. The responsiveness of different APTT reagents to mild factor VIII, IX and XI deficiencies. Int J Lab Hematol. 2011;33:154–8. doi: 10.1111/j.1751-553X.2010.01261.x. [DOI] [PubMed] [Google Scholar]
- 41.Chitlur M, Sorensen B, Rivard GE, et al. Standardization of thromboelastography: a report from the TEG-ROTEM working group. Haemophilia. [Accessed on 15/06/2014]. Available at: http://www.ncbi.nlm.nih.gov/pubmed/21323795. [DOI] [PubMed]
- 42.Ghosh K, Shetty S, Kulkarni B. Correlation of thromboelastographic patterns with clinical presentation and rationale for use of antifibrinolytics in severe haemophilia patients. Haemophilia. 2007;13:734–9. doi: 10.1111/j.1365-2516.2007.01544.x. [DOI] [PubMed] [Google Scholar]
- 43.Lang T, Bauters A, Braun SL, et al. Multi-centre investigation on reference ranges for ROTEM thromboelastometry. Blood Coagul Fibrinolysis. 2005;16:301–10. doi: 10.1097/01.mbc.0000169225.31173.19. [DOI] [PubMed] [Google Scholar]
- 44.Van Veen JJ, Gatt A, Bowyer AE, et al. Calibrated automated thrombin generation and modified thromboelastometry in haemophilia A. Thromb Res. 2009;123:895–901. doi: 10.1016/j.thromres.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 45.Chitlur M, Warrier I, Rajpurkar M, et al. Thromboelastography in children with coagulation factor deficiencies. Br J Haematol. 2008;142:250–6. doi: 10.1111/j.1365-2141.2008.07063.x. [DOI] [PubMed] [Google Scholar]
- 46.Chitlur M, Rivard GE, Lillicrap D, et al. Recommendations for performing thromboelastography/thromboelastometry in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12:103–6. doi: 10.1111/jth.12458. [DOI] [PubMed] [Google Scholar]