

Abstract
Background
The migratory locust, Locusta migratoria manilensis, is an immensely destructive agricultural pest that forms a devastating and voracious gregarious phase. The fungal insect pathogen, Metarhizium acridum, is a specialized locust pathogen that has been used as a potent mycoinsecticide for locust control. Little, however, is known about locust immune tissue, i.e. fat body and hemocyte, responses to challenge by this fungus.
Methods
RNA-seq (RNA sequencing) technology were applied to comparatively examine the different roles of locust fat body and hemocytes, the two major contributors to the insect immune response, in defense against M. acridum. According to the sequence identity to homologies of other species explored immune response genes, immune related unigenes were screened in all transcriptome wide range from locust and the differential expressed genes were identified in these two tissues, respectively.
Results
Analysis of differentially expressed locust genes revealed 4660 and 138 up-regulated, and 1647 and 23 down-regulated transcripts in the fat body and hemocytes, respectively after inoculation with M. acridum spores. GO (Gene Ontology) enrichment analysis showed membrane biogenesis related proteins and effector proteins significantly differentially expressed in hemocytes, while the expression of energy metabolism and development related transcripts were enriched in the fat body after fungal infection. A total of 470 immune related unigenes were identified, including members of the three major insect immune pathways, i.e. Toll, Imd (immune deficiency) and JAK/STAT (janus kinase/signal transduction and activator of transcription). Of these, 58 and three were differentially expressed in the insect fat body or hemocytes after infection, respectively. Of differential expressed transcripts post challenge, 43 were found in both the fat body and hemocytes, including the LmLys4 lysozyme, representing a microbial cell wall targeting enzyme.
Conclusions
These data indicate that locust fat body and hemocytes adopt different strategies in response to M. acridum infection. Fat body gene expression after M. acridum challenge appears to function mainly through activation of innate immune related genes, energy metabolism and development related genes. Hemocyte responses attempt to limit fungal infection primarily through regulation of membrane related genes and activation of cellular immune responses and release of humoral immune factors.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-2089-9) contains supplementary material, which is available to authorized users.
Keywords: Locust, Locusta migratoria, Insect immunity, Fat body, Hemocyte, Entomopathogen, Fungal virulence, Metarhizium acridum
Background
The migratory locust, Locusta migratoria manilensis, undergoes a striking behavioral phase transition, from a solitary, essentially harmless animal to a seemingly endless cloud of voracious, traveling swarms that devour the vegetation in their path. Both significant historical and modern accounts of their rampaging effects have been recorded. Continued outbreaks and infestations in Madagascar, the Red Sea coast, and in the Caucasus and Central Asia are but several recent examples [1]. Invertebrates, including locusts, lack the ability to produce true antibodies as part of adaptive immune responses, and instead rely on innate mechanisms for mitigating microbial infections, consisting of cellular and humoral responses [2]. Humoral factors include the production of oxygen and nitrogen free radicals, anti-microbial peptides (AMPs), and enzyme cascades, e.g. the prophenoloxidase (PPO) pathway, the latter of which produce cytotoxic quinones and mediate coagulation and melanization responses upon pathogen detection [3]. Many humoral compounds are produced and secreted into the hemocoel by the fat body, the main insect organ of intermediate metabolism [3]. The fat body produces most of the proteins and metabolites found in the hemolymph, and acts as the central controller of the synthesis and utilization of energy reserves, i.e. glycogen and lipids [4]. This organ also functions to sequester, and release upon appropriate (hormonal) signal, proteins or other molecules required for morphogenesis, egg maturation, and lipid/hormone transport, examples of which include growth factors, vitellogenins, and lipophorins, respectively [5].
Insects also have macrophage-equivalent cells termed hemocytes that circulate in the hemolymph and are capable of secreting a number of humoral immune factors. One of the main activities of hemocytes is to phagocytose foreign cells and material. Hemocyte aggregation seeks to isolate and entrap foreign materials within the hemocoel, resulting in nodule formation and encapsulation that is coupled with melanization via PPO activation, thus linking aspects of the humoral and cellular responses [6]. Insect hemocytes are produced by stem cells that are mesodermally located, and differentiate into a variety of different morphologically and functionally distinct lineages. Depending upon the insect, a combination of a number of the most common types of hemocytes, i.e. granulocytes, prohemocytes, plasmatocytes, spherulocytes, and/or oenocytoids are produced [7]. One mechanism of hemocytes action occurs via recognition of foreign microbial targets by pattern recognition receptors (PPRs) that respond to pathogen-associated molecular pattern (PAMP) molecules found on microorganisms. Significant information concerning insect immune systems is known for dipterans, i.e. the Drosophila model system as well as various mosquito species [8, 9], hymenopteran honey bees, Apis mellifera [10], and more recently the coleopteran (beetles) insect Tribolium castaneum [11]. A variety of immune related genes have been shown to be expressed in insect hemocytes and fat bodies. In the mosquito, Anopheles gambia, 1053 genes were found to be predominantly expressed in adult female hemocytes [12], with 13 and 44 immune related genes were differentially transcribed in A. gambiae hemocytes after challenged by Escherichia coli or Micrococcus luteus [13]. In the mosquito species, Aedes aegypti and Armigeres subalbatus, 169 and 103 immune related EST clusters were identified in hemocytes, with a small subset, 11 and 16 genes, differentially expressed after bacterial inoculation, respectively [14]. In the tsetse fly, Glossinia morsitans, 60 putative immunity-pathway-related genes were identified in the fat body [15], and 80 putative immune clusters were identified in the fat body of Antheraea mylitta after challenged by Escherichia coli [16]. Whole body transcriptional responses of several insects, including of the corn borer, Ostrinia furnacalis and the whitefly, Bemesia tabaci to infection by B. bassiana have been reported [17, 18]. Although changes in the expression of various immune-related genes were noted, such whole organism approaches are likely to obscure significant portions of the immune response, and information concerning the reactions of specific immune-related tissues is lacking. In addition, far less is known concerning both general and specific microbial immune responses of hemimetabolous orthopterans that include locusts, than their holometabolous counterparts.
Insect pathogenic fungi, especially Beauveria and Metarhizium species, have long been considered as potential pest biological control agents, with several commercial products currently available worldwide [19, 20]. Entomogenous fungi have been suggested to engage in a co-evolutionary arms race with their insect hosts [21]. These agents pose less environmental risk than chemical insecticides and are compatible with organic and sustainable farming practices, notably available in developing countries. While many entomopathogenic fungi are broad host range insect pathogens, e.g. B. bassiana and M. roberstsii, several lineages have evolved high but restricted virulence towards certain insect hosts [22, 23]. M. acridum is one such restricted host-range species, particularly effective against orthopteran insects and has been used in both Africa and Asia as an effective agent as part of Integrated Pest Management (IPM) programs for locust control [24]. Infection occurs via attachment of fungal spores (conidia) to host surfaces, followed by germination and penetration of the insect cuticle [25]. As penetrating hyphae reach the hemocoel, they produce single celled structures that are capable of evading various immune reactions including hemocytes and fat body activated antimicrobial responses [26, 27]. Death of the host typically occurs within 3–7 days, after which the fungus sporulates on the dead insect, producing cells capable of infecting another round of hosts. A number of genes implicated in M. acridum virulence have been characterized, and intriguingly, addition of a single esterase gene can expand the host range of M. acridum to certain Leptidoperans [28–30].
Here, we explored the transcriptional responses of critical host immune tissues, namely the hemocytes and the fat body, of L. migratoria, to fungal infection by M. acridum. Transcriptomic analysis revealed that hemocytes and the fat body adopt distinct strategies in response to infection by the fungus. A relatively small number of genes were affected in hemocytes, which appeared to elevate the expression of pathways for the release of reactive nitrogen intermediates (RNI), and alter aspects of membrane, potentially to facilitate phagocytosis or engulfment pathways involved in defense against fungi. In contrast, a larger set of genes was activated in the fat body including those affecting known classical antimicrobial pathways, e.g. Toll and JAK/STAT. In addition, altered expression of cell differentiation and maturation genes, and changes in basic energy metabolism were noted, perhaps linked to an increase in metabolic energy needed in attempts to thwart the infection. Analysis of conserved immune pathways led to their identification in L. migratoria (~470 transcripts). Among them, 59 (13 %) and three (6.5 %) showed significant differential expression in the fat body and hemocytes, respectively, after M. acridum infection. These results reveal the transcriptional responses of the fat body and hemocytes in defense against a locust specific entomopathogenic fungus, and detail immune related genes in the transcriptome of the Orthopteran, L. migratoria manilensis.
Methods
Insects, fungal strains, and inoculation protocol
Adult males of the migratory locust, Locusta migratoria manilensis (Orthoptera: Acrididae), reared until 5 days after final ecdysis were used in all experiments. Locusts were maintained in metal cages at 30 ± 3 °C with 70–75 % relative humidity and a photoperiod of 16 h light, 8 h dark, and supplied with fresh wheat shoots, wheat bran (supplemented with dried brewer’s yeast) and water as described [31]. The fungal strain M. acridum CQMa102, was isolated from the yellow-spined bamboo locust, Ceracris kiangsu Tsai, by the Genetic Engineering Center of Chongqing University. The fungus was grown on one-quarter strength Sabouraud dextrose agar (SDA) for 15 days at 28 °C, after which conidia were harvested and suspended in cottonseed oil. Mycelia were removed by filtration through sterile lens paper. The concentration of spores was determined using a Neubauer haemocytometer. Healthy locusts were selected and inoculated with 5 μl of conidial suspensions adjusted to 1 × 108 conidia as described [32]. Control insects were treated with the same volume of cottonseed oil. Treated insects were reared in individual cages. Assay were performed using 20 locusts and each assay was repeated three times.
Sample preparation, library construction and sequencing
Locusts were collected after 8, 16, 24, and 32 h post infection, respectively. For each time course, ~30 infected and 30 control male adults were placed on ice to anesthetize them and the fat bodies were dissected in a Petri dish on ice in locust physiological saline solution (LoPS, 150 mM NaCl, 10 mM KCl, 4 mM CaCl2, 2 mM MgCl2, 4 mM NaHCO3, 5 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulphonic acid pH 7.2, 0.1 % Ficoll). Hemolymph were collected from locusts at the same time as described [33]. The dissected fat bodies and hemocyte were immediately transferred to mortars containing liquid nitrogen and homogenized, followed by RNA isolation using the Trizol Reagent (Invitrogen) according to the manufacturer’s instructions. The RNA samples were further digested with ten units of DNase I (Takara, China) for 1 h at 37 °C to remove residual genomic DNA. The quantity and quality of the RNA samples were examined using a Nanodrop ND-1000 spectrophotometer (LabTech, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, California, USA). Select RNA samples from different time courses derived from the same treatment and tissue were pooled in equal proportions to construct cDNA libraries using TruSeq RNA Sample Prep Kit v2 (Illumina, USA) following manufacturer’s instructions. Briefly, poly(A) mRNA from 1 μg of total RNA was isolated using oligo(dT)-conjugated magnetic beads. Purified mRNAs were fragmented (200 nt to 700 nt) and reverse transcribed into cDNA using Super Script II Reverse Transcriptase (Invitrogen, USA). Second-strand cDNA was synthesized in reaction mixtures containing 1× buffer, dNTPs, RNase H, and DNA polymerase I. Short fragments were purified using the Agencourt® AMPure® XP beads (Beckman Coulter Inc., Beverly, MA, USA) and resolved with EB buffer for end repair and single nucleotide A (adenine) tailing. Fragments were then connected with sequencing adapters, and enriched by 15 cycles of PCR amplification to obtain adequate fragments for the final cDNA library. The quantification and qualification of the cDNA library were assessed by an Agilent 2100 Bioanalyzer and ABI Step One Plus Real-Time PCR System, and sequenced on the Illumina HiSeq™ 2000 pair-end system (Illumina, USA). Illumina sequencing was performed at the Beijing Genomics Institute (BGI-Shenzhen, China).
Assembly and annotation of transcriptomes
All raw transcriptome data generated from Illumina sequencing were deposited in the SRA database (NCBI) with accession number SRX1036497 (fat body control), SRX1036507 (fat body infected), SRX1036511 (hemocyte control) and SRX1036517 (hemocyte infected). Before assembling the clean reads, the raw reads were preprocessed using filter-fq software. The raw reads containing only adaptor, sequences, reads with >5 % unknown nucleotides, and low quality reads (reads containing more than 20 % bases with Q-value ≤10) were removed. The obtained clean read datasets were assembled using Trinity (release 20130225) [34] under default parameters, except for min_kmer_cov and min_glue set at three. The four SRA datasets in our experiment were assembled separately at the beginning. Another 37 locust SRA datasets were downloaded from the NCBI. Among these downloaded SRA datasets, those who have same sequencing lengths were assembled separately similarly. These 41 partially assembled datasets then were combined to assemble together according to the following processes. The Trinity-based assembled unigenes from each sample were further processed using the Clustering software TGICL platform [35], to identify sequence splicing and redundancy, resulting in data containing the non-redundant unigenes of maximum sequence length. These resultant unigene datasets from each sample were then assembled into a unique “All-unigene sequence” dataset using the TGICL platform. The All-unigene sequences were then separated into two classes: clusters (CL, collection of homologous unigenes, i.e. sharing >70 % sequence identity) and singletons (unigene).
The unigene sequence datasets were annotated using available protein databases including Nr (non-redundant protein databases), SwissProt, KEGG (Kyoto Encyclopedia of Genes and Genomes) and COG (Cluster of Orthologous Groups), using the blastx algorithm (http://www.ncbi.nlm.nih.gov/) with a cut-off E-value of 10−5 [36]. Provisional protein functional information was assigned from comparative annotation to the most similar protein in those databases.
Differentially expression and immune related unigene analysis
Fat body and hemocytes genes that were differentially expressed between control and infected populations were identified, respectively, using a table of counts constructed with fragments per kb per million fragments (FPKM) values, which adjusted the number of fragments by the total number of fragments mapped and the length of the gene [37, 38]. Difference gene expression (DEG) of fat body and hemcoyte between control and infection group were determined by using FPKM value under the standard of false discovery rate (FDR) <0.001 and an absolute value of the log2ratio >1.
The locust transcriptome database was used as the background to search for GO terms enriched within the DEG dataset using the hypergeometric test and a p-value <0.05 as the parameters for determining significantly enriched terms. Similarly, pathways significantly enriched with the DEG datasets were identified by mapping all differentially expressed genes to terms in the KEGG database using the hypergeometric test with a p-value <0.05.
Immune related genes were preliminary identified via screening using the BLASTX search algorithm against immune-related family members downloaded from the orthodb database (http://cegg.unige.ch/orthodb7), which included Insecta, waterflea and tick sequences. Searches were parameterized with a cut-off E-value of <10−5. Putative immunity-related genes were further analyzed by comparing their protein domains with the deduced protein domains of different family members. Protein domains are determined using Pfam (http://www.sanger.ac.uk/Software/Pfam/) and SMART (http://smart.embl.de/). Manual screening was further performed to verify all identified immune related genes, which were classified into different immune-gene related families.
Quantitative RT-PCR analyses
To validate the results of the DEG, the expression change between control and fungal infected groups were examined by qRT-PCR for all of immune-related differentially expressed genes in the fat body (58 total) and hemocyte (three total) transcriptomes. Specific primers were designed for each gene and are listed in Additional file 1: Table S1. Total RNA from each sample was extracted as described above. 1 μg total RNA was reverse-transcribed in a 20 μl reaction using the Primescript TM RT reagent kit (TaKaRa, China). qRT-PCR was conducted using the CFX96TM Real-Time System (Bio-Rad, Hercules, CA, USA) with SYBR green (TaKaRa, China) using the following cycling parameters: 95 °C for 3 min, and 40 cycles of 95 °C for 5 s, 60 °C for 15 s, followed by melting curve generation from 65 to 95 °C. Primers designed to the actin genes were used as a reference control, and nuclease-free water was used as a negative control. All protocols for qRT-PCR experiments are in accordance with the Minimum Information Required for Publication of Quantitative Real-Time PCR Experiments guidelines [39]. Ct value was calculated from the results of three biological replications. The relative expression levels of each gene was analyzed according to 2-ΔΔCT [ΔΔCt = ΔCt(test) - ΔCt (calibrator)] method [40].
Results
Illumina sequencing and read assembly
Locusts infected with spores of M. acridum and un-inoculated insects were used to generate cDNA libraries for examining insect responses to the fungal pathogen. The fat bodies and hemocytes of M. acridum-infected and untreated locusts were isolated and RNA purified from these tissues as detailed in the Methods section. Four cDNA libraries corresponding to control and fungal-treated, fat body and hemocyte derived tissues, respectively, were generated and sequenced using the Illumina platform. A total of 18.79 Gb of nucleotide sequences were obtained. Q20 percentages (sequencing error rate <1 %) and GC percentages for the different samples were as follows; (1) hemocyte (control), 95.56 and 40.31 %, (2) hemocyte treated, 94.19 and 40.23 %, (3) fat body (control), 93.89 and 41.55 %, and (4) fat body treated, 85.33 and 44.39 %, respectively. These GC percentages are in agreement with a previous report [41]. For assembly, 37 locust SRA datasets were downloaded from NCBI. After removal of adaptor sequences, ambiguous reads, and low quality reads (Q20 <20) from the combined (41 SRA) datasets, clean reads with same sequencing lengths were assembled separately, and then the resultant partial assembly further assembled together. In comparison with an analysis examining the separate assembly of our four datasets, the N50 and mean length of unigenes assembled from the combined SRAs dataset increased significantly, from 559–804 bp to 1607 bp and from 484–603 bp to 914 bp (Table 1), respectively. The total number of nucleotides (nt), mean nt length, N50 values, assembly into contigs, scaffolds, and the unigene sets for each condition/library is given in Table 1. These final mean lengths and N50 values are comparable or exceed that reported by others [42, 43]. Total assembled nucleotides was equal to 47.5G, representing a 22-fold coverage of the coding sequence space of the locust genome (~2.5G) [44], indicating a robust dataset and analysis pipeline for accurate sequence assembly and adequate transcriptome coverage. The final assembled sequences have been submitted to the NCBI Transcriptome Shotgun Assembly (TSA) Database under the accession #: GDIO00000000.
Table 1.
Summary for the Illumina sequencing and de novo assembly of Locusta migratoria manilensis transcriptome
| Sample | Total number | Total length (nt) | Mean length (nt) | N50 | |
|---|---|---|---|---|---|
| Contig | FB Control | 76,610 | 24,270,667 | 214 | 244 |
| FB Treated | 90,480 | 35,948,413 | 248 | 302 | |
| Hemo Control | 102,738 | 37,214,667 | 241 | 298 | |
| Hemo Treated | 96,108 | 36,214,648 | 247 | 310 | |
| Scaffold | FB Control | 78,784 | 25,901,123 | 329 | 442 |
| FB Treated | 104,204 | 37,589,106 | 361 | 519 | |
| Hemo Control | 104,946 | 39,524,954 | 377 | 587 | |
| Hemo Treated | 98,818 | 38,331,782 | 388 | 621 | |
| Unigene | FB Control | 44,235 | 21,396,869 | 484 | 559 |
| FB Treated | 58,552 | 31,524,499 | 538 | 663 | |
| Hemo Control | 56,995 | 33,350,180 | 585 | 764 | |
| Hemo Treated | 53,979 | 32,541,902 | 603 | 804 | |
| Together with SRA | 50,809 | 46,450,242 | 914 | 1607 |
Gene identification, functional annotation and classification
Among the 50,809 unigenes, approximately 22,698 were annotated. Within each cDNA dataset, 22,496, 19,009, 16,502 and 9781 unigenes could be provisionally annotated by searching against the Nr (non-redundant protein databases), SwissProt, KEGG (Kyoto Encyclopedia of Genes and Genomes) and COG (Cluster of Orthologous Groups), respectively. Searches performed using the Nr database resulted in the largest proportion of provisional annotations, producing hits for 44.3 % of all distinct sequences. Analysis of the E-value distribution for those sequences for which a hit could be identified in the Nr database, showed a relatively even distribution across the range parameters used, i.e. 10−5 >E-value >0 (Fig. 1a). Overall similarity for the majority of identified sequences (~70 %) was greater than 40 %, with the remainder (~30 %) showing similarity values ranging from 17 to 40 % (Fig. 1b). The species distribution for ~40 % of the best-matching sequences were seven other main insects, with the red flour beetle, Tribolium castaneum (11.7 %), and the human body louse, Pediculus humanus corporis (9.5 %), representing the top two insect species to which best-matching hits were seen (Fig. 1c). Approximately 58 % of the best-matching sequences were distributed to other organisms, e.g. Apis mellifera (2.4 %).
Fig. 1.
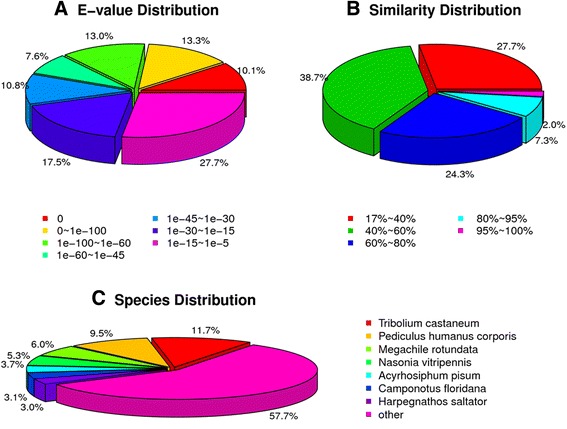
Summary of transcriptome analysis. a E-value distribution of BLAST hits for each unique sequence. b Similarity distribution of the top BLAST hits each sequence. c Species distribution shown as a percentage of the total homologous sequences with an E-value >e−25 using the first BLAST search hit for each sequence in the analysis
Unigene sequences were mapped using the international standardized gene functional classification (GO) system, which offers a dynamic-updated controlled vocabulary and a strictly defined concept to comprehensively describe properties of genes and their products in any organism. Annotated genes were binned into the three GO ontologies: molecular function, cellular component and biological process. Of the unigene set, 11,308 were annotated to at least one GO term. Among these, 8538 (75.5 %), 6281 (55.54 %), 9203 (81.38 %) unigenes were grouped into categories comprising biological process, cellular component and molecular function, respectively (Fig. 2). In the biological process, the most abundant subcategories were cellular processes (7064) and metabolic processes (5676). In the cellular component category, 348 unigenes were involved in immune system processes. The cell/cell part categories had 5308 unigenes assigned to them. Within the molecular function category, catalytic activity (enzymes, 5809) and binding (5565) were the two most abundant transcript annotation subcategories (Fig. 2).
Fig. 2.
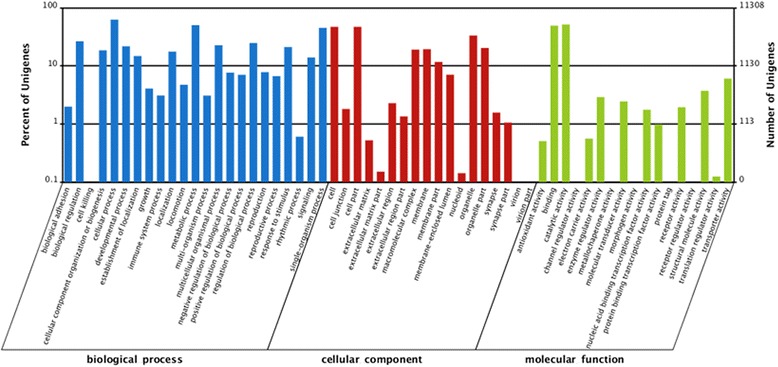
GO annotation of the overall unigene dataset. The total locust transcriptome dataset (9781 unigenes) were classified into biological process, cellular component, and molecular function subcategories
COGs were delineated by comparing protein sequences encoded in complete genomes, representing major phylogenetic lineages. Each COG consists of individual proteins or groups of paralogs from at least 3 lineages and thus corresponds to an ancient conserved domain. In this study, 9781 unigenes were functionally grouped into the 25 COG categories. Of the total unigene set, 41 % identified within the COG annotation were found to belong to the subcategory of general function prediction (via the top hit). Translation, ribosomal structure and biogenesis were the second most abundant (1998 unigenes), followed by genes involved in transcription (1812 unigenes), replication, recombination and repair (1773 unigenes), and function unknown (1674). Among them, 2.3 % of unigenes were divided into the subcategory of defense mechanisms (Fig. 3).
Fig. 3.
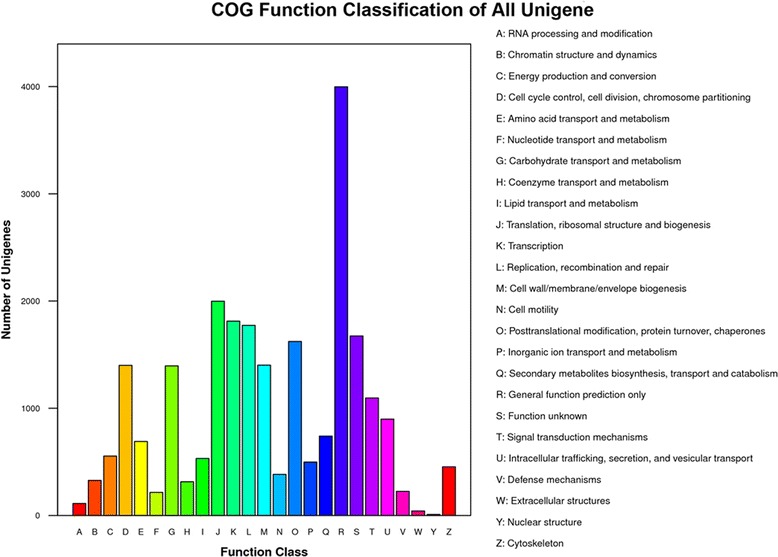
Analysis of clusters of orthologous groups of proteins (COGs). In all, 9781 unigenes were functionally grouped into the 25 COG categories and sub-catagories
Identification of differentially expressed genes in response to Metarhizium acridum infection
Pairwise comparisons of the transcriptome of locust fat body and hemocyte tissues untreated and infected by M. acridum were performed. After infection by M. acridum, a large alteration in the transcriptome of the locust fat body was seen, with 4660 genes up-regulated and 1674 genes down-regulated. In contrast, the hemocyte response was much smaller with 161 and 23 genes up- and down-regulated, respectively, after infection by the fungus. The amplitude of the signals, i.e. the extent of transcriptional activation or repression, also differed between the fat body and hemocytes upon exposure to the pathogen. The fold changes (log2 ratio) of gene expression in the fat body ranged from −13.92 to 15.90, whereas in hemocytes the range was from −4.81 to 9.81.
GO annotation analyses resulted in 128 (69.6 %) and 4700 (74.2 %) differential expressed unigenes that could be mapped to the term “biological process” in the hemocyte and fat body datasets, respectively (Fig. 4a & b). For hemocytes, enriched expressed genes including those involved in nitric oxide biosynthesis, cellular amino acid biosynthesis, cold acclimation, organic acid biosynthesis, carboxylic acid biosynthesis, cuticle development and the chitin-based cuticle molting cycle, small molecule biosynthesis (Table 2). In the fat body response, GO annotation indicated enrichment of transcripts involved in development and cell differentiation including; mRNA translation (e.g. negative regulation of oskar involved in germ-plasm formation), oocyte development, pole plasm assembly, cytoplasmic organization, and cell maturation (Table 2). Overlap between the DEGs (differential expressed genes) enriched in the fat body and hemocytes after exposure to M. acridum was small (Fig. 5a), with those GO annotated within cellular and metabolic processes having the maximum number differentially expressed unigenes, i.e. six. The six unigenes were: aspartate 1-decarboxylase, phosphoserine phosphatase, two proteophosphoglycan ppg4, protein tyrosine phosphatase and zinc finger protein 768 (Fig. 5b).
Fig. 4.
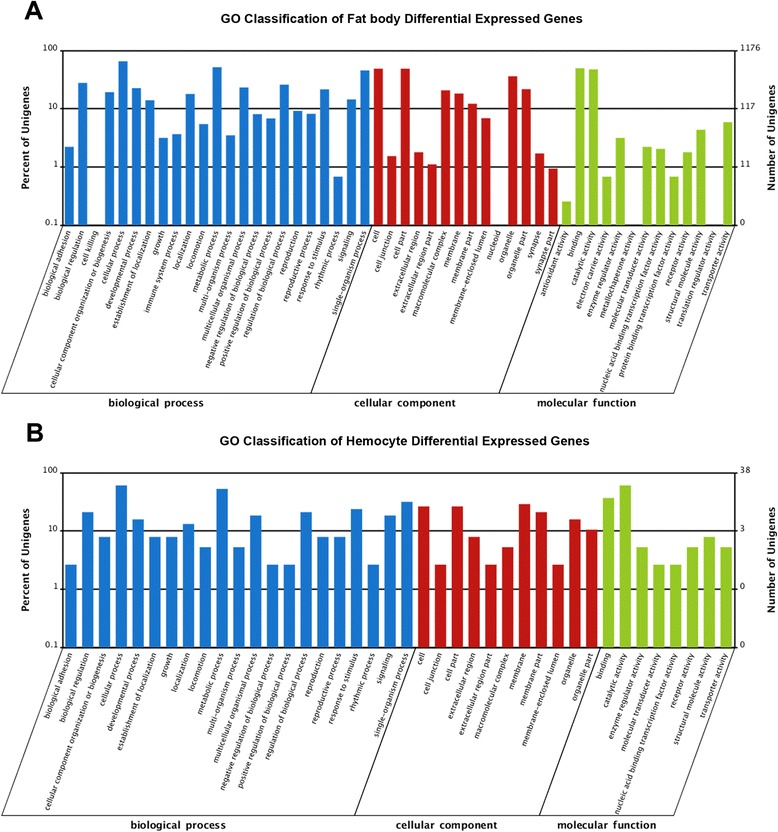
GO classification of differential expressed genes (DEGs) in the locust fat body (a) and in hemocytes (b) after M. acridum infection
Table 2.
GO enrichment analysis
| Gene ontology term | Cluster frequency | Genome frequency of use | Corrected P-value |
|---|---|---|---|
| Biological process | |||
| Hemocyte | |||
| Nitric oxide biosynthetic process | 2/29, 7 % | 4/8538, <0.01 % | 0.01231 |
| Cellular amino acid biosynthetic process | 4/29, 14 % | 89/8538, 1.0 % | 0.03972 |
| Cold acclimation | 2/29, 7 % | 7/8538, 0.1 % | 0.04282 |
| Nitric oxide metabolic process | 2/29, 7 % | 7/8538, 0.1 % | 0.04282 |
| Response to cold | 2/29, 7 % | 9 out of 8538 genes, 0.1 % | 0.07310 |
| Organic acid biosynthetic process | 4/29, 14 % | 128/8538, 2 % | 0.15838 |
| Carboxylic acid biosynthetic process | 4/29, 14 % | 128/8538, 2 % | 0.15838 |
| Cuticle development involved in chitin-based cuticle molting cycle | 2/29, 7 % | 18/8538, 0.2 % | 0.30486 |
| Cellular amino acid metabolic process | 5/29, 17 % | 265/8538, 3 % | 0.33062 |
| Small molecule biosynthetic process | 4/29, 14 % | 174/8538, 2.0 % | 0.49169 |
| Single-organism biosynthetic process | 4/29, 14 % | 183/8538, 2 % | 0.59005 |
| Fat body | |||
| Regulation of pole plasm oskar mRNA localization | 11/893, 1 % | 25/8538, 0.3 % | 0.03332 |
| Regulation of oocyte development | 11/893, 1 % | 27/8538, 0.3 % | 0.08004 |
| Negative regulation of oskar mRNA translation | 5/893, 1 % | 6/8538, 0.1 % | 0.13125 |
| Pole plasm assembly | 11/893, 1 % | 30/8538, 0.4 % | 0.24967 |
| Cytoplasm organization | 11/893, 1 % | 33/8538, 0.4 % | 0.65908 |
| Cell maturation | 23/893, 3 % | 104/8538, 1 % | 0.71693 |
| Oocyte anterior/posterior axis specification | 13/893, 2 % | 45/8538, 0.5 % | 0.95554 |
| Cellular component | |||
| Hemocyte | |||
| Integral to membrane | 8/17, 47 % | 893/6281, 14 % | 0.03580 |
| Intrinsic to membrane | 8/17, 47 % | 907/6281, 14 % | 0.03976 |
| Membrane | 11/17, 65 % | 2186/6281, 35 % | 0.34152 |
| Membrane part | 8/17, 47 % | 1322/6281, 21 % | 0.44314 |
| Extracellular region | 3/17, 18 % | 257/6281, 4 % | 0.90284 |
| Fat body | |||
| Intracellular | 548/659, 83 % | 4915/6281, 78 % | 0.19681 |
| Intracellular part | 523/659, 79 % | 4660/6281, 74 % | 0.22058 |
| Proton-transporting ATP synthase complex | 8/659, 1 % | 23/6281, 0.4 % | 0.56557 |
| Respiratory chain | 14/659, 2 % | 58/6281, 0.9 % | 0.73999 |
| Cytosolic small ribosomal subunit | 10/659, 2 % | 35/6281, 0.6 % | 0.83309 |
| Molecular function | |||
| Hemocyte | |||
| Nitric-oxide synthase activity | 2/31, 7 % | 3/9203, 0 % | 0.00213 |
| Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, NAD(P)H as one donor, and incorporation of one atom of oxygen | 2/31, 7 % | 4/9203, 0 % | 0.00426 |
| FMN binding | 2/31, 7 % | 10/9203, 0.1 % | 0.03158 |
| NADP binding | 2/31, 7 % | 18/9203, 0.2 % | 0.10560 |
| Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen | 3/31, 10 % | 79/9203, 0.9 % | 0.14961 |
| Transaminase activity | 2/31, 7 % | 27/9203, 0.3 % | 0.23774 |
| Transferase activity, transferring nitrogenous groups | 2/31, 7 % | 27/9203, 0.3 % | 0.23774 |
| Hydrolase activity, acting on ester bonds | 6/31, 19 % | 558/9203, 6 % | 0.63056 |
| Monooxygenase activity | 2/31, 7 % | 45/9203, 0.5 % | 0.64579 |
| Calmodulin binding | 2/31, 7 % | 46/9203, 0.5 % | 0.67373 |
| Structural constituent of cuticle | 2/31, 7 % | 49/9203, 0.5 % | 0.76074 |
| Phosphoric ester hydrolase activity | 4/31, 13 % | 280/9203, 3 % | 0.89824 |
| Fat body | |||
| mRNA 3′-UTR binding | 9/955, 1 % | 19/9203, 0.2 % | 0.02320 |
| O-methyltransferase activity | 5/955 genes, 1 % | 9/9203 genes, 0.1 % | 0.51422 |
| Stearoyl-CoA 9-desaturase activity | 3/955, 0.3 % | 3/9203, 0 % | 0.54600 |
| Acyl-CoA desaturase activity | 3/955, 0.3 % | 3/9203, 0 % | 0.54600 |
| Nucleic acid binding | 159/955, 17 % | 1241/9203, 14 % | 0.89676 |
| mRNA 5′-UTR binding | 5/955, 1 % | 10/9203, 0.1 % | 0.94087 |
Fig. 5.
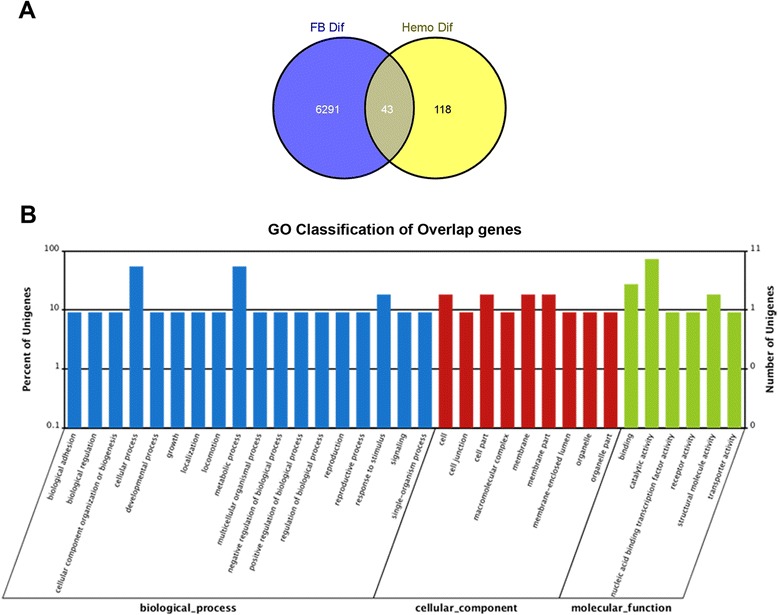
a Overlap between differentially expressed genes in the locust fat body and in hemocytes after M. acridum infection. b GO classification of fat body and hemocyte overlapping DEG dataset
Annotations of transcripts into the “cellular component” category resulted in 57 (31 %) and 2588 (41 %) unigenes placed into various subcategories from the hemocyte and fat body DEGs, respectively (Fig. 4a & b). In hemocytes, membrane proteins and processes dealing with membrane biogenesis were the most enriched differentially expressed genes (Table 2). In the fat body, general metabolism, including ATP (Adenosine triphosphate) synthase and respiratory chain enzymes, along with intracellular processes and ribosomal subunits were the top subcategories showing differential expression enrichment (Table 2). A small set of co-differentially expressed unigenes were identified which included aspartate 1-decarboxylase, zinc finger protein, and multidrug resistance protein 1, the former two of which were also identified in the development and cell differentiation GO analysis.
GO “molecular function” analysis could assign 1388 (22 %) and 48 (26 %) of unigenes derived from the fat body and hemocyte DGE sets, respectively (Fig. 4a & b). Within the molecular function, the most abundant subcategories in hemocyte DEG dataset were nitric oxide synthase, oxidoreductase, FMN (flavin mononucleotide) and NADP (nicotinamide adenine dinucleotide phosphate) binding proteins, transaminase, transferase, hydrolase, monooxygenase, calmodulin-binding, and cuticle structural proteins and enzymes. (Table 2). In the fat body, abundant GO terms subcategories were identified to include; mRNA 3′- and 5′-UTR binding, O-methyltransferase activity, stearoyl-CoA 9-desaturase activity, acyl-CoA desaturase activity, and nucleic acid binding (Table 2). The co-differentially expressed unigene set included functions within catalytic activity (8), binding (3) and structural molecule activity (2).
KEGG pathway enrichment analysis revealed genes homologous to those found in pathways corresponding to neural processes, e.g. Parkinson’s disease, and myotrophic lateral sclerosis (ALS), as well as mRNA surveillance, spliceosome, and oxidative phosphorylation as being the top five most abundantly populated differentially (control versus fungal infected) expressed pathways in the fat body dataset. KEGG analysis of the hemocyte DEG dataset indicated enrichment of genes involved in amino acid metabolism, e.g. arginine, proline, glycine, serine, threonine, and tyrosine. Co-differential expressed pathways found in both the fat body and hemocyte datasets included sugar metabolism and response, and immune and infection responses, and signal transduction pathways, e.g. protein tyrosine phosphatases (PTP, Unigene21232_All) that are part of the JAK/STAT pathway.
GO enrichment analysis and KEEG pathway analysis revealed that complex biological processes or metabolic pathways were affected in both hemocyte and fat body. Overall, two major categories of genes were activated in hemocyte after infection. The first involved pathways linked to release of humoral immune factors, e.g. nitric oxide synthesis related genes, including two nitric oxide synthase [45], GTP (Guanosine-5′-triphosphate) cyclohydrolase I [46] mitochondrial uncoupling protein 2 [47], and melanization related genes, like GTP cyclohydrolase 1 [48]. The second category encompassed cellular immune function, and included protein rhomboid, cdc42, lipopolysaccharide-induced tumor necrosis factor-alpha factor homolog, which are proteins involved in phagocytosis [49–51], etc. By contrast, some humoral innate immune related genes, like Caspase-1 precursor, were activated in the fat body (more detailed information is given in the Discussion section concerning identification of immune related genes). Meanwhile, energy metabolism and reproduction related genes were affected significantly in fat body.
Identification of immunity-related genes
A number of locust immune related genes expressed in the fat body and hemocyte were identified using NCBI-BLAST analysis combined with conserved domain comparisons. Initial analyses were set to identify all immue related genes present in the transcriptome data. A second analysis was subsequently performed examining the differential expressed, i.e. control versus fungal infected, immune related gene sets in fat body and hemocyte subdatasets. In total 470 immune-related transcripts were identified. Immune-related responses found in the fat body encompassed genes involved in: (1) cellular pathogen recognition pathways and humoral immune reactions, (2) immune signal modulation and signal transduction, and (3) effectors and related-activties released into hemolymph. In the dataset obtained, 36 immune related genes were up-regulated and 23 were down-regulated in the fat body after infection (Fig. 6). In contrast, only three immune related genes, one lysozyme and two involved in NOS (nitric oxide synthases) pathway, were identifed in the hemocytes after infection, all of which were up-regulated. Within the fat body immune related dataset, DEGs included pathogen recognition and binding proteins, e.g. peptidoglycan recognition proteins (PGRPs, 2/14), C-type lectins (3/14), scavenger receptor class B protein (SCRB, 3/15), and immunoglobulin-like cell adhesion molecules (dscam proteins, 5/99); innate immune activation and suppression factors, e.g., serine protease inhibitors (serpins, 7/36), and prophenoloxidases (2/10); antimicrobial proteins, e.g. lysozymes (3/7); oxidative and stress responses, e.g. peroxidase (Pox, 2/37), superoxide dismutases (SODs, 3/12), and peroxiredoxin (1/11); cell death and inflammation, e.g. caspases (6/18); viral response and RNAi (RNA interference), e.g. argonaute (2/9), and NOS pathways, e.g. NOS synthase (2/5). In addition, members of the JAK/STAT immune realted signal trasnduction pathways were found including: domeless (1/2), SOCS (suppressor of cytokine signaling, 5/14), and PIAS (protein inhibitor of activated STAT, 2/6); as well as members of the Toll pathway, e.g. Toll receptors (2/21), Tollip (Toll Interacting Protein, 1/2), Pellino (6/12); and of the IMD pathway, e.g. IMD (1/2). A summary of the immune related gene identified is given in Additional file 1: Table S1.
Fig. 6.
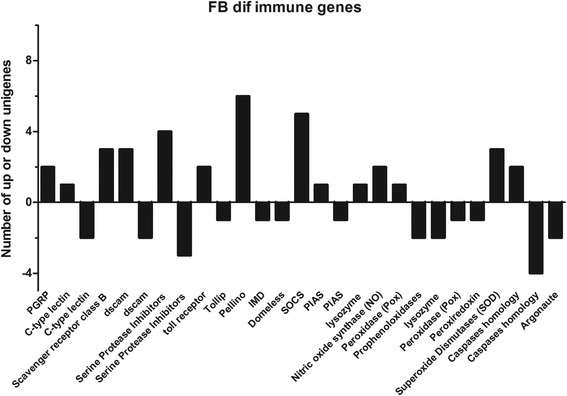
Functional classification of fat body DEGs in response to M. acridum infection
qRT-PCR analysis of immune-related genes
qRT-PCR (Real-Time fluorescence quantitative PCR) was used to validate the differential expression of the immune related genes in the transcriptomic data. Sixty genes, including the 58 immune related DEGs identified in the fat body analysis and the three found in the hemocyte dataset were examined (one unigene is codifferentially expressed in fat body and hemocyte). For two genes (Lys4, SCRB9), primers could not be designed due to the limited sequence lengths isolated. Overall, 50/58 (86.2 %) were consistent with respect to expression between the transcriptomic and q-RT-PCR data (Fig. 7, Additional file 2: Table S2). With respect to the fat body DEGs, 7/55 (13 %) were found to be inconsistent with respect to expression between the transcriptomic and q-RT-PCR data (Fig. 7, Additional file 2: Table S2). These included Dscam9 and Dscam 75, the serpins, SRPN23 and SRPN30, Pellino7 involved in the Toll pathway, Domeless2, part of the JAK/STAT pathway, and NOS5. OF the hemocyte DEGs, 1/2, namely the NOS1 gene, showed inconsistent results comparing the transcriptomic and the q-RT-PCR data. A summary of the main immune-related pathways and specific genes up- or down- regulated in the fat body and in hemocytes, respectively after M. acridum infection is given in Fig. 8.
Fig. 7.
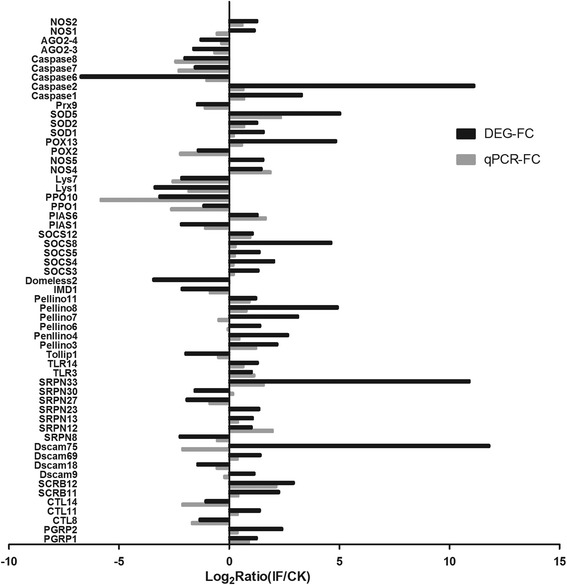
Q-RT-PCR analysis of DEGs and comparison to transcriptomics data
Fig. 8.
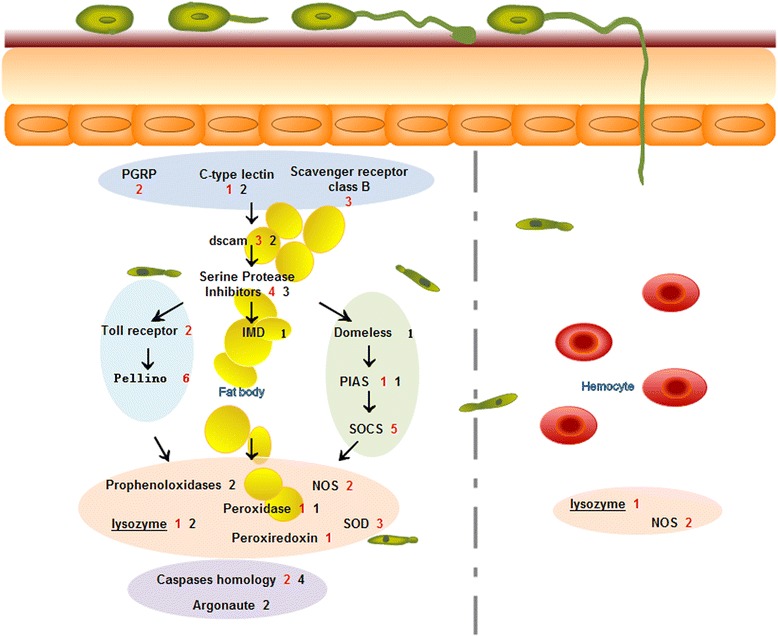
Summary of the major immune related DEGs identified in the fat body and in hemocytes to be up regulated (red) and down regulated (black) after M. acridum infection
Discussion
Insect responses to microbial pathogens begin at the cuticle and continue within the body of the organism with innate and induced responses. The insect fat body is the major organ mediating immune responses in insects, and it also governs organismal energy homeostasis including intermediary metabolism tissue, energy storage and utilization, and the synthesis of many hemolymph proteins and circulating metabolites [15]. Thus, changes in fat body gene expression in response to pathogens can result in a steep energy and/or reproductive cost [52]. Our results using the locust specific fungal pathogen, M. acridum showed similar changes in host gene expression broadly affecting reproductive and energy metabolism, effects consistent with other reports of such changes as a result or response to infection, acting as a potentially adaptive strategy to minimize or delay the spread of the infection [53]. In addition, phagocytic hemocytes, cells that freely float within the open circulatory system of insects, can consume foreign objects, including microbes, as well as initiate and/or contribute to encapsulation and melanization reactions that seek to limit the spread of invading microbes.
Limited reports [54], however, have focused on these two essential aspects of immune reactions, particularly examining specific tissue transcriptomic responses to pathogen attack. Here, we have examined the response of locust fat body and hemocytes to infection with the necrophytic insect pathogen, M. acridum. These data have allowed for the reconstruction of significant aspects of the locust immune pathways and the nature of the response to the fungal pathogen.
Microbial recognition
Carbohydrate moeities appear to be important microbial antigens recognized by the insect innate immune system. These include often repeating polysaccharide units found on microbial surface strcutures and glycoproteins, e.g. lipopolysaccharide (LPS) on gram negative bacteria, peptidoglycan on gram positive bacteria, and high mannose and other surface glucans on yeast and filamentous fungi [55, 56]. Recognition initiates insect innate immune response and this step is mediated by pattern recognition molecules (pathogen sensors) that recognize conserved molecular patterns found on pathogens but presumably lacking in the host [57]. A variety of pattern recognition molecules have been described including peptidoglycan recognition protein (PGRP), β-1,3-glucan recognition protein (βGRPs), gram-negative binding proteins (GNBPs), Calcium-dependent (C-type) lectins (CTLs), and scavenger receptors (SCRs) [58]. Recognition, e.g. PGRP family members capable of distinguishing between various invading bacteria, acts upstream of Toll and Imd pathways to mediate. PGRPs can be categorized into short (PGRP-S) and long (PGRP-L) members, although all of these proteins have at least one conserved N-acetylmuramyl-alanine amidase-like domain [59].
Various insects have different PGRP repertiores; Drosophila has 13 PGRP genes encoding 19 proteins (due to alternative splicing) [60], mosquito (Anopheles gambiae); 7 PGRP genes encoding 9 proteins [61], and the silkworm, Bombyx mori, has 12 distinct PGRP genes [62]. Here, we identified 14 putative PGRP transcripts. Among them, two unigenes, PGRP-SA and PGRP-LC-like, were upregulated in the fat body in response to M. acridum infection. PGRP-SA is an essential component for activating the Toll pathway in Drosophilia [63]. PGRP-LC activates the Imd pathway [64]. These data indicate that in response to M. acridum both the Toll and Imd pathways have the potential to be activated.
The βGRP/GNBP proteins are another pattern recognition protein family found in most insects [65], including Drosophila (three genes) [66], A. gambiae (seven genes) [61], the honey bee, Apis mellifera (two genes) [10], tobacco hookworm, Manduca sexta (two genes) [67, 68], and in the beetle, Tribolium castaneum (three genes) [11]. βGRP proteins have high affinity for β-1,3-glucans found in fungal cell walls, while GNBPs bind to gram-negative or -positive bacteria [66]. All these members of this family contain a conserved N-terminal β-1,3-glucan-recognition domain for the detection of pathogens or parasites. Here, we found five GNBP genes (LmGNBP1-5) in the overall transcriptomic data, however none of these were differentially expressed after M. acridum infection in either the fat body or hemocytes. Our sequence alignment revealed that LmGNBP1 and 5 are identical to GNBP1 as named in Wang et al. [69] and LmGNBP3 is identical to GNBP2 [69] which was also the same as βGRP as reported by Zheng et al. [70]. In the Wang et al. report, LmGNBP1 and 5 (termed GNBP1) was initially expressed in hemocytes and was induced in the fat body 6 and 9 h after conidial injection into the hemocoel (note that this infection prtocol is different than ours in which infection was topical, i.e. followed the “natural route). We did not see any significant differential expression of LmGNBP1 and 5 although both LmGNBP1 and 5 are highly expressed (high FPKM values = 1619.8831 and 2318.7453, respectively) in heomcytes, with low expression in the fat body. Only a slight increase in expression of these genes were seen in the fat body after infection (FPKM from 11.26 to 16.05 and 4.4 to 4.7, respectively). These data suggest that LmGNBP1 and 5 are constitutively expressed in hemocytes, with only minor expression in the fat body consistant with the overall results of Wang et al. [69], althoug we did not see appreciable induction in the fat body. As noted above, this can be due to the different infection methods used. Regarding LmGNBP2 (termed βGRP in the Zheng et al. report [70], and GNBP3 in the Wang et al. report [69]), this gene was induced in hemocytes 8 and 12 h after inoculation of conidia, but no differences were seen earlier (4 h) or later (24, 48 and 72 h) post infection (as measured by q RT-PCR) in Zheng et al. paper. However, LmGNBP2 was detected in hemocyte in the absence of infection [70]. In our research, LmGNBP2 (= βGRP = GNBP3) simmilarly showed high expression in hemocytes and low expression in the fat body. As we did not examine intermediate time points, it is possible that a transient increase in LMGNBP2 was missed in our dataset. However, other have also reported constitutive but no significant differential expression of LmGNBP2 in any tissues [69], consistent with our results.
CTLs are a large family of carbohydrate binding proteins [71] that contribute to a number of inverberate immune responses, including microbial clearance [72], hemocyte nodule formation [73], activation of prophenoloxidase [74] and opsonisation [75]. In our study, 14 CTLs were identified in the transcriptome of the locust. Within the fat body differentially expressed gene set, LmCTL8 and LmCTL14 were significantly down regulated, whereas LmCTL9 was up-regulated after fungal challenge.
Galectins are a phylogenetically ancient lectin family with evolutionary conserved carbohydrate binding domains [62]. Galectin-like proteins have been identified in D. melanogaster (DmGALEs) and A. gambiae (AgGALEs). In vitro experiments indicate that DmGALEs can bind to ß-galactoside sugar [76], and that these proteins may play functions via facilitation of microbial recognition and/or phagocytosis [77]. Here, we identified five galectin-like proteins in the overall locust transcriptome, however, none of them appeared to be differentially expressed in either the fat body or in hemocytes after M. acridum infection.
Scavenger receptors are cell surface glycoproteins with structurally diverse transmembrane multidomains, divided into at least eight subfamilies (classes A to H, with A, B, and C, representing the major subfamilies) [78]. Class A scavenger receptors (SCRAs) are thought to function in phagocytic recognition of unoposonized and opsonsonnized microorganisms [79]. Class B SCRs participate in the phagocytosis of microbes [80], apoptotic-cell binding [81], etc. Class C SCRs are transmembrane or secreted multidomain proteins characterized by two complement-control protein (CCP) domains followed by a MAM domain (meprin A5 antigen and RPTP Mu), and usually a somatomedin-B-like (BO) domain. Drosophila has four class C SCRs subtypes (types I-IV), three of which has been shown to be involved in phagocytosis and innate immunity [82]. Our overall transcriptome dataset contained four unigenes corresponding to class A SCRs, 15 class B SCRs, and five class C SCRs. None of these genes, however, were found to be differentially expressed in either the fat body or in hemocytes after expsoure to M. acridum.
Immunoglobulin-like genes
The Down syndrome cell adhesion molecule (DSCAM) is a immunoglobulin (Ig)-superfamily member with conserved architecture containing variable Ig and transmembrane domains [83]. Dscam is encoded by a single gene in Drosophilia but it can generate more than 18,000 isoforms via alternative splicing in Drosophila immune-competent cells [84]. DSCAM deficiency correlates with reduced phagocytotic uptake of bacteria [84] and differential splice variants of Dscam during exposure to different bacterial strain in A. gambiae has been reported [85] indicated that the complexity of dscam and its function in pathogen recognition. In the overall locust transcriptome dataset, we found ~100 Dscam unigenes (designated as DSCAM1-100), however, these likely represent splice variants. Among these, six unigenes showed significant differential expression in the locust fat body after M. acridum infection; three of which were upregualted (Dscam9, 69, and 76), with the latter >11-fold, whereas the other three unigenes were down regulated in the locust fat body (Dscam14, 18, and 71).
Modulation
Once microbial pathogens are recognized by various receptor or recognition factors, different signalling cascades are activated via amplifaction or supressed via dampening. This typically occurs via activation/repression of extracellular proteases, via interplay between serine proteases and (serine) protease inhibitors (serpins). Clip domain serine proteases (CLIPs) are a large family of proteases that contain disulfide knotted clip domain(s), unique to arthropods. These domains are coupled to the active, C-terminal serine protease domain [86]. CLIPs are involved in melanization [87] and stimulation of the Toll pathway via activation of the phenoloxidase cascade by acting on the Toll-ligand Spätzle and other downstream proteins [88, 89]. Drosophila has 37 CLIP genes, Anopheles 41, Bombyx 15 and Apis 18. In L. migratoria, we ientified 34 potential CLIP unigenes, however, none were significantly differentially expressed after fungal infection.
Serpins (serine protease inhibitors) are a superfamily of proteins found in all higher eukaryotes as well as some viruses [90] that function through a unique suicide substrate-like inhibitory mechanism [91]. Serpins, typically composed of 350–400 amino acid residues, usually have a reactive center loop (RCL) located 30–40 residues from the C-terminal end that functions to bind the active site of specific proteases mimicking a substrate. Once an RCL moeity binds to its target protease, the protease can cleave the serpin at the scissile bond resulting in a covalent link of the serpin to the protease, thus blocking its activity [91]. Thirty-six serpin (srpn) transcripts were identified in the overall transcriptome dataset. Of these four were up-regulated and three down-regulated in the fat body after M. acridum infection. No srpn transcripts showed any significant differential expression in hemocytes after fungal infection.
Signal transduction pathways: Toll, Imd, and JAK/STAT
After the extracellular signal derived from pathogen recognition has been modulated, and concomittant with activation of extracellualr defense pathways, the immune signal is transduced to target cells that activate cellular responses including the production of antimicrobial compounds and effectors. A number of immune related signal transduction pathways are present in insects including the Toll, Imd, and JAK/STAT pathways [10]. The Toll pathway is initiated by the input from PGRP/GNBP recognition of epitopes on the cell surfaces of bacteria or fungi [92]. Recognition (binding) activates extracellular serine protease cascades via cleavage of the extracellular cytokine pro-Spätzle to mature Spätzle. Mature Spätzle (C-terminal 106 amino acids) then binds to Toll receptors on insect cell surfaces, resulting in the recruitment (inside the cell) of the Tube/Myd88 complex that acts to stimulate the Pelle kinase [3]. Pelle phosphroylates Cactus, an inhibitory factor, resulting in its dissociation from the Cactus/Dorsal/Dif complex [93]. Dorsal and Dif are then free to translocate from the cytoplasm into the nucleus to activate the transcription of additional defense responses including the production of antimicrobial peptides. This is achieved via the activities of a host of additional proteins including Tollip, Pellino, TRAF2 (TNF receptor associated factor-2), and ECSIT (evolutionarily conserved signaling intermediate in Toll pathway), [94–96].
Spätzle shares high structural similarity with cystine-knot superfamily proteins, e.g. nerve growth factor (NGF) and Coagulogen [97]. Both D. melanogaster and A. gambiae contain six Spätzle genes, whereas B. mori has only three orthologs. However, thus far, it appears that only one Spätzle ortholog participates in immunity in Drosophila [98], and one Spätzle gene has been shown to be up regulated after bacterial infection in A. gambiae. In this study, four distinct Spätzle transcripts were identified, but none showed any significant differential expression in either the fat body or in hemocytes after fungal challenge. Toll and Toll-like receptors (TLR) are evolutionarily conserved proteins found from insects to mammals [99]. Toll/Toll-like receptors numbers vary from nine in Drosophila, to ten in A. gambiae, fourteen in B, mori, and five in Apis mellifera [62]. In L. migratoria manilensis, we have identified two Toll and twenty-one Toll-like receptor unigenes in the transcriptome dataset. Amongst these, expresssion of one Toll receptor was down-regulated, and expression of two Toll-like receptors were up-regulated in the locust fat body post fungal challenge. No changes were seen for any Toll/Toll-like receptors in hemocytes after fungal infection. Of downstream componenet of the Toll pathway, one MyD88 (death domain containing myeloid differentiation factor 88) unigene was identified in the transcriptome dataset. Tube and Pelle are two additional proteins that share homology with MyD88 proteins (include Death domains). In our study, no Tube transcript was identified while two Pelle unigenes were found in the overall transcriptome dataset. None of the these genes, i.e. MyD88/Pelle, showed any differential expression in the fat body or in hemocytes after exposure to M. acridum. Twelve Pellino unigene transcripts were found in the overall locust transcriptome dataset, with expression of six Pellino unigenes up regulated in the locust fat body after M. acridum infection. Four Cactus and two Rel family transcription factor Dorsal/Dif unigenes were identified in the transcriptome dataset, none of which showed significant differential expression in the presence of M. acridum. Additonal intercellular components identified included Tollip (two unigenes), TRAF2 (TNF receptor associated factor-2) (two unigenes) and ECSIT (a single unigene). Amongst these, only one Tollip unigene was found to be significantly up regulated in infected locust fat bodies. Intruigingly, manipulation of the Toll system via genetic engineering of the insect pathogenic fungus B. bassiana to express a Drosophila serpin resulted in increased viruelnce towards a Lepidopteran host [100].
The Imd pathway has been shown to be induced by bacterial mesodiaminopimelic acid (DAP)-type peptidoglycans as well as their fungal counterparts, with weaker activation by lysine (Lys)-type peptidoglycan. Imd activation proceeds via epitope recognition by the transmembrane receptor PGRP-LC or by the receptor PGRP-LE [101]. Activated receptors then bind to the Imd protein which in turn interacts with the FADD (FAS-associated death-domain) protein. FADD associates with the apical caspase death-related Ced-3/Nedd2-like protein (DREDD) leading to the cleavage of Imd by DREDD. Proteolyzed Imd interacts with the Drosophila inhibitor of apoptosis-2 (dIAP-2) protein [102], which results in recruitment of downstream components, i.e. TAK1 (Transforming growth factor beta activated kinase-1) [103] and its adaptor TAB2 (TGF-beta activated kinase) [104]. Once recruited, TAK1 activates the IkB-Kinase (IKK) complex, resulting in the phosphorylation of the NF-kB protein (Relish) [105]. Further proteolytic cleavage of Relish by DREDD, results in a protein that can be translocated into the nucleus leading to transcriptional induction of the expression of defense compounds including antimicoribal peptides and other effectors [106, 107]. All essential components of the Imd pathway were identified in L. migratoria manilensis indicating that the Imd pathway is conserved in Orthoptera. In addition, three unigenes coding for Caspar proteins that act to suppress immune activation by blocking nuclear translocation of Relish were identified in the locust transcriptome dataset. Among this pathway, only IMD1 showed significant down-regulation in infected locust fat bodies.
The JAK/STAT pathway is known to respond to viral infections [108] and tissue damage [109]. Upon infection, hemocytes release cytokines, e.g. Unpaired-3 (Upd-3), that bind to the Domeless receptor [109] resulting in activation of JAK kinases (Janus kinase, e.g. encoded by the Hopscotch gene). Activated JAKs phosphorylates cytosolic STATs (signal transducers and activators of transcription) promoting their translocation to the nucleus where they act as transcriptional activators. With the exception of one component, Upd, all members of this pathway, including a single Hopscotch and two STAT unigenes, were identified in the locust transcriptome dataset. However, Upd has also not been found in A. mellifera, A. gambiae, T. castaneum, and B. mori, indicating that it may be a Drosophila specific ligand for the JAK/STAT Pathway. Two unigene transcripts for Domeless were identified in the locust, and one of them was significantly down regulated in the fat body after M. acridum infection. A number of additional JAK/STAT interacting proteins were identified. These included SOCS and PIAS genes. In this study, fourteen SOCS unigenes were identified, five of which were up regulated in the locust fat body after fungal challenge. Six unigene transcripts corresponding to PIAS genes were identifed, one of which (LmPIAS1) up regulated, whereas another (LmPIAS6) was down regulated in the locust fat body after M. acridum infection.
Effectors
As part of the insect microbial defense response, pathogen recognition and signal modulation and transduction lead to the expression of various effectors that target the invading organism. These effectors stimulate phenoloxidase-dependent melanization, cellular apoptosis, and the production of antimicrobial compounds and peptides. PPO exists as an inactive zymogen in hemolymph. Recognition and regulation by serine proteases as described above, leads to PPO activation (conversion of PPO to PO) by an ultimate protease [110]. PO hydroxylates and oxidizes monophenols to quinones, resulting in several physiological consequences including toxicity to microbes, stimulation of melanin synthesis, faciliatation of the sequestreation of parasites, and wound healing [111]. Our overall transcriptome dataset contained ten PPO unigens, two of which were significantly down regulated in the locust fat body after M. acridum infection. Lysozyme (chicken or conventional type, C-type) is one of three classes (A, B, & C) of lysozymes found in the animal kingdom, with only the C-type found primarily in the Arthropoda and the phylum, Chordata. All lysozymes are characterized by their ability to hydrolyze the β-(1,4)-glycosidic bond between the alternating N-acetylmuramic acid (NAM) and N-acetylglucosamine (NAG) residues of peptidoglycan, the predominant cell wall polymer found in bacteria [112]. In this study, seven c-type lysozyme unigenes were identified in the L. migratoria transcriptome. Within the fat body response to M. acridum infection, two lysozyme unigenes were down-regulated and one up-regulated after fungal exposure. The latter lysozyme unigene was also up-regulated in hemocytes post-fungal infection. AMPs can be classified into three main groups; (a) proline, histidine or other amino acids-rich peptides, (b) linear peptides with α-helical conformations (e.g. cecropins and magainins), and (c) cyclic or open-ended cyclic peptides with pairs of cysteine disulfide bonds (e.g. defensins and protegrin). Unigenes corresponding to a defensin and a diptericin were identified in the overall transcriptome library, however, no differential expression of any of these was found in either the fat body or in hemocytes after M. acridum infection. This result is in accordance with previous report [69]. Oxygen-derived free radicals and nitric oxide are known to play important roles, both directly and/or as signalling molecules, during pathogen induced immune responses. For example, local production of free radicals is a critical component of acute-phase oxidative defense that targets invading microbes [113]. The production of these free radicals involved a variety of enzymes including, NOS, NADPH oxidases (NOX), peroxidases (POX), glutathione oxidases (GTX), superoxide dismutases (SOD), catalases, thioredoxins, thioredoxin reductases, and peroxiredoxins. In the overall transcriptome dataset we identified five NOS, seven NOX, 37 POX, 12 SOD, three catalase, 19 thioredoxin reductase, and 11 peroxiredoxin unigenes. Of these, two NOS unigene showed up regulation in the fat body, with two other NOS unigenes up regulated in hemocytes post-fungal infection. In addition, one POX and three SOD unigene were up-regulated whereas another POX and one peroxiredoxin unigene were down-regulated in the fat body.
Other immune molecules
Caspases (cysteine aspartate-specific proteinases) both initiate and execute cellular apoptosis via cleavage of target proteins that lead to cell death. However, caspases are also involved in immune responses in non-apoptotic mechanisms, e.g. the Drosophila DmDredd and DmIAP2 gene products are essential for Imd signaling [104, 114]. Caspases are also involved in insect antiviral responses [115]. Our transcriptome analysis of L. migratoria manilensis identified 20 caspase unigenes, five of which were down regulated and two of which were up regulated in the fat body of infected locusts. RNAi pathways play important roles not in normal development and in the targeted of foreign nucleic acids (primarily viral). Key components of the RNAi pathway include Dicer, enzyme that cleave RNA into siRNA (Small interfering RNA), and the RNA-induced silencing complex (RISC) that includes argonaute, which captures the siRNA, and targets gene transcription. Within the locust overall transcriptome dataset, four Dicer and nine argonaute unigenes were identified. Two argonaute unigenes were identified as being down regulated in the fat body of infected locusts, with no transcripts of any RNAi components identified found to be significantly differential expressed in hemocytes post M. acridum infection. However, glucose dehydrogenase was found to be a locust differential expressed protein in hemocyte in response to M. acridum that has been shown to be involved in encapsulation [116], and protein spinster homolog one induced caspase-independent autophagic cell death, factors which might facilitates removal of dead hemocyte after infection.
Conclusion
Significant aspects of the locust physiology, including the main immune pathways were identified in the overall transcriptome analysis performed. By examining the reponse to infection of the two major organs or tissues involved in the immune response, namely the fat body and hemocytes, a set of differentially expressed genes were identified in each tissue. As M. acridum is a specific fungal pathogen of locusts, these data indicate that the locust attempts to mount a challegene to the fungal infection. The most significant responses, in terms of changes in gene expression levels, were found in the fat body, with hemocytes showing altered expression of only three genes (LmLys4, LmNOS1, LmNOS2). This may not be too surprising, as hemocytes are general scavengers, primed to target any invading microbe, depedning upon proper stimulation. In contrast, the fat body is capable of responding to specific threats via induction and signaling pathways to turn on the production of specific effectors, e.g. lysozyme and free radical production. Locust pathways for melanization, phagocytosis, and encapsulation were identified, however none of these were enough to thwart the fungus, indicating that it has evolved mechanisms for overcoming these defenses. These data establish the groundwork for further exploration of locust immune responses and the identification of potential targets for locust control.
Acknowledgements
This work was financially supported by the the High Technology Research and Development Program (863 Program) of China (No. 2011AA10A204) and the Natural Science Foundation of China (No. 30971913).
Data deposition
The sequences reported in this paper have been deposited as raw reads in the GenBank SRA database (accession number.: SRX1036497, SRX1036507, SRX1036511 and SRX1036517). This Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GDIO00000000. The version described in this paper is the first version, GDIO00000000.
Abbreviations
- ALS
Myotrophic lateral sclerosis
- AMPs
Anti-microbial peptides
- ATP
Adenosine triphosphate
- BO
Somatomedin-B-like
- Caspases
Cysteine aspartate-specific proteinases
- CCP
Complement-control protein
- CLIPs
Clip domain serine proteases
- COG
Cluster of orthologous groups
- CTLs
Calcium-dependent (C-type) lectins
- DEGs
Differential expressed genes
- dIAP-2
Drosophila inhibitor of apoptosis-2
- DREDD
Death-related Ced-3/Nedd2-like protein
- DSCAM
Down syndrome cell adhesion molecule
- ECSIT
Evolutionarily conserved signaling intermediate in Toll pathway
- EST
Expressed sequenced tag
- FADD
FAS-associated death-domain
- FDR
False discovery rate
- FMN
Flavin mononucleotide
- FPKM
Fragments per kb per million fragments
- GNBPs
Gram-negative binding proteins
- GO
Gene ontology
- GTP
Guanosine-5′-triphosphate
- GTX
Glutathione oxidases
- IKK
IkB-Kinase
- Imd
Immune deficiency
- IPM
Integrated pest management
- JAK/STAT
Janus kinase/signal transduction and activator of transcription
- KEGG
Kyoto Encyclopedia of genes and genomes
- LoPS
Locust physiological saline solution
- LPS
Lipopolysaccharide
- MAM
Meprin A5 antigen and RPTP Mu
- NADP
Nicotinamide adenine dinucleotide phosphate
- NAG
N-acetylglucosamine
- NAM
N-acetylmuramic acid
- NOS
Nitric oxide synthases
- NOX
NADPH oxidases
- Nr
Non-redundant protein databases
- PAMP
Pathogen-associated molecular pattern
- PGRPs
Peptidoglycan recognition proteins
- Pox
Peroxidase
- PPO
Prophenoloxidase
- PPRs
Pattern recognition receptors
- PTP
Protein tyrosine phosphatases
- qRT-PCR
Real-time fluorescence quantitative PCR
- RCL
Reactive center loop
- RISC
RNA-induced silencing complex
- RNAi
RNA interference
- RNI
Reactive intermediates nitron
- SCRAs
Class A scavenger receptors
- SCRB
Scavenger receptor class B protein
- SCRs
Scavenger receptors
- SDA
Sabouraud dextrose agar
- serpins
Serine protease inhibitors
- siRNA
Small interfering RNA
- SOCS
Suppressor of cytokine signaling
- SODs
Superoxide dismutases
- SRA
Sequence read archive
- TAB2
TGF-beta activated kinase 1
- TAK1
Transforming growth factor beta activated kinase-1
- TOLLIP
Toll interacting protein
- TRAF2
TNF receptor associated factor-2
- Upd-3
Unpaired-3
- βGRP
β-1,3-glucan recognition protein
Additional files
Summary of the putative immunity-related unigenes identified in Locusta migratoria manilensis. (DOCX 65 kb)
qRT-PCR primers. (DOCX 16 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XY conceived of the study and revised the mansucript. ZW perfomed the bioinfomatic analysis, qRT-PCR validation mainly and drafted the manuscript. CJ participated in data analysis and qRT-PCR validation partially. NK revised the manuscript. ZZ collected the materials and prepared the samples for RNA-seq. LS analyzed partial data. All authors have read and approved the final manuscript.
Contributor Information
Wei Zhang, Email: zhangwei976@126.com.
Jianhong Chen, Email: chenjianhong777@163.com.
Nemat O. Keyhani, Email: keyhani@ufl.edu
Zhengyi Zhang, Email: zhangzhengyijilin@126.com.
Sai Li, Email: sklisai@163.com.
Yuxian Xia, Email: yuxianxia@cqu.edu.cn.
References
- 1.Situation update: locust crisis in Madagascar FAO. [http://www.fao.org/fileadmin/user_upload/emergencies/docs/2014-05-08_MAG_Locust_Crisis_Situation_Update_EN.pdf].
- 2.Nappi AJ, Ottaviani E. Cytotoxicity and cytotoxic molecules in invertebrates. Bioessays. 2000;22(5):469–480. doi: 10.1002/(SICI)1521-1878(200005)22:5<469::AID-BIES9>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 3.Tsakas S, Marmaras V. Insect immunity and its signalling: an overview. ISJ. 2010;7:228–238. [Google Scholar]
- 4.Arrese EL, Soulages JL. Insect fat body: energy, metabolism, and regulation. Annu Rev Entomol. 2010;55:207. doi: 10.1146/annurev-ento-112408-085356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mirth CK, Riddiford LM. Size assessment and growth control: how adult size is determined in insects. Bioessays. 2007;29(4):344–355. doi: 10.1002/bies.20552. [DOI] [PubMed] [Google Scholar]
- 6.Mavrouli MD, Tsakas S, Theodorou GL, Lampropoulou M, Marmaras VJ. MAP kinases mediate phagocytosis and melanization via prophenoloxidase activation in medfly hemocytes. Biochim Biophys Acta (BBA) - Mol Cell Res. 2005;1744(2):145–156. doi: 10.1016/j.bbamcr.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 7.Hartenstein V. Blood cells and blood cell development in the animal kingdom. Annu Rev Cell Dev Biol. 2006;22:677–712. doi: 10.1146/annurev.cellbio.22.010605.093317. [DOI] [PubMed] [Google Scholar]
- 8.Irving P, Troxler L, Heuer TS, Belvin M, Kopczynski C, Reichhart J-M, Hoffmann JA, Hetru C. A genome-wide analysis of immune responses in Drosophila. Proc Natl Acad Sci. 2001;98(26):15119–15124. doi: 10.1073/pnas.261573998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waterhouse RM, Kriventseva EV, Meister S, Xi Z, Alvarez KS, Bartholomay LC, Barillas-Mury C, Bian G, Blandin S, Christensen BM. Evolutionary dynamics of immune-related genes and pathways in disease-vector mosquitoes. Science. 2007;316(5832):1738–1743. doi: 10.1126/science.1139862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evans J, Aronstein K, Chen Y, Hetru C, Imler JL, Jiang H, Kanost M, Thompson G, Zou Z, Hultmark D. Immune pathways and defence mechanisms in honey bees Apis mellifera. Insect Mol Biol. 2006;15(5):645–656. doi: 10.1111/j.1365-2583.2006.00682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zou Z, Evans JD, Lu Z, Zhao P, Williams M, Sumathipala N, Hetru C, Hultmark D, Jiang H. Comparative genomic analysis of the Tribolium immune system. Genome Biol. 2007;8(8):R177. doi: 10.1186/gb-2007-8-8-r177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pinto SB, Lombardo F, Koutsos AC, Waterhouse RM, McKay K, An C, Ramakrishnan C, Kafatos FC, Michel K. Discovery of plasmodium modulators by genome-wide analysis of circulating hemocytes in Anopheles gambiae. Proc Natl Acad Sci. 2009;106(50):21270–21275. doi: 10.1073/pnas.0909463106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baton LA, Robertson A, Warr E, Strand MR, Dimopoulos G. Genome-wide transcriptomic profiling of Anopheles gambiae hemocytes reveals pathogen-specific signatures upon bacterial challenge and Plasmodium berghei infection. BMC Genomics. 2009;10(1):257. doi: 10.1186/1471-2164-10-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartholomay LC, Cho W-L, Rocheleau TA, Boyle JP, Beck ET, Fuchs JF, Liss P, Rusch M, Butler KM, Wu RC-C. Description of the transcriptomes of immune response-activated hemocytes from the mosquito vectors Aedes aegypti and Armigeres subalbatus. Infect Immun. 2004;72(7):4114–4126. doi: 10.1128/IAI.72.7.4114-4126.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Attardo G, Strickler‐Dinglasan P, Perkin S, Caler E, Bonaldo M, Soares M, El‐Sayeed N, Aksoy S. Analysis of fat body transcriptome from the adult tsetse fly, Glossina morsitans morsitans. Insect Mol Biol. 2006;15(4):411–424. doi: 10.1111/j.1365-2583.2006.00649.x. [DOI] [PubMed] [Google Scholar]
- 16.Gandhe AS, Arunkumar K, John SH, Nagaraju J. Analysis of bacteria-challenged wild silkmoth, Antheraea mylitta (lepidoptera) transcriptome reveals potential immune genes. BMC Genomics. 2006;7(1):184. doi: 10.1186/1471-2164-7-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Shen D, Zhou F, Wang G, An C. Identification of immunity-related genes in Ostrinia furnacalis against entomopathogenic fungi by RNA-Seq analysis. PLoS One. 2014;9(1):e86436. doi: 10.1371/journal.pone.0086436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia J, Zhang C-R, Zhang S, Li F-F, Feng M-G, Wang X-W, Liu S-S. Analysis of whitefly transcriptional responses to Beauveria bassiana infection reveals new insights into insect-fungus interactions. PLoS One. 2013;8(7):e68185. doi: 10.1371/journal.pone.0068185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ortiz-Urquiza A, Luo Z, Keyhani NO. Improving mycoinsecticides for insect biological control. Appl Microbiol Biotechnol. 2015;99(3):1057–1068. doi: 10.1007/s00253-014-6270-x. [DOI] [PubMed] [Google Scholar]
- 20.Glare T, Caradus J, Gelernter W, Jackson T, Keyhani N, Kohl J, Marrone P, Morin L, Stewart A. Have biopesticides come of age? Trends Biotechnol. 2012;30(5):250–258. doi: 10.1016/j.tibtech.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 21.Pedrini N, Ortiz-Urquiza A, Huarte-Bonnet C, Fan Y, Juárez MP, Keyhani NO. Tenebrionid secretions and a fungal benzoquinone oxidoreductase form competing components of an arms race between a host and pathogen. Proc Natl Acad Sci. 2015;112(28):E3651–E3660. doi: 10.1073/pnas.1504552112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao QA, Jin K, Ying SH, Zhang YJ, Xiao GH, Shang YF, Duan ZB, Hu XA, Xie XQ, Zhou G, et al. Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum. PLoS Genet. 2011;7(1):e1001264. doi: 10.1371/journal.pgen.1001264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao G, Ying SH, Zheng P, Wang ZL, Zhang S, Xie XQ, Shang Y, St Leger RJ, Zhao GP, Wang C, et al. Genomic perspectives on the evolution of fungal entomopathogenicity in Beauveria bassiana. Sci Rep. 2012;2:483. doi: 10.1038/srep00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang CS, Leger RJS. Developmental and transcriptional responses to host and nonhost cuticles by the specific locust pathogen Metarhizium anisopliae var. acridum. Eukaryot Cell. 2005;4(5):937–947. doi: 10.1128/EC.4.5.937-947.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ortiz-Urquiza A, Keyhani NO. Action on the surface: entomopathogenic fungi versus the insect cuticle. Insects. 2013;4:357–374. doi: 10.3390/insects4030357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wanchoo A, Lewis MW, Keyhani NO. Lectin mapping reveals stage-specific display of surface carbohydrates in in vitro and haemolymph-derived cells of the entomopathogenic fungus Beauveria bassiana. Microbiol-Sgm. 2009;155:3121–3133. doi: 10.1099/mic.0.029157-0. [DOI] [PubMed] [Google Scholar]
- 27.Kurtti TJ, Keyhani NO. Intracellular infection of tick cell lines by the entomopathogenic fungus Metarhizium anisopliae. Microbiol-Sgm. 2008;154:1700–1709. doi: 10.1099/mic.0.2008/016667-0. [DOI] [PubMed] [Google Scholar]
- 28.Jin K, Han LR, Xia YX. MaMk1, a FUS3/KSS1-type mitogen-activated protein kinase gene, is required for appressorium formation, and insect cuticle penetration of the entomopathogenic fungus Metarhizium acridum. J Invertebr Pathol. 2014;115:68–75. doi: 10.1016/j.jip.2013.10.014. [DOI] [PubMed] [Google Scholar]
- 29.Luo S, He M, Cao Y, Xia Y. The tetraspanin gene MaPls1 contributes to virulence by affecting germination, appressorial function and enzymes for cuticle degradation in the entomopathogenic fungus, Metarhizium acridum. Environ Microbiol. 2013;15(11):2966–79. doi: 10.1111/1462-2920.12166. [DOI] [PubMed] [Google Scholar]
- 30.Wang SB, Fang WG, Wang CS, Leger RJS. Insertion of an esterase gene into a specific locust pathogen (Metarhizium acridum) enables it to infect caterpillars. PLoS Pathog. 2011;7(6):e1002097. doi: 10.1371/journal.ppat.1002097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gillespie JP, Bateman R, Charnley AK. Role of cuticle-degrading proteases in the virulence of metarhizium spp. for the desert locust, Schistocerca gregaria. J Invertebr Pathol. 1998;71(2):128–137. doi: 10.1006/jipa.1997.4733. [DOI] [PubMed] [Google Scholar]
- 32.Bateman R, Carey M, Moore D, Prior C. The enhanced infectivity of Metarhizium flavoviride in oil formulations to desert locusts at low humidities. Ann Appl Biol. 1993;122(1):145–152. doi: 10.1111/j.1744-7348.1993.tb04022.x. [DOI] [Google Scholar]
- 33.Gillespie JP, Burnett C, Charnley AK. The immune response of the desert locust Schistocerca gregaria during mycosis of the entomopathogenic fungus, Metarhizium anisopliae var acridum. J Insect Physiol. 2000;46(4):429–437. doi: 10.1016/S0022-1910(99)00128-6. [DOI] [PubMed] [Google Scholar]
- 34.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, Lee Y, White J, Cheung F, Parvizi B. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19(5):651–652. doi: 10.1093/bioinformatics/btg034. [DOI] [PubMed] [Google Scholar]
- 36.Zhang F, Guo H, Zheng H, Zhou T, Zhou Y, Wang S, Fang R, Qian W, Chen X. Massively parallel pyrosequencing-based transcriptome analyses of small brown planthopper (Laodelphax striatellus), a vector insect transmitting rice stripe virus (RSV) BMC Genomics. 2010;11(1):303. doi: 10.1186/1471-2164-11-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonizzoni M, Dunn WA, Campbell CL, Olson KE, Marinotti O, James AA. Complex modulation of the Aedes aegypti transcriptome in response to dengue virus infection. PLoS One. 2012;7(11):e50512. doi: 10.1371/journal.pone.0050512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55(4):611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 40.Pfaffl MW. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001;29(9):e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu S, Wei W, Chu Y, Zhang L, Shen J, An C. De Novo transcriptome analysis of wing development-related signaling pathways in Locusta migratoria Manilensis and Ostrinia furnacalis (Guenée) 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Meng X, Liu C, Gao H, Zhang Y, Liu Z. Cys-loop ligand-gated ion channel gene discovery in the Locusta migratoria manilensis through the neuron transcriptome. Gene. 2015;561(2):276–282. doi: 10.1016/j.gene.2015.02.048. [DOI] [PubMed] [Google Scholar]
- 43.Chen S, Yang P, Jiang F, Wei Y, Ma Z, Kang L. De novo analysis of transcriptome dynamics in the migratory locust during the development of phase traits. PLoS One. 2010;5(12):e15633. doi: 10.1371/journal.pone.0015633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Fang X, Yang P, Jiang X, Jiang F, Zhao D, et al. The locust genome provides insight into swarm formation and long-distance flight. Nat Commun. 2014;5. [DOI] [PMC free article] [PubMed]
- 45.Nappi AJ, Vass E, Frey F, Carton Y. Nitric oxide involvement in Drosophila immunity. Nitric Oxide. 2000;4(4):423–430. doi: 10.1006/niox.2000.0294. [DOI] [PubMed] [Google Scholar]
- 46.He A, Rosazza JP. GTP cyclohydrolase I: purification, characterization, and effects of inhibition on nitric oxide synthase in nocardia species. Appl Environ Microbiol. 2003;69(12):7507–7513. doi: 10.1128/AEM.69.12.7507-7513.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kizaki T, Suzuki K, Hitomi Y, Taniguchi N, Saitoh D, Watanabe K, Onoé K, Day NK, Good RA, Ohno H. Uncoupling protein 2 plays an important role in nitric oxide production of lipopolysaccharide-stimulated macrophages. Proc Natl Acad Sci. 2002;99(14):9392–9397. doi: 10.1073/pnas.142206299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tang H. Regulation and function of the melanization reaction in Drosophila. Fly. 2009;3(1):105–111. doi: 10.4161/fly.3.1.7747. [DOI] [PubMed] [Google Scholar]
- 49.Cox D, Chang P, Zhang Q, Reddy PG, Bokoch GM, Greenberg S. Requirements for both Rac1 and Cdc42 in membrane ruffling and phagocytosis in leukocytes. J Exp Med. 1997;186(9):1487–1494. doi: 10.1084/jem.186.9.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baxt LA, Baker RP, Singh U, Urban S. An Entamoeba histolytica rhomboid protease with atypical specificity cleaves a surface lectin involved in phagocytosis and immune evasion. Genes Dev. 2008;22(12):1636–1646. doi: 10.1101/gad.1667708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lombardo F, Ghani Y, Kafatos FC, Christophides GK. Comprehensive genetic dissection of the hemocyte immune response in the malaria mosquito Anopheles gambiae. PLoS Pathog. 2013;9(1):e1003145. doi: 10.1371/journal.ppat.1003145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ahmed AM, Hurd H. Immune stimulation and malaria infection impose reproductive costs in Anopheles gambiae via follicular apoptosis. Microbes Infect. 2006;8(2):308–315. doi: 10.1016/j.micinf.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 53.Hurd H. Host fecundity reduction: a strategy for damage limitation? Trends Parasitol. 2001;17(8):363–368. doi: 10.1016/S1471-4922(01)01927-4. [DOI] [PubMed] [Google Scholar]
- 54.Kounatidis I, Ligoxygakis P. Drosophila as a model system to unravel the layers of innate immunity to infection. Open Biol. 2012;2(5):120075. doi: 10.1098/rsob.120075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Franc NC, White K. Innate recognition systems in insect immunity and development: new approaches in Drosophila. Microbes Infect. 2000;2(3):243–250. doi: 10.1016/S1286-4579(00)00304-X. [DOI] [PubMed] [Google Scholar]
- 56.Fujita T. Evolution of the lectin–complement pathway and its role in innate immunity. Nat Rev Immunol. 2002;2(5):346–353. doi: 10.1038/nri800. [DOI] [PubMed] [Google Scholar]
- 57.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 58.Hultmark D. Drosophila immunity: paths and patterns. Curr Opin Immunol. 2003;15(1):12–19. doi: 10.1016/S0952-7915(02)00005-5. [DOI] [PubMed] [Google Scholar]
- 59.Kurata S. Peptidoglycan recognition proteins in Drosophila immunity. Dev Comp Immunol. 2014;42(1):36–41. doi: 10.1016/j.dci.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Werner T, Liu G, Kang D, Ekengren S, Steiner H, Hultmark D. A family of peptidoglycan recognition proteins in the fruit fly Drosophila melanogaster. Proc Natl Acad Sci. 2000;97(25):13772–13777. doi: 10.1073/pnas.97.25.13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Christophides GK, Zdobnov E, Barillas-Mury C, Birney E, Blandin S, Blass C, Brey PT, Collins FH, Danielli A, Dimopoulos G. Immunity-related genes and gene families in Anopheles gambiae. Science. 2002;298(5591):159–165. doi: 10.1126/science.1077136. [DOI] [PubMed] [Google Scholar]
- 62.Tanaka H, Ishibashi J, Fujita K, Nakajima Y, Sagisaka A, Tomimoto K, Suzuki N, Yoshiyama M, Kaneko Y, Iwasaki T. A genome-wide analysis of genes and gene families involved in innate immunity of Bombyx mori. Insect Biochem Mol Biol. 2008;38(12):1087–1110. doi: 10.1016/j.ibmb.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 63.Michel T, Reichhart J-M, Hoffmann JA, Royet J. Drosophila Toll is activated by Gram-positive bacteria through a circulating peptidoglycan recognition protein. Nature. 2001;414(6865):756–759. doi: 10.1038/414756a. [DOI] [PubMed] [Google Scholar]
- 64.Choe K-M, Werner T, Stöven S, Hultmark D, Anderson KV. Requirement for a peptidoglycan recognition protein (PGRP) in Relish activation and antibacterial immune responses in Drosophila. Science. 2002;296(5566):359–362. doi: 10.1126/science.1070216. [DOI] [PubMed] [Google Scholar]
- 65.Zhang R, Cho HY, Kim HS, Ma YG, Osaki T, Kawabata S-i, Söderhäll K, Lee BL. Characterization and properties of a 1, 3-β-d-glucan pattern recognition protein of Tenebrio molitor larvae that is specifically degraded by serine protease during prophenoloxidase activation. J Biol Chem. 2003;278(43):42072–42079. doi: 10.1074/jbc.M307475200. [DOI] [PubMed] [Google Scholar]
- 66.Kim Y-S, Ryu J-H, Han S-J, Choi K-H, Nam K-B, Jang I-H, Lemaitre B, Brey PT, Lee W-J. Gram-negative bacteria-binding protein, a pattern recognition receptor for lipopolysaccharide and β-1, 3-glucan that mediates the signaling for the induction of innate immune genes in Drosophila melanogaster cells. J Biol Chem. 2000;275(42):32721–32727. doi: 10.1074/jbc.M003934200. [DOI] [PubMed] [Google Scholar]
- 67.Ma C, Kanost MR. A β1,3-glucan recognition protein from an insect, Manduca sexta, agglutinates microorganisms and activates the Phenoloxidase cascade. J Biol Chem. 2000;275(11):7505–7514. doi: 10.1074/jbc.275.11.7505. [DOI] [PubMed] [Google Scholar]
- 68.Jiang HB, Ma CC, Lu ZQ, Kanost MR. B-1,3-Glucan recognition protein-2 (beta GRP-2) from Manduca sexta: an acute-phase protein that binds beta-1,3-glucan and lipoteichoic acid to aggregate fungi and bacteria and stimulate prophenoloxidase activation. Insect Biochem Mol Biol. 2004;34(1):89–100. doi: 10.1016/j.ibmb.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 69.Wang Y, Yang P, Cui F, Kang L. Altered immunity in crowded locust reduced fungal (Metarhizium anisopliae) pathogenesis. PLoS Pathog. 2013;9:e1003102. doi: 10.1371/journal.ppat.1003102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zheng X, Xia Y. β-1, 3-Glucan recognition protein (βGRP) is essential for resistance against fungal pathogen and opportunistic pathogenic gut bacteria in Locusta migratoria manilensis. Dev Comp Immunol. 2012;36(3):602–609. doi: 10.1016/j.dci.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 71.Drickamer K, Fadden AJ. Biochemical Society Symposia: 2002. London: Portland on behalf of The Biochemical Society; 1999; 2002. Genomic analysis of C-type lectins; pp. 59–72. [DOI] [PubMed] [Google Scholar]
- 72.Yu X-Q, Kanost MR. Manduca sexta lipopolysaccharide-specific immulectin-2 protects larvae from bacterial infection. Dev Comp Immunol. 2003;27(3):189–196. doi: 10.1016/S0145-305X(02)00099-X. [DOI] [PubMed] [Google Scholar]
- 73.Koizumi N, Imamura M, Kadotani T, Yaoi K, Iwahana H, Sato R. The lipopolysaccharide-binding protein participating in hemocyte nodule formation in the silkworm Bombyx mori is a novel member of the C-type lectin superfamily with two different tandem carbohydrate-recognition domains. FEBS Lett. 1999;443(2):139–143. doi: 10.1016/S0014-5793(98)01701-3. [DOI] [PubMed] [Google Scholar]
- 74.Yu X-Q, Kanost MR. Immulectin-2, a lipopolysaccharide-specific lectin from an insect, Manduca sexta, is induced in response to gram-negative bacteria. J Biol Chem. 2000;275(48):37373–37381. doi: 10.1074/jbc.M003021200. [DOI] [PubMed] [Google Scholar]
- 75.Jomori T, Natori S. Function of the lipopolysaccharide-binding protein of Periplaneta americana as an opsonin. FEBS Lett. 1992;296(3):283–286. doi: 10.1016/0014-5793(92)80305-Z. [DOI] [PubMed] [Google Scholar]
- 76.Pace KE, Baum LG. Insect galectins: roles in immunity and development. Glycoconj J. 2002;19(7–9):607–614. doi: 10.1023/B:GLYC.0000014092.86763.2f. [DOI] [PubMed] [Google Scholar]
- 77.Mey A, Leffler H, Hmama Z, Normier G, Revillard J-P. The animal lectin galectin-3 interacts with bacterial lipopolysaccharides via two independent sites. J Immunol. 1996;156(4):1572–1577. [PubMed] [Google Scholar]
- 78.Murphy JE, Tedbury PR, Homer-Vanniasinkam S, Walker JH, Ponnambalam S. Biochemistry and cell biology of mammalian scavenger receptors. Atherosclerosis. 2005;182(1):1–15. doi: 10.1016/j.atherosclerosis.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 79.Pierini LM. Uptake of serum‐opsonized Francisella tularensis by macrophages can be mediated by class A scavenger receptors. Cell Microbiol. 2006;8(8):1361–1370. doi: 10.1111/j.1462-5822.2006.00719.x. [DOI] [PubMed] [Google Scholar]
- 80.Agaisse H, Burrack LS, Philips JA, Rubin EJ, Perrimon N, Higgins DE. Genome-wide RNAi screen for host factors required for intracellular bacterial infection. Science. 2005;309(5738):1248–1251. doi: 10.1126/science.1116008. [DOI] [PubMed] [Google Scholar]
- 81.Franc NC, Heitzler P, White K. Requirement for croquemort in phagocytosis of apoptotic cells in Drosophila. Science. 1999;284(5422):1991–1994. doi: 10.1126/science.284.5422.1991. [DOI] [PubMed] [Google Scholar]
- 82.Rämet M, Pearson A, Manfruelli P, Li X, Koziel H, Göbel V, Chung E, Krieger M, Ezekowitz RAB. Drosophila scavenger receptor CI is a pattern recognition receptor for bacteria. Immunity. 2001;15(6):1027–1038. doi: 10.1016/S1074-7613(01)00249-7. [DOI] [PubMed] [Google Scholar]
- 83.Schmucker D, Clemens JC, Shu H, Worby CA, Xiao J, Muda M, Dixon JE, Zipursky SL. Drosophila Dscam is an axon guidance receptor exhibiting extraordinary molecular diversity. Cell. 2000;101(6):671–684. doi: 10.1016/S0092-8674(00)80878-8. [DOI] [PubMed] [Google Scholar]
- 84.Watson FL, Püttmann-Holgado R, Thomas F, Lamar DL, Hughes M, Kondo M, Rebel VI, Schmucker D. Extensive diversity of Ig-superfamily proteins in the immune system of insects. Science. 2005;309(5742):1874–1878. doi: 10.1126/science.1116887. [DOI] [PubMed] [Google Scholar]
- 85.Dong Y, Taylor HE, Dimopoulos G. AgDscam, a hypervariable immunoglobulin domain-containing receptor of the Anopheles gambiae innate immune system. PLoS Biol. 2006;4(7):e229. doi: 10.1371/journal.pbio.0040229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jiang H, Kanost MR. The clip-domain family of serine proteinases in arthropods. Insect Biochem Mol Biol. 2000;30(2):95–105. doi: 10.1016/S0965-1748(99)00113-7. [DOI] [PubMed] [Google Scholar]
- 87.Volz J, Müller HM, Zdanowicz A, Kafatos FC, Osta MA. A genetic module regulates the melanization response of Anopheles to Plasmodium. Cell Microbiol. 2006;8(9):1392–1405. doi: 10.1111/j.1462-5822.2006.00718.x. [DOI] [PubMed] [Google Scholar]
- 88.Kambris Z, Brun S, Jang I-H, Nam H-J, Romeo Y, Takahashi K, Lee W-J, Ueda R, Lemaitre B. Drosophila immunity: a large-scale in vivo RNAi screen identifies five serine proteases required for toll activation. Curr Biol. 2006;16(8):808–813. doi: 10.1016/j.cub.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 89.Jang I-H, Chosa N, Kim S-H, Nam H-J, Lemaitre B, Ochiai M, Kambris Z, Brun S, Hashimoto C, Ashida M. A spätzle-processing enzyme required for toll signaling activation in Drosophila innate immunity. Dev Cell. 2006;10(1):45–55. doi: 10.1016/j.devcel.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 90.Gettins PG. Serpin structure, mechanism, and function. Chem Rev. 2002;102(12):4751–4804. doi: 10.1021/cr010170+. [DOI] [PubMed] [Google Scholar]
- 91.Potempa J, Korzus E, Travis J. The serpin superfamily of proteinase inhibitors: structure, function, and regulation. J Biol Chem-Paper Edition. 1994;269(23):15957–15960. [PubMed] [Google Scholar]
- 92.Aggrawal K, Silverman N. Peptidoglycan recognition in Drosophila. Biochem Soc Trans. 2007;35(Pt 6):1496–1500. doi: 10.1042/BST0351496. [DOI] [PubMed] [Google Scholar]
- 93.Feldhaar H, Gross R. Immune reactions of insects on bacterial pathogens and mutualists. Microbes Infect. 2008;10(9):1082–1088. doi: 10.1016/j.micinf.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 94.Lemaitre B, Hoffmann J. The host defense of Drosophila melanogaster. Annu Rev Immunol. 2007;25:697–743. doi: 10.1146/annurev.immunol.25.022106.141615. [DOI] [PubMed] [Google Scholar]
- 95.Cha G-H, Cho KS, Lee JH, Kim M, Kim E, Park J, Lee SB, Chung J. Discrete functions of TRAF1 and TRAF2 in Drosophila melanogaster mediated by c-Jun N-terminal kinase and NF-κB-dependent signaling pathways. Mol Cell Biol. 2003;23(22):7982–7991. doi: 10.1128/MCB.23.22.7982-7991.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kopp E, Medzhitov R, Carothers J, Xiao C, Douglas I, Janeway CA, Ghosh S. ECSIT is an evolutionarily conserved intermediate in the Toll/IL-1 signal transduction pathway. Genes Dev. 1999;13(16):2059–2071. doi: 10.1101/gad.13.16.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mizuguchi K, Parker JS, Blundell TL, Gay NJ. Getting knotted: a model for the structure and activation of Spätzle. Trends Biochem Sci. 1998;23(7):239–242. doi: 10.1016/S0968-0004(98)01216-X. [DOI] [PubMed] [Google Scholar]
- 98.Weber AN, Tauszig-Delamasure S, Hoffmann JA, Lelièvre E, Gascan H, Ray KP, Morse MA, Imler J-L, Gay NJ. Binding of the Drosophila cytokine Spätzle to Toll is direct and establishes signaling. Nat Immunol. 2003;4(8):794–800. doi: 10.1038/ni955. [DOI] [PubMed] [Google Scholar]
- 99.Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF. A family of human receptors structurally related to Drosophila toll. Proc Natl Acad Sci. 1998;95(2):588–593. doi: 10.1073/pnas.95.2.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang L, Keyhani NO, Tang G, Tian C, Lu R, Wang X, Pei Y, Fan Y. Expression of a toll signaling regulator serpin in a mycoinsecticide for increased virulence. Appl Environ Microbiol. 2014;80(15):4531–4539. doi: 10.1128/AEM.01197-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Leulier F, Vidal S, Saigo K, Ueda R, Lemaitre B. Inducible expression of double-stranded RNA reveals a role for dFADD in the regulation of the antibacterial response in Drosophila adults. Curr Biol. 2002;12(12):996–1000. doi: 10.1016/S0960-9822(02)00873-4. [DOI] [PubMed] [Google Scholar]
- 102.Paquette N, Broemer M, Aggarwal K, Chen L, Husson M, Ertürk-Hasdemir D, Reichhart J-M, Meier P, Silverman N. Caspase-mediated cleavage, IAP binding, and Ubiquitination: linking three mechanisms crucial for drosophila NF-κB signaling. Mol Cell. 2010;37(2):172–182. doi: 10.1016/j.molcel.2009.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vidal S, Khush RS, Leulier F, Tzou P, Nakamura M, Lemaitre B. Mutations in the Drosophila dTAK1 gene reveal a conserved function for MAPKKKs in the control of rel/NF-κB-dependent innate immune responses. Genes Dev. 2001;15(15):1900–1912. doi: 10.1101/gad.203301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kleino A, Valanne S, Ulvila J, Kallio J, Myllymäki H, Enwald H, Stöven S, Poidevin M, Ueda R, Hultmark D. Inhibitor of apoptosis 2 and TAK1‐binding protein are components of the Drosophila Imd pathway. EMBO J. 2005;24(19):3423–3434. doi: 10.1038/sj.emboj.7600807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hedengren M, Dushay MS, Ando I, Ekengren S, Wihlborg M, Hultmark D. Relish a central factor in the control of humoral but not cellular immunity in Drosophila. Mol Cell. 1999;4(5):827–837. doi: 10.1016/S1097-2765(00)80392-5. [DOI] [PubMed] [Google Scholar]
- 106.Ertürk-Hasdemir D, Broemer M, Leulier F, Lane WS, Paquette N, Hwang D, Kim C-H, Stöven S, Meier P, Silverman N. Two roles for the Drosophila IKK complex in the activation of Relish and the induction of antimicrobial peptide genes. Proc Natl Acad Sci. 2009;106(24):9779–9784. doi: 10.1073/pnas.0812022106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stöven S, Silverman N, Junell A, Hedengren-Olcott M, Erturk D, Engström Y, Maniatis T, Hultmark D. Caspase-mediated processing of the Drosophila NF-κB factor Relish. Proc Natl Acad Sci. 2003;100(10):5991–5996. doi: 10.1073/pnas.1035902100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dostert C, Jouanguy E, Irving P, Troxler L, Galiana-Arnoux D, Hetru C, Hoffmann JA, Imler J-L. The Jak-STAT signaling pathway is required but not sufficient for the antiviral response of drosophila. Nat Immunol. 2005;6(9):946–953. doi: 10.1038/ni1237. [DOI] [PubMed] [Google Scholar]
- 109.Agaisse H, Petersen U-M, Boutros M, Mathey-Prevot B, Perrimon N. Signaling role of hemocytes in Drosophila JAK/STAT-dependent response to septic injury. Dev Cell. 2003;5(3):441–450. doi: 10.1016/S1534-5807(03)00244-2. [DOI] [PubMed] [Google Scholar]
- 110.Kawabata T, Yasuhara Y, Ochiai M, Matsuura S, Ashida M. Molecular cloning of insect pro-phenol oxidase: a copper-containing protein homologous to arthropod hemocyanin. Proc Natl Acad Sci. 1995;92(17):7774–7778. doi: 10.1073/pnas.92.17.7774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cerenius L, Lee BL, Söderhäll K. The proPO-system: pros and cons for its role in invertebrate immunity. Trends Immunol. 2008;29(6):263–271. doi: 10.1016/j.it.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 112.Fukamizo T. Chitinolytic enzymes catalysis, substrate binding, and their application. Curr Protein Pept Sci. 2000;1(1):105–124. doi: 10.2174/1389203003381450. [DOI] [PubMed] [Google Scholar]
- 113.Kumar S, Barillas-Mury C. Ookinete-induced midgut peroxidases detonate the time bomb in anopheline mosquitoes. Insect Biochem Mol Biol. 2005;35(7):721–727. doi: 10.1016/j.ibmb.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 114.Leulier F, Rodriguez A, Khush RS, Abrams JM, Lemaitre B. The Drosophila caspase Dredd is required to resist Gram‐negative bacterial infection. EMBO Rep. 2000;1(4):353–358. doi: 10.1093/embo-reports/kvd073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Clem R. Role of apoptosis in infection. Heidelberg: Springer; 2005. The role of apoptosis in defense against baculovirus infection in insects; pp. 113–129. [DOI] [PubMed] [Google Scholar]
- 116.Cox-Foster DL, Stehr JE. Induction and localization of FAD-glucose dehydrogenase (GLD) during encapsulation of abiotic implants in Manduca sexta larvae. J Insect Physiol. 1994;40(3):235–249. doi: 10.1016/0022-1910(94)90047-7. [DOI] [Google Scholar]