

Abstract
In this article we review recent advances made in the pathophysiology, diagnosis, and treatment of inhalation injury. Historically, the diagnosis of inhalation injury has relied on nonspecific clinical exam findings and bronchoscopic evidence. The development of a grading system and the use of modalities such as chest computed tomography may allow for a more nuanced evaluation of inhalation injury and enhanced ability to prognosticate. Supportive respiratory care remains essential in managing inhalation injury. Adjuncts still lacking definitive evidence of efficacy include bronchodilators, mucolytic agents, inhaled anticoagulants, nonconventional ventilator modes, prone positioning, and extracorporeal membrane oxygenation. Recent research focusing on molecular mechanisms involved in inhalation injury has increased the number of potential therapies.
Introduction
Despite important advances in the care of patients with inhalation injury, which continues to be largely supportive, morbidity and mortality remain high [1]. Inhalation injury can feature supraglottic thermal injury, chemical irritation of the respiratory tract, systemic toxicity due to agents such as carbon monoxide (CO) and cyanide, or a combination of these insults. The resultant inflammatory response may cause higher fluid resuscitation volumes, progressive pulmonary dysfunction, prolonged ventilator days, increased risk of pneumonia, and acute respiratory distress syndrome (ARDS) [2, 3].
In this review we describe the recent advances made in our understanding of the pathophysiology of inhalation injury, diagnostic criteria and injury severity, complications, current treatment options, and future avenues of research.
Pathophysiology
Inhalation injury complicates burns in approximately 10 to 20 % of patients and significantly increases morbidity and mortality [2–5]. Other factors associated with a significant effect on mortality include burn size and age [6, 7] and the incidence of inhalation injury is correlated with an increase in both these factors [6, 7]. Inhalation injury has also been found to be an independent predictor of mortality in burn patients [8] and worsens survival even among patients with similar age and burn size [8]. Thermal airway injury is generally limited to supraglottic structures, whereas injury to the lower airway is chemical in nature. In the setting of steam, however, the injury is pervasive, causing damage to both upper airways and direct thermal injury to the lungs [9]. The degree of inhalation injury is variable and is dependent on several factors: the gas components inhaled, the presence of particulate matter (soot), the magnitude of the exposure, and individual host factors such as underlying lung disease and inability to flee the incident.
Historically, it was speculated that the combustion of certain materials, such as noncommercial polyurethane foam, resulted in the formation of a neurotoxin [10]; however, newer material testing methods revealed most smoke toxicity can be explained by a small number of toxic gases exerting their effects through asphyxiation, systemic toxicity, or direct effects on respiratory tissue [11]. Many products of combustion, such as carbon dioxide, function as simple asphyxiants by displacing oxygen at the alveolar level. This is further exacerbated by the hypoxic fire environment. CO functions systemically as an asphyxiant by (1) competitively displacing oxygen from hemoglobin and (2) binding to cytochrome oxidase at the mitochondrial level. By contrast, hydrogen cyanide binds only to cytochrome oxidase [1].
Other combustion products, such as halogen acids, formaldehyde, and unsaturated aldehydes (for example, acrolein), function as respiratory irritants. The chemical irritation causes denuding of the respiratory mucosa, leading to sloughing within the airways, and induces the host inflammatory response. Furthermore, the chemical injury stimulates vasomotor and sensory nerve endings to produce neuropeptides. Preclinical studies have shown these neuropeptides can induce an inflammatory response [12]. Substance P and calcitonin gene-related peptide are two suggested neuropeptides inducing tissue injury after inhalation injury [12, 13]. Lange et al. [13] found antagonists to calcitonin gene-related peptide and substance P attenuated the fluid shifts/inflammation in an ovine model subjected to smoke and inhalation injury. These neuropeptides then induce bronchoconstriction and nitric oxide synthase (NOS) to generate reactive oxygen species (ROS) [14]. As described by Kraneveld and Nijkamp [14], these neuropeptides can function as tachykinins, inducing a robust inflammatory response with the downstream effects of bronchoconstriction, increased vascular permeability, and vasodilation. Furthermore, tachykinins like substance P and neurokinin A can modulate immune cells and stimulate neutrophil and eosinophil chemotaxis [14].
Overall, these factors potentiate local cellular damage and the loss of hypoxic pulmonary vasoconstriction. The loss of hypoxic pulmonary vasoconstriction causes bronchial blood flow to increase by a factor of 10 within 20 min of inhalation injury. ROS may also induce mitochondrial dysfunction and cellular apoptosis [15]. Tissue factor expressed by damaged respiratory epithelial cells and alveolar macrophages initiates the extrinsic coagulation cascade, disrupting pro- and anti-coagulant alveolar homeostasis. We found, for example, that smoke inhalation injury contributes to a hypercoagulable state in the lung by inducing plasminogen activator inhibitor 1 and stabilizing its mRNA [16].
In addition, the increased bronchial blood flow delivers activated polymorphonuclear leukocytes and cytokines to the lung, potentiating the host inflammatory response. The loss of an intact bronchial epithelium and the effects of ROS result in a loss of plasma proteins and fluid from the intravascular space into the alveoli and bronchioles [17]. The transvascular shift of protein causes exudate and cast formation within the airways, leading to alveolar collapse or complete occlusion of the airways [17]. Experimental measures to decrease bronchial blood flow show attenuation of airway obstruction, pulmonary edema, and improved oxygenation [18]. These processes - loss of hypoxic vasoconstriction, increased blood flow to injured lung segments, decreased ventilation of collapsed segments - contribute to ventilation-perfusion mismatch as a primary mechanism of hypoxemia following the smoke inhalation injury [19]. Atelectasis, dysfunction of the immune system, and mechanical ventilation, in turn, predispose to pneumonia as a common complication of inhalation injury.
Many studies are now being completed to better understand the role pro- and anti-inflammatory mediators, or other immune modulators, play in patient outcomes. Albright et al. [1] demonstrated a graded increase in inflammatory cytokines from bronchoalveolar lavage fluid (IL-4, IL-6, IL-9, IL-15, interferon-gamma, granulocyte macrophage colony-stimulating factor, monocyte chemoattractant protein-1) that correlates with the severity of inhalation injury noted on bronchoscopic evaluation (grades 3 or 4 versus 1 or 2). They also found a significant shift from a macrophage-predominant population of cells in lavage fluid to one dominated by neutrophils [1]. This is thought to contribute to the later immune dysfunction, bacterial overgrowth, and pneumonia [1]. The source of the cytokines identified in inhalation is thought to be secondary to complement activation by heat denatured proteins [20]. The stimulation of the complement cascade releases histamine, resulting in xanthine oxidase upregulation and ROS formation [20, 21].
Davis et al. [22] showed several plasma immune mediators were associated with increased inhalation injury severity, even after adjusting for age and percentage of total body surface area burned. IL-1 receptor antagonist (IL-1RA), an anti-inflammatory immune mediator, had the strongest correlation with injury severity and outcome measures, including mortality [22]. The authors also found a much lower IL-1β to IL-1RA ratio in patients with inhalation injury who died. Given that IL-1β is an essential component of the host defense, they hypothesized that insufficient IL-1β or excessive IL-1RA results in systemic immune dysfunction.
The formation of ROS, such as superoxide anions (O2−), hydrogen peroxide (H2O2), and hydroxyl radicals (OH−; the most unstable and reactive), appears to play a major role in numerous injury models [23]. Under normal circumstances the body has compensatory antioxidant mechanisms to mitigate the effects of ROS. Following reperfusion or injury, however, there is a large burst of ROS, which overwhelm the body’s protective measures. As a result, the ROS can lead to cell injury through neutrophil attraction and cytokine production. Indeed, evidence of oxidative stress is found in plasma and lung tissue following smoke inhalation injury [24]. IL-8, a potent chemokine, has been suggested to play a vital role in the initiation and progression of lung inflammation after smoke inhalation [25]. NOS-dependent formation of ROS has been studied in inhalation injury. Activated neutrophils produce large quantities of superoxide that combine with nitric oxide to produce peroxynitrite, which can damage DNA [20]. Peroxynitrite and the resultant DNA damage stimulates poly-(ADP ribose) polymerase-1 (PARP-1), a nuclear repair enzyme and coactivator of NF-ĸB, which can deplete ATP and produce cell damage [26, 27]. Combined, these factors contribute to an increase in IL-8 [27], induce NOS, neutrophil chemotaxis, and increase ROS production [20]. For this reason, heparin/acetylcysteine combinations are being utilized as ROS scavengers in inhalation injury [20].
Diagnosis
Classically, the diagnosis of inhalation injury was subjective and made on the basis of clinical findings. When evaluating a patient with suspected inhalation injury, a clinician first reviews the history and reported mechanism to determine the likelihood of an inhalation injury. Pertinent information includes exposure to flame, smoke, or chemicals (industrial and household), duration of exposure, exposure in an enclosed space, and loss of consciousness or disability. Pertinent physical exam findings include facial burns, singed facial or nasal hair, soot or carbonaceous material on the face or in the sputum, and signs of airway obstruction including stridor, edema, or mucosal damage [3]. Older patients, and those with more extensive burns, are at increased risk of inhalation injury because of prolonged exposure to the fire environment [8].
There are several modalities for confirming inhalation injury to include fiberoptic bronchoscopy (FOB), chest computed tomography (CT), carboxyhemoglobin measurement, radionuclide imaging with 133Xenon, and pulmonary function testing. Many of these modalities lack sensitivity, are invasive, or are subject to significant variability between institutions. In studies by Shirani et al. [8], the following gradation of morbidity and mortality risk was seen in order of increasing risk: (1) patients without inhalation injury; (2) patients with inhalation injury by 133Xenon scan only, but not by FOB; and (3) patients with inhalation injury by FOB. Also, presence of inhalation injury on FOB predicted risk of acute lung injury and the need for increased fluid resuscitation volumes. More recent studies have found a significant correlation between the severity of inhalation injury on FOB and mortality [28].
There are several difficulties in diagnosing the presence and severity of inhalation injuries. Although several laboratories have developed dose–response models of inhalation injury in large animals [29], the characteristics of the material inhaled are important in determining the degree of respiratory failure. In addition, differences in the individual host inflammatory response may lead to a heterogeneous clinical presentation [30]. FOB is unable to assess distal airways and respiratory bronchioles; therefore, damage to this portion of the lung has been proposed as an explanation for the discordance between bronchoscopic severity of injury and mortality. Despite these limitations, FOB continues to be the standard technique used to assess the presence and severity of inhalation injury. Its relative ease and availability allows the initial diagnosis to be made (Fig. 1), and allows the inhalation injury to be followed serially (Figs. 2 and 3).
Fig. 1.
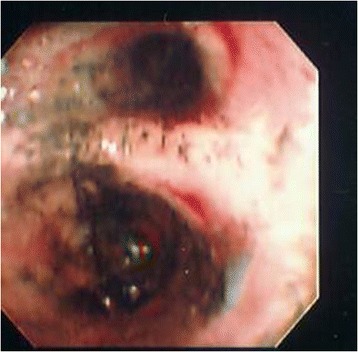
Fiberoptic bronchoscopy of patient on post-burn day 0
Fig. 2.
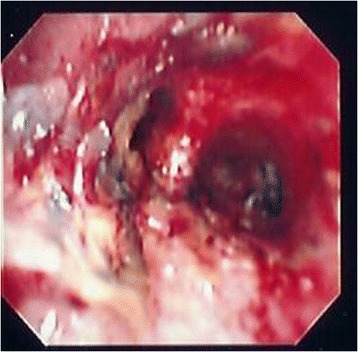
Fiberoptic bronchoscopy of patient on post-burn day 4
Fig. 3.
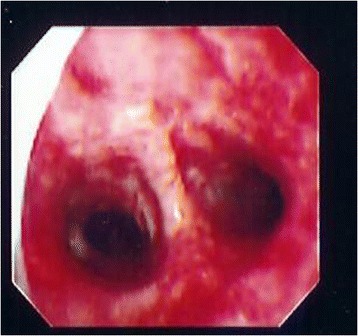
Fiberoptic bronchoscopy of patient on post-burn day 10
Given the lack of a widely standardized and validated method for scoring inhalation injury severity, Woodson [30] has proposed a large multicenter study to create such a scoring system to allow for more reliable prognostic estimations. To date, no such study has been done, though one is currently underway based on clinical, radiographic, bronchoscopic, and biochemical parameters (ClinicalTrials.gov identifier NCT01194024).
Multiple studies have demonstrated that inhalation injury is a graded phenomenon with severity correlating with outcome. The Abbreviated Injury Score grading scale for inhalation injury on bronchoscopy has been shown to correlate with an increase in mortality as well impaired gas exchange [1, 28, 31]. This scale is shown in Table 1. Endorf and Gamelli [3] found that patients with more severe inhalation injury on initial bronchoscopy (grades 2, 3, 4) had worse survival rates than patients with lower scores (grades 0 or 1) (P = 0.03). They also noted the highest-grade inhalation injuries were not necessarily associated with an increased fluid requirement, contrary to prior data. Lastly, they found patients with an arterial partial pressure of oxygen (PaO2)/fraction of inspired oxygen (FiO2) ratio <350 upon presentation had a statistically significant increase in fluid resuscitation needs compared with patients with a ratio >350 (P = 0.03) [3]. Ryan et al. [32] have stated that, at this time, the most reliable indicator of the impact of inhalation injury is the PaO2/FiO2 ratio after the resuscitation has started. This is based on a retrospective review by Hassan et al. [28] of 105 patients admitted with inhalation injury. They assessed respiratory function by using the PaO2/FiO2 ratio from 0 to 192 h after injury. Their study showed a significant difference (P < 0.01) in PaO2/FiO2 ratios between patients who died (mean PaO2/FiO2 ratio 20.17) and those who survived (mean PaO2/FiO2 ratio 32.24). Ultimately, they propose to use PaO2/FiO2 ratio as a predictor of survival once the initial burn resuscitation has been completed and a full response to injury is able to be mounted [28]. Similarly, Cancio et al. [33] found that the mean alveolar-arterial oxygen gradient during the first 2 days was an independent predictor of mortality in mechanically ventilated burn patients. It is important to note that PaO2/FiO2 can be arbitrarily high or low depending on the choice of ventilator mode. In addition, PaO2/FiO2 may be affected by the volume of resuscitation. Therefore, we do not use PaO2/FiO2 as a basis for diagnosis of inhalation injury, but use it to trend the patient’s oxygenation and potential need for nonconventional ventilation.
Table 1.
Abbreviated Injury Score grading scale for inhalation injury on bronchoscopy [1]
| Grade | Class | Description |
|---|---|---|
| 0 | No injury | Absence of carbonaceous deposits, erythema, edema, bronchorrhea, or obstruction |
| 1 | Mild injury | Minor or patchy areas of erythema, carbonaceous deposits, bronchorrhea, or bronchial obstruction |
| 2 | Moderate injury | Moderate degree of erythema, carbonaceous deposits, bronchorrhea, or bronchial obstruction |
| 3 | Severe injury | Severe inflammation with friability, copious carbonaceous deposits, bronchorrhea, or obstruction |
| 4 | Massive injury | Evidence of mucosal sloughing, necrosis, endoluminal obstruction |
Other means of evaluating the severity of inhalation injury include chest CT. First, a scoring system for severity of CT scan findings has been developed [29]. Our group studied 25 patients with inhalation injury and 19 patients without inhalation injury who received a chest CT within 24 h of admission [34]. The severity of radiographic findings was calculated by looking at 1-cm axial slices from the chest CT and these were scored by adding the highest radiologist’s score (RADS) for each quadrant. The RADS scoring system is shown in Table 2, and the various RADS findings are shown in Fig. 4. Our group assessed a composite endpoint of pneumonia, acute lung injury/ARDS, and death. We found that the detection of inhalation injury on bronchoscopy was associated with an 8.3-fold increase in the composite endpoint. A high RADS score (>8 per slice) in addition to a positive bronchoscopy was associated with a 12.7-fold increase, thus showing the potential for chest CT to complement bronchoscopy in detecting clinically significant inhalation injury [34].
Table 2.
Radiologist’s scoring table (RADS score) for inhalation injury [34]
| Finding | Score |
|---|---|
| Normal | 0 |
| Increased interstitial markings | 1 |
| Ground glass opacification | 2 |
| Consolidation | 3 |
Fig. 4.
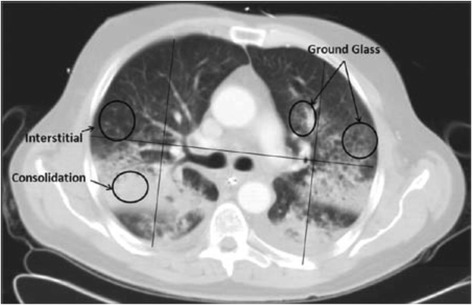
Example of radiologist’s score findings in chest computed tomography scan slice [34]
Second, Yamamura et al. [35] used CT imaging to measure bronchial wall thickness 2 cm distal to the tracheal bifurcation in patients who had sustained an inhalation injury. The authors noted a statistically significant correlation between bronchial wall thickness and the development of pneumonia, total number of ventilator days, and ICU length of stay. They also found that a bronchial wall thickness value of >3.0 mm predicted the development of pneumonia with a sensitivity of 79 % and specificity of 96 %. Interestingly, this study was not able to replicate the association between Abbreviated Injury Score bronchoscopic scoring and clinical outcomes as described above [35].
A third approach to using the CT scan is virtual bronchoscopy. A three-dimensional reconstructed image is presented in such a way that the viewer navigates through the lung as if using a bronchoscope. We found that virtual bronchoscopy agrees best with FOB in the detection of airway narrowing, and less so in the detection of blistering or necrosis [36].
Problems with using chest CT as part of a diagnostic algorithm for inhalation injury include determining the optimal timing of the test and how to interpret abnormal radiographic findings in the setting of a negative bronchoscopy. Putman et al. [37] found that chest radiography on admission was rarely helpful in determining the presence or severity of inhalation injury, but its use is helpful as a baseline for determining future changes.
Respiratory support
Given the limited availability of targeted therapies for inhalation injury, one of the fundamental tenets is supportive respiratory care. This includes aggressive pulmonary toilet and mechanical ventilation when indicated. It should be noted that approximately 20 to 33 % of patients hospitalized with inhalation injury experience some degree of upper airway obstruction due to pharyngeal edema that can progress rapidly [38]. As thermal injury increases airway edema and can lead to airway obstruction, early intubation is favored [4]. This is of particular concern in patients who receive large amounts of intravenous fluids during resuscitation. Generally speaking, the most experienced clinician in airway management should perform endotracheal intubation with the largest available, age-appropriate endotracheal tube for patients with suspected or impending upper airway obstruction in the setting of inhalation injury. One study suggests prophylactic intubation can decrease mortality related to pulmonary-related death in patients with inhalation injury [39].
Maintaining bronchial hygiene is paramount in patients who have suffered inhalation injury. Early ambulation, chest physiotherapy, airway suctioning, and therapeutic bronchoscopy are adjunctive tools [38]. Reper et al. [40] demonstrated that intrapulmonary percussive ventilation administered through a face mask to spontaneously breathing patients with smoke inhalation injury, hypoxia, and persistent atelectasis can result in a significant improvement in PaO2/FiO2 ratio.
A low threshold should be maintained for intubation and mechanical ventilation in inhalation injury due to the progressive nature of the airway edema. Interestingly, a study by Mackie et al. [41] showed an increased use of mechanical ventilation in patients at a Dutch burn center from 1997 to 2006 (76 %) compared with 1987 to 1996 (38 %) despite a decrease in the incidence of inhalation injury (34 % versus 27 %). The authors hypothesized that this was related to the institution of Advanced Trauma Life Support principles in the mid-1990s in the Netherlands. Mackie [42] also suggested mechanical ventilation may be a significant contributor to mortality in burn patients independent of inhalation injury. He proposed increased intrathoracic pressure from positive pressure ventilation led to decreased venous return, followed by decreased cardiac and urine output. The typical clinical reaction is to increase intravenous fluid administration, resulting in higher volumes of infused fluid, a known risk factor for adverse outcomes in burn patients [42].
Multiple challenges, to include concern for ventilator-induced lung injury in patients with inhalation injury, have led to the use of unconventional ventilator modes [43].
Conventional mechanical ventilation is limited in the patient with inhalation injury. In a patient with fibrin casts, extensive chest wall thermal injuries, or high volumes of resuscitative fluid maintaining the recommended tidal volumes of less than 7 ml/kg body weight and plateau pressures of less than 30 cm water [44], can prove difficult with conventional techniques. Therefore, in order to apply lung-protective ventilation in patients with inhalation injury, nonconventional ventilator modes are employed.
High-frequency percussive ventilation (HFPV) was first described in patients with inhalation injury as a means of assisting with clearance of sloughed respiratory mucosa and plugs, as well as decreasing iatrogenic barotrauma and the incidence of pulmonary infection [45]. Further studies have demonstrated benefits from using HFPV prophylactically (that is, not as a salvage mode) in both adult [46, 47] and pediatric populations [48]. Our group performed a randomized controlled trial to compare HFPV versus conventional low tidal volume (LTV) ventilation [49]. While we detected no difference in ventilator-free days between patients randomized to HFPV compared with LTV ventilation, there was a statistically significant increase in the PaO2/FiO2 ratio for the HFPV cohort on days 0 to 3 (Fig. 5). We also found that less patients in the HFPV cohort required conversion to a rescue mode of ventilation compared with the LTV ventilation cohort [49]. Also, HFPV was associated with a decrease in the incidence of pneumonia from 45 to 26 % (P < 0.005) and resulted in an improvement in survival [46]. Although HFPV cannot reverse the effects of inhalation injury, it can improve the clearance of secretions, provide positive pressure throughout the ventilator cycle, allow for lower airway pressures, and increase functional reserve capacity [46].
Fig. 5.
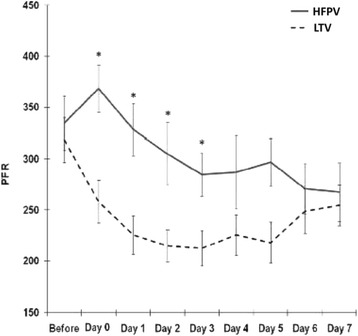
Comparison of the PaO2/FiO2 ratio over time between high frequency percussive ventilation (HFPV) and low-tidal volume ventilation (LTV) (asterisks denote P < 0.05) [49]
Interestingly, a recent retrospective study by Sousse et al. [50] compared high tidal volume (HTV) ventilation (15 ± 3 ml/kg, n = 190) with LTV ventilation (9 ± 3 ml/kg, n = 501) in pediatric patients suffering from inhalation injury. Patients on HTV had fewer days on the ventilator (P < 0.005), increased maximum peak inspiratory pressure (P < 0.02), and plateau pressures (P < 0.02) compared with those on LTV ventilation. Furthermore, the incidence of atelectasis and ARDs was significantly lower in the cohort receiving HTV ventilation (P < 0.0001 and P < 0.02, respectively). However, the HTV ventilation group were not without complications and had a significantly higher rate of pneumothorax compared with the LTV ventilation group (P < 0.03). For pediatric patients suffering from inhalation injury, HTV ventilation may be better than traditional LTV ventilation [50]. The mechanisms for this observation are unclear, but this study, much like the previous study, suggests that we must be cautious when extrapolating LTV ventilation to all patient populations, especially in those with different pathophysiologies. A randomized controlled trial may be necessary to tease out the true impact of these divergent strategies. Our group also looked at airway pressure release ventilation (APRV) in a prospective animal model study. We found that PaO2/FiO2 ratios were initially lower in pigs with inhalation injury on APRV compared with conventional mechanical ventilation, although this equilibrated at 48 h. Higher mean airway pressures were necessary to maintain oxygenation in APRV, and, in the end, no survival difference was seen between APRV and conventional mechanical ventilation [51].
Other nonventilator adjuncts to consider for inhalation injury include prone positioning and extracorporeal membrane oxygenation (ECMO). Our group showed that prone positioning led to a statistically significant increase in PaO2/FiO2 ratio in patients with inhalation injury and refractory ARDS whose initial ratio was an average of 87 ± 38 [52].
A systematic review and meta-analysis on the use of ECMO in inhalation injury was limited by number of studies and total patients available. There was a tendency towards increased survival in burn patients with acute hypoxemic respiratory failure treated with ECMO. ECMO use of <200 h was correlated with higher survival compared with time >200 h. There was no improvement in survival if ECMO was initiated once the PaO2/FiO2 ratio was <60 [53].
Targeted therapies
Bronchodilators
Bronchodilators have been used in inhalation injury to decrease airflow resistance and improve dynamic compliance. β2-adrenergic agonists such as albuterol and salbutamol have been studied in both sheep and humans. Ovine studies of smoke inhalation injury have shown that both nebulized epinephrine and albuterol decrease airway pressure by smooth muscle relaxation and increase PaO2/FiO2 ratio [54, 55] by limiting the degree of bronchospasm. In addition, epinephrine decreases blood flow to injured/obstructed airways, thus improving V/Q matching.
Muscarinic receptor antagonists such as tiotropium have been studied as well. The parasympathetic response, mediated via muscarinic receptors in the lung, causes smooth muscle constriction within the airways, release of cytokines, and stimulation of submucosal glands [2]. Therefore, by inhibiting these effects, airway pressures are decreased and mucus secretion and cytokine expression are reduced [56]. Jonkam et al. demonstrated in an ovine model that tiotropium improved PaO2/FiO2 ratios and decreased peak airway pressures in the first 24 h following inhalation injury [56].
There is also evidence that both beta agonists and muscarinic receptor antagonists may decrease the host inflammatory response. Additionally, both muscarinic and adrenergic receptors are found on respiratory epithelial gland cells and may impact regeneration and healing following injury. Jacob et al. [57] showed in an ovine model that albuterol/tiotropium resulted in an increase in bronchial ciliated duct and submucosal gland cell proliferation following smoke inhalation and burn injury. In healthy human volunteer and animal studies, epinephrine has been shown to decrease tumor necrosis factor-alpha levels and potentiate IL-10 (a cytokine inhibitor) after lipopolysaccharide stimulation [58, 59].
Mucolytic agents
N-acetylcysteine (NAC) is a powerful mucolytic and may have a role in mitigating ROS damage as it is a precursor of glutathione and a strong reducing agent. While it may aid in breaking up thick airway secretions, it is also an airway irritant and may produce bronchoconstriction; therefore, patients are frequently pre-dosed with a bronchodilating agent [38]. NAC has been proven to be effective in combination with aerosolized heparin for the treatment of inhalation injury in animal studies [60].
Anticoagulants
Inhaled anticoagulants have been used to ameliorate the formation of fibrin casts, which contribute to airway obstruction following inhalation injury. This became a prevalent treatment after Desai et al. [61] demonstrated its utility in a pediatric inhalation injury population. However, in a subsequent retrospective review by Holt et al. [62], a cohort of 150 patients with inhalation injury showed no significant improvement in clinical outcomes in patients treated with inhaled heparin and acetylcysteine. This retrospective study allowed for the institution of nebulized heparin every 4 h for up to 7 days at the attending physician's discretion, and it is unclear whether significant selection bias impacted results [62]. There has been at least one case report of coagulopathy in a patient receiving nebulized heparin and acetylcysteine for inhalation injury [63]. However, Yip et al. [64] demonstrated that nebulized heparin does not increase the risk for pulmonary or systemic bleeding.
Miller et al. [65] found in a retrospective study that patients with inhalation injury who received nebulized heparin and NAC in addition to albuterol experienced a survival benefit with a number needed to treat of 2.73. A multi-center randomized controlled trial by Glas et al. [66] is currently underway to assess nebulized heparin versus placebo in inhalation injury.
Enkhbaatar et al. [67] used a combination of aerosolized heparin and recombinant human antithrombin in an ovine model of cutaneous burn and smoke inhalation. They found the two agents resulted in better lung compliance, less pulmonary edema, and less airway obstruction than controls. Interestingly, neither agent used alone had the same effect [67]. Using this same injury model, they demonstrated that a fibrinolytic agent, tissue plasminogen activator, decreased pulmonary edema, airway obstruction and airway pressures and improved gas exchange [68].
A systematic review of inhaled anticoagulants, including heparin, heparinoids, antithrombin, and fibrinolytics, in inhalation injury confirmed improved survival and decreased morbidity in preclinical and clinical studies [69]. Additionally, anticoagulants may have a systemic role in mitigating host inflammatory response. Combined burn and smoke inhalation injury is associated with myocardial impairment similar to septic cardiomyopathy. Rehberg et al. [70] demonstrated a decrease in the inflammatory changes underlying myocardial dysfunction and improvement in contractility in an ovine model following administration of recombinant human antithrombin.
Anti-inflammatory agents
More specific therapies to mitigate the host inflammatory response and the positive feedback loop introduced through neutrophil migration into the airway and production of ROS and peroxynitrite (ONOO−) is an area of intense interest [71]. Neutralization of peroxynitrite with peroxynitrite decomposition catalysts has been demonstrated to be cytoprotective and provide beneficial effects in an ovine model of smoke inhalation injury. Hamahata et al. [72] demonstrated that peroxynitrite decomposition catalyst delivery into the bronchial artery of sheep subjected to burns and inhalation injury attenuated pulmonary damage when compared with a control group that received saline. Additionally, in an animal and human in vitro model of smoke inhalation injury, Perng et al. [25] demonstrated that NOS-mediated activation of the host inflammatory response was attenuated by inhibiting a specific signaling pathway (adenosine-monophosphate-activated protein kinase).
Systemic toxicities
CO has an affinity for hemoglobin 200 to 250 times greater than oxygen and exposure results in hypoxia and ischemia. Unlike inhalation injury, CO has deleterious effects at the level of hemoglobin and more specifically the ability for oxygen delivery. CO acts to displace oxygen from hemoglobin (forming carboxyhemoglobin (COHb)) and binds to cytochrome c oxidase. COHb shifts the oxygen dissociation curve to the left, ultimately leading to decreased oxygen delivery at the tissue level and interfering with cellular respiration at the mitochondrial level [73]. Symptoms of CO toxicity include confusion, stupor, coma, seizures, and myocardial infarction [74]. CO diagnosis requires the use of a Co-oximeter (not available in every blood gas lab), since elevated COHb levels may be present despite normal PaO2 and oxygen saturation readings. Available since 2005, newer, non-invasive CO-oximetry monitors permit more rapid diagnosis [75]. CO poisoning is associated with an increased risk of mortality even at long-term follow-up (median of 7.6 years) [74]. Complications of CO poisoning include persistent and delayed neurologic sequelae and myocardial injury, as well as functional effects on leukocytes, platelets, and vascular endothelium [73]. Treatment of CO poisoning involves providing 100 % oxygen, which shortens the half-life of COHb to about 45 min.
Hyperbaric oxygen therapy (HBO) has been used to treat CO poisoning and can further reduce the COHb half-life to about 20 min. The benefits of HBO were evident in a study of 75 patients with acute CO poisoning. Three treatment sessions were administered within 24 h and neuropsychological tests were administered at various points throughout the study. They found the rates of cognitive sequelae were reduced at 6 weeks and 12 months after CO poisoning in patients with three HBO sessions [76]. It should be pointed out that the theoretical basis for HBO up to 24 h after exposure is to facilitate the clearance of CO from cytochrome c oxidase in the brain, rather than to increase its clearance from hemoglobin in the blood. Logistical factors have limited the utilization of HBO. A systematic review found that not enough evidence exists at this point to determine definitively whether HBO reduces adverse neurologic outcomes after CO poisoning [77]. Continued advancements in HBO technology combined with increased ICU accessibility will likely result in the generation of more clinical studies in this area.
The gaseous form of cyanide, hydrogen cyanide (HCN), is formed in fire atmospheres from the thermal decomposition of nitrogen-containing polymers, both natural (wool, silk, and paper), and synthetic (nylon and polyvinyl chloride). The significance of HCN in fire environments is unclear [11]. The lack of a rapid and reliable test to detect cyanide poisoning limits our understanding of the role of HCN in inhalation injury [78]. Additionally, symptoms can mimic CO poisoning. Dumestre et al. [79] found that most burn centers do not test for cyanide poisoning on admission and do not administer an antidote on the basis of clinical suspicion alone. Lactate has been suggested as a marker for severity of cyanide poisoning without other comorbidities [80], but its role in inhalation injury is less clear in a population at risk for CO poisoning and with coexisting hypovolemic shock. Hydroxocobalamin, the most commonly available antidote (sold as Cyanokit®), binds to HCN to form cyanocobalamin, which is nontoxic and excreted in the urine. The standard dose of 5 g is infused intravenously over 15 min. A second dose of 5 g can be administered in patients with severe toxicity or poor clinical response. It is generally regarded as safe. Red discoloration of the skin and urine is common, which may interfere with colorimetric assays.
Sodium nitrite (300 mg) and sodium thiosulfate (12.5 g) are also commercially available (sold as Nithiodote™). Prior to 2007, when hydroxocobalamin became available, sodium nitrite and sodium thiosulfate were used primarily for cyanide poisoning despite limited data as to their efficacy. In 2012, Bebarta et al. [81] evaluated sodium thiosulfate versus hydroxocobalamin in a swine model of severe cyanide poisoning. They found that sodium thiosulfate failed to reverse cyanide-induced cardiovascular collapse. Further, sodium thiosulfate was not found to be effective when added to hydroxocobalamin. Hydroxocobalamin alone was found to be effective for severe cyanide toxicity. Treatment with nitrites carries significant risk of hypotension and methemoglobinemia, which can further jeopardize tissue oxygen delivery.
Complications from inhalation injuryInhalation injury can be divided into anatomic levels and the mechanism of injury - direct thermal injury to the upper airways or chemical injury to the subglottic region and tracheobronchial tree [82–84]. The associated complications vary with the level of injury and are also effected by intubation, infection, and chronic inflammation [82]. In addition, the complications from injury may be acute or delayed. Pneumonia and airway obstruction are early complications of inhalation injury and have been well described in the literature [4, 8]. However, there is a paucity of data on the long-term or delayed complications from inhalation injury [84].
Pneumonia
The most common complication following inhalation injury is respiratory tract infection [85]. Thermal injury activates the host inflammatory response which, when coupled with direct pulmonary injury, places the respiratory system at risk for infection. There is also evidence inhalation injury damages ciliated cells and causes them to detach from the airway epithelium [86]. Coupled with the exfoliation of airway epithelium by chemical irritation, the loss of ciliated cells impairs pulmonary immune function [85, 86]. Surfactant production is also impaired [87] as is mucociliary transport secondary to damage to airway epithelium [88]. The development of respiratory tract infection is also effected by decreased function of pulmonary macrophages [89]. Once the diagnosis of pneumonia is made, empiric antibiotics should be immediately administered. The antibiotic regimen should then be tailored based on the final sputum culture.
At this institution, 1,058 burn patients were evaluated with 35 % diagnosed with inhalation injury via bronchoscopy or 133Xenon lung scan [8]. Of these patients, 38 % developed pneumonia compared with 8.8 % in those without inhalation injury. These authors reported an estimated 20 % increase in mortality with burns and concomitant inhalation injury; mortality increased to 60 % with the development of pneumonia. They found inhalation injury and pneumonia to be independent risk factors for mortality [8].
Airway obstruction
With direct injury to airway epithelium and fluid shifts, upper airway obstruction and pulmonary edema can occur [83]. Airway obstruction is further exacerbated by large fluid resuscitations and should be avoided [83]. Approximately one-third to one-fifth of patients with inhalation injury suffer from acute airway obstruction due to injury to supraglottic structures [38]. These patients require a secure airway either by intubation or tracheostomy [90]. Musosal edema usually peaks around 24 h post-burn and slowly improves over the following several days [90, 91].
From intubation, certain acute complications are known - barotrauma and suction-related injuries - which place the patient more at risk for hospital-acquired pneumonia [90]. Delayed consequences of intubation include tracheomalacia, subglottic stenosis, or innominate fistula [38, 43, 90, 92]. Complications associated with tracheostomies include bleeding, tube malposition, tracheal ulcerations, and tracheitis [91].
Subglottic stenosis and other complications
Direct thermal injury below the vocal cords is unusual given the heat dissipation that occurs in the upper airways [93]. It is the particulate matter from the smoke inhalation and inhalation of steam that contributes significantly to the inflammatory cascade below the larynx [84, 91, 93, 94] and the formation of scar tissue or polyps.
Endobronchial polyps have been reported as both acute and delayed consequences of inhalation injury [84]. The etiology of polyps has been attributed to the epithelialization and fibrous replacement of granulation tissue after damage to the mucosal surfaces. Prevalence is unknown and development can be acute or delayed [84].
A retrospective review by Yang et al. [95] evaluated the incidence of tracheal stenosis in 1,878 burn patients. They found 0.36 % (seven patients) developed tracheal stenosis with five of them having FOB-confirmed inhalation injury (5.5 %). The average time to development was 7 months post-burn. Six patients required intubation for either respiratory distress or prophylaxis. They found prolonged intubation, the presence of inhalation injury, repeated intubates, and neck scar contractures impacted the development of tracheal stenosis [95]. Given the delayed development of stenosis, patients are at risk even after discharge and the true rate is unknown as patients can be symptom free. Other studies report a higher rate of tracheal stenosis in patients with inhalation injury (24 % [96] and 53 % [97]), and predominates in those who underwent intubation [98].
Other complications associated with inhalation injury are bronchiectasis [82, 99, 100], bronchiolitis obliterans [100], vocal cord fixation or fusion [82, 101], and dysphonia [102]. In order to identify and monitor the development of these complications, long-term follow-up (pulmonary function testing and FOB) is necessary [82, 98].
Conclusion
Inhalation injury remains a significant cause of morbidity and mortality in thermally injured patients. Treatment of inhalation injury remains largely supportive. Recent research has led to substantial gains in the understanding of the molecular pathophysiology of inhalation injury. These advances as well as preclinical studies on targeted therapies provide hope for reversal of specific mechanisms of morbidity and improvement in outcomes.
Acknowledgments
This research was supported in part by appointment of one of the authors (LCC) to the Knowledge Preservation Program at the US Army Institute of Surgical Research, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the US Department of Energy and the US Army Medical Research and Material Command. The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the Department of the Army, Department of the Air Force, or the Department of Defense.
Abbreviations
- APRV
Airway pressure release ventilation
- ARDS
Acute respiratory distress syndrome
- CO
Carbon monoxide
- COHb
Carboxyhemoglobin
- CT
Computed tomography
- ECMO
Extracorporeal membrane oxygenation
- FiO2
Fraction of inspired oxygen
- FOB
Fiberoptic bronchoscopy
- HBO
Hyperbaric oxygen therapy
- HCN
Hydrogen cyanide
- HFPV
High-frequency percussive ventilation
- HTV
High tidal volume
- IL
Interleukin
- IL-1RA
IL-1 Receptor antagonist
- LTV
Low tidal volume
- NAC
N-acetylcysteine
- NOS
Nitric oxide synthase
- PaO2
Arterial partial pressure of oxygen
- RADS
Radiologist’s score
- ROS
Reactive oxygen species
Footnotes
Competing interests
LCC received funding from Percussionaire, Inc. to speak at the Bird Institute, Sandpoint, ID (2013). The author’s institution (USAISR) has received research support in the form of equipment loans from Maquet Medical Systems, Novalung GmbH, Alung Technologies, Inc. and Oridion Capnography, Inc.
Authors’ contributions
PFW completed the literature search and drafted the manuscript. MFB edited the manuscript and assisted with writing the manuscript. LAW, IRD, JBL, and NLB assisted with editing and manuscript outline. LCC helped edit, give oversight, and write the manuscript. KKC conceived the review, participated in its design and helped to draft the manuscript. All authors read and approved the final manuscript.
References
- 1.Albright JM, Davis CS, Bird MD, Ramirez L, Kim H, Burnham EL, et al. The acute pulmonary inflammatory response to the graded severity of smoke inhalation injury. Crit Care Med. 2012;40:1113–21. doi: 10.1097/CCM.0b013e3182374a67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dries DJ, Endorf FW. Inhalation injury: epidemiology, pathology, treatment strategies. Scand J Trauma Resusc Emerg Med. 2013;21:31. doi: 10.1186/1757-7241-21-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Endorf FW, Gamelli RL. Inhalation injury, pulmonary perturbations, and fluid resuscitation. J Burn Care Res. 2007;28:80–3. doi: 10.1097/BCR.0B013E31802C889F. [DOI] [PubMed] [Google Scholar]
- 4.Palmieri TL. Inhalation injury: research progress and needs. J Burn Care Res. 2007;28:549–54. doi: 10.1097/BCR.0B013E318093DEF0. [DOI] [PubMed] [Google Scholar]
- 5.You K, Yang HT, Kym D, Yoon J, Haejun Y, Cho YS, et al. Inhalation injury in burn patients: establishing the link between diagnosis and prognosis. Burns. 2014;40:1470–5. doi: 10.1016/j.burns.2014.09.015. [DOI] [PubMed] [Google Scholar]
- 6.El-Helbawy RH, Ghareeb FM. Inhalation injury as a prognostic factor for mortality in burn patients. Ann Burns Fire Disasters. 2011;24:82–8. [PMC free article] [PubMed] [Google Scholar]
- 7.Smith DL, Cairns BA, Ramadan F, Dalston JS, Fakhry SM, Rutledge R, et al. Effect of inhalation injury, burn size, and age on mortality: a study of 1447 consecutive burn patients. J Trauma. 1994;37:655–9. doi: 10.1097/00005373-199410000-00021. [DOI] [PubMed] [Google Scholar]
- 8.Shirani KZ, Pruitt BA, Jr, Mason AD., Jr The influence of inhalation injury and pneumonia on burn mortality. Ann Surg. 1987;205:82–7. doi: 10.1097/00000658-198701000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moritz AR, Henriques FC, McLean R. The effects of inhaled heat on the air passages and lungs: an experimental investigation. Am J Pathol. 1945;21:311–31. [PMC free article] [PubMed] [Google Scholar]
- 10.Petajan JH, Voorhees KJ, Packham SC, Baldwin RC, Einhorn IN, Grunnet ML, et al. Extreme toxicity from combustion products of a fire-retarded polyurethane foam. Science. 1975;187:742–4. doi: 10.1126/science.1114319. [DOI] [PubMed] [Google Scholar]
- 11.Gann RG, Averill JD, Butler KM, Jones WW, Mulholland GW, Neviaser JL, et al. International Study of the Sublethal Effects of Fire Smoke on Survivability and Health (SEFS): Phase I Final Report. NIST Technical Note 1439. 2001. http://fire.nist.gov/bfrlpubs/fire01/PDF/f01080.pdf.
- 12.Fontan JJ, Cortright DN, Krause JE, Velloff CR, Karpitskyi VV, Carver TW, Jr, et al. Substance P and neurokinin-1 receptor expression by intrinsic airway neurons in the rat. Am J Physiol Lung Cell Mol Physiol. 2000;278:L344–55. doi: 10.1152/ajplung.2000.278.2.L344. [DOI] [PubMed] [Google Scholar]
- 13.Lange M, Enkhbaatar P, Traber DL, Cox RA, Jacob S, Mathew BP, et al. Role of calcitonin gene-related peptide (CGRP) in ovine burn and smoke inhalation injury. J Appl Physiol. 2009;107:176–84. doi: 10.1152/japplphysiol.00094.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kraneveld AD, Nijkamp FP. Tachykinins and neuro-immune interactions in asthma. Int Immunopharmacol. 2001;1:1629–50. doi: 10.1016/S1567-5769(01)00099-6. [DOI] [PubMed] [Google Scholar]
- 15.Sureshbabu A, Bhandari V. Targeting mitochondrial dysfunction in lung diseases: emphasis on mitophagy. Front Physiol. 2013;4:384. doi: 10.3389/fphys.2013.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Midde KK, Batchinsky AI, Cancio LC, Shetty S, Komissarov AA, Florova G, et al. Wood bark smoke induces lung and pleural plasminogen activator inhibitor 1 and stabilizes its mRNA in porcine lung cells. Shock. 2011;36:128–37. doi: 10.1097/SHK.0b013e31821d60a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murakami K, Traber DL. Pathophysiological basis of smoke inhalation injury. News Physiol Sci. 2003;18:125–9. doi: 10.1152/nips.01427.2002. [DOI] [PubMed] [Google Scholar]
- 18.Morita N, Enkhbaatar P, Maybauer DM, Maybauer MO, Westphal M, Murakami K, et al. Impact of bronchial circulation on bronchial exudates following combined burn and smoke inhalation injury in sheep. Burns. 2011;37:465–73. doi: 10.1016/j.burns.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cancio LC, Batchinsky AI, Dubick MA, Park MS, Black IH, Gomez R, et al. Inhalation injury: pathophysiology and clinical care proceedings of a symposium conducted at the Trauma Institute of San Antonio, San Antonio, TX, USA on 28 March 2006. Burns. 2007;33:681–92. doi: 10.1016/j.burns.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 20.Traber DLHD, Enkhbaatar P, Maybauer MO, Maybauer DM. The pathophysiology of inhalation injury. In: Herndon DN, editor. Total burn care. 2. Philadelphia: Saunders; 2012. pp. 219–28. [Google Scholar]
- 21.Buehner M, Pamplin J, Studer L, Hughes RL, King BT, Graybill JC, et al. Oxalate nephropathy after continuous infusion of high-dose vitamin C as an adjunct to burn resuscitation. J Burn Care Res. 2015 doi: 10.1097/BCR.0000000000000233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis CS, Janus SE, Mosier MJ, Carter SR, Gibbs JT, Ramirez L, et al. Inhalation injury severity and systemic immune perturbations in burned adults. Ann Surg. 2013;257:1137–46. doi: 10.1097/SLA.0b013e318275f424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weyker PD, Webb CA, Kiamanesh D, Flynn BC. Lung ischemia reperfusion injury: a bench-to-bedside review. Semin Cardiothorac Vasc Anesth. 2013;17:28–43. doi: 10.1177/1089253212458329. [DOI] [PubMed] [Google Scholar]
- 24.Park MS, Cancio LC, Jordan BS, Brinkley WW, Rivera VR, Dubick MA. Assessment of oxidative stress in lungs from sheep after inhalation of wood smoke. Toxicology. 2004;195:97–112. doi: 10.1016/j.tox.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 25.Perng DW, Chang TM, Wang JY, Lee CC, Lu SH, Shyue SK, et al. Inflammatory role of AMP-activated protein kinase signaling in an experimental model of toxic smoke inhalation injury. Crit Care Med. 2013;41:120–32. doi: 10.1097/CCM.0b013e318265f653. [DOI] [PubMed] [Google Scholar]
- 26.Espinoza LA, Smulson ME, Chen Z. Prolonged poly(ADP-ribose) polymerase-1 activity regulates JP-8-induced sustained cytokine expression in alveolar macrophages. Free Radic Biol Med. 2007;42:1430–40. doi: 10.1016/j.freeradbiomed.2007.01.043. [DOI] [PubMed] [Google Scholar]
- 27.Lange M, Connelly R, Traber DL, Hamahata A, Nakano Y, Esechie A, et al. Time course of nitric oxide synthases, nitrosative stress, and poly(ADP ribosylation) in an ovine sepsis model. Crit Care. 2010;14:R129. doi: 10.1186/cc9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hassan Z, Wong JK, Bush J, Bayat A, Dunn KW. Assessing the severity of inhalation injuries in adults. Burns. 2010;36:212–6. doi: 10.1016/j.burns.2009.06.205. [DOI] [PubMed] [Google Scholar]
- 29.Park MS, Cancio LC, Batchinsky AI, McCarthy MJ, Jordan BS, Brinkley WW, et al. Assessment of severity of ovine smoke inhalation injury by analysis of computed tomographic scans. J Trauma. 2003;55:417–27. doi: 10.1097/01.TA.0000083609.24440.7F. [DOI] [PubMed] [Google Scholar]
- 30.Woodson LC. Diagnosis and grading of inhalation injury. J Burn Care Res. 2009;30:143–5. doi: 10.1097/BCR.0b013e3181923b71. [DOI] [PubMed] [Google Scholar]
- 31.Mosier MJ, Pham TN, Park DR, Simmons J, Klein MB, Gibran NS. Predictive value of bronchoscopy in assessing the severity of inhalation injury. J Burn Care Res. 2012;33:65–73. doi: 10.1097/BCR.0b013e318234d92f. [DOI] [PubMed] [Google Scholar]
- 32.Ryan CM, Fagan SP, Goverman J, Sheridan RL. Grading inhalation injury by admission bronchoscopy. Crit Care Med. 2012;40:1345–6. doi: 10.1097/CCM.0b013e31823c8b2f. [DOI] [PubMed] [Google Scholar]
- 33.Cancio LC, Galvez E, Jr, Turner CE, Kypreos NG, Parker A, Holcomb JB. Base deficit and alveolar-arterial gradient during resuscitation contribute independently but modestly to the prediction of mortality after burn injury. J Burn Care Res. 2006;27:289–96. doi: 10.1097/01.BCR.0000216457.25875.F4. [DOI] [PubMed] [Google Scholar]
- 34.Oh JS, Chung KK, Allen A, Batchinsky AI, Huzar T, King BT, et al. Admission chest CT complements fiberoptic bronchoscopy in prediction of adverse outcomes in thermally injured patients. J Burn Care Res. 2012;33:532–8. doi: 10.1097/BCR.0b013e318237455f. [DOI] [PubMed] [Google Scholar]
- 35.Yamamura H, Kaga S, Kaneda K, Mizobata Y. Chest computed tomography performed on admission helps predict the severity of smoke-inhalation injury. Crit Care. 2013;17:R95. doi: 10.1186/cc12740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwon HP, Zanders TB, Regn DD, Burkett SE, Ward JA, Nguyen R, et al. Comparison of virtual bronchoscopy to fiber-optic bronchoscopy for assessment of inhalation injury severity. Burns. 2014;40:1308–15. doi: 10.1016/j.burns.2014.06.007. [DOI] [PubMed] [Google Scholar]
- 37.Putman CE, Loke J, Matthay RA, Ravin CE. Radiographic manifestations of acute smoke inhalation. AJR Am J Roentgenol. 1977;129:865–70. doi: 10.2214/ajr.129.5.865. [DOI] [PubMed] [Google Scholar]
- 38.Mlcak RP, Suman OE, Herndon DN. Respiratory management of inhalation injury. Burns. 2007;33:2–13. doi: 10.1016/j.burns.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 39.Venus B, Matsuda T, Copiozo JB, Mathru M. Prophylactic intubation and continuous positive airway pressure in the management of inhalation injury in burn victims. Crit Care Med. 1981;9:519–23. doi: 10.1097/00003246-198107000-00004. [DOI] [PubMed] [Google Scholar]
- 40.Reper P, van Looy K. Chest physiotherapy using intrapulmonary percussive ventilation to treat persistent atelectasis in hypoxic patients after smoke inhalation. Burns. 2013;39:192–3. doi: 10.1016/j.burns.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 41.Mackie DP, van Dehn F, Knape P, Breederveld RS, Boer C. Increase in early mechanical ventilation of burn patients: an effect of current emergency trauma management? J Trauma. 2011;70:611–5. doi: 10.1097/TA.0b013e31821067aa. [DOI] [PubMed] [Google Scholar]
- 42.Mackie DP. Inhalation injury or mechanical ventilation: which is the true killer in burn patients? Burns. 2013;39:1329–30. doi: 10.1016/j.burns.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 43.Fitzpatrick JC, Cioffi WG., Jr Ventilatory support following burns and smoke-inhalation injury. Respir Care Clin N Am. 1997;3:21–49. [PubMed] [Google Scholar]
- 44.Petrucci N, Iacovelli W. Lung protective ventilation strategy for the acute respiratory distress syndrome. Cochrane Database Syst Rev. 2007:Cd003844. doi:10.1002/14651858.CD003844.pub3. [DOI] [PubMed]
- 45.Cioffi WG, Graves TA, McManus WF, Pruitt BA., Jr High-frequency percussive ventilation in patients with inhalation injury. J Trauma. 1989;29:350–4. doi: 10.1097/00005373-198903000-00012. [DOI] [PubMed] [Google Scholar]
- 46.Cioffi WG, Jr, Rue LW, 3rd, Graves TA, McManus WF, Mason AD, Jr, Pruitt BA., Jr Prophylactic use of high-frequency percussive ventilation in patients with inhalation injury. Ann Surg. 1991;213:575–80. doi: 10.1097/00000658-199106000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hall JJ, Hunt JL, Arnoldo BD, Purdue GF. Use of high-frequency percussive ventilation in inhalation injuries. J Burn Care Res. 2007;28:396–400. doi: 10.1097/BCR.0B013E318053D2D6. [DOI] [PubMed] [Google Scholar]
- 48.Cortiella J, Mlcak R, Herndon D. High frequency percussive ventilation in pediatric patients with inhalation injury. J Burn Care Rehabil. 1999;20:232–5. doi: 10.1097/00004630-199905000-00014. [DOI] [PubMed] [Google Scholar]
- 49.Chung KK, Wolf SE, Renz EM, Allan PF, Aden JK, Merrill GA, et al. High-frequency percussive ventilation and low tidal volume ventilation in burns: a randomized controlled trial. Crit Care Med. 2010;38:1970–7. doi: 10.1097/CCM.0b013e3181cd0f87. [DOI] [PubMed] [Google Scholar]
- 50.Sousse LE, Herndon DN, Andersen CR, Ali A, Benjamin NC, Granchi T, et al. High tidal volume decreases adult respiratory distress syndrome, atelectasis, and ventilator days compared with low tidal volume in pediatric burned patients with inhalation injury. J Am Coll Surg. 2015;220:570–8. doi: 10.1016/j.jamcollsurg.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Batchinsky AI, Burkett SE, Zanders TB, Chung KK, Regn DD, Jordan BS, et al. Comparison of airway pressure release ventilation to conventional mechanical ventilation in the early management of smoke inhalation injury in swine. Crit Care Med. 2011;39:2314–21. doi: 10.1097/CCM.0b013e318225b5b3. [DOI] [PubMed] [Google Scholar]
- 52.Hale DF, Cannon JW, Batchinsky AI, Cancio LC, Aden JK, White CE, et al. Prone positioning improves oxygenation in adult burn patients with severe acute respiratory distress syndrome. J Trauma Acute Care Surg. 2012;72:1634–9. doi: 10.1097/TA.0b013e318247cd4f. [DOI] [PubMed] [Google Scholar]
- 53.Asmussen S, Maybauer DM, Fraser JF, Jennings K, George S, Keiralla A, et al. Extracorporeal membrane oxygenation in burn and smoke inhalation injury. Burns. 2013;39:429–35. doi: 10.1016/j.burns.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 54.Lange M, Hamahata A, Traber DL, Cox RA, Kulp GA, Nakano Y, et al. Preclinical evaluation of epinephrine nebulization to reduce airway hyperemia and improve oxygenation after smoke inhalation injury. Crit Care Med. 2011;39:718–24. doi: 10.1097/CCM.0b013e318207ec52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Palmieri TL, Enkhbaatar P, Bayliss R, Traber LD, Cox RA, Hawkins HK, et al. Continuous nebulized albuterol attenuates acute lung injury in an ovine model of combined burn and smoke inhalation. Crit Care Med. 2006;34:1719–24. doi: 10.1097/01.CCM.0000217472.97524.0E. [DOI] [PubMed] [Google Scholar]
- 56.Jonkam C, Zhu Y, Jacob S, Rehberg S, Kraft E, Hamahata A, et al. Muscarinic receptor antagonist therapy improves acute pulmonary dysfunction after smoke inhalation injury in sheep. Crit Care Med. 2010;38:2339–44. doi: 10.1097/CCM.0b013e3181f8557b. [DOI] [PubMed] [Google Scholar]
- 57.Jacob S, Zhu Y, Jonkam C, Asmussen S, Traber L, Herndon DN, et al. Effect of bronchodilators on bronchial gland cell proliferation after inhalation and burn injury in sheep. J Burn Care Res. 2013;34:386–93. doi: 10.1097/BCR.0b013e31826fc51e. [DOI] [PubMed] [Google Scholar]
- 58.van der Poll T, Coyle SM, Barbosa K, Braxton CC, Lowry SF. Epinephrine inhibits tumor necrosis factor-alpha and potentiates interleukin 10 production during human endotoxemia. J Clin Invest. 1996;97:713–9. doi: 10.1172/JCI118469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang H, Kim YK, Govindarajan A, Baba A, Binnie M, Marco Ranieri V, et al. Effect of adrenoreceptors on endotoxin-induced cytokines and lipid peroxidation in lung explants. Am J Respir Crit Care Med. 1999;160:1703–10. doi: 10.1164/ajrccm.160.5.9903068. [DOI] [PubMed] [Google Scholar]
- 60.Brown M, Desai M, Traber LD, Herndon DN, Traber DL. Dimethylsulfoxide with heparin in the treatment of smoke inhalation injury. J Burn Care Rehabil. 1988;9:22–5. doi: 10.1097/00004630-198801000-00007. [DOI] [PubMed] [Google Scholar]
- 61.Desai MH, Mlcak R, Richardson J, Nichols R, Herndon DN. Reduction in mortality in pediatric patients with inhalation injury with aerosolized heparin/N-acetylcystine [correction of acetylcystine] therapy. J Burn Care Rehabil. 1998;19:210–2. doi: 10.1097/00004630-199805000-00004. [DOI] [PubMed] [Google Scholar]
- 62.Holt J, Saffle JR, Morris SE, Cochran A. Use of inhaled heparin/N-acetylcystine in inhalation injury: does it help? J Burn Care Res. 2008;29:192–5. doi: 10.1097/BCR.0b013e31815f596b. [DOI] [PubMed] [Google Scholar]
- 63.Chopra A, Burkey B, Calaman S. A case report of clinically significant coagulopathy associated with aerosolized heparin and acetylcysteine therapy for inhalation injury. Burns. 2011;37:e73–5. doi: 10.1016/j.burns.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 64.Yip LY, Lim YF, Chan HN. Safety and potential anticoagulant effects of nebulised heparin in burns patients with inhalational injury at Singapore General Hospital Burns Centre. Burns. 2011;37:1154–60. doi: 10.1016/j.burns.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 65.Miller AC, Rivero A, Ziad S, Smith DJ, Elamin EM. Influence of nebulized unfractionated heparin and N-acetylcysteine in acute lung injury after smoke inhalation injury. J Burn Care Res. 2009;30:249–56. doi: 10.1097/BCR.0b013e318198a268. [DOI] [PubMed] [Google Scholar]
- 66.Glas GJ, Muller J, Binnekade JM, Cleffken B, Colpaert K, Dixon B, et al. HEPBURN - investigating the efficacy and safety of nebulized heparin versus placebo in burn patients with inhalation trauma: study protocol for a multi-center randomized controlled trial. Trials. 2014;15:91. doi: 10.1186/1745-6215-15-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Enkhbaatar P, Cox RA, Traber LD, Westphal M, Aimalohi E, Morita N, et al. Aerosolized anticoagulants ameliorate acute lung injury in sheep after exposure to burn and smoke inhalation. Crit Care Med. 2007;35:2805–10. doi: 10.1097/01.CCM.0000291647.18329.83. [DOI] [PubMed] [Google Scholar]
- 68.Enkhbaatar P, Murakami K, Cox R, Westphal M, Morita N, Brantley K, et al. Aerosolized tissue plasminogen inhibitor improves pulmonary function in sheep with burn and smoke inhalation. Shock. 2004;22:70–5. doi: 10.1097/01.shk.0000129201.38588.85. [DOI] [PubMed] [Google Scholar]
- 69.Miller AC, Elamin EM, Suffredini AF. Inhaled anticoagulation regimens for the treatment of smoke inhalation-associated acute lung injury: a systematic review. Crit Care Med. 2014;42:413–9. doi: 10.1097/CCM.0b013e3182a645e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rehberg S, Yamamoto Y, Bartha E, Sousse LE, Jonkam C, Zhu Y, et al. Antithrombin attenuates myocardial dysfunction and reverses systemic fluid accumulation following burn and smoke inhalation injury: a randomized, controlled, experimental study. Crit Care. 2013;17:R86. doi: 10.1186/cc12712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lange M, Szabo C, Enkhbaatar P, Connelly R, Horvath E, Hamahata A, et al. Beneficial pulmonary effects of a metalloporphyrinic peroxynitrite decomposition catalyst in burn and smoke inhalation injury. Am J Physiol Lung Cell Mol Physiol. 2011;300:L167–75. doi: 10.1152/ajplung.00277.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hamahata A, Enkhbaatar P, Lange M, Yamaki T, Nakazawa H, Nozaki M, et al. Administration of a peroxynitrite decomposition catalyst into the bronchial artery attenuates pulmonary dysfunction after smoke inhalation and burn injury in sheep. Shock. 2012;38:543–8. doi: 10.1097/SHK.0b013e31826e9c54. [DOI] [PubMed] [Google Scholar]
- 73.Kealey GP. Carbon monoxide toxicity. J Burn Care Res. 2009;30:146–7. doi: 10.1097/BCR.0b013e3181923b81. [DOI] [PubMed] [Google Scholar]
- 74.Henry CR, Satran D, Lindgren B, Adkinson C, Nicholson CI, Henry TD. Myocardial injury and long-term mortality following moderate to severe carbon monoxide poisoning. JAMA. 2006;295:398–402. doi: 10.1001/jama.295.4.398. [DOI] [PubMed] [Google Scholar]
- 75.Hampson NB. Noninvasive pulse CO-oximetry expedites evaluation and management of patients with carbon monoxide poisoning. Am J Emerg Med. 2012;30:2021–4. doi: 10.1016/j.ajem.2012.03.026. [DOI] [PubMed] [Google Scholar]
- 76.Weaver LK. Hyperbaric oxygen therapy for carbon monoxide poisoning. Undersea Hyperb Med. 2014;41:339–54. [PubMed] [Google Scholar]
- 77.Buckley NA, Juurlink DN, Isbister G, Bennett MH, Lavonas EJ. Hyperbaric oxygen for carbon monoxide poisoning. Cochrane Database Syst Rev. 2011:Cd002041. doi:10.1002/14651858.CD002041.pub3. [DOI] [PMC free article] [PubMed]
- 78.Erdman AR. Is hydroxocobalamin safe and effective for smoke inhalation? Searching for guidance in the haze. Ann Emerg Med. 2007;49:814–6. doi: 10.1016/j.annemergmed.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 79.Dumestre D, Nickerson D. Use of cyanide antidotes in burn patients with suspected inhalation injuries in North America: a cross-sectional survey. J Burn Care Res. 2014;35:e112–7. doi: 10.1097/BCR.0b013e31829b3868. [DOI] [PubMed] [Google Scholar]
- 80.Baud FJ, Borron SW, Megarbane B, Trout H, Lapostolle F, Vicaut E, et al. Value of lactic acidosis in the assessment of the severity of acute cyanide poisoning. Crit Care Med. 2002;30:2044–50. doi: 10.1097/00003246-200209000-00015. [DOI] [PubMed] [Google Scholar]
- 81.Bebarta VS, Pitotti RL, Dixon P, Lairet JR, Bush A, Tanen DA. Hydroxocobalamin versus sodium thiosulfate for the treatment of acute cyanide toxicity in a swine (Sus scrofa) model. Ann Emerg Med. 2012;59:532–9. doi: 10.1016/j.annemergmed.2012.01.022. [DOI] [PubMed] [Google Scholar]
- 82.Cancio LC. Airway management and smoke inhalation injury in the burn patient. Clin Plastic Surg. 2009;36:555–67. doi: 10.1016/j.cps.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 83.Moylan JA, Chan CK. Inhalation injury - an increasing problem. Ann Surg. 1978;188:34–7. doi: 10.1097/00000658-197807000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shin B, Kim M, Yoo H, Kim SJ, Lee JE, Jeon K. Tracheobronchial polyps following thermal inhalation injury. Tuberc Respir Dis (Seoul) 2014;76:237–9. doi: 10.4046/trd.2014.76.5.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pruitt BA, Jr, Erickson DR, Morris A. Progressive pulmonary insufficiency and other pulmonary complications of thermal injury. J Trauma. 1975;15:369–79. doi: 10.1097/00005373-197505000-00001. [DOI] [PubMed] [Google Scholar]
- 86.Jacob S, Kraft R, Zhu Y, Jacob RK, Herndon DN, Traber DL, et al. Acute secretory cell toxicity and epithelial exfoliation after smoke inhalation injury in sheep: an electron and light microscopic study. Toxicol Mech Methods. 2010;20:504–9. doi: 10.3109/15376516.2010.511302. [DOI] [PubMed] [Google Scholar]
- 87.Nieman GF, Clark WR, Jr, Wax SD, Webb SR. The effect of smoke inhalation on pulmonary surfactant. Ann Surg. 1980;191:171–81. doi: 10.1097/00000658-198002000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Loke J, Paul E, Virgulto JA, Smith GJ. Rabbit lung after acute smoke inhalation. Cellular responses and scanning electron microscopy. Arch Surg. 1984;119:956–9. doi: 10.1001/archsurg.1984.01390200074017. [DOI] [PubMed] [Google Scholar]
- 89.Sherwin RP, Richters V. Lung capillary permeability. Nitrogen dioxide exposure and leakage of tritiated serum. Arch Internal Med. 1971;128:61–8. doi: 10.1001/archinte.1971.00310190065007. [DOI] [PubMed] [Google Scholar]
- 90.Cochran A. Inhalation injury and endotracheal intubation. J Burn Care Res. 2009;30:190–1. doi: 10.1097/BCR.0b013e3181923eb4. [DOI] [PubMed] [Google Scholar]
- 91.Carrougher GJ. Inhalation injury. AACN Clin Issues Crit Care Nursing. 1993;4:367–77. [PubMed] [Google Scholar]
- 92.Kadilak PR, Vanasse S, Sheridan RL. Favorable short- and long-term outcomes of prolonged translaryngeal intubation in critically ill children. J Burn Care Rehabil. 2004;25:262–5. doi: 10.1097/01.BCR.0000124786.68570.7C. [DOI] [PubMed] [Google Scholar]
- 93.Toon MH, Maybauer MO, Greenwood JE, Maybauer DM, Fraser JF. Management of acute smoke inhalation injury. Crit Care Resusc. 2010;12:53–61. [PubMed] [Google Scholar]
- 94.Fang-Gang N, Yang C, Yu-Xuan Q, Yan-Hua R, Wei-Li D, Cheng W, et al. Laryngeal morphologic changes and epidemiology in patients with inhalation injury: a retrospective study. Burns. 2015 doi: 10.1016/j.burns.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 95.Yang JY, Yang WG, Chang LY, Chuang SS. Symptomatic tracheal stenosis in burns. Burns. 1999;25:72–80. doi: 10.1016/S0305-4179(98)00112-0. [DOI] [PubMed] [Google Scholar]
- 96.Jones WG, Madden M, Finkelstein J, Yurt RW, Goodwin CW. Tracheostomies in burn patients. Ann Surg. 1989;209:471–4. doi: 10.1097/00000658-198904000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lund T, Goodwin CW, McManus WF, Shirani KZ, Stallings RJ, Mason AD, Jr, et al. Upper airway sequelae in burn patients requiring endotracheal intubation or tracheostomy. Ann Surg. 1985;201:374–82. doi: 10.1097/00000658-198503000-00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gaissert HA, Lofgren RH, Grillo HC. Upper airway compromise after inhalation injury. Complex strictures of the larynx and trachea and their management. Ann Surg. 1993;218:672–8. doi: 10.1097/00000658-199321850-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mahut B, Delacourt C, de Blic J, Mani TM, Scheinmann P. Bronchiectasis in a child after acrolein inhalation. Chest. 1993;104:1286–7. doi: 10.1378/chest.104.4.1286. [DOI] [PubMed] [Google Scholar]
- 100.Tasaka S, Kanazawa M, Mori M, Fujishima S, Ishizaka A, Yamasawa F, et al. Long-term course of bronchiectasis and bronchiolitis obliterans as late complication of smoke inhalation. Respiration. 1995;62:40–2. doi: 10.1159/000196386. [DOI] [PubMed] [Google Scholar]
- 101.Cobley TD, Hart WJ, Baldwin DL, Burd DA. Complete fusion of the vocal cords; an unusual case. Burns. 1999;25:361–3. doi: 10.1016/S0305-4179(98)00185-5. [DOI] [PubMed] [Google Scholar]
- 102.Casper JK, Clark WR, Kelley RT, Colton RH. Laryngeal and phonatory status after burn/inhalation injury: a long term follow-up study. J Burn Care Rehabil. 2002;23:235–43. doi: 10.1097/00004630-200207000-00003. [DOI] [PubMed] [Google Scholar]