

Abstract
Glycosaminoglycans (GAGs) have been shown to bind to a wide variety of microbial pathogens, including viruses, bacteria, parasites, and fungi in vitro. GAGs are thought to promote pathogenesis by facilitating pathogen attachment, invasion, or evasion of host defense mechanisms. However, the role of GAGs in infectious disease has not been extensively studied in vivo and therefore their pathophysiological significance and functions are largely unknown. Here we describe methods to directly investigate the role of GAGs in infections in vivo using mouse models of bacterial lung and corneal infection. The overall experimental strategy is to establish the importance and specificity of GAGs, define the essential structural features of GAGs, and identify a biological activity of GAGs that promotes pathogenesis.
Keywords: Heparan sulfate, Chondroitin sulfate, Proteoglycan, Syndecan, Pneumonia, Keratitis, Cathelicidin, Antimicrobial peptide, Host defense
1 Introduction
Despite significant improvements in hygienic conditions and prophylactic and therapeutic interventions, infectious diseases continue to be a major global health problem. Infections killed approximately 9.3 million people worldwide in 2010, accounting for 18 % of all global deaths [1]. Lower respiratory infections, in particular, are associated with high mortality (fourth leading cause of death) and killed approximately 2.8 million people worldwide in 2010 [1]. Several infectious diseases are also associated with significant morbidity. For example, corneal infections afflict approximately 500,000 patients globally [2] and can lead to reduced visual acuity, irreversible scarring, and blindness [3–5]. Furthermore, infections often exacerbate and dysregulate the host’s inflammatory response, resulting in serious acute and chronic inflammatory complications [6–9]. In addition, infection by several pathogens can lead to malignant disease, such as gastric cancer by Helicobacter pylori [10], cervical cancer by human papillomavirus virus (HPV) [11], and liver cancer by hepatitis B and C viruses (HBV and HCV) [12]. A major gap in our scientific knowledge centers on how pathogens interact with host components and modulate or subvert their activities to promote pathogenesis in vivo.
Glycosaminoglycans (GAGs) have been shown to interact with a wide variety of pathogens, including viruses, bacteria, parasites, and fungi [13–15]. GAG–pathogen interactions have been implicated in many steps of pathogenesis, including host cell attachment and invasion, infection of neighboring cells, and dissemination and infection of distant tissues [14, 16]. In cell-based assays, many viruses, including HSV [17], HPV [18], HBV [19], HCV [20], and enterovirus [21], have been shown to bind to cell surface heparan sulfate (HS) and utilize HS as a receptor for their initial attachment to host cells. Several bacteria, such as H. pylori [22], Pseudomonas aeruginosa [23], and Borrelia burgdorferi [24], similarly bind to cell surface HS for their attachment. HS interactions have also been proposed to promote host cell invasion of intracellular pathogens, such as HSV [25], Neisseria gonorrhoeae [26], and Listeria monocytogenes [27], and to facilitate the dissemination of Mycobacterium tuberculosis [28] and replication of Toxoplasma gondii [29]. Furthermore, several bacterial pathogens have been shown to induce the release of dermatan sulfate (DS) from the extracellular matrix (ECM) [30] or HS from the cell surface [31–34] and exploit the ability of solubilized GAGs to counteract cationic antimicrobial factors or neutrophil-mediated host defense mechanisms. In addition, several pathogens have been shown to subvert HS to prevent detection by immune mechanisms [35, 36]. Altogether, these data suggest that GAG–pathogen interactions and the ability of pathogens to subvert GAG functions are important virulence mechanisms for a wide variety of microbes.
GAGs are unbranched polysaccharides composed of repeating disaccharide units. GAGs include HS, heparin, chondroitin sulfate (CS), DS, keratan sulfate (KS), and hyaluronan (HA), each with unique disaccharide units and chemical linkages. Except for HA, all GAGs in vivo are found covalently conjugated to specific core proteins as proteoglycans, and expressed ubiquitously on the cell surface, in the extracellular matrix (ECM), and in intracellular compartments. Biosynthesis of GAGs on proteoglycans is initiated with the assembly of a tetrasaccharide linkage region, which is attached to specific Ser residues in core proteins. An unmodified GAG precursor is polymerized and then extensively modified in the Golgi. For example, in HS biosynthesis, the unmodified HS precursor is sequentially modified by N-deacetylase N-sulfotransferases (NDSTs), C5 epimerase, 2-O-sulfotransferase (2OST), 6OSTs, and 3OSTs [37, 38]. These reactions do not go to completion, resulting in a highly heterogeneous mature HS chain. Importantly, the unique and complex sulfation patterns of GAGs enable them to bind specifically to many biomolecules and regulate diverse biological processes [39–41]. For example, efficient FGF-2 binding by HS requires N-sulfated glucosamine and 2-O-sulfated iduronic acid in a decasaccharide sequence, whereas antithrombin III binding by HS and heparin requires a central trisulfated (N-, 6-O-, and 3-O-sulfates) glucosamine residue in a minimal pentasaccharide sequence [42]. Several GAG–pathogen interactions are also dictated by GAG modifications. For instance, HCV envelope glycoproteins E1 and E2 require both and N- and 6-O-sulfate groups for efficient interaction with HS [43], whereas Chlamydia trachomatis OmcB interacts with 6-O-sulfated HS domains [44]. These observations suggest that microbes subvert specific GAG modifications to promote their pathogenesis, but whether GAG modifications are indeed important in vivo has yet to be determined. In fact, our knowledge of the role of GAGs in infections is mostly derived from cell-based experiments performed in vitro, and their physiological significance, relevance, and function in infectious diseases have yet to be determined.
There are diverse animal models to study the role of GAGs in infections in vivo, but each has its own advantages and disadvantages. Studies using invertebrates, such as Caenorhabditis elegans and Drosophila melanogaster, and lower vertebrates, such as Danio rerio (zebrafish), are simple and cost-effective, and have yielded valuable mechanistic information about host–pathogen interactions and innate immune responses to infections [45–47]. Mutant organisms lacking various GAGs, GAG modification enzymes, and proteoglycans have also been generated, and methods to specifically knockdown the expression of certain genes are established [48–50]. However, the lower organisms lack particular organs (e.g., lungs) and the structure and function of some organs do not closely resemble those of humans. The invertebrates also lack adaptive immunity.
Larger mammalian species, such as rabbits, dogs, and monkeys, have also been used and they too have generated much significant information about pathogenic and host defense mechanisms in vivo. Several drawbacks of these mammalian models include a relative slow rate of reproduction, high cost of maintenance, lack of specific experimental reagents to precisely determine molecular mechanisms, and ethical issues. Rodent models, in particular mouse models, are used frequently because of their small size, relative rapid reproduction cycle, relative cost-effectiveness, ease of handling, and abundant availability of specific experimental tools, including various transgenic mouse lines in which a particular gene is overexpressed or has been ablated globally or in a cell-specific manner. The availability of many inbred mouse strains (e.g., C57BL/6, BALB/c) also allows researchers to study genetically identical cohorts and reduces experimental variability from genetic variations. Furthermore, mice are readily amenable to experimental prophylactic and therapeutic approaches, and their immune system is well characterized. However, mice are not humans, and results from mouse studies should also be interpreted with caution when relating to human diseases. Regardless, for the above reasons, mice are currently the most frequently used animals to study mechanisms of various human diseases in vivo. Here we describe experimental approaches to study the role of GAGs in mouse models of bacterial lung and corneal infections. A method to investigate the role of GAGs in bacterial killing by innate antimicrobial factors is also described. The primary focus is on HS because a large number of microbes have been proposed to subvert HS and HSPGs for their pathogenesis, but the methods described below can be readily adapted to study the role of other GAGs and proteoglycans.
2 Materials
2.1 Intranasal Lung Infection Assay
Mice: Mice are used at the age of 5–10 weeks (see Note 1). Inbred wild-type (Wt) mice are available from several vendors (see Note 2).
Tryptic soy broth (TSB) and tryptic soy agar (TSA): Powder stocks, premade solutions, and premade plates are available from several vendors. When using powder stocks, sterilize resuspended broth by autoclaving.
Bacteria: S. aureus strain 8325-4 (see Note 3). Strain 8325-4 can be stored short term on TSA slants or plates at 4 °C, or long term in 40 % glycerol/TSB at −80 °C.
Inoculation materials: Micropipette and tips are from general supply vendors. Sterilize tips by autoclaving.
Surgical tools: Fine scissors and forceps. Sterilize surgical tools by autoclaving.
Phosphate-buffered saline (PBS), pH 7.4: Premade tablets and concentrated stock solutions are available from several commercial sources. Sterilize PBS by autoclaving or filtering through a sterile 0.22 μm filter.
Tissue straining medium: Dulbecco’s modified Eagle’s medium (DMEM) with 10 % fetal bovine serum (FBS). Do not add antibiotics to the medium (i.e., penicillin and streptomycin).
Cell strainer (70 μm mesh size), plunger from a 5 ml syringe, polystyrene petri dishes (i.e., ones for pouring bacterial plates, not for cell culture), and polypropylene microcentrifuge tubes are from general supply vendors. Sterilize microfuge tubes by autoclaving; others are sold as sterile supplies.
TSB containing 0.1 % (v/v) Triton X-100: Mix autoclaved TSB with Triton X-100 and filter sterilize.
Spectrophotometer: A conventional spectrophotometer and disposable plastic cuvettes are used to measure the turbidity of bacterial suspensions.
2.2 Effect of GAGs and GAG Antagonists in Bacterial Lung and Corneal Infection
GAGs: Purified GAGs are available from several commercial sources (see Note 4). Make concentrated stock solutions in autoclaved deionized water or neutral buffer (e.g., PBS) and store at 4 °C for short-term storage or at −80 °C for long-term storage.
GAG antagonists: Many general inhibitors of GAGs are available from commercial sources. These include the cationic compounds such as protamine and surfen, and polysulfated anionic compounds such as carrageenans and suramin, among others [14, 51]. Make concentrated stock solutions in autoclaved deionized water or neutral buffer and store at 4 °C for short-term storage or at −80 °C for long-term storage.
2.3 Effects of GAG Lyases and GAG Derivatives in Bacterial Lung and Corneal Infection
GAG lyases: Heparinase I, II, and III, chondroitinases A, B, C, AC, and ABC, hyaluronidase, and keratanase are available from commercial sources. Make concentrated stock solutions in autoclaved neutral buffer and store small aliquots at −80 °C. Do not repeat freeze-thaw.
GAG derivatives: Desulfated heparin compounds, oversulfated heparin, heparin oligosaccharides, CS oligosaccharides, oversulfated CS, DS oligosaccharides and oversulfated DS and HA oligosaccharides, among others are available from commercial sources (see Note 5). Make concentrated stock solutions in autoclaved deionized water or neutral buffer and store at 4 °C for short-term storage or at −80 °C for long-term storage.
2.4 Use of Transgenic Knockout (KO) Mice in Intranasal Lung and Corneal Infection
Global KO mouse lines: Several global KO mouse lines lacking genes for specific GAG modification enzymes or proteoglycan core proteins have been published. For HS modification enzymes, mice lacking Ndst2 [52], Ndst3 [53], Hs6st2 [54], or Hs3st1 [55] are viable. Global ablation of other HS modification enzymes results in either embryonic (e.g., Ext1, HS6st1) or perinatal (e.g., Ndst1, Glce, Hs2st) lethality. For HSPGs, global KO mice lacking syndecan-1 (Sdc1) [31, 56], Sdc3 [57], Sdc4 [58], glypican-1 (Gpc1) [59], Gpc3 [60], Gpc4 [61], serglycin (Prg1) [62], or collagen XVIII (Col18a1) [63] are viable. Global ablation of other HSPG core protein genes are either embryonic lethal (e.g., agrin, perlecan) or currently not available (e.g., syndecan-2). Contact the corresponding principal investigator for availability of these mice (see Note 6).
Conditional KO mouse lines: Several conditional KO lines for GAG modification enzymes have been published. For HS, mice harboring a floxed construct of Ext1 [64], Ndst1 [65], Hs2st [66], or Hs6st1 [66] have been generated and ablated in various cell types by crossing with cell-specific Cre reporter lines. These floxed conditional mice can be crossed with other Cre reporter mice to ablate GAG modification enzymes in certain cells or tissues. Contact the corresponding principal investigator for availability of these mice.
2.5 Scarified Corneal Infection
Sterile 29 G syringe needles.
Stereomicroscope.
Fine scissors and forceps. Sterilize by autoclaving.
TSB containing 0.1 % (v/v) Triton X-100. Sterilize by filtering.
Microfuge tubes. Sterilize by autoclaving.
GAGs, GAG antagonists, GAG lyases, and GAG derivatives. Prepare as described in Subheadings 2.2 and 2.3.
2.6 Antimicrobial Peptide Killing Assay
Bacteria: P. aeruginosa strain PAO1. PAO1 can be stored short term in a TSA slant or plate at 4 °C or long term in 40 % glycerol/TSB at −80 °C.
LL-37. Make concentrated stock solutions in deionized water or neutral buffer and store at 4 °C for short-term storage or at −80 °C for long-term storage.
PBS, TSB, TSA plates.
Microcentrifuge tubes. Sterilize by autoclaving.
GAGs and GAG derivatives. Prepare as described in Subheadings 2.2 and 2.3.
3 Methods
We describe here procedures of mouse models of S. aureus lung and corneal infection. These general methods can also be used to study pathogenic mechanisms of other microbial pathogens proposed to subvert GAGs for their pathogenesis. We also describe approaches to adapt these procedures to determine the significance of GAGs and to identify essential GAG modifications, and to characterize how GAGs modulate antimicrobial factors. The role of GAGs in vivo is initially probed by exogenous administration of GAG antagonists or purified GAGs. If GAGs promote infection, addition of GAG antagonists will inhibit pathogenesis and result in reduced tissue bacterial burden and other parameters of infection. General GAG biosynthesis inhibitors (e.g., xyloside, chlorate) have been used in studies in vitro, but these are not recommended for use in vivo because of their strong toxicity. Exogenous administration of excess GAGs should also reduce the tissue bacterial burden by inhibiting bacterial attachment if the GAG under study binds to the pathogen and facilitates attachment. On the other hand, if the GAG under study promotes pathogenesis by inhibiting host defense, then administration of the particular GAG should enhance bacterial virulence by interfering with bacterial eradication. An example of the latter mechanism is shown where addition of HS or heparin, but not CS-A or heparosan, promotes S. aureus corneal infection (Fig. 1b).
Fig. 1.
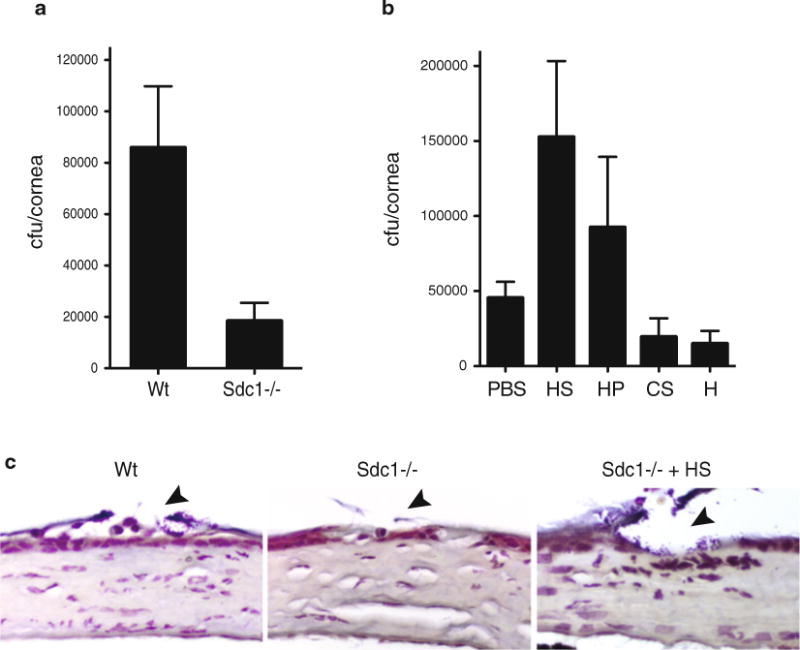
Syndecan-1 promotes S. aureus corneal infection in an HS-dependent manner. (a) Corneas of anesthetized Wt and Sdc1−/− mice on the BALB/c background were scratched with a 29 G needle and infected topically with 1 × 109 cfu of S. aureus strain 8325-4. The corneal bacterial burden was quantified at 10 h postinfection. Data shown are mean ± S.E. (n=9 in Wt and n=6 in Sdc1−/− group). (b) Scarified Wt and Sdc1−/− corneas were infected with 1 × 109 cfu of 8325-4 with or without 200 ng of HS or heparin (HP), or 500 ng of CS-A (CS) or heparosan (H), and the corneal bacterial burden was quantified at 10 h postinfection. Data shown are mean ± S.E. (n=11 in PBS, n=10 in HS, n=7 in HP, n=4 in CS, and n=5 in H group). (c) Paraffin-embedded eye sections of infected Wt and Sdc1−/− mice were Gram stained (arrowhead indicates injured areas). Note the increased number of Gram-positive cocci in Wt cornea infected with S. aureus only and Sdc1−/− cornea co-infected with S. aureus and HS compared to Sdc1−/− cornea infected with S. aureus only
GAG lyases and GAG derivatives are used to determine the essential structural features of GAGs that promote infection. Several GAG lyases selectively digest certain GAGs or regions in GAGs. For example, bacterial heparinase I and III digest sulfated and low sulfated regions of HS, respectively [67], thus allowing determination of whether sulfated or low sulfated HS domains are important in infection. Many selectively modified or size-defined GAG derivatives are also available, and these reagents are used to determine essential GAG modifications and minimum active size of GAGs. For example, if N-sulfate groups of HS/heparin promote infection by inhibiting host defense mechanisms, bacterial virulence will be enhanced upon addition of intact heparin or 2-O- or 6-O-desulfated heparin, but not N-desulfated heparin.
The response of Wt and KO mice lacking genes for certain GAG biosynthesis and modification enzymes, or proteoglycan core proteins is compared to establish the physiological significance and relevance of certain GAGs, GAG modifications, and proteoglycans in infections. An example is shown where Sdc1−/− mice significantly resist S. aureus corneal infection relative to Wt mice (Fig. 1a), indicating that syndecan-1 is an important HSPG that promotes S. aureus pathogenesis in the cornea. Furthermore, S. aureus does not bind to syndecan-1 [34] and addition of exogenous HS or heparin markedly increases bacterial infection in the injured Sdc1−/− cornea (Fig. 1b, c), suggesting that HS chains of syndecan-1 promotes S. aureus corneal infection by interfering with host defense mechanisms.
Lastly, a method to explore the underlying biological mechanisms of how pathogens subvert GAGs is described. The in vivo studies should suggest whether GAGs are promoting infection by serving as an attachment site, facilitating dissemination, or inhibiting host defense mechanisms. Because many studies have examined the role of GAGs as attachment sites for viruses [68–70], bacteria [24, 71, 72], and parasites [73, 74], and microbial binding and attachment assays are established, these will not be discussed in this review. Instead, we describe a method to study whether GAGs interfere with the antibacterial activity of cationic antimicrobial peptides. Several studies suggest that pathogens not only subvert GAGs as attachment sites, but also as soluble effectors that counteract cationic antimicrobial peptides [31, 75–79]. Several potent antimicrobial peptides with broad activity towards many microbes have been identified, including the human cathelicidin LL-37 [80]. Here, we describe a method to measure the ability of HS to specifically inhibit the killing of P. aeruginosa by LL-37 (Fig. 2).
Fig. 2.
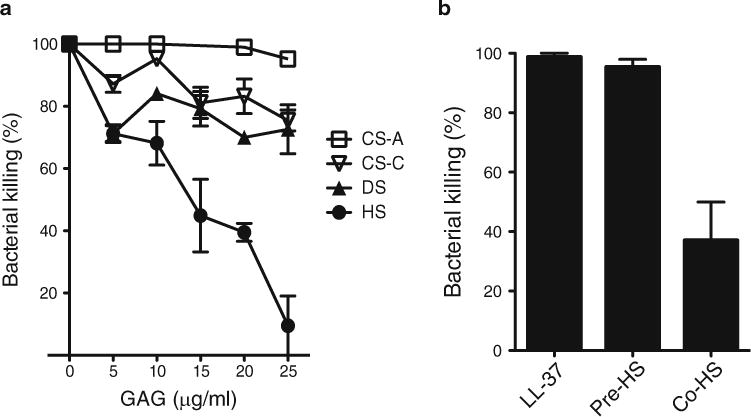
HS specifically inhibits the killing of P. aeruginosa by LL-37. (a) P. aeruginosa (103 cfu) was incubated with LL-37 (3 μg/ml) in 30 μl PBS for 2 h at 37 °C in the absence or presence of increasing doses of CS-A, CS-C, DS, or HS. Bacterial killing was determined by plating out serial dilutions. Data shown are mean ± S.E. (n=4 in each group). Note the significantly increased inhibitory activity of HS at doses ≥15 μg/ml compared to other GAGs. (b) P. aeruginosa was incubated with LL-37 (3 μg/ml) (LL-37 group), preincubated with HS (20 μg/ml) for 30 min, washed free of HS, and then incubated with LL-37 (Pre-HS group), or co-incubated with LL-37 and HS (Co-HS group) for 2 h at 37 °C in a microfuge tube. Bacterial killing was determined by plating out serial dilutions. Data shown are mean ± S.E. (n=4 in each group). This experiment shows that HS does not inhibit LL-37 activity by binding to the bacteria, but rather by directly binding to LL-37 and inhibiting its antibacterial activity
3.1 Intranasal Lung Infection Assay
Preparation of infectious inoculum. Grow 10 μl of S. aureus (strain 8325-4) from the glycerol stock overnight in 5 ml TSB at 37 °C with agitation. The next day, dilute the overnight culture and regrow 3–5 ml of the overnight culture in 30 ml TSB to mid-log growth phase (OD 600 nm: ~0.7) (see Note 7). Estimate the bacterial concentration by turbidity (i.e., based on the predetermined growth curve). Wash sufficient number of bacteria needed for the experiment by centrifuging at 10,000 × g for 5 min, resuspending bacteria in PBS, and centrifuging at 10,000 × g for 5 min. Discard supernatant and resuspend the bacterial pellet in PBS to the desired concentration. Plate out serial dilutions of the inoculum on TSA plates. Count number of colonies on the following day to determine the exact infectious dose. We generally infect with 107–109 cfu of S. aureus in 20 μl PBS per mouse.
Anesthetize mice with a mixture of ketamine (80–100 mg/kg) and xylazine (5–10 mg/kg) via intraperitoneal (i.p.) injection (see Note 8). Pinch a toe and see that there is no withdrawal reaction to confirm that a stable plane of anesthesia has been achieved to minimize variations in the inhalation of the infectious dose.
Slowly infect each nostril with 10 μl of the infectious inoculum using a micropipette (see Note 9). You are infecting too fast if you see expulsion of the inoculum from the nose (i.e., bubbles forming through the nostrils).
At various time points, euthanize mice by anesthesia followed by cervical dislocation. Carefully isolate the whole lung with fine scissors and forceps (see Note 10).
Weigh lungs and place in a 100 mm petri dish with 3 ml of DMEM with 10 % FBS (see Note 11). In the petri dish, strain lungs through a 70 μm filter using a plunger of a 5 ml polypropylene syringe. Wash the strainer once with 1 ml DMEM with 10 % FBS to remove loosely attached strained tissues.
Transfer 1–2 ml of the strained tissue mixture to microcentrifuge tubes and centrifuge at 10,000 × g for 10 min. Discard supernatant and resuspend pellet in 500 μl TSB containing 0.1 % Triton X-100.
Incubate for 30 min at room temperature with vigorous vortexing every 10 min to lyse host cells and to recover both intracellular and extracellular bacteria. S. aureus and most bacteria are not lysed by 0.1 % Triton X-100.
Prepare serial dilutions of the detergent extract in TSB and plate onto TSA plates.
Incubate overnight at 37 °C and count the colonies the following day. Based on the dilutions at steps 6 and 8, back calculate the bacterial burden per mg of lung tissue.
3.2 Effects of GAGs and GAG Antagonists in Intranasal Lung Infection (See Note 12)
To assess the effects of exogenous GAGs, dilute GAGs in PBS to the desired concentration. The dose of GAGs should be chosen based on preliminary titration experiments. For example, in studies examining the effects of heparin on intranasal P. aeruginosa lung infection [31], the dose of 35 ng per mouse was chosen based on preliminary experiments testing the ability of 10–300 ng of heparin to enhance P. aeruginosa lung virulence.
When co-administering GAGs with bacteria, resuspend washed bacteria in step 1 of Subheading 3.1 in the GAG solution. Proceed to steps 2–9 of Subheading 3.1 to determine the effects of exogenous GAGs on the lung bacterial burden. A PBS only control should be included in the assay as well as several other GAGs to serve as specificity controls.
The effects of treating mice with GAGs before or after infection can also be examined using this method. However, because intranasal infection of adult mice must be performed under anesthesia, any treatment before or after infection requires additional anesthesia. In these experiments, a control group treated identically with vehicle should be included.
To assess the effects of GAG antagonists, such as protamine, dilute antagonists in PBS to the desired concentration and resuspend the washed bacteria in step 1 of Subheading 3.1 in the GAG antagonist solution. Proceed to steps 2–9 of Subheading 3.1 to determine the effects of GAG antagonists on the lung bacterial burden (see Note 12).
3.3 Effects of GAGs Lyases and GAG Derivatives in Intranasal Lung Infection (See Note 12)
Dilute GAG lyases in PBS to the desired concentration. The effective dose should be selected based on preliminary titration experiments. In studies examining the effects of heparinase II and chondroitinase ABC on intranasal P. aeruginosa infection, we found that 0.3 mU per mouse of heparinase II and chondroitinase ABC effectively removes HS and CS from the surface of airway epithelial cells. Moreover, we found that heparinase II significantly attenuates bacterial virulence in newborn lungs, whereas chondroitinase ABC had no effect, indicating that HS specifically promotes P. aeruginosa lung infection in newborn mice [31].
Resuspend the washed bacteria in step 1 of Subheading 3.1 in the GAG lyase solution. Proceed to steps 2–9 of Subheading 3.1 to determine the effects of GAG lyases on the lung bacterial burden.
To assess the effects of GAG derivatives, such as desulfated heparin compounds and HS oligosaccharides, dilute the derivatives in PBS to the desired concentration and resuspend the washed bacteria in step 1 of Subheading 3.1 in this solution. Proceed to steps 2–9 of Subheading 3.1 to determine the effects of exogenous GAG derivatives on the lung bacterial burden (see Note 13).
3.4 Use of Transgenic KO Mice in Intranasal Lung Infection
To compare the response of Wt and KO mice lacking certain GAGs, GAG modification enzymes, or proteoglycans to i ntranasal bacterial infection, proceed to steps 1–9 of Subheading 3.1 using the control Wt mice and experimental KO mice (see Note 10 to assess other parameters of infection).
3.5 Scarified Corneal Infection Assay
Prepare mid-log growth phase S. aureus (strain 8325-4) as described in Subheading 3.1, step 1. We generally infect an eye with 107–109 cfu in 5 μl PBS. Plate out serial dilutions of the inoculum on TSA plates. Count number of colonies on the following day to determine the exact infectious dose.
Anesthetize mice as described in Subheading 3.1, step 2.
A single vertical scratch is made with a 29 G needle in one of the corneas without penetrating beyond the superficial stroma under a dissecting microscope (see Note 14). The other eye serves as an uninjured control.
Carefully infect the scarified cornea topically with an inoculum of up to 5 μl using a micropipette. This volume will fill the entire ocular surface, but uninjured regions in the cornea and conjunctiva are not infected even with a high bacterial dose. Avoid volumes larger than 5 μl as they may not be retained by the surface tension of the ocular surface and spill over, resulting in variable results.
Treat mice with an analgesic, such as buprenorphine (0.1 mg/kg, subcutaneously, once). This is required by the IACUC to alleviate pain from the scratch injury.
At various time points, euthanize mice by anesthesia followed by cervical dislocation. Enucleate eyes with fine forceps.
Isolate the cornea from the sclera with fine scissors and forceps under a stereomicroscope (i.e., a basic dissecting microscope) and place the isolated corneas in microfuge tubes with 200 μl TSB containing 0.1 % Triton X-100.
Incubate for 30 min at room temperature with vigorous vortexing every 10 min.
Prepare serial dilutions of the detergent extract and plate onto TSA plates. Incubate overnight at 37 °C and count the colonies the following day to determine the bacterial burden per cornea (see Note 15).
To examine the effects of GAGs, GAG antagonists, GAG derivatives, or GAG lyases, dilute the test reagents in PBS to the desired concentration and resuspend the washed bacteria in this solution (see Note 16). The effective dose of the reagents should be determined in preliminary titration experiments. Proceed to steps 2–9, Subheading 3.5 (above) to determine the effects of the reagents on the corneal bacterial burden. Results from studies examining the effects of HS, heparin, CS-A, and heparosan on S. aureus corneal infection in Sdc1−/− mice are shown (Fig. 1b).
To compare the response of Wt and KO mice lacking certain GAGs, GAG modification enzymes, or proteoglycans, proceed to steps 1–9, Subheading 3.5 (above) using control Wt mice and experimental KO mice. Results from studies comparing the response of Wt and Sdc1−/− mice on the BALB/c background to S. aureus corneal infection are shown (Fig. 1a).
3.6 Antimicrobial Peptide Killing Assay
Grow 10 μl of P. aeruginosa (strain PAO1) from the glycerol stock in 5 ml TSB overnight at 37 °C with agitation. The next day, dilute the overnight culture and regrow 3–5 ml of the overnight culture in 30 ml TSB to mid-log growth phase. Estimate the bacterial concentration by turbidity (i.e., based on the predetermined growth curve), spin down sufficient number of bacteria needed for the assay at 10,000 × g for 5 min, wash bacteria with PBS, and resuspend the washed bacteria to the desired concentration in PBS. Plate out serial dilutions of the resuspended bacteria onto TSA plates and count the number of colonies the following day to determine the exact bacterial concentration.
Dilute LL-37 (see Note 17) to the desired concentration in PBS. LL-37 has been reported to effectively kill P. aeruginosa at the dose range of 0.1–10 μg/ml [81, 82].
Dilute GAGs and GAG derivatives to the desired concentration in PBS.
Incubate bacteria and LL-37 in the absence or presence of GAGs and GAG derivatives for 2 h at 37 °C.
Make serial dilutions and plate out onto TSA plates. Incubate overnight and count the colonies the following day to determine the proportion of bacteria killed by LL-37. HS, but not CS-A, CS-C, or DS, potently inhibits the killing of P. aeruginosa by LL-37 at doses ≥15 μg/ml (Fig. 2a).
Acknowledgments
We would like to thank past and current members of the Park laboratory for developing essential reagents and constantly refining the described procedures. This work was supported by NIH grants R01 EY021765 and R01 HL107472.
Footnotes
All animal experiments must be approved by the local Institutional Animal Care and Use Committee (IACUC) and comply with federal guidelines for research with experimental animals.
C57BL/6 and BALB/c are the most frequently used inbred strains in mouse models of infectious disease. However, there are strain-specific differences in the susceptibility to infection. For example, BALB/c mice are highly susceptible to S. aureus corneal infection, whereas C57BL/6 mice are relatively less susceptible [83, 84]. Another note of caution is that the BL/6 strains from JAX and Charles River have different origins, hence they are genetically different and the designation “BL/6J” is used for Wt mice on the C57BL/6 background from JAX. Also, when comparing the response of Wt and KO mice, one should use Wt littermates obtained from het crosses of the KO line unless the KO line has been backcrossed ≥10 times onto a particular background (i.e., congenic).
We frequently use S. aureus strain 8325-4 for our studies because methods to genetically manipulate this laboratory strain are established. However, other S. aureus strains, including clinical isolates, can also be genetically modified and they are available from ATCC. In fact, it is important to confirm key data with at least two different strains to exclude the possibility of strain-specific effects for any pathogen under study.
The tissue source of GAGs should be considered. For example, most commercial HS is isolated from bovine or porcine tissues. Although we found that porcine HS and heparin potently enhance bacterial infection in mouse corneas (Fig. 1 and [34]) and lung [31], they may not be active in other tissue compartments.
Methods to chemically desulfate heparin are established and can be performed in-house [85–87]. Alternatively, GAGs from mutant CHO cells lacking certain GAGs and GAG modifications [68, 88] can be isolated and tested in the infection assays.
When using global or conditional KO mice to establish the teoglycan, the off-target effects of the mutation must be carefully considered. For example, ablation of Hs2st in endothelial cells increases N- and 6-O-sulfation of endothelial cell HS and results in enhanced neutrophil infiltration [89]. Increased or decreased innate immune responses may have profound effects on the outcome of infection.
The expression level of S. aureus virulence factors is regulated by growth phase [90]. In general, adhesin expression is high during early to mid-log growth, whereas exotoxin expression is high during late-log to stationary growth. The in vivo virulence of each bacterial species and strains at different growth phases should be determined in pilot studies.
An alternate anesthetic to ketamine/xylazine is isoflurane. Isoflurane anesthesia (2–4 %) is given by inhalation, with scavenging by either house vacuum or fume hood. Recovery from isoflurane anesthesia is faster than ketamine/xylazine, thus it is possible to increase the volume of the infectious inoculum up to ~50 μl in the intranasal lung infection assay.
Other routes of lung infection are inhalation, intratracheal, and peroral. Each route has its own advantages and disadvantages. Mice are obligate nose breathers; therefore, intranasal administration under anesthesia leads to lung infection and not gastric infection. However, deposition of bacteria can be variable because the inoculum is given in small volumes to avoid drowning of anesthetized mice. There is less experimental variability in intratracheal administration, but this method is invasive. The inhalation method is not invasive, does not require anesthesia, and uniform dosing can be achieved, but this method requires costly aerosol exposure systems and additional protective measures to protect personnel performing the assay and in the vicinity. The peroral approach mimics oropharyngeal aspiration, which is the route that causes aspiration pneumonia, and the procedure is simple. However, deposition of bacteria is asymmetric and nonuniform. We use the intranasal method because it is simple and cost-effective, and the drawbacks associated with variable dosing can be overcome with experience.
The procedure described in Subheading 3.1 measures the lung bacterial burden, but this method can be easily adapted to assess other key parameters of lung infection. For example, prior to isolation of lung lobes (Subheading 3.1, step 4), lungs can be lavaged with 1–3 ml of PBS to collect bronchoalveolar lavage (BAL) fluids [31]. BAL fluids can be used to assess inflammatory parameters, such as total protein (measure of lung injury, edema) by Bradford or BCA (kits available from Bio-Rad), cytokine levels by ELISA (Biolegend, Peprotech, R&D Systems), and leukocyte infiltration by differentially staining cytospun slides for leukocyte subsets by Giemsa (Fisher). Lungs can also be inflated, fixed, and processed for histopathological analyses [31]. Lung sections can be stained with hematoxylin and eosin to assess inflammation or Gram’s solution to visualize bacteria (see Fig. 1c), or immunostained to examine the expression of specific inflammatory factors (e.g., cytokines) and accumulation of leukocyte subsets (e.g., neutrophils, macrophages, lymphocytes). Lung lobes can also be homogenized to prepare total lung homogenates, which can be used to measure mRNA and protein levels of molecules relevant to infection.
Straining lung tissues in DMEM with 10 % FBS prevents nonspecific adhesion of bacteria to plastic surfaces.
Off-target and adverse effects of test compounds (e.g., GAGs and derivatives, GAG antagonists, GAG lyases) on the bacteria and host should be determined. For adverse effects on the host, parameters such as weight loss, blood leukocyte counts (by CBC analysis), tissue injury (by histopathology or serum chemistry), and BAL total protein should be assessed in mice administered with test compounds only. The effects of test compounds on bacterial growth and viability should also be determined. For example, protamine has antibacterial activity at high concentrations in vitro (≥50 μg/ml) and it can also induce allergic inflammatory responses in vivo [91].
An alternate route to intranasal administration of test compounds is intravenous (i.v.) injection through the tail vein. However, because larger amounts of test compounds are required and i.v. administration is associated with unforeseen systemic effects, we recommend the local, intranasal route for the lung infection assay and local, topical route for the corneal infection assay.
Injury to the corneal epithelium is required to establish infection because intact corneas are highly resistant to infection. The degree of infection can be controlled by the number and size of scratches.
The procedure described in Subheading 3.5 measures the corneal bacterial burden. However, this method can be easily adapted to measure other parameters of corneal infection [34]. For example, ocular surface fluids from infected mice can be collected by consecutively incubating with 5 μl of 1 % N-acetylcysteine in PBS for 5 min (to break mucous layer of tear film) and 5 μl of PBS. The recovered ocular surface fluid can be used to measure levels of inflammatory (e.g., cytokines) and host defense (e.g., antimicrobials) factors. The infected corneas can also be processed for histopathological analyses. Eye sections can be stained with hematoxylin-eosin, Gram’s solution (see Fig. 1c), or immunostained for inflammatory factors or leukocyte subsets. Corneal homogenates can also be used to measure mRNA and protein levels of molecules relevant to infection. However, because of the small size, several corneas will have to be pooled to obtain sufficient amounts for these studies.
Mice can also be treated with GAGs or GAG-related reagents before or after infection. However, pre- or posttreatment requires additional anesthesia to prevent blinking during topical administration of these reagents.
The bacterial killing assay described here is for the human cathelicidin LL-37, but this method can be used to determine the effects of GAGs on other antimicrobial peptides, such as α- and β-defensins and other cathelicidins (CRAMP, PR-39) [31].
References
- 1.Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilhelmus KR. Indecision about corticosteroids for bacterial keratitis: an evidence-based update. Ophthalmology. 2002;109:835–842. doi: 10.1016/s0161-6420(02)00963-6. [DOI] [PubMed] [Google Scholar]
- 3.Bourcier T, Thomas F, Borderie V, Chaumeil C, Laroche L. Bacterial keratitis: predisposing factors, clinical and microbiological review of 300 cases. Br J Ophthalmol. 2003;87:834–838. doi: 10.1136/bjo.87.7.834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Limberg MB. A review of bacterial keratitis and bacterial conjunctivitis. Am J Ophthalmol. 1991;112:2S–9S. [PubMed] [Google Scholar]
- 5.Jett BD, Gilmore MS. Host-parasite interactions in Staphylococcus aureus keratitis. DNA Cell Biol. 2002;21:397–404. doi: 10.1089/10445490260099683. [DOI] [PubMed] [Google Scholar]
- 6.Busse WW, Lemanske RF, Jr, Gern JE. Role of viral respiratory infections in asthma and asthma exacerbations. Lancet. 2010;376:826–834. doi: 10.1016/S0140-6736(10)61380-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abusriwil H, Stockley RA. The interaction of host and pathogen factors in chronic obstructive pulmonary disease exacerbations and their role in tissue damage. Proc Am Thorac Soc. 2007;4:611–617. doi: 10.1513/pats.200706-065TH. [DOI] [PubMed] [Google Scholar]
- 8.Folkesson A, Jelsbak L, Yang L, et al. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat Rev Microbiol. 2012;10:841–851. doi: 10.1038/nrmicro2907. [DOI] [PubMed] [Google Scholar]
- 9.Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369:840–851. doi: 10.1056/NEJMra1208623. [DOI] [PubMed] [Google Scholar]
- 10.Cover TL, Blaser MJ. Helicobacter pylori in health and disease. Gastroenterology. 2009;136:1863–1873. doi: 10.1053/j.gastro.2009.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bzhalava D, Guan P, Franceschi S, Dillner J, Clifford G. A systematic review of the prevalence of mucosal and cutaneous human papillomavirus types. Virology. 2013;445:224–231. doi: 10.1016/j.virol.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 12.Rehermann B. Pathogenesis of chronic viral hepatitis: differential roles of T cells and NK cells. Nat Med. 2013;19:859–868. doi: 10.1038/nm.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rostand KS, Esko JD. Microbial adherence to and invasion through proteoglycans. Infect Immun. 1997;65:1–8. doi: 10.1128/iai.65.1.1-8.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartlett AH, Park PW. Proteoglycans in host-pathogen interactions: molecular mechanisms and therapeutic implications. Expert Rev Mol Med. 2010;12:e5. doi: 10.1017/S1462399409001367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spillmann D. Heparan sulfate: anchor for viral intruders? Biochimie. 2001;83:811–817. doi: 10.1016/s0300-9084(01)01290-1. [DOI] [PubMed] [Google Scholar]
- 16.Teng YH, Aquino RS, Park PW. Molecular functions of syndecan-1 in disease. Matrix Biol. 2012;31:3–16. doi: 10.1016/j.matbio.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shukla D, Liu J, Blaiklock P, et al. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell. 1999;99:13–22. doi: 10.1016/s0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 18.Johnson KM, Kines RC, Roberts JN, Lowy DR, Schiller JT, Day PM. Role of heparan sulfate in attachment to and infection of the murine female genital tract by human papillomavirus. J Virol. 2009;83:2067–2074. doi: 10.1128/JVI.02190-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leistner CM, Gruen-Bernhard S, Glebe D. Role of glycosaminoglycans for binding and infection of hepatitis B virus. Cell Microbiol. 2008;10:122–133. doi: 10.1111/j.1462-5822.2007.01023.x. [DOI] [PubMed] [Google Scholar]
- 20.Shi Q, Jiang J, Luo G. Syndecan-1 serves as the major receptor for attachment of hepatitis C virus to the surfaces of hepatocytes. J Virol. 2013;87:6866–6875. doi: 10.1128/JVI.03475-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan CW, Poh CL, Sam IC, Chan YF. Enterovirus 71 uses cell surface heparan sulfate glycosaminoglycan as an attachment receptor. J Virol. 2013;87:611–620. doi: 10.1128/JVI.02226-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guzman-Murillo MA, Ruiz-Bustos E, Ho B, Ascencio F. Involvement of the heparan sulphate-binding proteins of Helicobacter pylori in its adherence to HeLa S3 and Kato III cell lines. J Med Microbiol. 2001;50:320–329. doi: 10.1099/0022-1317-50-4-320. [DOI] [PubMed] [Google Scholar]
- 23.Bucior I, Mostov K, Engel JN. Pseudomonas aeruginosa-mediated damage requires distinct receptors at the apical and basolateral surfaces of the polarized epithelium. Infect Immun. 2010;78:939–953. doi: 10.1128/IAI.01215-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Isaacs RD. Borrelia burgdorferi bind to epithelial proteoglycan. J Clin Invest. 1994;93:809–819. doi: 10.1172/JCI117035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Donnell CD, Tiwari V, Oh MJ, Shukla D. A role for heparan sulfate 3-O-sulfotransferase isoform 2 in herpes simplex virus type 1 entry and spread. Virology. 2006;346:452–459. doi: 10.1016/j.virol.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 26.Freissler E, Meyer auf der Heyde A, David G, Meyer TF, Dehio C. Syndecan-1 and syndecan-4 can mediate the invasion of OpaHSPG-expressing Neisseria gonorrhoeae into epithelial cells. Cell Microbiol. 2000;2:69–82. doi: 10.1046/j.1462-5822.2000.00036.x. [DOI] [PubMed] [Google Scholar]
- 27.Alvarez-Dominguez C, Vasquez-Boland J, Carrasco-Marin E, Lopez-Mato P, Leyva-Cobian F. Host cell heparan sulfate proteoglycans mediate attachment and entry of Listeria monocytogenes, and the listerial surface protein ActA is involved in heparan sulfate receptor recognition. Infect Immun. 1997;65:78–88. doi: 10.1128/iai.65.1.78-88.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pethe K, Alonso S, Biet F, et al. The heparin-binding haemagglutinin of M. tuberculosis is required for extrapulmonary dissemination. Nature. 2001;412:190–194. doi: 10.1038/35084083. [DOI] [PubMed] [Google Scholar]
- 29.Bishop JR, Crawford BE, Esko JD. Cell surface heparan sulfate promotes replication of Toxoplasma gondii. Infect Immun. 2005;73:5395–5401. doi: 10.1128/IAI.73.9.5395-5401.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmidtchen A, Frick I, Björck L. Dermatan sulfate is released by proteinases of common pathogenic bacteria and inactivates antibacterial alpha-defensin. Mol Microbiol. 2001;39:708–713. doi: 10.1046/j.1365-2958.2001.02251.x. [DOI] [PubMed] [Google Scholar]
- 31.Park PW, Pier GB, Hinkes MT, Bernfield M. Exploitation of syndecan-1 shedding by Pseudomonas aeruginosa enhances virulence. Nature. 2001;411:98–102. doi: 10.1038/35075100. [DOI] [PubMed] [Google Scholar]
- 32.Park PW, Foster TJ, Nishi E, Duncan SJ, Klagsbrun M, Chen Y. Activation of syndecan-1 ectodomain shedding by Staphylococcus aureus alpha-toxin and beta-toxin. J Biol Chem. 2004;279:251–258. doi: 10.1074/jbc.M308537200. [DOI] [PubMed] [Google Scholar]
- 33.Chen Y, Bennett A, Hayashida A, Hollingshead S, Park PW. Streptococcus pneumoniae sheds syndecan-1 ectodomains via ZmpC, a metalloproteinase virulence factor. J Biol Chem. 2005;282:159–167. doi: 10.1074/jbc.M608542200. [DOI] [PubMed] [Google Scholar]
- 34.Hayashida A, Amano S, Park PW. Syndecan-1 promotes Staphylococcus aureus corneal infection by counteracting neutrophil-mediated host defense. J Biol Chem. 2011;285:3288–3297. doi: 10.1074/jbc.M110.185165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dubreuil JD, Giudice GD, Rappuoli R. Helicobacter pylori interactions with host serum and extracellular matrix proteins: potential role in the infectious process. Microbiol Mol Biol Rev. 2002;66:617–629. doi: 10.1128/MMBR.66.4.617-629.2002. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duensing TD, Wing JS, van Putten JPM. Sulfated polysaccharide-directed recruitment of mammalian host proteins: a novel strategy in microbial pathogenesis. Infect Immun. 1999;67:4463–4468. doi: 10.1128/iai.67.9.4463-4468.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- 38.Lindahl U, Kusche-Gullberg M, Kjellén L. Regulated diversity of heparan sulfate. J Biol Chem. 1998;273:24979–24982. doi: 10.1074/jbc.273.39.24979. [DOI] [PubMed] [Google Scholar]
- 39.Perrimon N, Bernfield M. Specificities of heparan sulphate proteoglycans in developmental processes. Nature. 2000;404:725–728. doi: 10.1038/35008000. [DOI] [PubMed] [Google Scholar]
- 40.Funderburgh JL. Keratan sulfate: structure, biosynthesis, and function. Glycobiology. 2000;10:951–958. doi: 10.1093/glycob/10.10.951. [DOI] [PubMed] [Google Scholar]
- 41.Mikami T, Kitagawa H. Biosynthesis and function of chondroitin sulfate. Biochim Biophys Acta. 2013;1830:4719–4733. doi: 10.1016/j.bbagen.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 42.Whitelock JM, Iozzo RV. Heparan sulfate: a complex polymer charged with biological activity. Chem Rev. 2005;105:2745–2764. doi: 10.1021/cr010213m. [DOI] [PubMed] [Google Scholar]
- 43.Kobayashi F, Yamada S, Taguwa S, et al. Specific interaction of the envelope glycoproteins E1 and E2 with liver heparan sulfate involved in the tissue tropismatic infection by hepatitis C virus. Glycoconj J. 2012;29:211–220. doi: 10.1007/s10719-012-9388-z. [DOI] [PubMed] [Google Scholar]
- 44.Fechtner T, Stallmann S, Moelleken K, Meyer KL, Hegemann JH. Characterization of the interaction between the chlamydial adhesin OmcB and the human host cell. J Bacteriol. 2013;195:5323–5333. doi: 10.1128/JB.00780-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Callaghan D, Vergunst A. Non-mammalian animal models to study infectious disease: worms or fly fishing? Curr Opin Microbiol. 2010;13:79–85. doi: 10.1016/j.mib.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 46.Dorer MS, Isberg RR. Non-vertebrate hosts in the analysis of host-pathogen interactions. Microbes Infect. 2006;8:1637–1646. doi: 10.1016/j.micinf.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 47.Ferrandon D, Imler JL, Hetru C, Hoffmann JA. The Drosophila systemic immune response: sensing and signalling during bacterial and fungal infections. Nat Rev Immunol. 2007;7:862–874. doi: 10.1038/nri2194. [DOI] [PubMed] [Google Scholar]
- 48.Lee JS, Chien CB. When sugars guide axons: insights from heparan sulphate proteoglycan mutants. Nat Rev Genet. 2004;5:923–935. doi: 10.1038/nrg1490. [DOI] [PubMed] [Google Scholar]
- 49.Nakato H, Kimata K. Heparan sulfate fine structure and specificity of proteoglycan functions. Biochim Biophys Acta. 2002;1573:312–318. doi: 10.1016/s0304-4165(02)00398-7. [DOI] [PubMed] [Google Scholar]
- 50.Nishihara S. Glycosyltransferases and transporters that contribute to proteoglycan synthesis in Drosophila: Identification and functional analyses using the heritable and inducible RNAi system. Methods Enzymol. 2010;480:323–351. doi: 10.1016/S0076-6879(10)80015-1. [DOI] [PubMed] [Google Scholar]
- 51.Brown JR, Crawford BE, Esko JD. Glycan antagonists and inhibitors: a fount for drug discovery. Crit Rev Biochem Mol Biol. 2007;42:481–515. doi: 10.1080/10409230701751611. [DOI] [PubMed] [Google Scholar]
- 52.Forsberg E, Pejler G, Ringvall M, et al. Abnormal mast cells in mice deficient in a heparin-synthesizing enzyme. Nature. 1999;400:773–776. doi: 10.1038/23488. [DOI] [PubMed] [Google Scholar]
- 53.Pallerla SR, Lawrence R, Lewejohann L, et al. Altered heparan sulfate structure in mice with deleted NDST3 gene function. J Biol Chem. 2008;283:16885–16894. doi: 10.1074/jbc.M709774200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sugaya N, Habuchi H, Nagai N, Ashikari-Hada S, Kimata K. 6-O-sulfation of heparan sulfate differentially regulates various fibroblast growth factor-dependent signalings in culture. J Biol Chem. 2008;283:10366–10376. doi: 10.1074/jbc.M705948200. [DOI] [PubMed] [Google Scholar]
- 55.Shworak NW, HajMohammadi S, de Agostini AI, Rosenberg RD. Mice deficient in heparan sulfate 3-O-sulfotransferase-1: normal hemostasis with unexpected perinatal phenotypes. Glycoconj J. 2002;19:355–361. doi: 10.1023/A:1025377206600. [DOI] [PubMed] [Google Scholar]
- 56.Alexander CM, Reichsman F, Hinkes MT, et al. Syndecan-1 is required for Wnt-1-induced mammary tumorigenesis in mice. Nat Genet. 2000;25:329–332. doi: 10.1038/77108. [DOI] [PubMed] [Google Scholar]
- 57.Reizes O, Lincecum J, Wang Z, et al. Transgenic expression of syndecan-1 uncovers a physiological control of feeding behavior by syndecan-3. Cell. 2001;106:105–116. doi: 10.1016/s0092-8674(01)00415-9. [DOI] [PubMed] [Google Scholar]
- 58.Echtermeyer F, Streit M, Wilcox-Adelman S, et al. Delayed wound repair and impaired angiogenesis in mice lacking syndecan-4. J Clin Invest. 2001;107:R9–R14. doi: 10.1172/JCI10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jen YH, Musacchio M, Lander AD. Glypican-1 controls brain size through regulation of fibroblast growth factor signaling in early neurogenesis. Neural Dev. 2009;4:33. doi: 10.1186/1749-8104-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cano-Gauci DF, Song HH, Yang H, et al. Glypican-3-deficient mice exhibit developmental overgrowth and some of the abnormalities typical of Simpson-Golabi-Behmel syndrome. J Cell Biol. 1999;146:255–264. doi: 10.1083/jcb.146.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Allen NJ, Bennett ML, Foo LC, et al. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature. 2012;486:410–414. doi: 10.1038/nature11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abrink M, Grujic M, Pejler G. Serglycin is essential for maturation of mast cell secretory granule. J Biol Chem. 2004;279:40897–40905. doi: 10.1074/jbc.M405856200. [DOI] [PubMed] [Google Scholar]
- 63.Li Q, Olsen BR. Increased angiogenic response in aortic explants of collagen XVIII/endostatin-null mice. Am J Pathol. 2004;165:415–424. doi: 10.1016/S0002-9440(10)63307-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Inatani M, Irie F, Plump AS, Tessier-Lavigne M, Yamaguchi Y. Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science. 2003;302:1044–1046. doi: 10.1126/science.1090497. [DOI] [PubMed] [Google Scholar]
- 65.Wang L, Fuster M, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol. 2005;6:902–910. doi: 10.1038/ni1233. [DOI] [PubMed] [Google Scholar]
- 66.Stanford KI, Wang L, Castagnola J, et al. Heparan sulfate 2-O-sulfotransferase is required for triglyceride-rich lipoprotein clearance. J Biol Chem. 2010;285:286–294. doi: 10.1074/jbc.M109.063701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu D, Shriver Z, Venkataraman G, El Shabrawi Y, Sasisekharan R. Tumor cell surface heparan sulfate as cryptic promoters or inhibitors of tumor growth and metastasis. Proc Natl Acad Sci U S A. 2002;99:568–573. doi: 10.1073/pnas.012578299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Avirutnan P, Zhang L, Punyadee N, et al. Secreted NS1 of dengue virus attaches to the surface of cells via interactions with heparan sulfate and chondroitin sulfate E. PLoS Pathog. 2007;3:1798–1812. doi: 10.1371/journal.ppat.0030183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schowalter RM, Pastrana DV, Buck CB. Glycosaminoglycans and sialylated glycans sequentially facilitate Merkel cell polyomavirus infectious entry. PLoS Pathog. 2011;7:e1002161. doi: 10.1371/journal.ppat.1002161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hu YP, Lin SY, Huang CY, et al. Synthesis of 3-O-sulfonated heparan sulfate octasaccharides that inhibit the herpes simplex virus type 1 host-cell interaction. Nat Chem. 2011;3:557–563. doi: 10.1038/nchem.1073. [DOI] [PubMed] [Google Scholar]
- 71.Bucior I, Pielage JF, Engel JN. Pseudomonas aeruginosa pili and flagella mediate distinct binding and signaling events at the apical and basolateral surface of airway epithelium. PLoS Pathog. 2012;8:e1002616. doi: 10.1371/journal.ppat.1002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yabushita H, Noguchi Y, Habuchi H, et al. Effects of chemically modified heparin on Chlamydia trachomatis serovar L2 infection of eukaryotic cells in culture. Glycobiology. 2002;12:345–351. doi: 10.1093/glycob/12.5.345. [DOI] [PubMed] [Google Scholar]
- 73.Love DC, Esko JD, Mosser DM. A heparin-binding activity on Leishmania amastigotes which mediates adhesion to cellular proteoglycans. J Cell Biol. 1993;123:759–766. doi: 10.1083/jcb.123.3.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Oliveira FO, Jr, Alves CR, Calvet CM, et al. Trypanosoma cruzi heparin-binding proteins and the nature of the host cell heparan sulfate-binding domain. Microb Pathog. 2008;44:329–338. doi: 10.1016/j.micpath.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 75.Kaneider NC, Djanani A, Wiedermann CJ. Heparan sulfate proteoglycan-involving immunomodulation by cathelicidin antimicrobial peptides LL-37 and PR-39. ScientificWorldJournal. 2007;7:1832–1838. doi: 10.1100/tsw.2007.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baranska-Rybak W, Sonesson A, Nowicki R, Schmidtchen A. Glycosaminoglycans inhibit the antibacterial activity of LL-37 in biological fluids. J Antimicrob Chemother. 2006;57:260–265. doi: 10.1093/jac/dki460. [DOI] [PubMed] [Google Scholar]
- 77.Bergsson G, Reeves EP, McNally P, et al. LL-37 complexation with glycosaminoglycans in cystic fibrosis lungs inhibits antimicrobial activity, which can be restored by hypertonic saline. J Immunol. 2009;183:543–551. doi: 10.4049/jimmunol.0803959. [DOI] [PubMed] [Google Scholar]
- 78.Wu H, Monroe DM, Church FC. Characterization of the glycosaminoglycan-binding region of lactoferrin. Arch Biochem Biophys. 1995;317:85–92. doi: 10.1006/abbi.1995.1139. [DOI] [PubMed] [Google Scholar]
- 79.Zou S, Magura CE, Hurley WL. Heparin-binding properties of lactoferrin and lysozyme. Comp Biochem Physiol B. 1992;103:889–895. doi: 10.1016/0305-0491(92)90210-i. [DOI] [PubMed] [Google Scholar]
- 80.Zanetti M. The role of cathelicidins in the innate host defenses of mammals. Curr Issues Mol Biol. 2005;7:179–196. [PubMed] [Google Scholar]
- 81.Travis SM, Anderson NN, Forsyth WR, et al. Bactericidal activity of mammalian cathelicidin-derived peptides. Infect Immun. 2000;68:2748–2755. doi: 10.1128/iai.68.5.2748-2755.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schmidtchen A, Frick IM, Andersson E, Tapper H, Bjorck L. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol Microbiol. 2002;46:157–168. doi: 10.1046/j.1365-2958.2002.03146.x. [DOI] [PubMed] [Google Scholar]
- 83.Hume EB, Cole N, Khan S, et al. A Staphylococcus aureus mouse keratitis topical infection model: cytokine balance in different strains of mice. Immunol Cell Biol. 2005;83:294–300. doi: 10.1111/j.1440-1711.2005.01326.x. [DOI] [PubMed] [Google Scholar]
- 84.Girgis DO, Sloop GD, Reed JM, O’Callaghan RJ. Susceptibility of aged mice to Staphylococcus aureus keratitis. Curr Eye Res. 2004;29:269–275. doi: 10.1080/02713680490516783. [DOI] [PubMed] [Google Scholar]
- 85.Inoue Y, Nagasawa K. Selective N-desulfation of heparin with dimethyl sulfoxide containing water or methanol. Carbohydr Res. 1976;46:87–95. doi: 10.1016/s0008-6215(00)83533-8. [DOI] [PubMed] [Google Scholar]
- 86.Ishihara M, Kariya Y, Kikuchi H, Minamisawa T, Yoshida K. Importance of 2-O-sulfate groups of uronate residues in heparin for activation of FGF-1 and FGF-2. J Biochem. 1997;121:345–349. doi: 10.1093/oxfordjournals.jbchem.a021593. [DOI] [PubMed] [Google Scholar]
- 87.Kariya Y, Kyogashima M, Suzuki K, et al. Preparation of completely 6-O-desulfated heparin and its ability to enhance activity of basic fibroblast growth factor. J Biol Chem. 2000;275:25949–25958. doi: 10.1074/jbc.M004140200. [DOI] [PubMed] [Google Scholar]
- 88.Zhang L, Lawrence R, Frazier BA, Esko JD. CHO glycosylation mutants: proteoglycans. Methods Enzymol. 2006;416:205–221. doi: 10.1016/S0076-6879(06)16013-9. [DOI] [PubMed] [Google Scholar]
- 89.Axelsson J, Xu D, Kang BN, et al. Inactivation of heparan sulfate 2-O-sulfotransferase accentuates neutrophil infiltration during acute inflammation in mice. Blood. 2012;120:1742–1751. doi: 10.1182/blood-2012-03-417139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Novick RP. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol Microbiol. 2003;48:1429–1449. doi: 10.1046/j.1365-2958.2003.03526.x. [DOI] [PubMed] [Google Scholar]
- 91.Porsche R, Brenner ZR. Allergy to protamine sulfate. Heart Lung. 1999;28:418–428. doi: 10.1016/s0147-9563(99)70031-2. [DOI] [PubMed] [Google Scholar]