

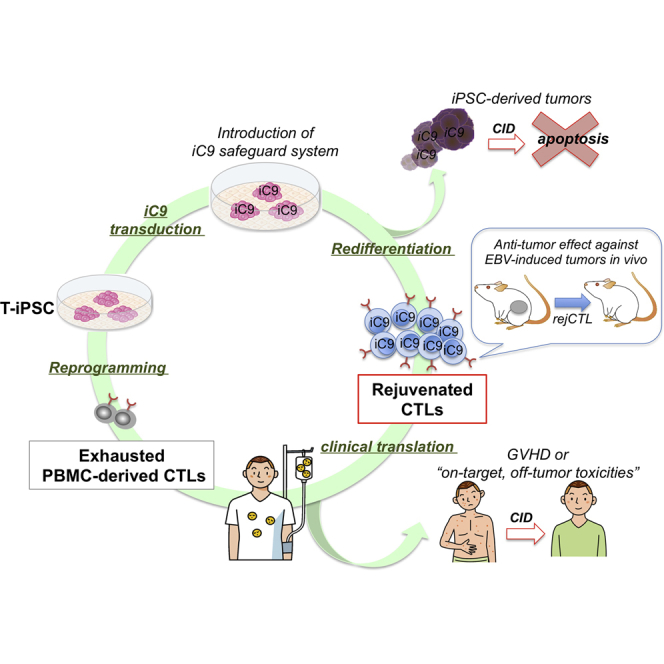
Summary
The discovery of induced pluripotent stem cells (iPSCs) has created promising new avenues for therapies in regenerative medicine. However, the tumorigenic potential of undifferentiated iPSCs is a major safety concern for clinical translation. To address this issue, we demonstrated the efficacy of suicide gene therapy by introducing inducible caspase-9 (iC9) into iPSCs. Activation of iC9 with a specific chemical inducer of dimerization (CID) initiates a caspase cascade that eliminates iPSCs and tumors originated from iPSCs. We introduced this iC9/CID safeguard system into a previously reported iPSC-derived, rejuvenated cytotoxic T lymphocyte (rejCTL) therapy model and confirmed that we can generate rejCTLs from iPSCs expressing high levels of iC9 without disturbing antigen-specific killing activity. iC9-expressing rejCTLs exert antitumor effects in vivo. The system efficiently and safely induces apoptosis in these rejCTLs. These results unite to suggest that the iC9/CID safeguard system is a promising tool for future iPSC-mediated approaches to clinical therapy.
Graphical Abstract
Highlights
-
•
iPSC-derived rejuvenated CTLs are effective against EBV-induced tumors in vivo
-
•
Rejuvenated CTLs are implemented with an inducible caspase-9 (iC9)-based suicide system
-
•
Upon induction, the iC9 system efficiently leads to apoptosis in rejuvenated CTLs
-
•
The iC9-based system provides a safeguard for future iPSC-mediated cell therapy
In this article, Nakauchi and colleagues show that iPSC-derived rejuvenated CTLs (rejCTLs) implemented with an inducible caspase-9 (iC9)-based suicide system are effective against EBV-induced tumors in vivo. Upon induction, the iC9-based system efficiently and safely leads to apoptosis in these rejCTLs in vitro and in vivo and provides a safeguard for future iPSC-mediated cell therapy.
Introduction
Human induced pluripotent stem cells (iPSCs) can unlimitedly self-renew and differentiate into various cell types (Takahashi et al., 2007). Their pluripotency makes iPSCs a promising tool for therapy in a wide range of diseases at present refractory to treatment (Inoue et al., 2014). Recent studies, however, reported the tumorigenic potential of contaminated undifferentiated iPSCs and the malignant transformation of differentiated iPSCs (Lee et al., 2013a, Nori et al., 2015). The tumorigenic risks of iPSCs could be reduced by several strategies, such as sorting out undifferentiated cells with antibodies targeting surface-displayed biomarkers (Tang et al., 2011), killing undifferentiated cells with cytotoxic antibodies (Choo et al., 2008), or elimination of remaining undifferentiated pluripotent cells with chemical inhibitors (Ben-David et al., 2013, Lee et al., 2013b). However, these strategies may not suffice to lower risk to acceptable levels, because the tumorigenic risk of iPSC-based cell therapy arises not just from contamination with undifferentiated iPSCs but also from other unexpected events associated with long-term culture for reprogramming and redifferentiation. There is always a chance of unexpected issues associated with first-in-human clinical studies.
Because suicide systems can be designed not to evoke cross-resistance to conventional agents, they can potentiate therapy—efficiently inducing apoptosis in transduced cells—without increasing toxicity. However, many suicide systems have drawbacks, proving less clinically effective than desired. HSV-TK, the gene encoding herpes simplex virus thymidine kinase, with ganciclovir (GCV), for example, is a well-known suicide gene system used as a safety switch for adoptive T cell therapy or cancer treatment (Ciceri et al., 2005, Ciceri et al., 2007). In fact, recent reports indicate that the HSV-TK/GCV approach effectively removes tumorigenic cells among murine iPSCs (Chen et al., 2013, Lim et al., 2013). This system is nonetheless potentially limited as a safeguard system, because HSV-TK targets DNA synthesis in a cell-cycle-dependent manner and can kill only fast-dividing cells. This may leave populations of slowly dividing cells intact, with resistance to treatment. Cell killing may require many days and is usually incomplete, potentially delaying clinical benefit (Brenner et al., 2013, Hoyos et al., 2012, Leen et al., 2014). Its prodrug, GCV, may cause side effects such as renal dysfunction, liver dysfunction, and pancytopenia. Moreover, because HSV-TK is immunogenic, humoral immunity may reduce its efficacy (Berger et al., 2006, Traversari et al., 2007).
On the other hand, inducible caspase-9 (iC9), encoded by a suicide gene engineered from human caspase-9 (CASP9), is not immunogenic, and can kill transduced cells in a cell-cycle-independent manner. iC9 is a fusion protein engineered by replacing the caspase recruitment domain (CARD) with a mutated FK506-binding protein (FKBP12) to allow conditional dimerization (Straathof et al., 2005b). In the presence of a chemical inducer of dimerization (CID; AP1903 or its functionally identical analog AP20187) (Clackson et al., 1998), dimerized iC9 directly activates intracytoplasmic caspase-3, bypassing activation of the mitochondrial apoptotic pathway, swiftly triggering apoptosis in transduced cells (Di Stasi et al., 2011). In a clinical study, infused iC9-transduced donor T cells underwent rapid apoptosis after CID treatment, and graft-versus-host disease (GVHD) symptoms receded dramatically without relapse of leukemia (Di Stasi et al., 2011, Zhou et al., 2014). iC9 is also a promising tool for cancer treatment, because its mesenchymal stromal cell-based delivery may effectively target non-small-cell lung cancer cells for elimination (Ando et al., 2014). Therefore, we decided to use iC9 to safeguard against problems during clinical and translational investigation of iPSC-based cell therapy.
We succeeded in generating iPSCs from antigen-specific cytotoxic T lymphocytes (CTLs) in HIV1-infected patients (Nishimura et al., 2013). These T cell-derived iPSCs (T-iPSCs) were then redifferentiated into HIV1-specific CTLs. CTLs continuously exposed to viral or tumor antigens, with long-term expansion, may become exhausted (Klebanoff et al., 2006, Wherry, 2011). Whereas fully “rejuvenated” CTLs (rejCTLs) derived from T-iPSCs have higher proliferative capacity, younger memory phenotype, and longer telomeres than the original patient-derived CTLs (Nishimura et al., 2013), the actual in vivo efficacy of rejCTLs has not been tested formally. Moreover, some engineered T cell immunotherapies incurred GVHD after donor lymphocyte infusion, or “on-target, off-tumor toxicities” (Fedorov et al., 2013, Gargett and Brown, 2014) that might also be seen with iPSC-derived T cell therapy. With clinical trials in prospect, we explored the effectiveness and safety of an iC9 safeguard system using these CTLs. We here document that rejCTLs are effective against Epstein-Barr virus (EBV)-induced tumors in vivo and that these rejCTLs expressing iC9 can be eliminated by activation of this system in vivo, showing that the iC9 safeguard system both is practically useful and exhibits enhanced safety in the CTL therapy model. These results may facilitate broad application of this safeguard system in other branches of iPSC-derived regenerative medicine.
Results
Activation of iC9 with CID Effectively Induces Apoptosis in iPSCs
To assess the efficacy of suicide gene therapy based on an iC9 system, we first cloned iC9 and mCherry into a lentiviral vector carrying either Ubc or EF1α promoters (Figure 1A). However, we could not obtain high titers of iC9-containing lentivirus. We hypothesized that the effect of iC9 during lentiviral packaging disturbed 293T cells with high-titer lentivirus production. The caspase inhibitor qVD-Oph completely blocks iC9-induced cell death (Ando et al., 2014). We therefore added qVD-Oph to the medium to protect 293T cells during lentivirus production. This greatly enhanced lentivirus production (up to 3.91 × 102- to 3.35 × 103-fold versus untreated cells; Figure S1A).
Figure 1.

iC9 Activation with CID Induces Robust Apoptosis of iPSCs
(A) Schematic representation of two lentiviral iC9 bicistronic vectors containing Ubc or EF1α promoters. Both vectors contain the suicide gene iC9, cleavable 2A-like sequence, and mCherry as a selectable marker.
(B) T-iPSC lines (EBV-iPSC and HIV1-iPSC) and cell lines TKDA3-4 and TKCBSeV9 were transduced with lentiviral Ubc-iC9. These established four Ubc-iC9-iPSC lines and four nontransduced iPSC lines were treated with CID (80 nM), and apoptosis was measured 24 hr later by flow cytometry for annexin V/7-AAD marking. Data are representative of three independent triplicate experiments. Error bars represent ± SD. ∗∗∗∗p < 0.0001 by two-way ANOVA. ns, not significant.
(C) NT-EBV-iPSCs and iC9-EBV-iPSCs were plated; on the following day, both iPSCs were left untreated or were treated with CID. On days 1, 3, and 7 after CID treatment, iPSCs in each dish were stained with methylene blue. Data are representative of three independent experiments.
See also Figure S1.
We established two T-iPSC lines for adoptive T cell therapy from CTLs specific for either human immunodeficiency virus 1 or Epstein-Barr virus (HIV1-iPS and EBV-iPS) and transduced them with lentivirus-derived iC9. Expression of human pluripotency-associated genes in iC9-iPSCs was comparable to that in non-transduced iPSCs (NT-iPSCs) (Figure S1B). Pluripotency of iC9-iPSCs was confirmed by teratoma formation (Brivanlou et al., 2003). Six weeks after intratestis or subcutaneous injection of iC9-iPSCs into nonobese diabetic severe combined immunodeficient (NOD-Scid) mice, teratomas containing all three germ layers had formed (Figure S1C).
To evaluate the efficacy of this iC9 system in inducing apoptosis, binding of annexin V phycoerythrin (“annexin V”) and uptake of 7-amino-actinomycin D (7-AAD) by iC9-iPSCs were quantified by flow cytometry 24 hr after addition of CID. Over 95% of Ubc-iC9-transduced iPSCs (EBV-iPS: 94.43% ± 3.00%; HIV1-iPS: 95.0% ± 5.54%) and over 75% of EF1α-iC9-transduced iPSCs (EBV-iPS: 77.88% ± 11.17%; HIV1-iPS: 72.19% ± 11.99%) underwent apoptosis; in controls lacking the iC9 system, 10%–15% underwent apoptosis (Figure S1D). Ubc-iC9 induced apoptosis in iPSCs more powerfully than EF1α-iC9 (p = 0.0007, EBV-iPS; p < 0.0001, HIV1-iPS; two-way ANOVA). In addition, after sorting several times for iC9/mCherry expression, silencing of Ubc-iC9 was not so clearly seen as was silencing of EF1α-iC9. We therefore decided mainly to use Ubc-iC9-iPSCs in this study. Because proportional iC9-induced cell death varied among lung cancer cell lines (Ando et al., 2014), we compared apoptosis rates among four iPSC lines transduced with Ubc-iC9. All four lines exhibited equally high sensitivity (>95%) to this iC9/CID system (Figure 1B). Moreover, iC9-EBV-iPSCs lost their ability to form colonies for 7 days after treatment with CID (Figure 1C), and even after 14 days (data not shown). On the other hand, NT-EBV-iPSC colony formation was not affected (Figure 1C). These results suggest that activation of iC9 effectively induces apoptosis in iPSCs in vitro.
The iC9-CID System Can Debulk iPSC-Derived Teratomas
To examine the effects of CID treatment on established tumors, we injected 1.3 × 106 iC9-iPSCs (iC9-EBV-iPS or iC9-HIV1-iPS) or NT-iPSCs (NT-EBV-iPS or NT-HIV1-iPS) into testes of NOD-Scid mice and monitored tumor volume (TV) once a week. All host mice except for two mice (NT-EBV-iPS, n = 8; iC9-EBV-iPS, n = 12; NT-HIV1-iPS, n = 8; iC9-HIV1-iPS, n = 8) developed bilateral intratesticular tumors. On day 29, average TVs were 334.45 ± 122.30 mm3 (NT-EBV-iPS), 483.07 ± 191.88 mm3 (iC9-EBV-iPS), 674.79 ± 394.68 mm3 (NT-HIV1-iPS), and 589.81 ± 263.16 mm3 (iC9-HIV1-iPS). The host mice received intraperitoneally injected CID (Figure 2A, left), 50 μg/mouse, for 3 successive days (day 30 to day 32). TVs were again determined on day 36. Tumors shrank dramatically only in mice carrying iC9-iPSC-derived tumors, and not in mice that received iPSCs without the iC9 system. The tumor size ratio was calculated as (TVday 36 − TVday 0)/(TVday 29 − TVday 0). We could not measure tumor size long term because many mice died, which we ascribe to failure to tolerate invasive weekly operations to permit testis measurement. In addition, histopathologic study required that some mice be sacrificed. However, regrowth of the tumors was not observed in surviving mice on day 45 (NT-EBV-iPS, n = 4; iC9-EBV-iPS, n = 4; NT-HIV1-iPS, n = 4; iC9-HIV1-iPS, n = 7) (Figure 2A, left). The ratio averaged 0.3 in mice injected with iC9-EBV-iPSCs and treated with CID. Because these testes included cystic cavities and because testes themselves grow as their owners grow, tumor size ratios in this group did not reach zero. However, they averaged 1.2 in mice injected with iC9-iPSCs and not treated with CID (p = 0.0005 by two-way ANOVA) and 1.6 in mice injected with NT-iPSCs and treated with CID (p = 0.0001 by two-way ANOVA) (Figure 2A, right). Similar results were also obtained in mice injected with iC9-HIV1-iPSCs (Figure 2A). Histopathologic examination of the tumors revealed a typical pattern of testicular teratoma in the untreated mice (Figure 2B, upper), whereas the testes of mice injected with iC9-iPSCs and treated with CID contained multiple cystic cavities with intramural nuclear debris, residua of apoptosis (Figure 2B, lower). All these findings suggest that the iC9-CID system can drastically reduce iPSC-derived teratomas by inducing apoptosis.
Figure 2.

iC9 Can Debulk Teratomas Originated from iPSCs
(A) NT-iPSCs or iC9-iPSCs were injected into the testes of NOD-Scid mice (day 0). Around day 30, mice were divided into four groups (EBV-iPS: NT without CID [n = 4], NT with CID [n = 3], iC9 without CID [n = 6], and iC9 with CID [n = 6]; HIV1-iPS: NT without CID [n = 4], NT with CID [n = 3], iC9 without CID [n = 4], and iC9 with CID [n = 4]) with or without daily intraperitoneal injections of CID. The average tumor volume in each group from day 0 to day 45 is shown (left). Error bars represent ± SEM. ∗∗p < 0.01 and ∗∗∗p < 0.001 by two-way ANOVA. Using TVs on days 0, 29, and 36, the tumor size ratio was calculated (right). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 by two-way ANOVA.
(B) Tumors formed by iC9-HIV1-iPSCs in testes of male NOD-Scid mice were untreated or treated by CID (left). Representative H&E-stained sections of intratesticular tumors of untreated or CID-treated mice (middle). The scale bars represent 1 mm. A high-magnification view of the testis of a CID-treated mouse (right). Hematoxylinophilic debris marks cell loss ascribed to CID-induced apoptosis. The scale bar represents 100 μm.
High Expression of iC9 Neither Inhibits Redifferentiation into rejCTLs nor Affects Their Antigen Specificity and Killing Function
To address the effectiveness and safety of the iC9 safeguard system in virus-specific rejCTLs, we generated hematopoietic progenitor cells from iC9-iPSCs cultured on C3H10T1/2 feeder cells in the presence of vascular endothelial growth factor (VEGF) (Nishimura et al., 2013, Takayama et al., 2008). On day 14 of culture, iPSC-derived sacs were extracted. They contained many hematopoietic progenitor cell candidates (Figure 3A). On the following day, flow cytometry analysis of these cells confirmed that 85% of CD235a−, CD34+, and CD43+ cells expressed iC9/mCherry (Figure 3A). To induce T cell differentiation, we transferred these cells onto NOTCH ligand-expressing C3H10T1/2 feeder cells (Timmermans et al., 2009) in the presence of Fms-related tyrosine kinase 3 ligand (FLT-3L), stem cell factor (SCF), and interleukin 7 (IL-7) (Nishimura et al., 2013). After 28 days of culture, T lineage cells were harvested, mixed with irradiated peripheral blood mononuclear cells (PBMCs), and stimulated with IL-7, IL-15, and phytohemagglutinin.
Figure 3.

iC9-iPSCs Can Efficiently Differentiate into Virus-Specific CTLs
(A) iC9-HIV1-iPSC sacs on day 14 of culture on C3H10T1/2 feeder cells (upper). The scale bar represents 500 μm. One day after sac extraction, extracted hematopoietic progenitor cells were analyzed by flow cytometry. Flow cytometric analysis of iC9/mCherry expression by CD235a−, CD34+, and CD43+ gated cells (lower).
(B) Flow cytometric analysis of peptide-HLA multimer labeling/CD8 expression by iPSC-derived CTLs 14 days after the third stimulation. Expression of iC9/mCherry by these CTLs is shown. The plots are representative of at least five independent differentiation experiments.
(C) Quantitative PCR analysis to compare the expression of cell-lysis molecules in PB CD8+ T cells, original EBV-CTLs, rejT-NT-EBV, and rejT-iC9-EBV (left). The same analysis was also carried out in PB CD8+ T cells, rejT-NT-HIV1, and rejT-iC9-HIV1 (right). Individual PCR results were normalized against 18S rRNA. Data are presented as the mean of three independent experiments ± SD.
(D) IFN-γ production by original EBV-CTLs, rejT-NT-EBV, and rejT-iC9-EBV in the presence of LMP2 peptide was measured using ELISPOT. IFN-γ production by original HIV1-CTLs, rejT-NT-HIV1, and rejT-iC9-HIV1 in the presence of Nef peptide was measured similarly. Data are presented as the mean ± SD and are representative of three independent triplicate experiments.
(E) In vitro 51Cr-release assay of original EBV-CTLs, rejT-NT-EBV, and rejT-iC9-EBV (effectors) and EBV-transformed B lymphoblastoid cell lines (targets) (upper) and that of rejT-NT-HIV1 and rejT-iC9-HIV1 (effectors) and Nef-presenting LCLs (targets) (lower). Data are presented as the mean ± SD and are representative of at least three independent triplicate experiments.
See also Figure S2.
Finally, we generated rejuvenated EBV-specific CTLs from NT- and iC9-EBV-iPSCs (rejT-NT-EBV and rejT-iC9-EBV). More than 95% of rejT-NT-EBV and rejT-iC9-EBV bound to multimers of their respective target major histocompatibility complex-viral peptide complex; these CTLs also expressed CD8. Similarly, rejuvenated HIV1-specific CTLs from NT- and iC9-HIV1-iPSCs were successfully generated (rejT-NT-HIV1 and rejT-iC9-HIV1). rejT-iC9-EBV and rejT-iC9-HIV1 expressed high levels of iC9, with 95% and 99%, respectively, expressing iC9/mCherry (Figure 3B). To determine whether these redifferentiated cells expressed cytotoxicity-related molecules, we compared gene expression profiles for rejT-iC9-EBV and rejT-iC9-HIV1 with those for other cell lineages, including the original EBV-CTL, rejT-NT-EBV, rejT-NT-HIV1, and PB CD8+ T cells. rejT-iC9-EBV and rejT-iC9-HIV1 showed cytotoxic potential, with expression of GZMB, FASLG, and PRF1 (Figure 3C). The expression profiles for these CTLs were similar to those for the original EBV-CTL and PB CD8+ T cells.
Because chimeric antigen receptor-expressing T cells from iPSCs expressed only CD8α and very few cells expressed low amounts of CD8β (Themeli et al., 2013), we also examined the expression of CD8α and CD8β in rejT-iC9-EBV and rejT-iC9-HIV1. It should be noted that most of these iPSC-derived CTLs also expressed CD8α (95.8% and 87.7%, respectively), whereas only 3% expressed CD8β (Figure S2).
We next determined the specificity of EBV-CTLs and HIV1-CTLs with interferon (IFN) γ enzyme-linked immunospot (ELISPOT) assays after stimulation with LMP2 or Nef peptides, respectively. Our results indicated that the original EBV-CTL clone, rejT-NT-EBV, and rejT-iC9-EBV showed specific activity against LMP2 (respectively, 310 ± 26, 231 ± 13, and 227 ± 24 IFN-γ spot-forming cells [SFCs]/1,000). Similarly, all three HIV1-CTLs (original HIV1-CTL clone, rejT-NT-HIV1, and rejT-iC9-HIV1) were activated by Nef (respectively, 109 ± 34, 149 ± 60, and 197 ± 10 IFN-γ SFCs/1,000) (Figure 3D) and showed strong antigen-specific cytotoxicity with respect to Nef-presenting cells. Poor proliferation capacity precluded cytotoxicity assays using the original HIV1-CTL clone. At an effector:target (E:T) ratio of 40:1, rejT-iC9-HIV1 and rejT-NT-HIV1 killed Nef peptide-expressing target cells (49.1% and 52.2% specific 51Cr release, respectively), with minimal recognition of control target cells pulsed with irrelevant peptides (3.9% and −1.3% specific 51Cr release, respectively). On the other hand, cytotoxicity of rejT-iC9-EBV, rejT-NT-EBV, and even the original EBV-CTL clone with respect to LMP2-presenting cells was relatively weak. At an E:T ratio of 40:1, the original EBV-CTL clone, rejT-iC9-EBV, and rejT-NT-EBV killed histocompatibility leukocyte antigen (HLA) class I-matched target cells (17%, 7.4%, and 6.7% specific 51Cr release, respectively), with minimal recognition of HLA class I-mismatched control target cells (0.6%, −1.2%, and −0.2% specific 51Cr release, respectively) (Figure 3E). Our results demonstrated that rejCTLs derived from iC9-iPSCs are virus specific and exhibit cytotoxic activity against virus-infected cells. High expression of iC9 thus neither inhibits redifferentiation into rejCTLs nor affects antigen specificity and killing function.
Safety and Effectiveness of iC9-iPSC-Derived CTLs for Tumor Therapy In Vivo
To elucidate whether iC9-iPSC-derived CTL therapy is safe and rejT-iC9-EBV exerts antitumor effects in vivo, EBV lymphoblastoid cell lines (EBV-LCLs) transduced with lentiviral vector encoding a GFP-firefly luciferase fusion protein (GFP/FFLuc) (3 × 106 cells/mouse) were intraperitoneally engrafted into NOD-Scid mice. Light emission was monitored as an indicator of tumor growth. Once bioluminescence occurred and progressively increased (around 5 days after tumor inoculation), mice were divided into four groups. In the control group, no treatment was given (n = 4); the others were treated with rejT-iC9-EBV (n = 7), rejT-NT-EBV (n = 5), or original EBV-CTLs (n = 5) (10 × 106 per dose, three doses) followed by intraperitoneal injection of recombinant human IL-2, 1,000 U/mouse, three times a week (Quintarelli et al., 2007, Vera et al., 2009). As shown by bioluminescent imaging (Figures 4A–4C), tumor signals declined as expected in mice treated with original EBV-CTLs (0.41-fold, range 0.009–1.12). rejT-iC9-EBV or rejT-NT-EBV also suppressed tumor signals as well as or better than original EBV-CTLs (0.06-fold, range 0.009–0.13, rejT-iC9-EBV; 0.097-fold, range 0.012–0.32, rejT-NT-EBV) (Figure 4C), yielding a survival advantage in the group treated with rejT-iC9-EBV compared to the group treated with original EBV-CTLs (p = 0.04) (Figure 4D). Tumor signals regressed further in the groups treated with rejT-iC9-EBV or rejT-NT-EBV than in the untreated group (rejT-iC9-EBV, p < 0.0001; rejT-NT-EBV, p < 0.0001; original EBV-CTLs, p = 0.0001; one-way ANOVA) (Figure 4C), and resulted in a significant survival advantage compared to tumor-bearing mice in the untreated group (p = 0.0039, rejT-iC9-EBV; p = 0.0047, rejT-NT-EBV) (Figure 4D). Bioluminescence imaging additionally revealed that signaling fell off more rapidly in mice treated with rejT-iC9-EBV than in mice treated with rejT-NT-EBV (Figure 4B). In contrast, signaling progressively increased in mice without treatment (8.998-fold, range 3.99–16.3). These results suggest that iC9-iPSC-derived CTLs are safe and effective for tumor therapy in vivo and provide a survival benefit in tumor-bearing mice.
Figure 4.

In Vivo Antitumor Effect of iC9-iPSC-Derived EBV-CTLs
(A) NOD-Scid mice were inoculated intraperitoneally with HLA-A∗02-positive EBV-LCL cells labeled with GFP/FFluc and then treated either with control or EBV-CTL lines. Mice were divided into three groups that around 5 days later received rejT-iC9-EBV (n = 7), rejT-NT-EBV (n = 5), or original EBV-CTLs (n = 5). “No treatment” indicates that mice were injected with EBV-LCLs but not with CTLs (n = 4). Images of three representative mice from each group are shown.
(B) Total body flux (photons/s) for each mouse was quantified, and group averages were calculated. Error bars represent ± SEM. ∗p < 0.05 by one-way ANOVA comparing no treatment to rejT-iC9-EBV, rejT-NT-EBV, or original EBV-CTLs.
(C) Total tumor growth by day 21 after CTL infusions is represented as log10 signal change. Error bars represent ± SD. ∗∗∗∗p < 0.0001 and ∗∗∗p < 0.001 by one-way ANOVA.
(D) Kaplan-Meier survival curves for treated and control mice (rejT-iC9-EBV, n = 7; rejT-NT-EBV, n = 5; original EBV-CTLs, n = 5; no treatment, n = 4). ∗∗p < 0.01 and ∗p < 0.05 by the Gehan-Breslow-Wilcoxon test.
Induction of Apoptosis in iC9-iPSC-Derived CTLs by a Cascade of Caspase Activation In Vitro and In Vivo
To gain further insights into the effectiveness of the iC9 safeguard system, rejT-iC9-EBV and rejT-iC9-HIV1 were treated with CID. Annexin V binding and 7-AAD uptake were examined 24 hr later. Our results clearly demonstrated that CID induces apoptosis in iC9-iPSC-derived CTLs (rejT-iC9-HIV1, 96.81% ± 1.84%; rejT-iC9-EBV, 75.7% ± 2.0%) that is robust compared with that in untreated CTLs (rejT-iC9-HIV1, 17.03% ± 1.01%; rejT-iC9-EBV, 28.13% ± 0.91%; Figure 5A). In contrast, apoptosis was not increased in CTLs without the iC9 safeguard system (original EBV-CTL, rejT-NT-EBV, and rejT-NT-HIV1). To examine the effects of CID on caspase activation, apoptosis-related gene expression profiles were obtained for rejT-iC9-HIV1 and rejT-iC9-EBV 24 hr after CID treatment (although treatment meant that such cells were few) and were compared with similar profiles in cells without CID treatment. CASP9 was upregulated 5.9 ± 3.24-fold in rejT-iC9-HIV1 and 9.3 ± 1.9-fold in rejT-iC9-EBV 24 hr after CID treatment. Because caspase-9 activation induces the activation of caspase-3, the effector caspase, we also assessed CASP3 expression. CASP3 expression slightly decreased (1.01 ± 0.26-fold) in rejT-iC9-HIV1 but increased 3.43 ± 0.84-fold in rejT-iC9-EBV 24 hr after CID addition. Interestingly, expression of XIAP, an inhibitor of apoptosis, was higher in rejT-iC9-EBV (6.5 ± 0.12-fold) than in rejT-iC9-HIV1 (1.91-fold) (Figure S3). The difference in XIAP expression between rejT-iC9-EBV and rejT-iC9-HIV1 likely affects sensitivity to iC9.
Figure 5.

Induction of Apoptosis in iC9-iPSC-Derived CTLs by Activation of iC9
(A) Flow cytometric analysis of annexin V binding and 7-AAD uptake by rejT-NT-HIV1 and rejT-iC9-HIV1 24 hr after CID treatment (left). Original EBV-CTLs, rejT-NT-EBV, rejT-iC9-EBV, rejT-NT-HIV1, and rejT-iC9-HIV1 were left untreated or treated with CID. Apoptosis was assessed 24 hr after treatment. Data are representative of three independent triplicate experiments. Error bars represent ± SD. ∗∗∗∗p < 0.0001 by two-way ANOVA.
(B) In vivo bioluminescent imaging of rejT-iC9-EBV expressing FFluc. NOD-Scid mice inoculated intraperitoneally with EBV-LCL cells and with rejT-iC9-EBV cells received three doses of CID (50 μg) intraperitoneally (n = 4). Comparison mice received no CID (n = 3). Images of three representative mice from each group are shown.
(C) FFluc signal intensities after rejT-iC9-EBV cell transfer in each group. Error bars represent ± SEM. ∗p < 0.05 by unpaired Student’s t test (two-tailed).
(D) Study schema of in vivo rejT-iC9-mCherry detection in peripheral blood (upper). mCherry expression was quantified by flow cytometry (n = 4 per group). Representative data of three experiments are shown (lower).
(E) Schematic illustration of the iC9/CID safeguard system to protect patients receiving iPSC-derived CTL therapy.
See also Figure S3.
We finally assessed the efficacy of the iC9 safeguard system in vivo using an iC9-iPSC-derived CTL therapy model (Figure 5B). The iC9/CID safeguard system was also effective in vivo. EBV-LCLs (5 × 106 cells/mouse) were intraperitoneally engrafted into NOD-Scid mice and treated with GFP/FFluc-labeled rejT-iC9-EBV cells on day 0 (10 × 106 cells/mouse). Infused rejT-iC9-EBV cells localized and could be tracked by bioluminescence imaging (Figure 5B). Bioluminescent T cell signaling declined after injection of CID (n = 4), whereas the signal persisted in control mice without CID treatment (n = 3) on day 6 (p = 0.015 by unpaired Student’s t test [two-tailed]; Figures 5B and 5C). Additionally, EBV-LCLs (3 × 106 cells/mouse) were intraperitoneally engrafted into NOD-Scid mice and, after tumor establishment (day −7 to day −5), mice were treated with rejT-iC9-EBV cells (days 0 and 7). Flow cytometry was used to monitor mCherry expression in peripheral blood. Soon after mCherry-expressing CTLs could be detected (around day 8), the mice received intraperitoneally injected CID (50 μg/mouse) or PBS for 3 successive days (n = 4 each) (day 9 to day 11). Twenty-four hours after the last dose of CID, rejT-iC9-EBV cells could not be detected in CID-treated mice (0.6% ± 0.4%) (Figure 5D), whereas rejT-iC9-EBV cells were present in control mice (8.25% ± 4.0%; p = 0.01 by unpaired Student’s t test) (data not shown); this held true even 6 days after treatment. These results collectively led us to conclude that the iC9/CID safeguard system leads by a cascade of caspase activation to efficient apoptosis in iC9-iPSC-derived CTLs in vitro and in vivo, and that it will be a useful tool against acute GVHD or cytokine release syndrome following cell therapy and to limit “on-target, off-tumor toxicities” seen in some engineered T cell immunotherapy.
Discussion
Our studies highlight the potential clinical significance of the iC9 safeguard system. Recent studies introducing suicide gene-based safeguard systems into mouse or rhesus iPSCs report that teratoma formation was significantly delayed or inhibited only when treatment started before implantation or soon after iPSC injection (Chen et al., 2013, Lim et al., 2013, Wu et al., 2014, Zhong et al., 2011). In our study, this was not so for CID treatment; when started 30 days after iC9-iPSC implantation, it caused dramatic teratoma regression. These results indicate that our lentiviral iC9/CID safeguard system may be useful practically.
Looking toward the introduction of the iC9/CID safeguard system to clinical use, we applied this system to iPSC-derived (rejuvenated) CTL therapy to show how this system could actually work. Our results indicated that introduction of iC9 into iPSCs does not interfere with their pluripotency and differentiation ability. iC9-iPSCs could efficiently differentiate into virus-specific CTLs that exhibited continuously high iC9/mCherry expression. These iC9-iPSC-derived CTLs could be eliminated by CID treatment in vivo. EF1α-iC9 expression in rhesus iPSCs reportedly is downregulated during in vitro and in vivo differentiation due to DNA methylation within the promoter (Wu et al., 2014). We initially compared the percentages of apoptosis between Ubc-iC9-iPSCs and EF1α-iC9-iPSCs 24 hr after CID treatment. CID could induce apoptosis more strongly in Ubc-iC9-iPSCs than in EF1α-iC9-iPSCs. Ubc-iC9 expression levels were still high during and after differentiation into virus-specific CTLs, and CID also induced robust apoptosis of Ubc-iC9-iPSC-derived CTLs. These data suggest that the Ubc promoter provides stable expression of the iC9 transgene after iC9/mCherry sorting and is suitable for iPSC-derived cell therapy. To increase the efficacy of this iC9 system further, iC9-transduced iPSCs could be cloned and selected for maximum sensitivity for CID and a minimum number of mutations in the genome.
Most of these CTLs do not express CD8β. Despite relatively weak cytotoxicity for LMP2 antigen in vitro and their unconventional phenotype (CD8α+, CD8β−), these rejT-iC9-CTLs exerted a significant antitumor effect in mouse xenograft models. These rejCTLs may kill the target cells using nonconventional, natural killer (NK)-like killing mechanisms. This in vivo antitumor effect of rejT-iC9-CTLs is encouraging that adoptive T cell therapy using rejT-iC9-CTLs targeting tumors will be clinically feasible, especially EBV-associated tumors such as Hodgkin’s disease, nasal NK cell lymphoma, nasopharyngeal carcinoma, and EBV-associated posttransplantation lymphoproliferative disease.
If increased efficiency of this iC9/CID system is required, the proteasome inhibitor bortezomib can be used. Bortezomib and iC9/CID are strongly synergistic, enhancing apoptosis in lung cancer cells by blocking the degradation of active caspase-3 and caspase-9 (Ando et al., 2014). Of interest in this regard is that the E3 ubiquitin ligase XIAP, an inhibitor of apoptosis, binds to caspase-3 and targets it for proteasomal degradation (Albeck et al., 2008, Huang et al., 2001, Suzuki et al., 2001). Knockdown of XIAP enhances iC9-induced apoptosis, suggesting that bortezomib maximizes iC9-induced apoptosis (Ando et al., 2014). Given that bortezomib downregulates nuclear factor κB (Hideshima et al., 2009), which is a transcriptional target of XIAP (Stehlik et al., 1998), bortezomib likely downregulates XIAP expression.
As far as we know, tumor formation in a clinical study using lentivirus vectors has not been reported. However, in clinical studies, to examine integration sites of lentivirus-iC9 and choose clones most likely to be safe appears prudent. An alternative approach may be to insert the iC9 system into a “safe harbor” site, such as AAVS1 or ROSA26, through homologous recombination using highly efficient new genetic manipulation technologies involving zinc-finger nucleases (Hockemeyer et al., 2009), transcription activator-like effector nucleases (Boch and Bonas, 2010), or clustered regularly interspaced short palindromic repeats (Cong et al., 2013, Jinek et al., 2013, Mali et al., 2013).
Our concept of the iC9/CID safeguard system for iPSC-derived cell therapy can be presented as a schema (Figure 5E). This safeguard system can eliminate contaminating iPSCs, debulk tumors originated from iPSCs, stop cytokine release syndrome associated with iPSC-derived CTL therapy, and control “on-target, off-tumor toxicities”. It should be applicable to other cell therapies using iPSC-derived cells. The iC9 safeguard system can be an efficient and reliable approach to provide safety for future regenerative therapy and first-in-human cell therapy.
Experimental Procedures
Vector Construction and Virus Production
A retrovirus vector encoding iC9 was purchased from Addgene (pMSCV-F-del Casp9.IRES.GFP; 15567). Two iC9-expressing lentiviral vectors (Ubc-iC9 and EF1α-iC9) were generated from CS-CDF-CG-PRE (Yamaguchi et al., 2012). Additional details are provided in Supplemental Experimental Procedures.
Generation of iC9-iPSCs
The institutional regulation board for human ethics at the Institute of Medical Science, University of Tokyo approved the experimental protocol. PBMCs of an HLA-A∗02:01-positive healthy donor were used to generate an LMP2-specific CTL clone (Burrows et al., 1995). This clone was specific for the LMP2 peptide FLYALALLL that is presented by HLA-A∗02:01 (Lautscham et al., 2003). EBV-iPS was established from this clone after informed consent was obtained. HIV1-iPS is a previously established T-iPSC line from a Nef138-8 (wild-type; WT)-specific CTL line derived from an HLA-A24-positive patient chronically infected with HIV1 (Kawana-Tachikawa et al., 2002, Nishimura et al., 2013). To introduce lentivirus-derived iC9 into iPSCs, 1 × 105 iPSCs were plated on 24-well plates coated with Corning Matrigel Matrix and cultured in Essential 8 medium (GIBCO, Life Technology) for 24 hr. iPSCs were then transduced with lentiviral iC9 at an MOI of 20; medium was replaced with fresh medium 6 hr after transduction. iC9-transduced iPSCs were further cultured and iC9/mCherry-expressing cells were sorted by flow cytometry (FACSAria II; BD Biosciences). Additional details are provided in Supplemental Experimental Procedures.
T Cell Differentiation from T-iPSCs
To differentiate human iPSCs into hematopoietic cells, we slightly modified a published protocol (Nishimura et al., 2013, Takayama et al., 2008). Details are provided in Supplemental Experimental Procedures.
Measurement of Apoptosis
CID/AP20187 was purchased from Clontech as a B/B homodimerizer. Cells that bound annexin V or took up 7-AAD were quantified by flow cytometry (FACSAria II; BD Biosciences) using FlowJo software (Tree Star). Details are provided in Supplemental Experimental Procedures.
Teratoma Formation and In Vivo CID Treatment
Animal care was accorded with the guidance of The University of Tokyo for animal and recombinant DNA experiments. The Animal Care and Use Committee, Institute of Medical Science, University of Tokyo approved all procedures. iC9-iPSCs and NT-iPSCs (1.3 × 106 cells) suspended in Corning Matrigel Matrix containing the Rock inhibitor Y-27632 (10 mM) were injected into the testicular interstitium of NOD-Scid mice. Tumor growth was monitored by measuring testis size in three dimensions once a week. Thirty days after injection, CID administration (50 μg intraperitoneally daily for 3 successive days) was begun. Tumors with TV <150 mm3 on day 29 were excluded from analysis. Using TVs on days 0, 29, and 36, the tumor size ratio was calculated as (TVday 36 − TVday 0)/(TVday 29 − Tvday 0). Thirty-five days after injection, the mice were sacrificed and tumors were assessed as described above.
Antitumor Activity in In Vivo Model
To evaluate the antitumor effects of rejT-iC9-CTLs, HLA class I-matched EBV-LCLs transduced with a lentiviral vector encoding GFP/FFluc were sorted for GFP expression by flow cytometry. NOD-Scid mice were engrafted intraperitoneally with EBV-LCLs (3 × 106 cells/mouse) and tumor growth was monitored using a bioluminescence system. Once a progressive increase of bioluminescence occurred, usually 5 days after tumor inoculation, mice were treated intraperitoneally with three once-weekly doses of rejT-iC9-EBV, rejT-NT-EBV, or original EBV-CTLs (10 × 106 CTLs/mouse). The mice treated with EBV-CTLs received IL-2 (1,000 U) intraperitoneally three times a week (Vera et al., 2009). One additional control group received only tumor cells. Tumor burden was monitored by the Xenogen IVIS imaging system. Mice were injected intraperitoneally with D-luciferin (150 mg/kg), and light output was analyzed using Xenogen Living Image software version 2.50. The intensity of the signal was measured as total photon/s/cm2/steradian (p/s/cm2/sr).
In Vivo Elimination of iC9-iPSC-Derived CTLs
To examine whether iC9-iPSC-derived CTLs can be eliminated by this iC9/CID safeguard system in vivo, NOD-Scid mice were engrafted intraperitoneally with EBV-LCLs (5 × 106 cells/mouse) and received 10 × 106 rejT-iC9-EBV cells labeled with GFP/FFluc on day 0. Mice were treated with CID (50 μg intraperitoneally daily for 3 successive days (day 2 to day 4). Comparison mice received no CID. Mice were imaged as described above.
Detection of iC9-iPSC-Derived CTLs In Vivo
NOD-Scid mice inoculated intraperitoneally with HLA-A∗02-positive EBV-LCL cells labeled with GFP/FFluc were treated with 10 × 106 rejT-iC9-EBV cells on day 0 and day 7. After rejT-iC9-EBV cells were detected in peripheral blood (around 8 days after the first rejT-iC9-EBV administration), the mice received intraperitoneally injected CID, 50 μg/mouse, for 3 successive days. Control mice received three doses of PBS. Flow cytometry of peripheral blood was used to identify rejT-iC9-EBV cells (expressing mCherry).
Statistics
All data are presented as mean ± SD or SEM as stated in the figure legends. Results were analyzed by ANOVA or unpaired Student’s t test (two-tailed) as stated in the text. Comparison of survival curves was done using the Gehan-Breslow-Wilcoxon test. All statistical analyses were performed using Excel (Microsoft) and Prism (GraphPad Software) programs. Values of p <0.05 were considered significant.
Author Contributions
M.A. planned and performed the experiments and wrote the manuscript. T.N. helped in performing CTL redifferentiation experiments. S.Y. provided scientific discussion and technical support. T.Y. provided plasmids and helped in generating lentivirus vectors. A.K.-T. and J.A. performed the 51Cr-release assay. T.H., Y.O., and Y.N. performed pathological diagnosis of teratomas. K.N., M.O., and M.N. provided the Sendai virus. J.J.M. and S.R.B. provided the EBV-CTL clone. M.K.B. and S.T. provided scientific discussion. H.N. directed the study and wrote the manuscript.
Acknowledgments
We wish to thank Masataka Kasai and A.S. Knisely for critical reading of the manuscript; Reiko Ishida and Hideyuki Murayama for technical help with quantitative PCR; Yuji Yamazaki for FACS operation; and Cliona M. Rooney and Norihiro Watanabe for helpful discussions. The project was supported in part by a Grant-in-Aid for Japan Society for the Promotion of Science (JSPS) Fellows, the Japan Science and Technology Agency, a Health Labour Sciences Research Grant from the Ministry of Health, Labour and Welfare, the California Institute of Regenerative Medicine, the Virginia and D.K. Ludwig Fund for Cancer Research, and the National Health and Medical Research Council of Australia.
Published: August 27, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures and three figures and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2015.07.011.
Supplemental Information
References
- Albeck J.G., Burke J.M., Aldridge B.B., Zhang M., Lauffenburger D.A., Sorger P.K. Quantitative analysis of pathways controlling extrinsic apoptosis in single cells. Mol. Cell. 2008;30:11–25. doi: 10.1016/j.molcel.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando M., Hoyos V., Yagyu S., Tao W., Ramos C.A., Dotti G., Brenner M.K., Bouchier-Hayes L. Bortezomib sensitizes non-small cell lung cancer to mesenchymal stromal cell-delivered inducible caspase-9-mediated cytotoxicity. Cancer Gene Ther. 2014;21:472–482. doi: 10.1038/cgt.2014.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-David U., Gan Q.F., Golan-Lev T., Arora P., Yanuka O., Oren Y.S., Leikin-Frenkel A., Graf M., Garippa R., Boehringer M. Selective elimination of human pluripotent stem cells by an oleate synthesis inhibitor discovered in a high-throughput screen. Cell Stem Cell. 2013;12:167–179. doi: 10.1016/j.stem.2012.11.015. [DOI] [PubMed] [Google Scholar]
- Berger C., Flowers M.E., Warren E.H., Riddell S.R. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 2006;107:2294–2302. doi: 10.1182/blood-2005-08-3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boch J., Bonas U. Xanthomonas AvrBs3 family-type III effectors: discovery and function. Annu. Rev. Phytopathol. 2010;48:419–436. doi: 10.1146/annurev-phyto-080508-081936. [DOI] [PubMed] [Google Scholar]
- Brenner M.K., Gottschalk S., Leen A.M., Vera J.F. Is cancer gene therapy an empty suit? Lancet Oncol. 2013;14:e447–e456. doi: 10.1016/S1470-2045(13)70173-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brivanlou A.H., Gage F.H., Jaenisch R., Jessell T., Melton D., Rossant J. Stem cells. Setting standards for human embryonic stem cells. Science. 2003;300:913–916. doi: 10.1126/science.1082940. [DOI] [PubMed] [Google Scholar]
- Burrows S.R., Silins S.L., Moss D.J., Khanna R., Misko I.S., Argaet V.P. T cell receptor repertoire for a viral epitope in humans is diversified by tolerance to a background major histocompatibility complex antigen. J. Exp. Med. 1995;182:1703–1715. doi: 10.1084/jem.182.6.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F., Cai B., Gao Y., Yuan X., Cheng F., Wang T., Jiang M., Zhou Y., Lahn B.T., Li W., Xiang A.P. Suicide gene-mediated ablation of tumor-initiating mouse pluripotent stem cells. Biomaterials. 2013;34:1701–1711. doi: 10.1016/j.biomaterials.2012.11.018. [DOI] [PubMed] [Google Scholar]
- Choo A.B., Tan H.L., Ang S.N., Fong W.J., Chin A., Lo J., Zheng L., Hentze H., Philp R.J., Oh S.K., Yap M. Selection against undifferentiated human embryonic stem cells by a cytotoxic antibody recognizing podocalyxin-like protein-1. Stem Cells. 2008;26:1454–1463. doi: 10.1634/stemcells.2007-0576. [DOI] [PubMed] [Google Scholar]
- Ciceri F., Bonini C., Gallo-Stampino C., Bordignon C. Modulation of GvHD by suicide-gene transduced donor T lymphocytes: clinical applications in mismatched transplantation. Cytotherapy. 2005;7:144–149. doi: 10.1080/14653240510018136. [DOI] [PubMed] [Google Scholar]
- Ciceri F., Bonini C., Marktel S., Zappone E., Servida P., Bernardi M., Pescarollo A., Bondanza A., Peccatori J., Rossini S. Antitumor effects of HSV-TK-engineered donor lymphocytes after allogeneic stem-cell transplantation. Blood. 2007;109:4698–4707. doi: 10.1182/blood-2006-05-023416. [DOI] [PubMed] [Google Scholar]
- Clackson T., Yang W., Rozamus L.W., Hatada M., Amara J.F., Rollins C.T., Stevenson L.F., Magari S.R., Wood S.A., Courage N.L. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc. Natl. Acad. Sci. USA. 1998;95:10437–10442. doi: 10.1073/pnas.95.18.10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stasi A., Tey S.K., Dotti G., Fujita Y., Kennedy-Nasser A., Martinez C., Straathof K., Liu E., Durett A.G., Grilley B. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov V.D., Themeli M., Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 2013;5:215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargett T., Brown M.P. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014;5:235. doi: 10.3389/fphar.2014.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hideshima T., Ikeda H., Chauhan D., Okawa Y., Raje N., Podar K., Mitsiades C., Munshi N.C., Richardson P.G., Carrasco R.D., Anderson K.C. Bortezomib induces canonical nuclear factor-kappaB activation in multiple myeloma cells. Blood. 2009;114:1046–1052. doi: 10.1182/blood-2009-01-199604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D., Soldner F., Beard C., Gao Q., Mitalipova M., DeKelver R.C., Katibah G.E., Amora R., Boydston E.A., Zeitler B. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyos V., Savoldo B., Dotti G. Genetic modification of human T lymphocytes for the treatment of hematologic malignancies. Haematologica. 2012;97:1622–1631. doi: 10.3324/haematol.2012.064303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Park Y.C., Rich R.L., Segal D., Myszka D.G., Wu H. Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain. Cell. 2001;104:781–790. [PubMed] [Google Scholar]
- Inoue H., Nagata N., Kurokawa H., Yamanaka S. iPS cells: a game changer for future medicine. EMBO J. 2014;33:409–417. doi: 10.1002/embj.201387098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M., East A., Cheng A., Lin S., Ma E., Doudna J. RNA-programmed genome editing in human cells. eLife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawana-Tachikawa A., Tomizawa M., Nunoya J., Shioda T., Kato A., Nakayama E.E., Nakamura T., Nagai Y., Iwamoto A. An efficient and versatile mammalian viral vector system for major histocompatibility complex class I/peptide complexes. J. Virol. 2002;76:11982–11988. doi: 10.1128/JVI.76.23.11982-11988.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff C.A., Gattinoni L., Restifo N.P. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol. Rev. 2006;211:214–224. doi: 10.1111/j.0105-2896.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautscham G., Haigh T., Mayrhofer S., Taylor G., Croom-Carter D., Leese A., Gadola S., Cerundolo V., Rickinson A., Blake N. Identification of a TAP-independent, immunoproteasome-dependent CD8+ T-cell epitope in Epstein-Barr virus latent membrane protein 2. J. Virol. 2003;77:2757–2761. doi: 10.1128/JVI.77.4.2757-2761.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.S., Tang C., Rao M.S., Weissman I.L., Wu J.C. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 2013;19:998–1004. doi: 10.1038/nm.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.O., Moon S.H., Jeong H.C., Yi J.Y., Lee T.H., Shim S.H., Rhee Y.H., Lee S.H., Oh S.J., Lee M.Y. Inhibition of pluripotent stem cell-derived teratoma formation by small molecules. Proc. Natl. Acad. Sci. USA. 2013;110:E3281–E3290. doi: 10.1073/pnas.1303669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leen A.M., Heslop H.E., Brenner M.K. Antiviral T-cell therapy. Immunol. Rev. 2014;258:12–29. doi: 10.1111/imr.12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim T.T., Geisen C., Hesse M., Fleischmann B.K., Zimmermann K., Pfeifer A. Lentiviral vector mediated thymidine kinase expression in pluripotent stem cells enables removal of tumorigenic cells. PLoS ONE. 2013;8:e70543. doi: 10.1371/journal.pone.0070543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T., Kaneko S., Kawana-Tachikawa A., Tajima Y., Goto H., Zhu D., Nakayama-Hosoya K., Iriguchi S., Uemura Y., Shimizu T. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:114–126. doi: 10.1016/j.stem.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Nori S., Okada Y., Nishimura S., Sasaki T., Itakura G., Kobayashi Y., Renault-Mihara F., Shimizu A., Koya I., Yoshida R. Long-term safety issues of iPSC-based cell therapy in a spinal cord injury model: oncogenic transformation with epithelial-mesenchymal transition. Stem Cell Reports. 2015;4:360–373. doi: 10.1016/j.stemcr.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintarelli C., Vera J.F., Savoldo B., Giordano Attianese G.M., Pule M., Foster A.E., Heslop H.E., Rooney C.M., Brenner M.K., Dotti G. Co-expression of cytokine and suicide genes to enhance the activity and safety of tumor-specific cytotoxic T lymphocytes. Blood. 2007;110:2793–2802. doi: 10.1182/blood-2007-02-072843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehlik C., de Martin R., Kumabashiri I., Schmid J.A., Binder B.R., Lipp J. Nuclear factor (NF)-kappaB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J. Exp. Med. 1998;188:211–216. doi: 10.1084/jem.188.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straathof K.C., Pulè M.A., Yotnda P., Dotti G., Vanin E.F., Brenner M.K., Heslop H.E., Spencer D.M., Rooney C.M. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105:4247–4254. doi: 10.1182/blood-2004-11-4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y., Nakabayashi Y., Takahashi R. Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc. Natl. Acad. Sci. USA. 2001;98:8662–8667. doi: 10.1073/pnas.161506698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Takayama N., Nishikii H., Usui J., Tsukui H., Sawaguchi A., Hiroyama T., Eto K., Nakauchi H. Generation of functional platelets from human embryonic stem cells in vitro via ES-sacs, VEGF-promoted structures that concentrate hematopoietic progenitors. Blood. 2008;111:5298–5306. doi: 10.1182/blood-2007-10-117622. [DOI] [PubMed] [Google Scholar]
- Tang C., Lee A.S., Volkmer J.P., Sahoo D., Nag D., Mosley A.R., Inlay M.A., Ardehali R., Chavez S.L., Pera R.R. An antibody against SSEA-5 glycan on human pluripotent stem cells enables removal of teratoma-forming cells. Nat. Biotechnol. 2011;29:829–834. doi: 10.1038/nbt.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Themeli M., Kloss C.C., Ciriello G., Fedorov V.D., Perna F., Gonen M., Sadelain M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013;31:928–933. doi: 10.1038/nbt.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermans F., Velghe I., Vanwalleghem L., De Smedt M., Van Coppernolle S., Taghon T., Moore H.D., Leclercq G., Langerak A.W., Kerre T. Generation of T cells from human embryonic stem cell-derived hematopoietic zones. J. Immunol. 2009;182:6879–6888. doi: 10.4049/jimmunol.0803670. [DOI] [PubMed] [Google Scholar]
- Traversari C., Marktel S., Magnani Z., Mangia P., Russo V., Ciceri F., Bonini C., Bordignon C. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood. 2007;109:4708–4715. doi: 10.1182/blood-2006-04-015230. [DOI] [PubMed] [Google Scholar]
- Vera J.F., Hoyos V., Savoldo B., Quintarelli C., Giordano Attianese G.M., Leen A.M., Liu H., Foster A.E., Heslop H.E., Rooney C.M. Genetic manipulation of tumor-specific cytotoxic T lymphocytes to restore responsiveness to IL-7. Mol. Ther. 2009;17:880–888. doi: 10.1038/mt.2009.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry E.J. T cell exhaustion. Nat. Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- Wu C., Hong S.G., Winkler T., Spencer D.M., Jares A., Ichwan B., Nicolae A., Guo V., Larochelle A., Dunbar C.E. Development of an inducible caspase-9 safety switch for pluripotent stem cell-based therapies. Mol. Ther. Methods Clin. Dev. 2014;1:14053. doi: 10.1038/mtm.2014.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T., Hamanaka S., Kamiya A., Okabe M., Kawarai M., Wakiyama Y., Umino A., Hayama T., Sato H., Lee Y.S. Development of an all-in-one inducible lentiviral vector for gene specific analysis of reprogramming. PLoS ONE. 2012;7:e41007. doi: 10.1371/journal.pone.0041007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B., Watts K.L., Gori J.L., Wohlfahrt M.E., Enssle J., Adair J.E., Kiem H.P. Safeguarding nonhuman primate iPS cells with suicide genes. Mol. Ther. 2011;19:1667–1675. doi: 10.1038/mt.2011.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Di Stasi A., Tey S.K., Krance R.A., Martinez C., Leung K.S., Durett A.G., Wu M.F., Liu H., Leen A.M. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood. 2014;123:3895–3905. doi: 10.1182/blood-2014-01-551671. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.