

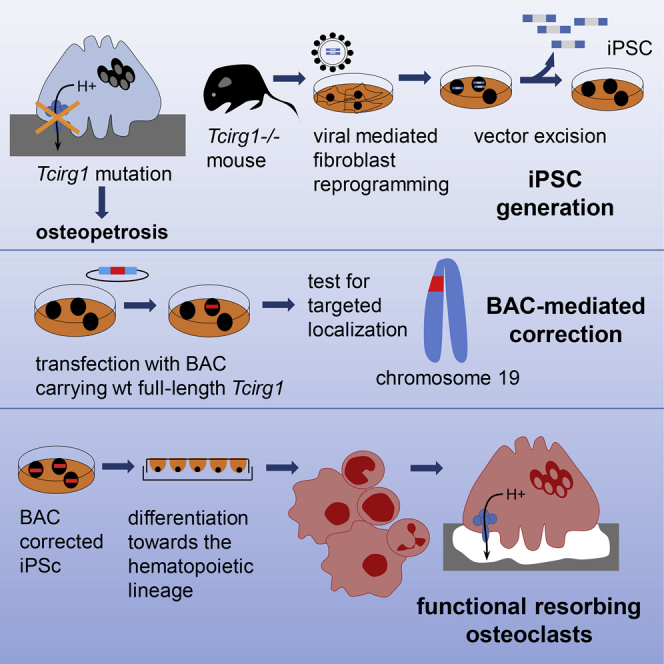
Summary
Autosomal recessive osteopetrosis is a human bone disease mainly caused by TCIRG1 gene mutations that prevent osteoclasts resorbing activity, recapitulated by the oc/oc mouse model. Bone marrow transplantation is the only available treatment, limited by the need for a matched donor. The use of induced pluripotent stem cells (iPSCs) as an unlimited source of autologous cells to generate gene corrected osteoclasts might represent a powerful alternative. We generated iPSCs from oc/oc mice, corrected the mutation using a BAC carrying the entire Tcirg1 gene locus as a template for homologous recombination, and induced hematopoietic differentiation. Similarly to physiologic fetal hematopoiesis, iPSC-derived CD41+ cells gradually gave rise to CD45+ cells, which comprised both mature myeloid cells and high proliferative potential colony-forming cells. Finally, we differentiated the gene corrected iPSC-derived myeloid cells into osteoclasts with rescued bone resorbing activity. These results are promising for a future translation into the human clinical setting.
Graphical Abstract
Highlights
-
•
iPSCs from oc/oc mice bearing Tcirg1 gene mutation were generated for the first time
-
•
A BAC-based approach corrects the Tcirg1 gene mutation
-
•
iPSCs differentiate similarly to physiologic fetal hematopoiesis
-
•
The osteopetrotic phenotype in osteoclasts from BAC-corrected iPSCs was rescued
In this article, Villa and colleagues present a multistep strategy by which iPSCs are generated from osteopetrotic mice, genetically corrected by homologous recombination using a BAC carrying the entire Tcirg1 gene locus, and differentiated toward hematopoietic early progenitors able to give rise to functional osteoclasts, rescuing the defective cellular phenotype.
Introduction
Bone is a highly specialized tissue under constant renewal that takes place through coordinated balanced destruction and reconstruction, respectively, played by osteoclasts and osteoblasts, known as bone remodeling (Teitelbaum and Ross, 2003). Osteoclasts are large fused multinucleated cells deriving from myeloid precursors belonging to the hematopoietic lineage. Defects in their resorbing activity lead to autosomal recessive osteopetrosis (ARO), a rare life-threatening bone disease that causes increased bone density, decreased bone strength with risk of multiple fractures, and progressive narrowing of the medullary cavity. In turn, these defects lead to anemia, thrombocytopenia, and compensatory extramedullary hematopoiesis and in some cases may associate to immune dysfunction (Sobacchi et al., 2013). The most frequently mutated gene is the T cell immune regulator 1 (TCIRG1), encoding the a3 subunit of the vacuolar-type proton transporting ATPase pump (Frattini et al., 2000), which mediates the acidification of the bone/osteoclast interface. The spontaneous mutant oc/oc mouse bears a deletion in the Tcirg1 gene and well mimics the clinical features of human osteopetrosis (Scimeca et al., 2000). To date, the only available treatment for ARO is the hematopoietic stem cell (HSC) transplantation (A.S. Schulz et al., 2013, abstract, 39th Annual Meeting of the European Group for Blood and Marrow Transplantation). However, several patients do not have access to grafts from HLA-matched siblings, a treatment that ensures the best 5-year disease-free survival rate (62% versus 40% with alternative donor transplantation including HLA-matched unrelated) (Orchard et al., 2015). To overcome the problem of donor availability, generation of autologous corrected HSCs followed by transplantation might represent a valid therapeutic option. HSC gene therapy is being utilized with efficacy in a number of genetic diseases (Aiuti et al., 2009, Aiuti et al., 2013, Biffi et al., 2013, Cavazzana-Calvo et al., 2010, Hacein-Bey-Abina et al., 2010). However, the use of integrating viral vectors to deliver wild-type (WT) cDNA does not allow to restore the integrity of the locus. For this reason, HSCs generated from iPSCs (Robinton and Daley, 2012, Takahashi and Yamanaka, 2006) might represent an alternative source of autologous donor cells, since iPSCs can be easily expanded in large quantities, manipulated to perform gene targeting, and then differentiated into corrected autologous HSCs. Initial experiments on gene targeting in human CD34+ cells by delivering artificial endonucleases and donor DNA template have recently been performed with success, providing a new promising technology (Genovese et al., 2014). Nevertheless, the relative low efficiency by which gene targeting occurs in long-term repopulating HSCs and progenitors may limit application of this technology to diseases in which there is a strong in vivo selective advantage of the corrected cells. Generation of iPSCs from patients who do not have access to an HLA-identical sibling donor could allow their site-specific genetic correction by homologous recombination, followed by differentiation toward hematopoietic progenitors and autologous transplantation.
In the present study, we present a step-by-step strategy by which gene-corrected osteopetrotic iPSCs were used to generate hematopoietic progenitor cells able to give rise in vitro to functional osteoclasts, thus providing a proof of principle for an autologous cell therapy approach to treat osteopetrosis and potentially other genetic blood disorders (Hanna et al., 2007).
Results
Derivation of Vector-free iPSCs from Osteopetrotic Mice
To obtain iPSCs from WT and oc/oc mouse fibroblasts, we used a third-generation, Cre-excisable lentiviral vector expressing a single cistron encoding for Oct4, Sox2, and Klf4 under the control of the spleen focus-forming virus promoter (sffv). Four weeks after a single transduction at low multiplicity of infection (MOI = 1), colonies with embryonic stem (ES) cell-like morphology (nine from WT and 19 from oc/oc fibroblasts) were picked and expanded. Total DNA was extracted from each clone to perform vector copy number (VCN) analysis. Three clones for each genotype (WT or oc/oc) with the lowest VCN and with a normal mouse chromosome complement (2n = 40) were selected for subsequent analysis. With the aim of obtaining viral vector-free cell lines, excision of the reprogramming lentiviral vector was achieved by transient expression of the Cre recombinase in the six clones. After sub-cloning, six lines with a VCN equal to zero were selected for further characterization: WT iPSCs 6.25.2, 6.25.39, and 6.25.68 (hereafter referred to as WT 2, WT 39, and WT 68, respectively) and oc/oc iPSCs 13.62, 13.65, and 16.74 (oc/oc 62, oc/oc 65, and oc/oc 74, respectively) (Figure 1A). The clones displayed stemness characteristics that persisted after the excision procedure. They expressed alkaline phosphatase (Figure 1B; Figure S1A) and other typical stemness markers as revealed by immunofluorescence (Figure 1C; Figure S1B) and RT-PCR (data not shown). Cytogenetic analysis showed a normal karyotype (Figure 1D; Figure S1C). All clones were bona fide pluripotent as assessed by their ability to differentiate toward the three germ layers in vitro (Figure 1E; data not shown) and in vivo (Figures 1F and S1D), forming teratomas upon subcutaneous injection into NSG mice.
Figure 1.

Generation and Characterization of iPSCs from WT and oc/oc Mice
(A) Experimental outline for the generation of WT and oc/oc iPSC clones with excised reprogramming vector (VCN = 0) and carrying the correct chromosome complement (see text for details).
(B) Expression of alkaline phosphatase on WT and oc/oc iPSC colonies.
(C) Expression of Oct4, Sox2, Nanog, and SSEA-1 on WT and oc/oc iPSCs as revealed by immunofluorescence. Nuclei are stained with DAPI. Scale bars, 100 μm.
(D) Karyotype analysis of WT and oc/oc iPSC clones.
(E) Generation of the three germ layers in vitro by differentiated WT and oc/oc iPSCs is revealed by immunofluorescence. Expression of Brachyury, AFP, and Nestin indicate formation of mesoderm, endoderm, and ectoderm, respectively. Scale bars, 50 μm.
(F) H&E staining of teratomas generated after subcutaneous injection of WT and oc/oc iPSC clones into NSG mice, demonstrating differentiation into the three germ layer derivatives (i, mesoderm-cartilage; ii, mesoderm-adipose tissue; iii, endoderm-glandular structure; iv, endoderm-glandular structures (intestinal paneth or pancreatic acinar cell-like); v, ectoderm-primitive and mature neuroepithelium; vi, ectoderm-primitive neuroepithelium). Scale bars, 50 μm.
See also Figure S1.
In conclusion, we were able to generate Tcirg−/− iPSC clones from oc/oc mice, with phenotypic and functional characteristics comparable to those displayed by iPSCs obtained from WT mice. All tested iPSC clones were free from the reprogramming vector, contained a normal set of chromosomes, and maintained stemness and pluripotency.
Efficient BAC-Mediated Tcirg1 Gene Correction in oc/oc iPSCs with Preserved Stemness and Pluripotency
The oc/oc iPSC lines were transfected with a Bacterial Artificial Chromosome (BAC) containing the full-length Tcirg1 gene, including its 5′- and 3′-flanking regions, with the aim of replacing one of the mutated alleles with the correct form. To select cells with BAC insertion, we engineered the donor vector to introduce a FRT-flanked neomycin resistance cassette (neo) into the intron 10 (Figure 2A). Transfected cells were cultured in the presence of neomycin and resistant iPSC colonies were picked and analyzed for the presence of the corrected Tcirg1 allele by PCR analysis on genomic DNA and for expression of the corrected transcript by RT-PCR (data not shown). iPSC clones that were positive for both PCRs were then subjected to fluorescence in situ hybridization (FISH) analysis using the BAC as probe to test for the correct sub-centromeric localization of the BAC on chromosome 19 and for screening of possible off-target integrations (Figures 2B and 2C). In the three clones named 13.62.18, 13.62.28, and 16.74.32 (hereafter referred to as 18-BAC, 28-BAC, and 32-BAC), whose expression of the corrected Tcirg1 gene is shown in Figure 2D, the BAC probe was localized only to chromosome 19 (Figure 2C), while other clones were found to have random integrations (Figure S2A). Despite these manipulations, 18-BAC, 28-BAC, and 32-BAC iPSC clones maintained a normal karyotype (Figure S2B) as well as their pluripotency characteristics, as shown by the expression of stemness markers (Figures S2C and S2D), the capacity to form embryoid bodies (EBs) containing the three germ layers, and the ability to generate teratomas in NSG mice (Figure S2E). Moreover, beating cardiomyocytes were obtained in all iPSC lines tested (data not shown). When injected in blastocysts, clone 32-BAC contributed to chimera formation (Figure S2F).
Figure 2.

Correction of Tcirg1 Mutation in oc/oc iPSCs
(A) Upper lane: schematic representation of the BAC used for the gene targeting, containing the 11-kb Tcirg1 locus plus its 5′ and 3′ flanking regions (55 and 78 kb, respectively), as well as CmR and HIS3 genes belonging to the BAC backbone. Middle and lower lanes: schematic representation of the 1.6-kb deletion in genomic DNA present in oc/oc mutants compared with the WT genomic region. oc/oc mice are homozygous for the mutation.
(B) Experimental outline for generating gene-corrected oc/oc iPSC clones (see text for details). Only clones containing and expressing corrected Tcirg1, as assessed by PCR on genomic DNA and by RT-PCR, were subjected to FISH and karyotype analyses.
(C) Representative metaphase spreads after FISH using the BAC as probe (red), which hybridizes with the BAC-inserted fragment as well as with the mutated form of Tcirg1. White arrows indicate the presence of the probe only on the two 19 chromosomes in clones 18-BAC, 28-BAC, and 32-BAC, in a sub-centromeric localization corresponding to the endogenous Tcirg1 location. Since these clones were previously screened by PCR for the presence of the correct Tcirg1 gene, the presence of two signals in the expected position suggests that at least one of the two alleles was targeted. Chromosomes were counterstained by DAPI (blue). Scale bars, 5 μm.
(D) RT-PCR analysis for detection of Tcirg1 expression in the WT 39 iPSC clone (used as a positive control), in the oc/oc 74 iPSC clone (negative control), and in the three corrected oc/oc iPSC clones, using the primers represented by red arrows in (A). Mouse Gapdh was used as housekeeping control.
(E) PCR analysis for detection of HIS3 and CmR backbone sequences into the genome of iPSC clones. First lane: the positive control showing the PCR product obtained from the BAC; second lane: the WT iPSC 39 clone used as a negative control; third and fourth lanes: 18-BAC and 32-BAC devoid of the tested backbone sequences. These sequences were absent also in 28-BAC, which was tested separately (data not shown). The last two lanes show a 168-bp fragment of the CmR and a 191-bp fragment of HIS3 gene obtained in two clones previously discarded due to the BAC insertion in other chromosomes.
See also Figure S2.
Because of the high degree of homology between the BAC and the endogenous Tcirg1 locus, we were unable to map the exact location of the recombination between these two sequences by standard Southern blot or PCR analysis. However, we reasoned that clones that underwent homologous recombination should not contain sequences belonging to the BAC backbone. To assess whether vector sequences were absent in BAC-corrected iPSC clones, we performed PCRs to detect a portion of chloramphenicol resistance (CmR) and the yeast HIS3 gene (Figure 2E), as an indirect evidence of targeted correction, as previously described (Howden et al., 2011). By this assay, we confirmed the absence of backbone sequences in the three selected clones, while these sequences were detected in clones where the BAC was mapped by FISH to other different portions of the genome. This suggests, although does not formally prove, that BAC integrated via homologous recombination in the targeted locus.
In conclusion, we successfully corrected Tcirg1 mutation in oc/oc iPSCs without affecting their stem cell characteristics.
BAC-Corrected iPSCs Can Be Successfully Differentiated into Hematopoietic Early Progenitor Cells
A multistep procedure was undertaken to generate hematopoietic progenitors from WT, oc/oc, and BAC-corrected oc/oc iPSC lines. Pluripotent cells were induced to form EBs through the “hanging-drop” method. Two days later, EBs were transferred into tissue-culture treated plates where they adhered in the presence of BMP4, VEGF, and growth factors inducing hematopoietic differentiation (interleukin-3 [IL-3], IL-6, SCF, Flt3-ligand, GM-CSF). Hematopoietic cell clusters appeared starting from day 8 of culture and progressively increased in number and size up to day 15 (data not shown). When immuno-magnetically purified cKit+ cells obtained from these cultures were plated in methylcellulose, they gave rise to erythroid, myeloid, and high proliferative potential mixed colonies containing monocytes, macrophages, granulocytes, and erythroblasts, indicating the presence of multi-potent and/or oligo-potent progenitors (Figure 3A). The presence of cells at different stages of the hematopoietic differentiation was further confirmed by fluorescence-activated cell sorting (FACS) analysis performed on pooled colonies (Figure 3A). The proportion of myeloid precursors was similar in colonies generated by WT and BAC-corrected iPSCs. Flow cytometry analysis performed prior to purification of cKit+ cells confirmed the presence of hematopoietic lineage cells in cultures originated from all iPSC clones, as revealed by the expression of the pan-hematopoietic marker CD45 in a proportion of the cells (Figure 3B). Most of the CD45+ cells co-expressed the CD11b myeloid cell marker, suggesting that this culture condition potentially supported the development of osteoclast progenitors, the relevant cell type in our model (Figure 3B).
Figure 3.

Differentiation of iPSCs toward the Hematopoietic Lineage
(A) Methylcellulose colony assays were performed at the end of the hematopoietic differentiation procedure with cKit+ cells purified from cultures initiated by WT, oc/oc, or oc/oc BAC-corrected iPSCs. Pictures on the left represent erythroid (BFU-E), myeloid (CFU-GM and CFU-M), and mixed (CFU-GEMM) colonies obtained from differentiated WT iPSCs; pictures on the right show CFU-GM colonies from one oc/oc and one corrected iPSC clone (scale bars, 400 μm). Colony assays were performed with all nine clones, for a total of 16–30 independent replicates/group (WT, oc/oc, and BAC-corrected). Once scored, some individual colonies were cytospun and stained with May-Grunwald and Giemsa. The insert shows a representative example of a cytospin preparation of a CFU-GEMM colony showing macrophages (M), monocytes (Mo), granulocytes (Gr), and erythrocytes (Er) (scale bar, 50 μm). In other experiments, FACS analysis was performed on pooled colonies to confirm the presence of TER119+ erythroid cells, CD11b+F4/80+ macrophages, TER119–CD11b+Ly6G+ polymorphonucleated (PMN) granulocytes. On the PMN– gate monoblasts, Pro-monocytes and monocytes were defined as CD31+Ly6C–, CD31+Ly6C+, and CD31–Ly6C+, respectively. Three clones for each group (WT, oc/oc and BAC-corrected) were used, and results are pooled (five to 15 independent replicates/group) and shown in the bar graph.
(B) Representative FACS analyses showing the expression of the CD45 pan-hematopoietic and the CD11b myeloid cell marker at day 12 of culture of WT, oc/oc, and corrected iPSC-derived EBs induced to differentiate toward the hematopoietic lineage. Numbers in the FACS plots indicate percentages among alive cells. Negative control is represented in the left contour plot.
(C) Upper panels: representative time course FACS analysis showing the expression of CD41 and CD45 hematopoietic markers at different time points of the differentiation protocol of one BAC-corrected iPSC clone. Upper contour plots show the control. Results from all cell lines are summarized and quantified in the bar graphs, using all nine clones. For each time point from three to 14 independent experiments were performed.
(D) The graph shows the percentage of CD11b+ cells within CD45+-gated cells in all iPSC clones. For each time point from three to 14 independent experiments were performed.
Error bars in all bar graphs indicate SEM. Significance of differences was determined by Mann-Whitney test. ∗p < 0.03; ∗∗p < 0.005.
We next performed flow cytometry analysis at different time points during hematopoietic differentiation of iPSC to identify the optimal cell-culture length to obtain the highest yield of CD45+ cells to be further differentiated into osteoclasts, without losing early progenitors. Interestingly, time course analysis revealed an in vitro differentiation pattern reminiscent of physiologic fetal hematopoiesis. CD41+ cells, including CD41hi and especially CD41low cells, which have been shown to include definitive HSCs in the mouse embryo (Robin et al., 2011), were abundantly present at day 8 and subsequently were gradually replaced by CD45+ cells, typical of adult-type hematopoiesis (Figure 3C). Importantly, all iPSCs gave rise to CD41+ and CD45+ cells with similar yield and kinetics. The maximum yields of myeloid CD45+CD11b+ cells were reached at days 10–12, with no major differences among the different clones, with the exception of a tendency to a slower kinetics of myeloid maturation from uncorrected oc/oc iPSC that is significant at day 10 (Figure 3D).
Overall, the gene correction procedure preserved the hematopoietic differentiation capacity of oc/oc-derived iPSCs.
Gene-Corrected oc/oc-Derived iPSCs Generate Functional Osteoclasts with Resorbing Activity
To demonstrate that gene-corrected iPSCs from oc/oc mice were functionally competent, hematopoietic cells generated by all iPSC lines were forced to differentiate in vitro into multinucleated osteoclasts and assayed for their capacity to resorb dentine, a specific test for osteoclast function. In detail, at the end of the hematopoietic differentiation procedure, CD45+ cells were purified by immuno-magnetic beads and cultured in the presence of M-CSF and SCF to further stimulate the proliferation of progenitor cells and to drive monocyte differentiation. Six days later, we replaced SCF with RANKL to induce the fusion of pre-osteoclasts and the activation of mature osteoclasts. The identity of osteoclast cells in culture was confirmed by the tartrate resistant alkaline phosphatase (TRAP) staining and the count of the nuclei (>3). iPSCs obtained from WT mice successfully generated TRAP+ multinucleated osteoclasts similar to those obtained from the spleen or BM of adult WT mice (Figure 4A, i–iii). Importantly, these osteoclasts were equally able to efficiently resorb dentine, indicating the ability of iPSCs to undergo terminal differentiation to a very specialized cell type (Figure 4B, i and ii). iPSCs derived from oc/oc mice were also able to differentiate into osteoclasts, which were phenotypically similar to those obtained from oc/oc mice fresh splenocytes (Figure 4A, iv and v). As expected, oc/oc osteoclasts, either spleen derived (data not shown) or iPSC derived, were unable to resorb dentine (Figure 4B, iii). We then performed the same experiment with hematopoietic cells obtained from gene-corrected oc/oc iPSCs, after removing the neo cassette from the 18-BAC iPSC clone by transient transfection of the Flp recombinase (thus obtaining the 18-BACf clone), to avoid possible interference of the neo sequence with the high expression of the Tcirg1 protein occurring in mature resorbing osteoclasts. Remarkably, we obtained a complete rescue of the capacity to resorb dentine (Figures 4A, vi, and 4B, iv), demonstrating that BAC-mediated correction of the mutated Tcirg1 was successful.
Figure 4.

Differentiated iPSCs Generate Functional Osteoclasts
(A) Images of TRAP+ osteoclasts derived from iPSCs (i, iv, and vi) and from fresh mouse tissues (ii and v, spleen; iii, BM). Osteoclasts are defined as giant, multinucleated TRAP+ cells. Results are representative of at least three independent experiments/group. Scale bars, 100 μm.
(B) Representative images of dentine discs stained with toluidine blue, showing the presence of resorption pits in dentine discs previously seeded with osteoclast precursors obtained from WT or BAC-corrected oc/oc iPSCs (i and iv, respectively) and from the BM of a WT mouse (ii), but not in discs seeded with the non-corrected oc/oc 62 iPSC clone (iii). Results are representative of at least three independent experiments/group. Scale bars, 100 μm.
Discussion
In this paper, we describe a multi-step process to produce gene-corrected hematopoietic cells including early progenitors and functional osteoclasts from iPSCs generated from a preclinical mouse model of ARO.
Autologous HSC gene therapy represents an emerging therapeutic opportunity for genetic diseases with hematological origin to avoid the requirement of compatible donors. However, viral vector-mediated gene correction of patient HSCs carry the risk of insertional mutagenesis (Nienhuis, 2013) and unregulated transgene expression. Gene correction by homologous recombination would represent a major step forward; however, the difficulty of ex vivo expanding and/or selecting a small fraction of gene-edited HSCs still represents a challenge. As an alternative to ex vivo HSCs, iPSCs represent an innovative source of donor cells. They are pluripotent stem cells derived from the reprogramming of somatic cells, so they have an easily accessible cell source, obtainable without invasive procedures. The feasibility to perform gene targeting is greater, for their propensity to homologous recombination and for the possibility of selecting and expanding clones where gene editing has occurred.
In the present study, we tested the feasibility of gene-correction technology using as donor a BAC engineered vector. Taking advantage of the large genomic region carried by this vector, the full-length form of Tcirg1 was inserted, correcting the large mutation that causes the phenotype in the mouse. Since in human ARO the TCIRG1 gene is mainly affected by point mutations located in different positions within the gene locus, the use of a BAC allows the utilization of the same donor vector for every type of mutations found in TCIRG1-dependent patients. In contrast, targeted genome editing using artificial nucleases would imply designing either a different donor template for each mutation, or a single cDNA template, thus not completely restoring the native locus including introns and regulatory sequences.
We generated iPSCs using a third-generation self-inactivating excisable lentiviral vector, which proved to be efficacious for obtaining vector-free pluripotent stem cells. More recent techniques, such as the use of the non-integrating Sendai virus, would allow production of iPSCs without all the in vitro manipulations required for Cre-recombinase excision, which include extensive sub-cloning and selection of vector-free and normal karyotype clones. All these procedures could be skipped thus avoiding the risk of introducing additional mutations. Importantly, if pre-natal tissues are used to generate patient-specific iPSCs, as proposed by others (Anchan et al., 2011), autologous iPSC-mediated cell therapy for ARO could be envisioned to treat patients at birth, if not earlier, to prevent bone as well as secondary neurological defects, which cannot be rescued if transplantation is not performed soon after birth. Moreover, iPSCs represent an unlimited supply of corrected cells for additional future use.
To evaluate the ability of WT and oc/oc corrected iPSCs to form functional osteoclasts, a protocol for the hematopoietic differentiation was set up, to obtain myeloid cells as well as HSCs/early progenitors. Various differentiation protocols have been described to obtain in vitro the hematopoietic lineage, but the only ones which provide long-term stem cells utilize the overexpression of additional transcription factors, such as Hoxb4, Lhx2, or the combination of Gata2, Gfi1b, cFos, and Etv6 (Hanna et al., 2007, Hanna et al., 2010, Takahashi and Yamanaka, 2006). The overexpression of these factors represents an additional manipulation and requires the use of integrating vectors for the stable expression of transgenes. For this reason, we opted to avoid the overexpression of supplementary transgenes, thus inducing differentiation in more physiological conditions. Indeed, in the presence of a hematopoietic cytokine cocktail the EB method induces first the mesoderm development at the expense of the other two germ layers, and then pushes toward the hematopoietic specification, likely through a hemogenic endothelium intermediate. All tested clones behaved similarly in terms of hematopoietic differentiation, with the exception of a slight delay in the kinetics of myeloid differentiation in oc/oc clones that was restored upon gene correction. Despite all the efforts, the scientific community has not yet reached the goal of reproducing in vitro the complex microenvironment made of cells, cytokines, and signals that sustains the correct HSC development. Accordingly, we were not able to demonstrate engraftment of our iPSC-derived hematopoietic progenitors in oc/oc recipient mice (F. Ficara, unpublished data). Further amelioration of the in vitro procedures aimed at obtaining a sufficient number of safe, functional HSCs in a timely manner from somatic cells going through a pluripotent intermediate is therefore needed before iPSC research can be translated in the clinical practice for the cure of diseases with hematological origin. Nevertheless, upon gene correction, oc/oc iPSCs differentiated into hematopoietic progenitors and then to mature osteoclasts able to efficiently resorb dentine in vitro, demonstrating their fully functional correction. Interestingly, it has been proposed that ARO infants could benefit from early transplantation of myeloid progenitors differentiated toward the osteoclast lineage (Cappariello et al., 2010). In the mouse, this approach has been shown to prevent defects in the formation of foramina, which the optic and acoustic nerves pass through, thus reducing visual and hearing defects. In this regard, autologous iPSCs could provide an unlimited source of autologous mature osteoclasts for repeated infusions.
Generation of iPSCs from patients who do not have access to an HLA-identical donor could allow their site-specific genetic correction by homologous recombination, followed by differentiation toward hematopoietic progenitors and autologous transplantation. Further optimization of protocols aimed at expanding in vitro hematopoietic progenitors is needed to allow the clinical application of this therapeutic approach to the clinical arena.
Experimental Procedures
Animals
C57BL/6J WT, congenic C57BL/6J-Ly5.1, B6C3Fe a/a Tcirg1 oc/J-Ly5.2 (oc/oc), and non-obese diabetic-scid IL2rγ(null) (NSG) mice were purchased from Charles River Laboratories or Jackson Laboratory. oc/oc-Ly5.1 mice were obtained after crossing of congenic Ly5.1 with oc/oc animals. Animal care and experimental procedures were performed in accordance with ethical rules by the institutional review board.
Cells
Mouse embryonic fibroblasts (MEFs) to be used as feeder layer were derived from WT embryonic day 13.5 embryos; WT iPSCs were generated from Ly5.1 MEFs; oc/oc iPSCs were derived from oc/oc-Ly5.1 mice fibroblasts obtained from tail biopsy. iPSC colonies were co-cultured with Mitomycin C (Sigma)-treated MEFs and passaged using Trypsin-EDTA 0.5% (Lonza) dissociation. They were cultured in ES medium, containing Knockout-DMEM (Gibco), 15% embryonic stem cell-qualified fetal bovine serum (ES-FBS, Life Technologies), 100 U/ml P/S (Lonza), 2 mM L-glutamine (Lonza), β-mercaptoethanol (Life Technologies), 1 × non-essential amino acids (100×, Life Technologies) supplemented with 103 U/ml leukemia inhibitor factor (LIF, Millipore). Medium was changed daily. All steps were performed maintaining cells at 37°C with 5% CO2.
Reprogramming Procedure
Fibroblasts were reprogrammed by an overnight infection at MOI = 1 in the presence of 4 ng/ml polybrene (Sigma) with a third-generation lentiviral vector carrying LoxP sites in the sinLTRs and expressing the Oct4, Sox2, and Klf4 reprogramming factors. The following day, the medium was replaced and after 48 hr cells were seeded onto MEF feeder layer with ES medium, which was changed daily. iPSC-like colonies were individually picked 4 weeks later, with a reprogramming efficiency of 0.01%. To excise the vector, iPSCs were transfected with plasmid DNA expressing the Cre recombinase cDNA in presence of Lipofectamine 2000 (Life Technologies) at a 1:3 ratio (pDNA:Lipofectamine 2000). After a week, iPSCs were subcloned at a limiting dilution to obtain clonal lines.
Gene Correction
The RP24-241G10 BAC (containing the entire WT Tcirg1 genomic region) belonging to the RPCI-24 C57BL/6J mouse BAC library was purchased from Children’s Hospital Oakland Research Institute (CHORI). A neo resistance cassette surrounded by FRT sites was inserted in intron 10 of the Tcirg1 gene (Gene Bridges). After an overnight BAC transfection in the presence of Lipofectamine 2000 on a MEF feeder layer, cells were transferred on DR4-resistant MEFs (Applied StemCell), and neomycin was added the following day. We obtained and successfully isolated neomycin resistant colonies in all experiments.
In order to avoid possible MEFs contamination in PCR analysis, all clones were purified with anti-SSEA-1 (stage-specific embryonic antigen-1) magnetic beads (Miltenyi Biotec) according to the manufacturer’s instructions and passaged three times on gelatin-coated plates before collecting genomic DNA and RNA for analysis.
For excision of the Neo resistance cassette from clone 13.62.18, the pCAG-Flpe plasmid (Addgene) was transfected in the presence of Lipofectamine 2000. Clones were cultured with or without neomycin and those that were not resistant were isolated and further expanded.
Alkaline Phosphatase Staining
Leucocyte Alkaline Phosphatase Kit (Sigma-Aldrich) was used, accordingly to kit instructions.
Chromosome Preparations and Analysis
Chromosome analysis was performed on slide preparation of cells grown on coverslips. Briefly, cell cultures were treated with colchicine (KaryoMAX colcemid solution, Life Technologies) at a final concentration of 0.1 μg/ml for 2 hr at 37°C. After hypotonic treatment with 0.075 M KCl and fixation in methanol:acetic acid (3:1 v/v), slides were air-dried and mounted with Eukitt mounting medium (Sigma-Aldrich). Chromosome counts and karyotype analyses were performed on metaphases stained with Vectashield mounting medium with DAPI (Vector Laboratories) for G-banding. Images were captured using an Olympus BX61 Research Microscope equipped with a cooled charge-coupled device (CCD) camera and analyzed with Applied Imaging Software CytoVision (CytoVision Master System with mouse karyotyping).
FISH Assay
The BAC RP24-241G10 (mapping to the proximal region of the mouse chromosome 19) was used as DNA probe and labeled via nick translation (Life Technologies), using Spectrum Orange-dUTP (Vysis). The labeled probe was resuspended in the hybridization buffer (50% formamide, 10% dextran sulfate, 1 × Denhart’s solution, 0.1% SDS, 40 mM Na2HPO4 [pH 6.8], 2 × saline sodium citrate [SSC]) containing 10× mouse Cot1 DNA (Life Technologies), denatured at 80°C for 10 min, and pre-annealed at 37°C for 20 min before hybridization. Slides were treated with Pepsin (0.004%) at 37°C for 30 s and dehydrated through the ethanol series before denaturation in 70% formamide/2 × SSC. Hybridization was done overnight at 37°C. Stringent washings were done in 50% formamide/2 × SSC at 42°C. Slides were mounted in Vectashield mounting medium with DAPI and then scored under an Olympus BX61 Research Microscope equipped with a cooled CCD camera. Images were captured and analyzed with Applied Imaging Software CytoVision (CytoVision Master System with Karyotyping and FISH). At least 20 images were analyzed for each clone.
In Vitro Germ Layer Differentiation
EBs were obtained by culturing 1,500 iPSCs in 20 μl hanging drops in ES medium without LIF for 2 days, before seeding on gelatin-coated coverslips in 24-well plates.
After 5 days of culture, an immunofluorescence was performed on part of the cells using antibodies against Alphafetoprotein (1:50, AFP, R&D Systems, MAB1368) and Brachyury (1:100, Abcam, ab20680). The remaining cells were treated with retinoic acid (PeproTech) to induce ectoderm differentiation. After 19 days of culture, immunofluorescence was performed using an antibody against Nestin (1:100, Abcam, ab11306). To further validate mesoderm differentiation, EBs were seeded at day 5 of differentiation and checked for cardiomyocyte beating activity starting from day 7.
Immunofluorescence
iPSCs or EBs were seeded on 0.1% gelatin-coated glass coverslips in 24-well culture dishes and fixed with 4% paraformaldehyde (PFA) in PBS for 15 or 20 min at room temperature, washed twice with PBS, and then permeabilized with 0.1% Triton X-100 in PBS for 5 min. After 1 hr in blocking solution (5% FBS in PBS), iPSCs were incubated with primary antibodies directed against Oct4 (1:200, Abcam, ab18976), Sox2 (1:200, Abcam, ab97959), Nanog (1:100, Novus Biologicals, NB100-58842) or SSEA-1 (1:100, Cell Signaling, MC480), and EBs with antibodies directed against AFP, Brachyury, or Nestin followed by repeated washing and incubation with secondary antibodies (goat anti-mouse immunoglobulin G (IgG) (H+L) or goat anti-rabbit IgG (H+L), Invitrogen). Nuclei were counterstained with DAPI and mounted with ProLong Gold antifade reagent (Life Technologies).
Teratoma Formation and Blastocyst Injection
5 × 106 iPSCs were subcutaneously injected in NSG mice. Two/three weeks later, mice were sacrificed, and dissected nodules were fixed with 4% formaline and embedded in paraffine for H&E staining. For the chimera assay, 32-BAC clone (passage 12 from BAC-mediated correction) was injected into albino C57BL/6 host blastocysts in the Mouse Mutant Core Facility of the Institute for Research in Biomedicine (IRB) in Barcelona. Chimerism was assessed after birth by the appearance of black coat color over the white background of the host pups.
Differentiation to the Hematopoietic Lineage
800-cell EBs were formed in hanging drops in culture medium without LIF. At day 2, EBs were transferred into tissue-culture plates, and the following cytokines were added to induce differentiation: 50 ng/ml m-BMP4, 30 ng/ml m-VEGF164 (both from R&D), 50 ng/ml m-SCF, 50 ng/ml h-Flt3L, 10 ng/ml m-IL-3, 10 ng/ml m-IL-6, and 10 ng/ml m-GM-CSF (all from PeproTech). At day 4 and 8, fresh medium was added with the same cytokine cocktail with the exception of m-BMP4. FACS analyses were performed from day 8 to 12 of culture in most experiments.
Colony Forming Cell Assay
At the end of the above described hematopoietic differentiation, progenitors were purified with anti-cKit magnetic beads (Miltenyi Biotec) and seeded into methylcellulose-containing medium (methoCult 3234; STEMCELL Technologies) in the presence of the following cytokines: 3 U/ml h-EPO (R&D), 20 ng/ml m-SCF, 20 ng/ml h-Flt3L, 10 ng/ml m-IL-3, 10 ng/ml m-IL-6, 10 ng/ml m-GM-CSF, and 10 ng/ml m-TPO (all purchased from PeproTech), and colonies were scored at day 10. Representative colonies were cytospun, and cell types were determined after May-Grunwald-Giemsa staining (Carlo Erba).
Osteoclast Differentiation
CD45+ cells were purified from iPSC-derived hematopoietic cells by magnetic beads (Miltenyi Biotec) and were cultured for a week in Minimal Essential Medium Alpha Modification (αMEM, Gibco), 10% FBS, 100 U/ml P/S, 2 mM L-glutamine in presence of 20 ng/ml m-SCF and 100 ng/ml m-M-CSF (PeproTech). Cells were subsequently transferred either on flat-bottomed 96-well tissue culture plates or on dentine slices (Osteosite, iDS, Pantec) and cultured in the same medium supplemented with 20 ng/ml m-M-CSF and 30 ng/ml m-RANKL (PeproTech). When osteoclasts were visible on tissue culture plates (after 5–10 days of culture), they were fixed and stained with tartrate resistance acid phosphatase (TRAP) assay (Sigma-Aldrich: acid phosphatase, leukocyte) accordingly to manufacturer’s instructions, while dentine slices were stained with 1% toluidine blue for 3 min and then washed in water. A similar protocol was pursued with oc/oc or WT mice freshly isolated splenocytes or BM cells, after purification of CD11b+ (instead of CD45+) cells by magnetic beads (Miltenyi Biotec).
Flow Cytometry
Staining of cells for FACS analysis was performed in PBS containing 2% fetal calf serum and 1 mM EDTA, with the following conjugated monoclonal antibodies (mAbs) obtained from either eBioscience or Pharmingen: CD11b (M1/70, 553312 or 53-0112-82), CD31 (390, 12-0311-83), pan-CD45 (30/F11, 12-0451-82), CD41 (eBioMWReg30, 48-0411), F4/80 (BM8, 25-4801-82), Ly6C (AL-21, 553104), Ly6G (RB6-8C5, 17-5931-81). LSR Fortessa or FACS Canto II flow cytometers equipped with Diva software (BD) were used for data acquisition and FlowJo software (Tree Star) was employed for data analysis.
Unstained and/or FMO (fluorescence minus one) samples were employed to set gates for FACS analysis.
Author Contributions
P.V, A.V., and F. Ficara conceived the project; P.V., A.V., F. Ficara, T.N., and S.M designed the experiments and analyzed the data; L.N. and A.L. designed and constructed the reprogramming lentiviral vector; T.N., S.M., M.P., M.E.C., L.C., M.L.F., F. Faggioli, C.R., S.S., D.S., E.S., S.M., C.B., A.L., C.S., and F. Ficara performed the experiments; S.M., T.N., and F. Ficara organized the figures and wrote the manuscript; all authors edited and commented on the manuscript.
Acknowledgments
The research leading to these results has received funding from the European Community’s Seventh Framework Programme under grant agreement n.602300 (SYBIL) to A.V., PRIN Project 20102M7T8X_003 to A.V., Ricerca Finalizzata from Ministero della salute RF-2009-1499,542 to A.V., Programma Nazionale per la Ricerca-Consiglio Nazionale delle Ricerche “Aging Program 2012-2014” to A.V. and P.V., PE-2011-02347329 to P.V., Lombardia Region - National Research Council of Italy (16/7/2012) Project “MbMM - Basic methodologies for innovation in the diagnosis and therapy of multi factorial diseases” (signed 25/7/2013) to P.V., Telethon (TIGET grant D2) EU (FP7 601958 SUPERSIST, ERC Advanced Grant 249845 TARGETINGGENETHERAPY) and the Italian Ministry of Health to L.N. F. Ficara was supported by a Marie Curie International Reintegration Grant (grant number PIRG6-GA-2009-256452). We thank Barbara Tondelli for the generation of the oc/oc mouse strain in the Ly5.1 genetic background.
Published: September 3, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures and two figures and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2015.08.005.
Supplemental Information
References
- Aiuti A., Cattaneo F., Galimberti S., Benninghoff U., Cassani B., Callegaro L., Scaramuzza S., Andolfi G., Mirolo M., Brigida I. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 2009;360:447–458. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- Aiuti A., Biasco L., Scaramuzza S., Ferrua F., Cicalese M.P., Baricordi C., Dionisio F., Calabria A., Giannelli S., Castiello M.C. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anchan R.M., Quaas P., Gerami-Naini B., Bartake H., Griffin A., Zhou Y., Day D., Eaton J.L., George L.L., Naber C. Amniocytes can serve a dual function as a source of iPS cells and feeder layers. Hum. Mol. Genet. 2011;20:962–974. doi: 10.1093/hmg/ddq542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi A., Montini E., Lorioli L., Cesani M., Fumagalli F., Plati T., Baldoli C., Martino S., Calabria A., Canale S. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- Cappariello A., Berardi A.C., Peruzzi B., Del Fattore A., Ugazio A., Bottazzo G.F., Teti A. Committed osteoclast precursors colonize the bone and improve the phenotype of a mouse model of autosomal recessive osteopetrosis. J. Bone Miner. Res. 2010;25:106–113. doi: 10.1359/jbmr.090715. [DOI] [PubMed] [Google Scholar]
- Cavazzana-Calvo M., Payen E., Negre O., Wang G., Hehir K., Fusil F., Down J., Denaro M., Brady T., Westerman K. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frattini A., Orchard P.J., Sobacchi C., Giliani S., Abinun M., Mattsson J.P., Keeling D.J., Andersson A.K., Wallbrandt P., Zecca L. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat. Genet. 2000;25:343–346. doi: 10.1038/77131. [DOI] [PubMed] [Google Scholar]
- Genovese P., Schiroli G., Escobar G., Di Tomaso T., Firrito C., Calabria A., Moi D., Mazzieri R., Bonini C., Holmes M.C. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S., Hauer J., Lim A., Picard C., Wang G.P., Berry C.C., Martinache C., Rieux-Laucat F., Latour S., Belohradsky B.H. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2010;363:355–364. doi: 10.1056/NEJMoa1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J., Wernig M., Markoulaki S., Sun C.W., Meissner A., Cassady J.P., Beard C., Brambrink T., Wu L.C., Townes T.M., Jaenisch R. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318:1920–1923. doi: 10.1126/science.1152092. [DOI] [PubMed] [Google Scholar]
- Hanna J.H., Saha K., Jaenisch R. Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell. 2010;143:508–525. doi: 10.1016/j.cell.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howden S.E., Gore A., Li Z., Fung H.L., Nisler B.S., Nie J., Chen G., McIntosh B.E., Gulbranson D.R., Diol N.R. Genetic correction and analysis of induced pluripotent stem cells from a patient with gyrate atrophy. Proc. Natl. Acad. Sci. USA. 2011;108:6537–6542. doi: 10.1073/pnas.1103388108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nienhuis A.W. Development of gene therapy for blood disorders: an update. Blood. 2013;122:1556–1564. doi: 10.1182/blood-2013-04-453209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard P.J., Fasth A.L., Le Rademacher J., He W., Boelens J.J., Horwitz E.M., Al-Seraihy A., Ayas M., Bonfim C.M., Boulad F. Hematopoietic stem cell transplantation for infantile osteopetrosis. Blood. 2015;126:270–276. doi: 10.1182/blood-2015-01-625541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin C., Ottersbach K., Boisset J.C., Oziemlak A., Dzierzak E. CD41 is developmentally regulated and differentially expressed on mouse hematopoietic stem cells. Blood. 2011;117:5088–5091. doi: 10.1182/blood-2011-01-329516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinton D.A., Daley G.Q. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481:295–305. doi: 10.1038/nature10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scimeca J.C., Franchi A., Trojani C., Parrinello H., Grosgeorge J., Robert C., Jaillon O., Poirier C., Gaudray P., Carle G.F. The gene encoding the mouse homologue of the human osteoclast-specific 116-kDa V-ATPase subunit bears a deletion in osteosclerotic (oc/oc) mutants. Bone. 2000;26:207–213. doi: 10.1016/s8756-3282(99)00278-1. [DOI] [PubMed] [Google Scholar]
- Sobacchi C., Schulz A., Coxon F.P., Villa A., Helfrich M.H. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat. Rev. Endocrinol. 2013;9:522–536. doi: 10.1038/nrendo.2013.137. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Teitelbaum S.L., Ross F.P. Genetic regulation of osteoclast development and function. Nat. Rev. Genet. 2003;4:638–649. doi: 10.1038/nrg1122. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.