

Summary
Adult skeletal muscle stem cells, or satellite cells (SCs), regenerate functional muscle following transplantation into injured or diseased tissue. To gain insight into human SC (huSC) biology, we analyzed transcriptome dynamics by RNA sequencing of prospectively isolated quiescent and activated huSCs. This analysis indicated that huSCs differentiate and lose proliferative potential when maintained in high-mitogen conditions ex vivo. Further analysis of gene expression revealed that p38 MAPK acts in a transcriptional network underlying huSC self-renewal. Activation of p38 signaling correlated with huSC differentiation, while inhibition of p38 reversibly prevented differentiation, enabling expansion of huSCs. When transplanted, expanded huSCs differentiated to generate chimeric muscle and engrafted as SCs in the sublaminar niche with a greater frequency than freshly isolated cells or cells cultured without p38 inhibition. These studies indicate characteristics of the huSC transcriptome that promote expansion ex vivo to allow enhanced functional engraftment of a defined population of self-renewing huSCs.
Graphical Abstract
Highlights
-
•
Prospective isolation of highly pure huSCs from diverse muscles
-
•
RNA sequencing resource for studying the huSC transcriptome
-
•
Core transcription factor regulatory network of huSC differentiation
-
•
Expanded huSCs that are genetically manipulable and self-renew in vivo
In this article, Rando and colleagues purify and study human skeletal muscle stem cells using RNA sequencing and cell transplantation. They show that p38 MAPK signaling is important for human muscle stem cell differentiation and that pharmacologic inhibition of p38 enables expansion of muscle stem cells capable of self-renewing after transplantation.
Introduction
In adult mammals, skeletal muscle is regenerated by a population of tissue-resident muscle stem cells, also known as satellite cells (SCs). Quiescent SCs in uninjured muscle are activated in response to injury or disease (Mauro, 1961). Upon activation, SCs undergo a proliferative expansion, yielding a pool of muscle progenitor cells that subsequently fuse to form functional multinucleate muscle fibers (Snow, 1978). In mice, transplanted SCs are capable of engrafting as constituents of multinucleate muscle fibers or as self-renewed muscle stem cells (Collins et al., 2005). When transplanted into dystrophin-deficient mdx mice, wild-type SC-derived muscle progenitors fuse to regenerating host fibers and restore dystrophin expression (Karpati et al., 1989, Partridge et al., 1989). Transplantation of allogeneic muscle progenitors or of genetically corrected autologous muscle progenitors is, therefore, a promising approach to treating inherited muscle diseases, such as Duchenne muscular dystrophy. However, owing to a limited understanding of human SC (huSC) biology, it is still unclear as to what extent findings from mouse studies will translate to human cell-based therapies.
A major barrier to the development of stem cell-based therapies is the inability to generate large numbers of transplantable stem cells with the potential to both self-renew and differentiate. In general, the contribution of donor SCs to muscle regeneration has been shown to correlate with the number of cells transplanted (Bosnakovski et al., 2008, Sacco et al., 2008). Although transplantation of SCs in association with donor muscle fibers has been shown to enhance engraftment efficiency (Collins et al., 2005, Hall et al., 2010), biopsies, surgical specimens, and post-mortem tissue donations are expected to yield few cells relative to the number that will be required for therapeutic huSC engraftment, and techniques for the growth and manipulation of progenitor cells ex vivo are, therefore, expected to be an important element of cell-based therapies. Culturing huSCs also will be important for therapies involving gene correction of autologous cells, which will need to be engineered, selected, and expanded ex vivo. These technical challenges are compounded by the loss of regenerative potential that occurs when SCs are grown in culture. In studies of mouse SCs, expansion in culture for as little as 3 days led to a 10-fold reduction in engraftment effiency (Montarras et al., 2005). An even more marked reduction in transplantation linked to cell culture was observed in comparisons of cultured and freshly isolated canine muscle progenitor cells (Parker et al., 2012). Although the transplantation efficiency of muscle progenitors from model organisms has been enhanced previously by manipulation of Notch signaling (Parker et al., 2012) or substrate elasticity (Gilbert et al., 2010), there are no techniques currently for the expansion of self-renewing huSCs ex vivo.
Our limited understanding of huSC biology can be attributed in part to the difficulty of obtaining tissue samples and of isolating a pure population of huSCs (Boldrin et al., 2010). By comparison, techniques for the prospective isolation of mouse SCs have identified a well-defined myogenic SC population and have enabled extensive characterization of the molecular regulation of their quiescence, activation, and differentiation (Bosnakovski et al., 2008, Collins et al., 2005, Fukada et al., 2007, Liu et al., 2013, Montarras et al., 2005, Sherwood et al., 2004). Previous studies have used prospective isolation of mononuclear cell types with distinct surface protein expression to define subsets of myogenic and non-myogenic cells in human muscle. These studies have found that a myogenic population resides within cells expressing CD56 (NCAM) (Bareja et al., 2014, Pisani et al., 2010a, Pisani et al., 2010b, Zheng et al., 2007). Zheng et al. have identified both CD56+CD34− and CD56+CD34+ subsets with myogenic potential. The CD56+CD34+ subset is thought to represent a myoendothelial population with the capacity to differentiate into myogenic, chondrogenic, or osteogenic lineages (Zheng et al., 2007). A similar study of myogenic potential within muscle-resident human cell populations showed that a CD56+CD34+ population of bipotent progenitors can give rise to both myogenic and adipogenic cell types in vitro (Pisani et al., 2010a). In a second study, the same authors confirmed that both CD56+CD34+ and CD56+CD34− cells have myogenic potential, but only the latter is restricted to a myogenic fate (Pisani et al., 2010b). More recently, Bareja et al. (2014) used CD34 as a negative selection marker for identification of a myogenic huSC within the CD56+ population.
Thus, there remains a lack of consensus regarding the identity of huSCs and further studies are required to assess the myogenic identity of prospectively isolated populations at the resolution of single cells. Notably, previous prospective isolation studies assessed the myogenic potential of purified cell populations by the analysis of human-specific gene expression in engrafted muscle tissue, but did not study their stem cell potential by evaluating the self-renewal of these populations in vivo. Nonetheless, the dual potential for self-renewal and differentiation remains the defining feature of stem cells. Therefore, further characterization of the identity and stem cell characteristics of huSCs is essential for developing insight into their mechanisms of self-renewal.
To further our understanding of huSC biology, we used prospective cell isolation, RNA sequencing (RNA-seq) analyses, and cell transplantation to study a defined population of human myogenic progenitors with the potential to self-renew. This information was leveraged to identify changes in the molecular phenotype and self-renewal potential within the purified huSC population. Specifically, we mapped a core transcription factor regulatory network of self-renewal, and we established an essential role for p38 mitogen-activated protein kinase (MAPK) in the regulation of huSC regenerative capacity akin to that observed in mouse. Reversible pharmacologic inhibition of p38 in cultured huSCs resulted in a gene expression program consistent with the promotion of huSC self-renewal, and it allowed for expansion of a population of huSCs ex vivo with enhanced self-renewal and engraftment potential.
Results
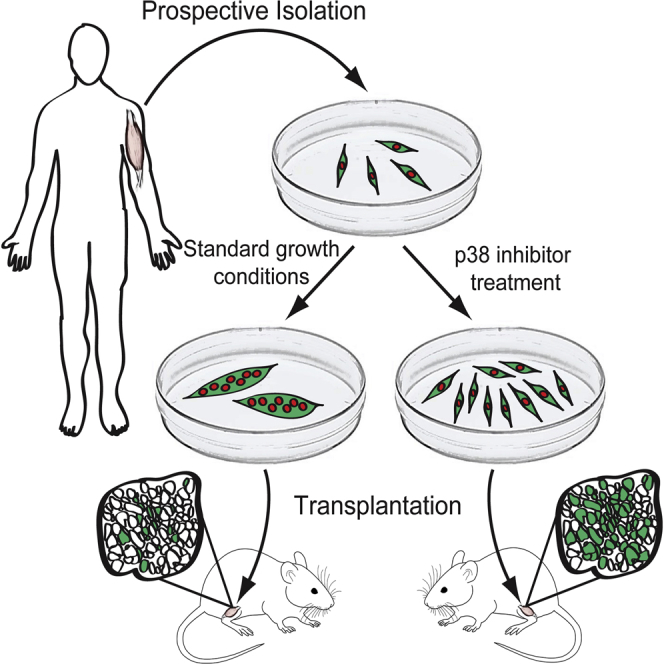
As the starting point of this study, we prospectively isolated huSCs from surgical specimens of skeletal muscle using fluorescence-activated cell sorting (FACS) to analyze cell-surface protein expression. To identify huSC surface markers, we sequentially screened known markers of mouse SCs and huSCs by analyzing extracellular protein expression in PAX7-expressing SCs associated with human muscle fiber explants (Figure S1A). Like mouse SCs, huSCs expressed β1-integrin (ITGB1), but did not express the endothelial marker CD31 or the hematopoietic marker CD45 (Sherwood et al., 2004). We observed that huSCs, in contrast to mouse SCs (Montarras et al., 2005, Sherwood et al., 2004), did not express detectable CD34. Furthermore, we found that the epidermal growth factor receptor (EGFR), which is expressed by mouse SCs (Golding et al., 2007) and exhibits a rapid decrease in expression early in the process of differentiation (Leroy et al., 2013), also was expressed by human fiber-associated SCs. Taken together, our studies defined huSCs as CD34−CD31−CD45− cells that can be further specified as a population that expresses ITGB1 and EGFR (Figures 1A and S1A). Using this surface protein signature, huSCs could be prospectively isolated from all muscles tested, including latissimus dorsi, serratus anterior, and rectus abdominis (Figure S1B).
Figure 1.

Identification and Prospective Isolation of HuSCs
(A) Cell-sorting scheme used to isolate huSCs by FACS for a representative specimen of latissimus dorsi muscle. Red gates indicate subpopulations containing huSCs. Numbers indicate percentage of total events falling within each gate; given error represents SD (n = 15).
(B) Immunofluorescence (IF) analysis of PAX7 expression in purified huSCs at low (top) and high (bottom) magnification 3 days after isolation. Cells were stained with antibodies against PAX7 and with DAPI to identify nuclei.
(C) qRT-PCR analysis of PAX7, PAX3, MYF5, MYOD, MYOG, and MEF2C mRNA levels in the huSC and huSC-depleted cell populations after a period of 7 days in culture is shown (n = 4). Error bars represent SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To analyze the purity of cells within the putative huSC population, we quantified the percentage of cells expressing PAX7 (Figure 1B), which serves as a marker of huSCs. This analysis revealed that 96% ± 2% (n = 8) of cells in the huSC population express PAX7 (Figure 1B), while no PAX7-expressing cells were observed in the remaining huSC-depleted population, which consisted of all cells expressing CD34, CD31, or CD45 (Figure S1C). These data indicate that the sorting strategy highly enriches myogenic cells. As an additional analysis of the myogenic identity of the sorted cell populations, single-cell-derived clones were analyzed for expression of myogenic genes (Figure S1D). Although 100% of cells in the putative huSC-derived population yielded clones that expressed PAX7, no PAX7-expressing clones were derived from the remaining huSC-depleted cells. The myogenic identity of purified huSCs also was evaluated by analyzing the expression of a suite of myogenic transcription factors relative to huSC-depleted cells (Figure 1C). In this analysis, cultured huSCs expressed significantly more PAX7, PAX3, MYF5, MYOD, MYOG, and MEF2C than the remaining huSC-depleted population. We also observed that a subset of activated huSC progeny fused to form multinucleate myotubes when cultured to ∼90% confluency (Figure S1E). In contrast, the huSC-depleted population of cells grown under the same conditions did not exhibit signs of myogenesis in culture (Figure S1F), further indicating that the purification strategy efficiently enriches myogenic cells.
We next aimed to characterize changes in huSCs during ex vivo culture, which, in mice, results in a marked reduction in the capacity for tissue regeneration and self-renewal upon subsequent transplantation (Gilbert et al., 2010, Montarras et al., 2005). For this analysis, we used a basal culture medium supplemented with fetal bovine serum (FBS) and with insulin, transferrin, and selenium (Rocheteau et al., 2012). Additional specific growth factors were not included to limit perturbations that might confound the interpretation of stem cell behavior ex vivo. To study the behavior of huSCs during their growth in culture, we first used time-lapse microscopy to observe purified cells beginning shortly after prospective isolation. These real-time studies of huSCs revealed that, upon isolation, initially quiescent cells undergo a process of activation in which they initially grow in size and become motile (Movie S1). HuSCs first divided, on average, 83.3 ± 14.1 hr post-isolation (Figure S1G). Activated huSC progeny undergo a second cell division that occurs, on average, 25.5 ± 5.7 hr later (Figure S1H). Interestingly, the time to first cell division and the time between the first and second cell divisions were considerably longer for huSCs than for mouse SCs studied in similar conditions (Rodgers et al., 2014).
To further characterize the behavior of huSCs ex vivo, we maintained SC cultures and analyzed their growth over a period of 3 weeks. Cells were serially passaged to maintain cultures below 40% of confluency. In four independent cultures, we observed that freshly isolated cells underwent a period of exponential growth between ∼4 and 9 days after isolation, typical of isolated murine SCs between ∼3 and 5 days after isolation. After the exponential growth phase, the huSCs entered a phase of linear growth kinetics (Figure S1I), and this pattern persisted for the remainder of the observed period in culture. Thus, huSCs give rise to a pool of proliferating progeny in culture, which as a population exhibits a progressive slowing of the growth rate.
To study the molecular regulation of the phenotypic transition that occurs in activated huSCs, and as a resource for future studies of huSC-mediated myogenesis, we generated transcriptome profiles using RNA-seq of both freshly purified, quiescent huSCs and activated huSCs, the latter having been cultured for 7 days (Figure 2A). We chose to analyze cells following 7 days in culture in order to capture the population in a fully activated state, knowing that at 7 days the population of cultured huSCs is in a phase of exponential growth (Figure S1I) and the majority of cells in the population has undergone at least one division (Figures S1G and S1H; Movie S1). Relative to activated cells, freshly isolated quiescent huSCs express transcripts, such as NFIA, NDRG2, and CHRDL2, which also are expressed in quiescent, undifferentiated murine SCs (Fukada et al., 2007, Liu et al., 2013; Figure 2B). Quiescent huSCs also express high levels of MYF5, which decrease in activated huSCs (Figure 2C). Consistent with our previous characterizations of mouse SCs activated in response to injury in vivo (Liu et al., 2013), activated huSCs upregulated genes associated with cell proliferation and myogenic differentiation: unbiased analysis of the genes that exhibited greater than 10-fold increased expression in activated huSCs relative to quiescent huSCs revealed enrichment for genes associated with the mitotic cell cycle (gene ontology [GO]:0000278; p = 6.5 × 10−9) and muscle organ development (GO:0007517; p = 1.5×10−10). Furthermore, cultured huSCs expressed differentiation-associated transcription factors such as MYOG and MEF2C (Figure 2D). Cultured huSCs as a population also upregulated genes required for myofibril assembly and muscle contraction (e.g., MYH3, CACNG1, and MYL2) (Figure S2A), supporting the conclusion that activated huSCs initiate a transcriptional program of myogenic differentiation even under high-mitogen growth conditions and sub-confluent cell density.
Figure 2.

Transcriptome Profiles of Prospectively Isolated Quiescent and Activated HuSCs
(A) Schematic shows RNA-seq analysis of prospectively isolated quiescent and ex vivo activated huSCs.
(B) Representative RNA-seq analysis shows quiescent (blue) and activated (red) muscle SCs isolated from latissimus dorsi muscle.
(C) RNA-seq analysis shows MYF5 expression in quiescent and activated huSCs.
(D) RNA-seq analysis shows expression of differentiation-associated transcription factors in quiescent and activated huSCs.
(E) Quantification of DUSP1 expression in quiescent and activated huSCs based on RNA-seq analyses is shown.
(F) Transcription factor regulatory network of huSCs constructed from analysis of differential gene expression in quiescent and activated huSCs (see Experimental Procedures for details). Highlighted in red are transcription factors with known associations with the p38 MAPK signaling pathway; highlighted in green are myogenic regulatory factors. The strength of evidence for a given interaction is reflected by the hue of the edge connecting two nodes, with darker edges indicating greater confidence. For RNA-seq data, reads from two biological replicates per condition are shown mapped to the reference genome. The number of sequenced fragments per kilobase of exon per million fragments mapped (FPKM) in each condition is shown for individual genes (n = 2 biological replicates per condition). Error bars represent SD.
As an initial application of our RNA-seq datasets for discovery of regulatory mechanisms in huSCs, we compared transcriptomes of quiescent huSCs to those of activated huSCs to identify loci that were differentially expressed. In quiescent huSCs, we identified 998 highly expressed genes, including 133 loci annotated as transcription factors, 66 as receptors, and 56 as signaling molecules by the PANTHER gene ontology database (Table S1). Importantly, several known quiescent rodent SC-associated markers, such as CALCR, CXCR4, and SPRY1, were identified in this analysis, suggesting that the method was reliable in its identification of enriched transcripts. The same analysis of transcripts enriched in activated huSCs highlighted 708 transcripts, including 29 annotated transcription factors, 40 receptors, and 28 signaling molecules (Table S1). Here again we found many interesting and unanticipated transcripts and were reassured to find several known important regulators of myogenic differentiation, including MYOD1, MYOG, and MEF2C.
We further analyzed our transcriptional profiling data to computationally define biological processes over-represented among lists of differentially expressed genes. With this analysis, we identified a number of significantly enriched (p < 0.05) biological processes and their associated genes in both quiescent and activated huSCs (Tables S2 and S3). In activated huSCs, we identified hundreds of genes associated with muscle contraction, muscle development, mitosis, and the cell cycle, among other processes. In quiescent huSCs, we identified numerous genes linked to transcriptional regulation, signal transduction, and cell-cycle control. Of particular interest, several important components of the Notch signaling pathway were identified as differentially expressed genes in this analysis. The Notch signaling pathway is a regulator of various aspects of rodent SC quiescence and activation, and Notch transcriptional activity is required for the maintenance of quiescence in mouse SCs (Bjornson et al., 2012, Conboy and Rando, 2002, Mourikis et al., 2012). In cultured huSCs, we identified in particular downregulation of expression of the Notch receptor NOTCH3 and of the canonical Notch target genes HEY1, HEY2, and HES1 (Table S3), supporting the model arising from studies of mouse SCs that Notch signaling plays criticial roles in the function of undifferentiated muscle progenitors. These analyses suggest that networks of transcribed genes with specific roles in quiescent and activated huSCs can be efficiently identified within RNA-seq data.
To take further advantage of these RNA-seq datasets of huSCs, we sought to identify signaling pathways governing processes of proliferative expansion, self-renewal, and differentiation in order to seek genes and pathways whose manipulation might be valuable for therapeutic applications of prospectively isolated huSCs. Our analysis of transcription in huSCs revealed that, among the genes that are differentially expressed by activated huSCs relative to quiescent huSCs, genes associated with MAPK signaling are significantly enriched (p = 1.2 × 10−3). Among these genes, we noted a 35-fold decrease upon huSC activation in culture in the expression of DUSP1 (Figure 2E), which encodes a dual-specificity phosphatase that dephosphorylates and, thus, inactivates p38 MAPK. This finding was of particular interest to us, given the implication of p38 MAPK signaling in the regulation of rodent muscle progenitor differentiation in the context of muscle regeneration (Lluís et al., 2006, Shi et al., 2010).
With our RNA-seq data, we constructed a core transcription factor network regulating the phenotypic changes upon culturing quiescent huSCs using published studies to identify relationships among known transcription factors that were computationally predicted to regulate genes that were differentially expressed by quiescent and activated huSCs (Figure 2F). In addition to the myogenic regulatory factors MYF5, MYOD1, MYOG, and MRF4, this network included a number of transcription factors regulated by p38, including HIF1A, PTEN, GATA4, SP3, SMAD3, NF-κB, VDR, AR, and MYC. To measure the extent of p38 activation in huSCs in vivo and in vitro, we analyzed the levels of phosphorylated (activated) p38 (p-p38). Although p-p38 was absent from quiescent SCs in histologic sections of human skeletal muscle, we observed robust p38 phosphorylation in activated huSCs in culture (Figure 3A). The activation of p38 in activated huSCs, as assessed by its phosphorylation, was independent of the density at which cells were cultured (Figures S2B and S2C). Taken together, these data support a model in which p38 MAPK is activated during ex vivo huSC growth, concurrent with a loss of proliferative potential and expression of differentiation-associated genes.
Figure 3.

Regulation of p38 MAPK Signaling Controls HuSC Fate
(A) IF analysis shows activated p-p38 in quiescent huSCs in vivo (top) and activated huSCs ex vivo (bottom).
(B) RNA-seq analysis of markers of terminal differentiation in control untreated (red) and p38i-treated (green) activated huSCs. Sequencing reads from two biological replicates per condition are shown mapped to the reference genome. The FPKM in each condition is shown for individual genes (n = 2 biological replicates per condition).
(C) Representative fluorescence microscopy analysis of EdU incorporation in control and p38i-treated huSCs cultured for 9 days. The percentage of huSCs in each condition incorporating EdU during a 1-hr pulse was quantified (n = 4). Error bars represent SD. ∗∗p < 0.01.
To determine whether the inhibition of p38 MAPK in activated huSCs could maintain their proliferative potential and prevent expression of genes for myogenic differentiation, we grew the cells in the presence of a small molecule p38 inhibitor (p38i; SB 203580) and analyzed both their transcriptional and functional properties. Though various p38 inhibitors have been developed, SB 203580 was selected for this study because its targeting specificity has been studied extensively (Cuenda and Cohen, 1999, Davies et al., 2000, Eyers et al., 1998, Gum et al., 1998). Given previous observations of dose-dependent cell growth inhibition by p38 inhibitors in transformed muscle progenitor cell lines (Jones et al., 2005), we first studied the effects of varying doses of p38 inhibitor on PAX7 expression and cell proliferation in activated huSCs. PAX7 expression, which is correlated with the undifferentiated muscle progenitor fate (Rocheteau et al., 2012), exhibited a dose-dependent increase as the concentration of p38i was increased from 0 to 10 μM, but did not significantly change at concentrations beyond 10 μM (Figure S3A). We next examined the effect of varying concentrations of p38i on the rate of proliferation of undifferentiated cells by analyzing huSCs cultured with or without inhibitor for only 3 days post-isolation. Although concentrations of p38i up to 10 μM showed equivalent rates of EdU incorporation, when cells were cultured with 20 μM p38i, the frequency of EdU incorporation was reduced significantly (Figure S3B). Because 10 μM p38i maximally induced expression of the stem cell marker PAX7 without significantly limiting cell proliferation, this drug concentration was used for all subsequent studies of cell fate.
To test for effective inhibition of p38 activity in SC cultures treated with 10 μM p38i, we quantified the amount of p38 target protein phosphorylation in control and p38i-treated cultures. These studies revealed marked decreases in the levels of phosphorylation of two p38 substrates, phopsho-HSP27 (Ser82) (Larsen et al., 1997) and phospho-GYS1 (Ser645) (Kuma et al., 2004), in the presence of p38i (Figures S3C–S3F). Consistent with a model in which p38 activity prevents myogenic lineage progression, p38i-treated cells expressed levels of MYOG, MYH3, and MYL2, for example, that were significantly reduced in comparison to untreated cells (Figure 3B). We observed similar suppression of MYOG expression by SB 203580 and an alternative, specific p38 inhibitor, SB 239063, further supporting the conclusion that pharmacologic manipulation of p38 in huSCs limits the expression of differentiation-associated genes (Figures S3G and S3H). Cells cultured in the presence of p38i also exhibited markedly diminished expression of MYOG and myosin heavy chain (MyHC) protein compared to controls (Figures S3I and S3J). Unbiased analysis of the set of genes that exhibited decreased expression due to p38i treatment revealed enrichment for genes associated with muscle contraction (GO:0006936; p = 3.7 × 10−15) and muscle organ development (p = 2.2 × 10−5). Although p38 inhibition blocked SC differentiation, we found that the expression of MYOD1 was not decreased in treated cells (Figure S3K), consistent with our findings in murine SCs of high levels of Myod1 expression in quiescent cells (Liu et al., 2013). The bulk of p38i-treated cultured huSCs also exhibited undiminished expression of the myogenic regulatory factor MYF5 relative to untreated cells (Figure S3L). Pharmacologic inhibition of p38 MAPK signaling thus maintained cultured huSCs in an undifferentiated state ex vivo.
As a first test of the ability of p38 inhibition to maintain the proliferative potential of huSCs as undifferentiated cells, we measured the proliferation index of huSCs cultured in the presence or absence of p38i for 9 days by monitoring incorporation of EdU. While only 4.4% ± 0.4% of untreated cells incorporated EdU during a 1-hr pulse, 27% ± 2% of p38i-treated cells incorporated EdU (Figure 3C). Analysis of metaphase spreads of p38i-treated huSCs revealed normal chromosome number in 100% of cells after ten passages (Figure S3M). Consistent with the observation of increased EdU incorporation in p38i-treated huSCs after 9 days in culture, expression of genes associated with DNA replication and cell division, including MKI67, NDC80, AURKB, and RFC4, was increased in p38i-treated cells relative to untreated cells in the analyses of RNA-seq data (Figure S3N). Growth curves of p38i-treated huSCs maintained in culture for 3 weeks demonstrated preservation of exponential growth kinetics and massive expansion of the treated huSC cultures in comparison to parallel untreated control cultures from the same donor (Figure S3O). Thus, blocking huSC differentiation by inhibiting p38 maintains cells in an undifferentiated state in which they continued to proliferate without evidence of genetic instability.
Although inhibition of p38 MAPK signaling prevented spontaneous differentiation of huSCs ex vivo, this effect was reversible. After 2 weeks of expansion in the presence of p38i, the drug was chased for a period of 5 days, during which we observed a significant increase in MYOG expression (Figure S4A). After the 5-day chase, we also observed terminal differentiation of expanded cells into multinucleate MyHC-expressing myotubes (Figure S4B). These data indicate that the inhibition of huSC differentiation by p38i is reversible and that cells expanded with p38i retain the ability to terminally differentiate.
To test directly the self-renewal and tissue regenerative potential of expanded huSCs in vivo, we analyzed cell engraftment upon xenotransplantation into immunodeficient mice. As adult stem cells, huSCs are expected to yield progeny that either regenerate muscle tissue or replenish the pool of self-renewing stem cells. In transplantation studies, we compared the p38i-treated activated huSCs to two control cell populations as follows: (1) freshly isolated, uncultured quiescent huSCs; and (2) activated huSCs grown in the absence of p38i. We first tested huSC regenerative potential by analyzing the expression of human-specific nuclear lamin A/C (LMNA) and human-specific ITGB1 following transplantation of huSCs into tibialis anterior muscles. Expression of human/mouse laminin also was used to visualize gross muscle morphology. We observed robust engraftment of human LMNA-labeled nuclei that correlated with the expression of human ITGB1 in muscle fibers (Figures 4A and 4B; Figure S4C). The p38i-treated activated huSCs engrafted to form human ITGB1-expressing chimeric muscle fibers with approximately 4-fold and 11-fold greater efficiency than freshly isolated quiescent huSCs and control activated huSCs, respectively (221 ± 20 chimeric fibers per muscle cross-section from p38i-treated activated huSCs versus 49 ± 14 from quiescent huSCs and 21 ± 13 from untreated activated huSCs; p < 0.001 in separate comparisons of each control condition to the p38i-treated condition) (Figure 4A). Furthermore, human-mouse chimeric muscle fibers formed by the engraftment of p38i-expanded huSCs expressed human dystrophin (DMD) (Figure S4D). We also analyzed expanded huSC engraftment by studying fusion of huSCs engineered ex vivo by adenoviral infection to express a GFP transgene, giving rise to chimeric GFP-expressing muscle fibers in transplanted NSG mice (Figures S4E and S4F). These analyses demonstrate as a proof-of-concept that the prospectively isolated and expanded cells are genetically manipulable and capable of expressing donor-derived DMD in chimeric fibers.
Figure 4.

Xenotransplantation Reveals Regenerative Potential of Expanded HuSCs
(A) Four weeks after transplantation, engraftment of freshly isolated quiescent huSCs, control activated huSCs, and p38i-treated activated huSCs was determined by IF analysis of human-specific ITGB1 expression (magenta); 3 × 104 cells were used for each transplant. Anti-laminin (green) detects laminin of mouse and human origin.
(B) Representative low-magnification (left) and high-magnification (right) images of engrafted p38i-treated activated huSCs 4 weeks after transplantation into mouse muscle. The yellow arrowhead identifies a sublaminar PAX7-expressing cell of human origin; the yellow arrow identifies a nearby sublaminar Pax7-expressing cell of mouse origin.
(C) Representative flow cytometric analysis of human-specific ITGB1 expression in the bulk population of mononuclear cells isolated from mouse muscle transplanted with 3 × 104 huSCs per transplant. Quantification of total mononuclear human ITGB1-expressing cells per transplanted muscle revealed a 3.6-fold and 7.7-fold increase in engraftment of p38i-treated huSCs relative to quiescent huSCs and untreated activated huSCs, respectively. ∗∗∗p < 0.001 comparing p38i-treated activated huSCs to control activated huSCs or quiescent huSCs (n = 4 per condition). Error bars represent SD.
To study the potential of transplanted huSCs to manifest a defining stem cell property by undergoing self-renewal to yield PAX7-expressing huSCs in vivo, we first examined the expression of PAX7 in nuclei of unfused human cells in recipient muscles. In this analysis, we observed PAX7-expressing nuclei of human origin that resided beneath the basal lamina (Figure 4B; Figures S4G and S4H). To compare quantitatively the potential of freshly isolated quiescent huSCs, control activated huSCs, and p38i-treated activated huSCs to engraft as mononuclear stem cells, we analyzed the abundance of unfused, donor human cells by flow cytometry. We initially confirmed that unfused donor cells expressed PAX7 as evidence that they had formed renewed huSCs (Figures S4I and S4J). Comparable to the analysis of fusion as an index of donor cell engraftment, p38i-treated activated huSCs had approximately 3.6-fold and 7.6-fold greater potential for engraftment as mononuclear SCs than quiescent huSCs and control activated huSCs, respectively (Figure 4C). Histological quantification of the number of human-derived, PAX7-expressing nuclei in cross-sections of transplanted muscle indicated a similar enhancement of engraftment by p38i-treated activated huSCs versus both quiescent huSCs and untreated activated huSCs. We, therefore, conclude that huSCs expanded in culture via p38 inhibition are capable of engrafting both as constituents of multinucleate muscle fibers and as self-renewed mononuclear huSCs.
The analysis of skeletal muscle regeneration and SC self-renewal following transplantation indicated that huSCs expanded in the presence of p38i exhibited tissue regenerative and self-renewal potential exceeding that of even freshly isolated quiescent huSCs. Given that quiescent huSCs are no more differentiated than p38i-treated cultured huSCs, the incongruent engraftment of these populations was surprising because it suggested that treating activated huSCs with p38i enhanced engraftment of the transplanted cells in part via a mechanism that was independent of blocking differentiation. We, therefore, sought to identify alternative mechanisms accounting for the enhanced self-renewal and tissue regenerative potential of p38i-treated huSCs using our RNA-seq analyses of these cell populations. In addition to the marked changes in gene expression noted previously (Figure 3; Table S4), we were intrigued to identify significant (p = 0.028) enrichment for genes associated with aging (GO:0007568) among the list of genes downregulated by p38 inhibition. This observation was particularly interesting in light of recent studies in which treatment of old mouse SCs with p38i was observed to rejuvenate aspects of their regenerative function, including their potential to regenerate damaged muscle following transplantation (Bernet et al., 2014, Cosgrove et al., 2014). We discovered six aging-associated genes with decreased expression in p38i-treated relative to untreated cultured huSCs: LIMS1, CRYAB, ENG, ENO3, LRP1, and SERPINE1. Of these, expression of CRYAB, ENG, LRP1, and SERPINE1 also was reduced by more than 50% in p38i-treated huSCs relative to quiescent huSCs. Based on these unbiased analyses of genome expression data, we propose that these genes in particular may function as important regulators of huSC self-renewal downstream of p38.
Discussion
In this study, we used a unique profile of surface markers to identify and prospectively isolate huSCs from surgical specimens. Importantly, these markers were observed to be expressed in situ by huSCs associated with whole human skeletal muscle fibers. Additionally, the marker profile used in this study was able to prospectively identify huSCs from each of three different muscles analyzed and from subjects of various ages. These markers, therefore, represent a reproducible and broadly applicable signature of huSCs. Additional analysis of huSC markers may allow simplification of the sorting strategy presented here. For instance, CD31 may redundantly identify CD34-expressing endothelial cells and may not be necessary to isolate a pure population of huSCs. As in previous studies (Bareja et al., 2014, Pisani et al., 2010b), we found that the predominant myogenic population did not express CD34. In contrast to previous studies, however, we found little evidence for a second myogenic population within the CD34-expressing cell population. Analysis of myogenic marker expression in single cells and clones revealed an absence of spontaneous myogenic activity within CD34+ cells. Future studies are required to determine what technical variables account for differences in the characterization of the myogenic potential of CD34-expressing progenitors in human muscle.
We report an RNA-seq-based characterization of huSCs, including prospectively isolated quiescent huSCs analyzed immediately after sorting. A number of observations stemming from our studies support the characterization of these cells as quiescent. Most notably, these cells were non-dividing upon isolation, as clearly manifested in our time-lapse microscopy studies. Indeed, although the isolation and culture in high-mitogen conditions induces these cells to activate, the huSCs typically did not undergo their first division until after more than 3 days in culture. The RNA-seq data provide additional confirmation of the quiescent nature of the prospectively isolated huSCs, which expressed low levels of numerous proliferation-related genes relative to the cultured huSCs. That these purified huSCs are functionally quiescent is not to say that they are identical to the huSCs in vivo. In fact, we know that the process of isolating the cells, which takes 5–6 hr on average and involves physical and enzymatic perturbations of the cell and its environment, is enough to induce biochemical changes within the huSCs. The extent of these differences, however, is difficult to assess given that the most thorough characterizations of the cells, such as RNA-seq, obviously cannot be performed on cells in vivo. Still, we know from several studies of SC quiescence in mouse that characterizations of FACS-purified cells can offer insight into the biology of SC quiescence in vivo (Cheung et al., 2012, Hausburg et al., 2015, Liu et al., 2013, Rodgers et al., 2014).
These studies add to a growing body of information related to the role of p38 MAPK signaling in skeletal muscle progenitor cells. Over a decade ago, a requirement for p38 in the regulation of rodent myoblast differentiation was defined (Cuenda and Cohen, 1999, Zetser et al., 1999). Based on these prior studies, the functions of p38 MAPK are clearly diverse and complex. p38 activation correlates with rodent SC activation, and pharmacologic inhibition of p38α/β can promote muscle progenitor cell-cycle exit (Jones et al., 2005). Intriguingly, and somewhat paradoxically, genetic absence of p38α, the dominant p38 isoform regulating the myogenic program in mice (Lovett et al., 2010), enhances proliferative expansion of the murine SC population postnatally and in response to injury, although p38α is not absolutely required for myogenic differentiation in vivo (Brien et al., 2013). That the p38α isoform is not required for differentiation may be due to redundant functions of p38β or ɣ isoforms, both of which are expressed in muscle and are capable of independently regulating muscle progenitor differentiation (Wang et al., 2008). Our studies suggest that, in humans, there is a switch that occurs in the activation of p38 as SCs are activated in culture, akin to the activation of p38 observed in mouse (Jones et al., 2005), going from an inactive unphosphorylated form in quiescent huSCs to an active phosphorylated form in cultured huSCs. Altogether, our data and previously published studies of p38 in rodent muscle progentiors support a model in which inhibition of p38 activity promotes exponential growth of progenitors by maintaining an undifferentiated fate. This model seems to be conserved between mice and humans. We further demonstrate that pharmacologic manipulation of p38 signaling can be leveraged for enhancement of both ex vivo expansion and subsequent in vivo engraftment of huSCs.
In our investigation, transplantation of huSCs expanded in the presence of p38i, generated more chimeric muscle fibers, and self-renewed huSCs in comparison to freshly isolated cells. What might account for the enhanced regenerative potential of expanded huSCs? One possibility is that the activated state of expanded cells relative to quiescent cells enabled the expanded cells to proliferate more during the course of muscle regeneration. In previous studies of cultured mouse SCs, this phenomenon may not have manifested as efficient engraftment because activation coincided with differentiation. By divorcing SC activation from differentiation, p38 inhibition may have revealed an increased engraftment potential of activated relative to quiescent cells. This possibility is particularly interesting given that we observed slower activation kinetics in purified huSCs relative to what has been reported for their mouse counterparts (Rodgers et al., 2014). An alternative explanation for the enhanced regenerative potential of the expanded huSCs is that p38 inhibition induces an epigenetic change in the cultured cells that promotes their engraftment potential. This hypothesis may relate to recent observations in aforementioned studies of mouse SCs, which showed an improved function in aged mouse SCs following treatment with a p38 inhibitor (Bernet et al., 2014, Cosgrove et al., 2014). The huSCs used in our study were isolated from donors with an age that greatly exceeds that of even aged mice. It is, therefore, plausible that human adult SCs exhibit a favorable response to treatment with p38i analogous to that observed in SCs from aged mice.
Although the mechanisms of this epigenetic restoration of SC function have not been elucidated, our analysis of the gene expression changes stimulated by p38 inhibition identified several intriguing candidate regulators, including ENG, CRYAB, and SERPINE1. Our current understanding of the functions of these proteins suggests a plausible role for each in the aging of huSCs. ENG encodes endoglin, a membrane glycoprotein important in TGF-β signal transduction (Cheifetz et al., 1992). Increased expression of endoglin has been associated with aging phenotypes within endothelial and myeloid lineages (Aristorena et al., 2014, Blanco and Bernabeu, 2011), and excessive TGF-β signaling has been linked to impaired self-renewal of mouse SCs in the context of aging and chronic disease (Biressi et al., 2014, Carlson et al., 2008). Alpha-crystallin B chain (CRYAB) is a small heat shock protein, decreased expression of which has been linked to improved outcomes following cardiac ischemia in aged mice (Benjamin et al., 2007). SERPINE1, also known as plasminogen activator inhibitor-1 (PAI-1), is upregulated in senescent human cells and in various tissues of progeroid mouse models, including skeletal muscle (Baker et al., 2011, Mu and Higgins, 1995). Remarkably, genetic or pharmacologic inhibition of PAI-1 has been shown to improve organ function and extend the lifespan of progeroid mice (Eren et al., 2014). Future studies will be important to define the specific role of these genes during aging and muscle regeneration as regulators of huSCs downstream of p38 MAPK signaling.
Experimental Procedures
Human Skeletal Muscle Specimens
Surgical specimens of human skeletal muscle were isolated in accordance with the Stanford Institutional Review Board. All experiments, unless otherwise noted, were performed using specimens of latissimus dorsi muscle. Additional details are included in the Supplemental Experimental Procedures.
HuSC Isolation
Human skeletal muscle tissue was first minced and incubated in collagenase II (750 U/ml, Worthington Biochemical) in Ham’s F10 medium supplemented with 10% horse serum (Invitrogen) and 1% penicillin-streptomycin (Omega Scientific) at 37°C for 90 min in a shaking water bath (60 rpm). The digested muscle was washed in F10 with 10% horse serum and digested further in a shaking water bath at 37°C for 30 min in collagenase II (100 U/ml) and dispase (2 U/ml, Gibco) in F10 with 10% horse serum. Subsequent steps in the isolation procedure are detailed in the Supplemental Experimental Procedures.
Human Muscle Fiber Explants and Immunofluorescence Analysis
Human skeletal muscle specimens were incubated in collagenase (500 U/ml) in Ham’s F10 supplemented with 10% horse serum and 1% penicillin-streptomycin for 80 min in a shaking water bath. Tissue was then triturated with a glass Pasteur pipet to separate single muscle fibers. Additional details are included in the Supplemental Experimental Procedures.
HuSC Culture
Slides or plates were coated with extracellular matrix protein (ECM, Sigma) at a concentration of 1:500 (v/v) in DMEM with 1% penicillin-streptomycin. Culture medium was a 1:1 mixture of DMEM:MCDB supplemented with 20% FBS (Omega Scientific), 1% insulin-transferrin-selenium (ITS, Invitrogen), and 1% penicillin-streptomycin. Four serum lots from two manufacturers (Omega Scientific and Atlanta Biologicals) were tested in assays of huSC growth and differentiation. Additional protocols for SC culture are presented in the Supplemental Experimental Procedures.
qRT-PCR
Freshly isolated huSCs or huSC-depleted cells were cultured for 7 days prior to analysis. For RNA isolation, cells were first rinsed twice in PBS. Cells were then lysed and RNA was isolated using an RNeasy Mini Kit (QIAGEN) according to the manufacturer’s instructions. Additional details are described in the Supplemental Experimental Procedures.
Statistical Analysis
All statistical tests, except those involving gene set enrichment analyses, were unpaired, two-tailed t tests performed using GraphPad Prism 6.0. All error bars represent SDs, unless otherwise noted. Data from each experiment represent statistics compiled from true biological replicates, defined here as experiments performed with similar, but entirely unique, biological reagents. In the context of the human studies presented here, including the RNA-seq analyses, replicate experiments were always performed using cells from distinct donors.
Acknowledgments
The authors gratefully acknowledge the assistance of Amanda Khuong in the Department of Cardiothoracic Surgery and of Ziming Weng, Keith Bettinger, and Nathan Hammond at the Stanford Center for Genomics and Personalized Medicine. This work was supported by the Glenn Foundation for Medical Research, fellowships from the NIH (F30 AG035521), and Stanford University School of Medicine Medical Scientist Training Program to G.W.C.; by grants from the Department of Veterans Affairs (Merit Review) to J.B.S.; and by grants from the Department of Veterans Affairs (Merit Review) and the NIH (P01 AG036695, R37 AG23806, and R01 AR062185) to T.A.R.
Published: September 3, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, four figures, four tables, and one movie and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2015.08.004.
Accession Numbers
The accession number for the RNA-seq data reported in this paper is European Nucleotide Archive (ENA): PRJEB10091 (http://www.ebi.ac.uk/ena/data/view/PRJEB10091).
Supplemental Information
The 30 most highly upregulated transcription factors, receptors, and signaling molecules, as defined by the PANTHER gene ontology database, derived from RNA-seq comparisons of quiescent and activated huSCs. Loci for which the average fragments per kilobase of exon per million fragments mapped (FPKM) was <5 were excluded.
The biological processes, as defined by the PANTHER gene ontology database, significantly enriched (p < 0.05) among genes upregulated in activated relative to quiescent huSCs. Gene set enrichment was analyzed using the DAVID bioinformatics platform. The associated genes are listed with each term. The analysis used those loci for which FPKM > 5 and fold change > 2.
The biological processes, as defined by the PANTHER gene ontology database, significantly enriched (p < 0.05) among genes upregulated in quiescent relative to activated huSCs. Gene set enrichment was analyzed using the DAVID bioinformatics platform. The associated genes are listed with each term. The analysis used those loci for which FPKM > 5 and fold change > 2.
The biological processes, as defined by the PANTHER gene ontology database, significantly enriched (p < 0.05) among genes with increased or decreased expression in p38i-treated huSCs relative to untreated huSCs. Gene set enrichment was analyzed using the DAVID bioinformatics platform. The associated genes are listed with each term. The analysis used those loci for which FPKM > 5 and fold change > 2.
The movie begins 12 hr (t = 0) post-isolation and continues for 90 hr (t = 90). Relative time is indicated in yellow.
{kind=link}
References
- Aristorena M., Blanco F.J., de Las Casas-Engel M., Ojeda-Fernandez L., Gallardo-Vara E., Corbi A., Botella L.M., Bernabeu C. Expression of endoglin isoforms in the myeloid lineage and their role during aging and macrophage polarization. J. Cell Sci. 2014;127:2723–2735. doi: 10.1242/jcs.143644. [DOI] [PubMed] [Google Scholar]
- Baker D.J., Wijshake T., Tchkonia T., LeBrasseur N.K., Childs B.G., van de Sluis B., Kirkland J.L., van Deursen J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bareja A., Holt J.A., Luo G., Chang C., Lin J., Hinken A.C., Freudenberg J.M., Kraus W.E., Evans W.J., Billin A.N. Human and mouse skeletal muscle stem cells: convergent and divergent mechanisms of myogenesis. PLoS ONE. 2014;9:e90398. doi: 10.1371/journal.pone.0090398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin I.J., Guo Y., Srinivasan S., Boudina S., Taylor R.P., Rajasekaran N.S., Gottlieb R., Wawrousek E.F., Abel E.D., Bolli R. CRYAB and HSPB2 deficiency alters cardiac metabolism and paradoxically confers protection against myocardial ischemia in aging mice. Am. J. Physiol. Heart Circ. Physiol. 2007;293:H3201–H3209. doi: 10.1152/ajpheart.01363.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernet J.D., Doles J.D., Hall J.K., Kelly Tanaka K., Carter T.A., Olwin B.B. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat. Med. 2014;20:265–271. doi: 10.1038/nm.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biressi S., Miyabara E.H., Gopinath S.D., Carlig P.M.M., Rando T.A. A Wnt-TGFβ2 axis induces a fibrogenic program in muscle stem cells from dystrophic mice. Sci. Transl. Med. 2014;6:267ra176. doi: 10.1126/scitranslmed.3008411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornson C.R.R., Cheung T.H., Liu L., Tripathi P.V., Steeper K.M., Rando T.A. Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells. 2012;30:232–242. doi: 10.1002/stem.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco F.J., Bernabeu C. Alternative splicing factor or splicing factor-2 plays a key role in intron retention of the endoglin gene during endothelial senescence. Aging Cell. 2011;10:896–907. doi: 10.1111/j.1474-9726.2011.00727.x. [DOI] [PubMed] [Google Scholar]
- Boldrin L., Muntoni F., Morgan J.E. Are human and mouse satellite cells really the same? J. Histochem. Cytochem. 2010;58:941–955. doi: 10.1369/jhc.2010.956201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosnakovski D., Xu Z., Li W., Thet S., Cleaver O., Perlingeiro R.C.R., Kyba M. Prospective isolation of skeletal muscle stem cells with a Pax7 reporter. Stem Cells. 2008;26:3194–3204. doi: 10.1634/stemcells.2007-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brien P., Pugazhendhi D., Woodhouse S., Oxley D., Pell J.M. p38α MAPK regulates adult muscle stem cell fate by restricting progenitor proliferation during postnatal growth and repair. Stem Cells. 2013;31:1597–1610. doi: 10.1002/stem.1399. [DOI] [PubMed] [Google Scholar]
- Carlson M.E., Hsu M., Conboy I.M. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature. 2008;454:528–532. doi: 10.1038/nature07034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheifetz S., Bellón T., Calés C., Vera S., Bernabeu C., Massagué J., Letarte M. Endoglin is a component of the transforming growth factor-beta receptor system in human endothelial cells. J. Biol. Chem. 1992;267:19027–19030. [PubMed] [Google Scholar]
- Cheung T.H., Quach N.L., Charville G.W., Liu L., Park L., Edalati A., Yoo B., Hoang P., Rando T.A. Maintenance of muscle stem-cell quiescence by microRNA-489. Nature. 2012;482:524–528. doi: 10.1038/nature10834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C.A., Olsen I., Zammit P.S., Heslop L., Petrie A., Partridge T.A., Morgan J.E. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell. 2005;122:289–301. doi: 10.1016/j.cell.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Conboy I.M., Rando T.A. The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev. Cell. 2002;3:397–409. doi: 10.1016/s1534-5807(02)00254-x. [DOI] [PubMed] [Google Scholar]
- Cosgrove B.D., Gilbert P.M., Porpiglia E., Mourkioti F., Lee S.P., Corbel S.Y., Llewellyn M.E., Delp S.L., Blau H.M. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat. Med. 2014;20:255–264. doi: 10.1038/nm.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenda A., Cohen P. Stress-activated protein kinase-2/p38 and a rapamycin-sensitive pathway are required for C2C12 myogenesis. J. Biol. Chem. 1999;274:4341–4346. doi: 10.1074/jbc.274.7.4341. [DOI] [PubMed] [Google Scholar]
- Davies S.P., Reddy H., Caivano M., Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren M., Boe A.E., Murphy S.B., Place A.T., Nagpal V., Morales-Nebreda L., Urich D., Quaggin S.E., Budinger G.R.S., Mutlu G.M. PAI-1-regulated extracellular proteolysis governs senescence and survival in Klotho mice. Proc. Natl. Acad. Sci. USA. 2014;111:7090–7095. doi: 10.1073/pnas.1321942111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyers P.A., Craxton M., Morrice N., Cohen P., Goedert M. Conversion of SB 203580-insensitive MAP kinase family members to drug-sensitive forms by a single amino-acid substitution. Chem. Biol. 1998;5:321–328. doi: 10.1016/s1074-5521(98)90170-3. [DOI] [PubMed] [Google Scholar]
- Fukada S., Uezumi A., Ikemoto M., Masuda S., Segawa M., Tanimura N., Yamamoto H., Miyagoe-Suzuki Y., Takeda S. Molecular signature of quiescent satellite cells in adult skeletal muscle. Stem Cells. 2007;25:2448–2459. doi: 10.1634/stemcells.2007-0019. [DOI] [PubMed] [Google Scholar]
- Gilbert P.M., Havenstrite K.L., Magnusson K.E.G., Sacco A., Leonardi N.A., Kraft P., Nguyen N.K., Thrun S., Lutolf M.P., Blau H.M. Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science. 2010;329:1078–1081. doi: 10.1126/science.1191035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding J.P., Calderbank E., Partridge T.A., Beauchamp J.R. Skeletal muscle stem cells express anti-apoptotic ErbB receptors during activation from quiescence. Exp. Cell Res. 2007;313:341–356. doi: 10.1016/j.yexcr.2006.10.019. [DOI] [PubMed] [Google Scholar]
- Gum R.J., McLaughlin M.M., Kumar S., Wang Z., Bower M.J., Lee J.C., Adams J.L., Livi G.P., Goldsmith E.J., Young P.R. Acquisition of sensitivity of stress-activated protein kinases to the p38 inhibitor, SB 203580, by alteration of one or more amino acids within the ATP binding pocket. J. Biol. Chem. 1998;273:15605–15610. doi: 10.1074/jbc.273.25.15605. [DOI] [PubMed] [Google Scholar]
- Hall J.K., Banks G.B., Chamberlain J.S., Olwin B.B. Prevention of muscle aging by myofiber-associated satellite cell transplantation. Sci. Transl. Med. 2010;2:57ra83. doi: 10.1126/scitranslmed.3001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausburg M.A., Doles J.D., Clement S.L., Cadwallader A.B., Hall M.N., Blackshear P.J., Lykke-Andersen J., Olwin B.B. Post-transcriptional regulation of satellite cell quiescence by TTP-mediated mRNA decay. eLife. 2015;4:e03390. doi: 10.7554/eLife.03390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones N.C., Tyner K.J., Nibarger L., Stanley H.M., Cornelison D.D.W., Fedorov Y.V., Olwin B.B. The p38α/β MAPK functions as a molecular switch to activate the quiescent satellite cell. J. Cell Biol. 2005;169:105–116. doi: 10.1083/jcb.200408066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpati G., Pouliot Y., Zubrzycka-Gaarn E., Carpenter S., Ray P.N., Worton R.G., Holland P. Dystrophin is expressed in mdx skeletal muscle fibers after normal myoblast implantation. Am. J. Pathol. 1989;135:27–32. [PMC free article] [PubMed] [Google Scholar]
- Kuma Y., Campbell D.G., Cuenda A. Identification of glycogen synthase as a new substrate for stress-activated protein kinase 2b/p38beta. Biochem. J. 2004;379:133–139. doi: 10.1042/BJ20031559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen J.K., Yamboliev I.A., Weber L.A., Gerthoffer W.T. Phosphorylation of the 27-kDa heat shock protein via p38 MAP kinase and MAPKAP kinase in smooth muscle. Am. J. Physiol. 1997;273:L930–L940. doi: 10.1152/ajplung.1997.273.5.L930. [DOI] [PubMed] [Google Scholar]
- Leroy M.C., Perroud J., Darbellay B., Bernheim L., Konig S. Epidermal growth factor receptor down-regulation triggers human myoblast differentiation. PLoS ONE. 2013;8:e71770. doi: 10.1371/journal.pone.0071770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Cheung T.H., Charville G.W., Hurgo B.M.C., Leavitt T., Shih J., Brunet A., Rando T.A. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013;4:189–204. doi: 10.1016/j.celrep.2013.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lluís F., Perdiguero E., Nebreda A.R., Muñoz-Cánoves P. Regulation of skeletal muscle gene expression by p38 MAP kinases. Trends Cell Biol. 2006;16:36–44. doi: 10.1016/j.tcb.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Lovett F.A., Cosgrove R.A., Gonzalez I., Pell J.M. Essential role for p38alpha MAPK but not p38gamma MAPK in Igf2 expression and myoblast differentiation. Endocrinology. 2010;151:4368–4380. doi: 10.1210/en.2010-0209. [DOI] [PubMed] [Google Scholar]
- Mauro A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montarras D., Morgan J., Collins C., Relaix F., Zaffran S., Cumano A., Partridge T., Buckingham M. Direct isolation of satellite cells for skeletal muscle regeneration. Science. 2005;309:2064–2067. doi: 10.1126/science.1114758. [DOI] [PubMed] [Google Scholar]
- Mourikis P., Sambasivan R., Castel D., Rocheteau P., Bizzarro V., Tajbakhsh S. A critical requirement for notch signaling in maintenance of the quiescent skeletal muscle stem cell state. Stem Cells. 2012;30:243–252. doi: 10.1002/stem.775. [DOI] [PubMed] [Google Scholar]
- Mu X.C., Higgins P.J. Differential growth state-dependent regulation of plasminogen activator inhibitor type-1 expression in senescent IMR-90 human diploid fibroblasts. J. Cell. Physiol. 1995;165:647–657. doi: 10.1002/jcp.1041650324. [DOI] [PubMed] [Google Scholar]
- Parker M.H., Loretz C., Tyler A.E., Duddy W.J., Hall J.K., Olwin B.B., Bernstein I.D., Storb R., Tapscott S.J. Activation of Notch signaling during ex vivo expansion maintains donor muscle cell engraftment. Stem Cells. 2012;30:2212–2220. doi: 10.1002/stem.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge T.A., Morgan J.E., Coulton G.R., Hoffman E.P., Kunkel L.M. Conversion of mdx myofibres from dystrophin-negative to -positive by injection of normal myoblasts. Nature. 1989;337:176–179. doi: 10.1038/337176a0. [DOI] [PubMed] [Google Scholar]
- Pisani D.F., Clement N., Loubat A., Plaisant M., Sacconi S., Kurzenne J.-Y., Desnuelle C., Dani C., Dechesne C.A. Hierarchization of myogenic and adipogenic progenitors within human skeletal muscle. Stem Cells. 2010;28:2182–2194. doi: 10.1002/stem.537. [DOI] [PubMed] [Google Scholar]
- Pisani D.F., Dechesne C.A., Sacconi S., Delplace S., Belmonte N., Cochet O., Clement N., Wdziekonski B., Villageois A.P., Butori C. Isolation of a highly myogenic CD34-negative subset of human skeletal muscle cells free of adipogenic potential. Stem Cells. 2010;28:753–764. doi: 10.1002/stem.317. [DOI] [PubMed] [Google Scholar]
- Rocheteau P., Gayraud-Morel B., Siegl-Cachedenier I., Blasco M.A., Tajbakhsh S. A subpopulation of adult skeletal muscle stem cells retains all template DNA strands after cell division. Cell. 2012;148:112–125. doi: 10.1016/j.cell.2011.11.049. [DOI] [PubMed] [Google Scholar]
- Rodgers J.T., King K.Y., Brett J.O., Cromie M.J., Charville G.W., Maguire K.K., Brunson C., Mastey N., Liu L., Tsai C.-R. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert) Nature. 2014;510:393–396. doi: 10.1038/nature13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco A., Doyonnas R., Kraft P., Vitorovic S., Blau H.M. Self-renewal and expansion of single transplanted muscle stem cells. Nature. 2008;456:502–506. doi: 10.1038/nature07384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood R.I., Christensen J.L., Conboy I.M., Conboy M.J., Rando T.A., Weissman I.L., Wagers A.J. Isolation of adult mouse myogenic progenitors: functional heterogeneity of cells within and engrafting skeletal muscle. Cell. 2004;119:543–554. doi: 10.1016/j.cell.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Shi H., Boadu E., Mercan F., Le A.M., Flach R.J.R., Zhang L., Tyner K.J., Olwin B.B., Bennett A.M. MAP kinase phosphatase-1 deficiency impairs skeletal muscle regeneration and exacerbates muscular dystrophy. FASEB J. 2010;24:2985–2997. doi: 10.1096/fj.09-150045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow M.H. An autoradiographic study of satellite cell differentiation into regenerating myotubes following transplantation of muscles in young rats. Cell Tissue Res. 1978;186:535–540. doi: 10.1007/BF00224941. [DOI] [PubMed] [Google Scholar]
- Wang H., Xu Q., Xiao F., Jiang Y., Wu Z. Involvement of the p38 mitogen-activated protein kinase α, β, and γ isoforms in myogenic differentiation. Mol. Biol. Cell. 2008;19:1519–1528. doi: 10.1091/mbc.E07-08-0817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetser A., Gredinger E., Bengal E. p38 mitogen-activated protein kinase pathway promotes skeletal muscle differentiation. Participation of the Mef2c transcription factor. J. Biol. Chem. 1999;274:5193–5200. doi: 10.1074/jbc.274.8.5193. [DOI] [PubMed] [Google Scholar]
- Zheng B., Cao B., Crisan M., Sun B., Li G., Logar A., Yap S., Pollett J.B., Drowley L., Cassino T. Prospective identification of myogenic endothelial cells in human skeletal muscle. Nat. Biotechnol. 2007;25:1025–1034. doi: 10.1038/nbt1334. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The 30 most highly upregulated transcription factors, receptors, and signaling molecules, as defined by the PANTHER gene ontology database, derived from RNA-seq comparisons of quiescent and activated huSCs. Loci for which the average fragments per kilobase of exon per million fragments mapped (FPKM) was <5 were excluded.
The biological processes, as defined by the PANTHER gene ontology database, significantly enriched (p < 0.05) among genes upregulated in activated relative to quiescent huSCs. Gene set enrichment was analyzed using the DAVID bioinformatics platform. The associated genes are listed with each term. The analysis used those loci for which FPKM > 5 and fold change > 2.
The biological processes, as defined by the PANTHER gene ontology database, significantly enriched (p < 0.05) among genes upregulated in quiescent relative to activated huSCs. Gene set enrichment was analyzed using the DAVID bioinformatics platform. The associated genes are listed with each term. The analysis used those loci for which FPKM > 5 and fold change > 2.
The biological processes, as defined by the PANTHER gene ontology database, significantly enriched (p < 0.05) among genes with increased or decreased expression in p38i-treated huSCs relative to untreated huSCs. Gene set enrichment was analyzed using the DAVID bioinformatics platform. The associated genes are listed with each term. The analysis used those loci for which FPKM > 5 and fold change > 2.
The movie begins 12 hr (t = 0) post-isolation and continues for 90 hr (t = 90). Relative time is indicated in yellow.