

Abstract
Transforming growth factor beta (TGF-β) is the most potent pro-fibrogenic cytokine and its expression is increased in almost all of fibrotic diseases. Although signaling through Smad pathway is believed to play a central role in TGF-β's fibrogenesis, emerging evidence indicates that reactive oxygen species (ROS) modulate TGF-β's signaling through different pathways including Smad pathway. TGF-β1 increases ROS production and suppresses antioxidant enzymes, leading to a redox imbalance. ROS, in turn, induce/activate TGF-β1 and mediate many of TGF-β's fibrogenic effects, forming a vicious cycle (see graphic flow chart on the right). Here, we review the current knowledge on the feed-forward mechanisms between TGF-β1 and ROS in the development of fibrosis. Therapeutics targeting TGF-β-induced and ROS-dependent cellular signaling represents a novel approach in the treatment of fibrotic disorders.
Keywords: TGF-β, Oxidative stress, Fibrosis, NADPH oxidases, PAI-1
Graphical abstract
Highlights
-
•
TGF-β1 is the most potent ubiquitous profibrogenic cytokine.
-
•
TGF- β 1 induces redox imbalance by ↑ ROS production and ↓ anti-oxidant defense system
-
•
Redox imbalance, in turn, activates latent TGF-β1 and induces TGF-β1 expression.
-
•
Redox imbalance also mediates many of TGF-β1’s profibrogenic effects
1. Introduction
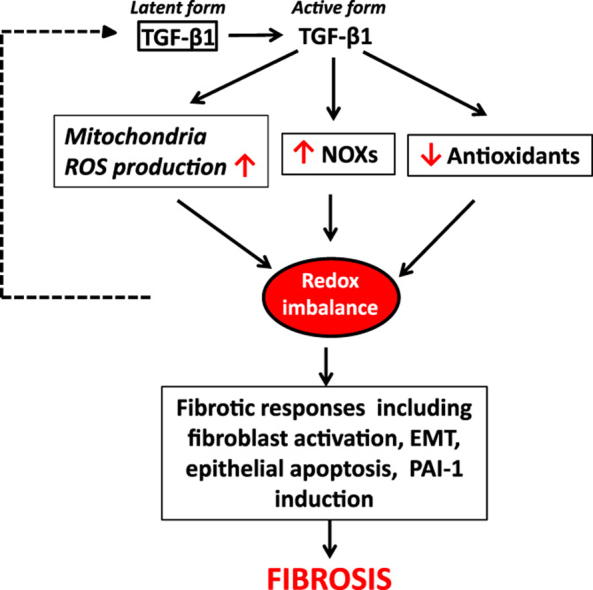
Transforming growth factor beta (TGF-β), the most potent and ubiquitous profibrogenic cytokine, plays a central role in the development of fibrosis involving almost all organ systems [1], [2], [3], [4]. Although TGF-β1 signaling through Smad pathway is believed to be responsible for the induction of many of TGF-β's responsive genes emerging evidence indicates that reactive oxygen (ROS) mediate TGF-β's signaling through different pathways including Smad pathway, mitogen activated protein kinase (MAPK) pathways, and Rho-GTPase pathway. TGF-β1 increases the production of ROS by impairing mitochondrial function and inducing NADPH oxidases (NOXs), mainly Nox4, a non-phagocytic NOX expressed by many different types of cells. TGF-β also suppresses antioxidant system including the synthesis of glutathione (GSH), the most abundant intracellular free thiol and an important antioxidant, and several other antioxidant enzymes, leading to oxidative stress or redox imbalance. Such a redox imbalance in turn induces/activates TGF-β1 and mediates TGF-β's fibrogenic effects [5]. In this review, we will focus on the mechanisms whereby TGF-β induces Nox4 and causes redox imbalance as well as the mechanisms whereby ROS activate/induce TGF-β and mediate TGF-β's fibrogenic effects. Suppression of TGF-β-induced ROS production may break this vicious cycle and have therapeutic potential for the treatment of fibrotic disorders.
2. TGF-β1 and fibrosis
Fibrosis is characterized by increased deposition of extracellular matrix (ECM) proteins in the interstitial, leading to stiffness and loss of organ architecture and function. Fibrosis affects almost all organ systems and accounts for 45% of disease-related death. Many cytokines/chemokines/growth factors contribute to the development of fibrosis; however, TGF-β is considered to be the most potent and ubiquitous profibrogenic cytokine. TGF-β mRNA and/or protein expression is increased in almost all fibrotic diseases involved in different organ systems and in experimental fibrosis models [6], [7], [8], [9], [10], [11], [12], [13], [14], [15], [16], [17], [18]. Overexpression of TGF-β induces [19], [20], [21], [22], [23], [24], [25], [26], [27], [28], [29] whereas administration of TGF-β binding proteins, anti-TGF-β antibody, or an inhibitor to TGF-β type I receptor ALK5 ameliorated fibrosis [24], [30], [31], [32], [33]. All these lines of evidence suggest that TGF-β plays a pivotal role in the development of fibrosis. Although it has been well documented that TGF-β exits its profibrogenic activity through activating fibroblasts, a recent study shows that epithelium-specific deletion of TGF-β receptor type II protects mice from bleomycin-induced lung fibrosis, suggesting that TGF-β signaling in epithelial cells is also important for its fibrogenic effects [34].
TGF-β, existing in three isoforms, TGFβ1, TGFβ2, and TGFβ3, is secreted by many different types of cells and involved in various cell functions including cell proliferation, differentiation, apoptosis, adhesion, and migration. Although all three isoforms are expressed in fibrotic tissues, the development of lung fibrosis is primarily attributed to TGFβ1 [35]. TGF-β1 signaling through Smad pathway, the canonical pathway, has been well described. Active TGF-β binds to type II receptor (TGFβR-II) on the cell membrane, which activates type I receptor (TGFβR-I), leading to phosphorylation of Smad2 and Smad3. Phosphorylated Smad2 and Smad3 then form a complex with common mediator Smad (Co-Smad), Smad4, which translocates to the nucleus and regulates gene transcription through binding to Smad binding elements present in the promoter of the target genes [36]. There are 7 mammalian type I receptors, termed ALK1-7 (activin receptor-like kinase 1-7) and TGF-β1 signaling mainly through ALK-5 [37]. Inhibitory Smads, Smad6 and Smad7, on the other hand, negatively regulate TGF-β signaling by binding to type I receptor or by competing with Smad2/3 for binding to Smad4 [38], [39], [40]. Numerous studies have shown that TGF-β1 plays a central role in the development of fibrosis in many tissues/organs under various pathological conditions through ALK-5 and Smad2/3 pathway [41], [42], [43], [44], [45], [46], [47]. The counter-regulatory role of Smad7 in the development of fibrosis has also been demonstrated in several cell types and in vivo [40], [48], [49]. Besides Smad pathways, studies have shown that TGF-β signaling through other non-canonical pathways, including mitogen activated protein kinase (MAPK) pathways, phosphatidylinositol-3-kinase (PI3K) pathway, and Rho-like GTPase pathways, are also critical for eliciting TGF-β's profibrogenic activity [25], [38], [50], [51], [52], [53], [54], [55], [56], [57], [58], [59], [60], [61]. TGF-β has been shown to activate MAPK pathways through TGF-β-activated kinase 1 (TAK1), a MAPK kinase kinase, or through Ras [38] in different types of cells [25], [51], [52], [53], [54], [55], [56], [57]. Importantly, it has been well documented that MAPK pathways are redox sensitivity, although the redox sensitivity molecules in these pathways remain to be identified. Rho-GTPases, a subfamily of small GTP-binding proteins within the Ras superfamily, regulates actin cytoskeleton, cell shape, adhesion, and migration. Rho-GTPase and its downstream effector ROCK have been shown to be involved in TGF-β-induced myofibroblast differentiation [61], [62], [63], [64], [65], [66], [67]. Interestingly, it has been reported that ROS derived from mitochondria, NADPH oxidases, or other sources activate RhoA [68], [69], [70], [71], [72], [73], [74] and that Nox4-derived ROS mediate TGF-β1-induced kidney myofibroblast differentiation through activating RhoA/Rho kinase pathway [61].
3. TGF-β induces redox imbalance
Redox imbalance or oxidative stress results from an increased production of reactive oxygen or nitrogen species (ROS/RNS) and/or reduced antioxidant capacity. ROS/RNS such as superoxide, hydrogen peroxide (H2O2), and nitric oxide (NO) are formed as a byproduct of the normal metabolism of oxygen and have important function in cell signaling and homeostasis. Overproduction of ROS/RNS, however, contributes to the pathophysiology of many diseases. Biological systems have developed superior antioxidant mechanisms, enzymatic and non-enzymatic, to scavenge or remove ROS/RNS generated during normal metabolism or under pathological conditions. The enzymatic system comprises mainly the superoxide dismutases (SODs), catalase, glutathione peroxidase (GPx), and peroxiredoxin; whereas non-enzymatic system includes glutathione (GSH), ascorbic acid, β-carotene, and α-tocopherol. TGF-β has been shown to increase ROS production and suppress antioxidant system and thereby induce oxidative stress or redox imbalance. Such a redox imbalance contributes importantly to TGF-β's pathophysiologic effects including fibrosis [75].
3.1. TGF-β increases mitochondrial ROS production
Mitochondria are the major source of ROS in cells. TGF-β1 has been shown to increase mitochondrial ROS production in different types of cells, which mediate TGF-β-induced cell apoptosis [76], [77], [78], [79], [80], senescence [81], [82], [83], epithelia–mesenchymal transition [84], fibrotic gene expression and myofibroblast differentiation [85], [86]; Ishikawa et al. reported that TGF-β increased ROS levels in the cytoplasm and mitochondria in mouse mammary epithelial cells (NMuMG) and decreased mitochondrial membrane potential [84]. Depletion of mitochondria (pseudo p0 cells), on the other hand, abrogated TGF-β-induced increase in intracellular ROS [84] The authors also showed that exogenously expressed mitochondrial thioredoxin inhibited TGF-β-induced expression of fibronectin and HMGA2, a central mediator of epithelial–mesenchymal transition (EMT), suggesting a novel mechanism involving mitochondria in TGF-β-mediated gene expression associated with EMT [84]. Different mechanisms whereby TGF-β stimulates mitochondrial ROS production have been proposed. Yoon et al showed that TGF-β induced a prolonged mitochondrial ROS production through decreasing complex IV activity in Mv1Lu cells, a mink lung epithelial cell line [81]. Using a specific inhibitor and siRNA to glycogen synthase kinase 3 (GSK3), Byun et al. further showed that phosphorylation of GSK α and β subunits may underlie the inhibition of complex IV activity and thereby mitochondrial ROS production by TGF-β1 [82]. Jain et al reported, on the other hand, that TGF-β increased ROS levels in both normal and fibrotic human lung fibroblasts by blocking complex III activity [85]. They further showed that treatment with mitochondrially targeted antioxidants or genetically disrupting mitochondrial complex III activity attenuated TGF-β-induced expression of profibrotic genes, including alpha smooth muscle actin (α-SMA) and connective tissue growth factor (CTGF), but had no significant effect on Smad phosphorylation or nuclear translocation, suggesting that mitochondria derived ROS mediate TGF-β's fibrogenic activity independent of Smad pathway [85]. TGF-β has also been shown to increase mitochondria oxygen consumption and ROS production through activating mammalian target of rapamycin (mTOR) pathway in mouse podocytes [87].
3.2. TGF-β increases ROS production through inducing NADPH oxidases
NAPDH oxidases (Noxs) are a group of heme-containing transmembrane proteins and important ROS producers for both phagocytic and non-phagocytic cells, although the biological functions of ROS generated in non-phagocytic cells are unclear at moment. Seven members have been identified in the Nox family: Nox1, Nox2, Nox3, Nox4, Nox5, Dual oxidase1 (Duox1), and Dual oxidase 2 (Duox2). TGF-β has been shown to induce the expression of several Nox enzymes including Nox 1, Nox2, and Nox4 in different types of cells [60], [80], [88], [89], [90], [91], [92], [93], [94], [95], [96], [97], [98], [99], [100], [101]. Of all Nox family members, the induction and role of Nox4 in TGF-β's fibrogenesis has been studied most [60], [90], [91], [92], [93], [94], [95], [96], [97], [98], [99], [100], [101]. Different from other Nox isoforms, Nox 4 is constitutively active and is expressed by many types of non-phagocytic cells including epithelial, endothelial, macrophage, and fibroblasts. Nox4 protein has been found to be associated with different cell compartments including endoplasmic reticulum (ER) [94], [102], [103], [104], perinuclear space [105], nucleus [94], [100], [106], and mitochondria [107], [108], [109]. A large body of literatures has shown that TGF-β induces Nox4 expression in various types of cells [80], [91], [93], [94], [95], [96], [97], [98], [99], [100], [101], [110] and that Nox4-derived ROS mediate TGF-β's fibrogenic effects, including fibroblast activation/myofibroblast differentiation [88], [92], [96], [111], [112], [113], epithelial and endothelial cell apoptosis [80], [91], [98], [114], epithelia–mesenchymal transition [101], and the expression of fibrotic/profibrotic genes [58], [88], [100], [115]. An increase in Nox4 expression has also been detected in fibrotic diseases including idiopathic pulmonary fibrosis (IPF), which correlates with increased expression of myofibroblast marker, α-SMA, further supporting the role of Nox4 in fibrotic diseases [111], [116], [117], [118].
Several pathways have been shown to be involved in the induction of Nox4 by TGF-β, including Smad pathway [92], [93], [101], [111], [119], [120], PI3K pathway [95], [119], MAPK pathways [112], [114], and RhoA/ROCK pathway [61]. Hecker et al. showed that TGF-β1 increased Nox4 expression with no effect on the expression of other Nox family members in human lung mesenchymal cells (hFLMCs) [111]. Treatment of hFLMCs with an inhibitor to TGF-β type I receptor (ALK-5) or transfection of cells with Smad3 siRNA almost completely blocked TGF-β-induced Nox4 expression and H2O2 production. In contrast, treatment of cells with inhibitors of to p38/ERK/JNK MAKP pathways had no significant effect on TGF-β-induced Nox4 expression [111], suggesting that Smad pathway not MAPK pathway, mediates the induction of Nox4 by TGF-β in lung mesenchymal cells. Using similar strategies, Yeh and Bondi also showed that Smad3 pathways were involved in Nox4 induction by TGF-β in fibroblasts [92], [120]. TGF-β induction of Nox4 in epithelial cells is also mediated through Smad pathway [101]. Boudreau et al. reported that TGF-β induced Nox4 expression and increased ROS production in human breast epithelial cells (MCF10A and MDA-MB-231); knockdown of Nox4 expression by siRNA or shRNA techniques significantly reduced TGF-β-induced ROS production and fibronectin mRNA expression [101]. Furthermore, they demonstrate that overexpression of constitutively active Smad3 increased whereas treatment with dominant-negative Smad3 or Smad3 inhibitor, SIS3 suppressed TGF-β-induced Nox4 expression [101]. However, conflicting results have been reported regarding whether Nox4-derived ROS is involved in Smad activation in TGF-β signaling process. Bondi et al. showed that TGF-β induces Nox2 and Nox4 in kidney myofibroblasts [92]. Treatment of fibroblasts with Nox inhibitor diphenyleneiodonium (DPI) had no significant effect on TGF-β-induced Smad3 phosphorylation but reduced TGF-β-stimulated α-SMA and fibronectin expression. Moreover, they showed that inhibition of Smad3 with inhibitor SIS3 reduced Nox4 expression and activity as well as TGF-β-induced fibrotic responses. These data further support the notion that Smad3 functions upstream of Nox4 and also suggest that Nox-derived ROS is not involved in Smad activation [92]. In contrast with above finding, Cucoranu et al. reported that TGF-β1 induced Nox4 in cardiac fibroblast; knockdown of Nox4 with Nox4siRNA blocked TGF-β-induced ROS production, α-SMA expression, and Smad2/3 phosphorylation, suggesting that Nox4-derived ROS mediate TGF-β1-induced Smad2/3 activation cardiac fibroblasts [96].
PI3K pathway has also been shown to be involved in the induction of Nox4 by TGF-β [95], [119], Michaeloudes et al. showed that TGF-β induced Nox4 but suppressed the expression of manganese-superoxide dismutase (Mn-SOD) and catalase, associated with an increase in ROS production and IL-6 expression in human airway smooth muscle cells [119]. Inhibition of Smad3 pathway by dominant negative technique or inhibition of PI3K pathway with LY294002 significantly reduced TGF-β1-induced Nox4 mRNA expression in these cells, suggesting that both Smad3 and PI3K pathways are involved in Nox4 induction by TGF-β in these cells [119]. Ismail et al. showed that hypoxia induced TGF-β, insulin-like growth factor binding protein-3 (IGFBP-3), and Nox4 and promoted proliferation in human pulmonary artery smooth muscle cells; anti-TGF-β antibody, on the other hand, attenuated hypoxia-mediated induction of Nox4, IGFBP-3, and cell proliferation [95]. They also showed that inhibition of PI3K pathway, but not Smad pathway, attenuated hypoxia-induced Nox4 and IGFBP-3 expression as well as cell proliferation [95]. Their results suggest that TGF-β mediated hypoxia-induced Nox4 expression and cell proliferation in human pulmonary artery smooth muscle cells through activation of PI3K pathway [95]. Using a pharmaceutical inhibitor and shRNA/siRNA techniques, Tobar et al. reported that JNK pathway, but not Smad pathway, was involved in TGF-β-induced Nox4 expression in human breast stromal cells [112]. Interestingly, Caja et al. showed that TGF-β induced Nox4 in liver tumor cells only when the cells were treated with ERK inhibitor PD98059, suggesting that ERK pathway suppresses Nox4 expression in these cells [114]. Taken together, these data suggest that TGF-β induces Nox4 gene expression in different types of cells through different signaling pathways.
Emerging evidence suggests that there is crosstalk between mitochondria and NADPH oxidases: mitochondria-derived ROS contribute to the increase in NOX expression in response to TGF-β whereas NOX-generated ROS cause mitochondria dysfunction and increase mitochondrial ROS production [121], [122], [123], [124]. It is reported that Nox4 expression is increased in mitochondria of aged mice; suppression of Nox4 expression with Nox4 shRNA decreases mitochondrial ROS level and improves mitochondrial function in vascular smooth muscle cells from aged mice, supporting the notion that a feed-forward relation between mitochondria and Nox4 in ROS production may underlie increased oxidative stress and cardiac dysfunction during aging [125]. Crosstalk between mitochondria and Nox enzyme has also been shown to mediate TGF-β's profibrogenic effect. Jain et al. showed that TGF-β induced the expression of profibrogenic proteins including α-SMA and CTGF through increasing mitochondrial ROS whereas mitochondria targeted antioxidant mitoQ not only suppressed TGF-β-induced expression of profibrotic genes but also the expression of Nox4 [85], suggesting a feed-forward interaction between mitochondria and Nox4 in TGF-β-induced ROS production.
3.3. Suppression of antioxidant system by TGF-β
Besides stimulation of ROS production, TGF-β can also induce redox imbalance by suppressing antioxidant system. Glutathione (GSH) is the most abundant intracellular free thiol and has multiple functions, including detoxification of electrophiles and synthesis of endogenous compounds such DNA. The most important function of GSH, however, is antioxidant defense. GSH can reduce hydrogen peroxide and lipid peroxides through GPx-catalyzed reactions and is also involved in reduction of oxidized protein thiols (cysteine residues) through glutaredoxin (Grx) catalyzed reactions. Importantly, GSH concentration decreases in various fibrotic diseases including cystic fibrosis [126], [127], chronic obstructive pulmonary disease (COPD) [128], acute respiratory distress syndrome (ARDS) [129], [130], [131], [132], [133], IPF [134], [135], [136], [137], [138], [139], [140], sarcoidosis [141], and chronic liver diseases [142], [143], [144], [145], [146], [147], [148]. GSH concentration is also decreased in experimental fibrosis models induced by different stimuli [25], [149], [150], [151], [152], [153], [154], [155], [156], [157], [158]. Although the mechanism leading to GSH depletion in fibrotic diseases remains unclear, emerging evidence suggests that increased TGF-β may contribute to this effect [25], [91], [159], [160], [161], [162], [163], [164], [165], [166], [167], [168]. De novo GSH synthesis is a two-step reaction catalyzed by glutamate cysteine ligase (GCL) and GSH synthase (GS). GCL is the rate limiting enzyme in de novo GSH synthesis and is composed of two subunits, the catalytically active heavy subunit (GCLC) and the modifier light subunit (GCLM). It has been reported that TGF-β suppresses the expression of GCLC genes and decreases GSH concentration in different types of cells in vitro [91], [159], [160], [161], [162], [163], [164], [165], [167], [168]. In a previous study, we showed that administration of AdTGF-β1223/225, an adenovirus expressing constitutively active TGF-β1, suppressed the expression of both GCLC and GCLM mRNAs and proteins, inhibited the GCL activity, and reduced GSH level in mouse lung tissue [25]. This was associated with induction of activating transcription factor 3 (ATF3), a transcriptional repressor involved in the regulation of GCLC [25], [167]. A decreased GCL gene expression is also reported in fibrotic diseases [140], [169]. Together, the data suggest that increased TGF-β expression may underlie the depletion of GSH observed in fibrotic diseases. As GSH is the most abundant intracellular free thiol and the ratio of GSH and glutathione disulfide (GSSG) determines cell redox status [170], a decrease in GSH concentration will lead to increased oxidative stress level or redox imbalance.
Superoxide, a highly reactive oxygen species, is generated under both physiological and pathological conditions, which is converted by superoxide dismutase (SOD) to hydrogen peroxide (H2O2) and then to H2O by catalase, glutathione peroxidase (GPx) or peroxiredoxin (Prx). Besides H2O2, GPx and Prx can also reduce lipid peroxides. Thioredoxin (Trx) and glutaredoxin (Grx), on the other hand, are involved in maintenance of the redox status of protein thiols [171]. In addition to inhibition of GCL gene expression and therefore GSH biosynthesis, TGF-β also suppresses the expression/activity of other antioxidant enzymes including SOD, catalase, and Grx in different types of cells/tissues [83], [91], [119], [172], [173], [174]. Extracellular superoxide dismutase (EC-SOD) is protective in several models of interstitial lung disease, including pulmonary fibrosis, and its expression is altered in the lung of IPF patient, although the underlying mechanism is unclear [175]. Cui et al. reported that TGF-β1 suppressed EC-SOD in cultured fibroblasts in vitro and in mouse lung tissue in vivo; overexpression of EC-SOD in mouse lung by adenovirus mediated gene transfer technique, on the other hand, blocked latent TGF-β1 activation and diminished subsequent fibrotic responses, suggesting an important role of EC-SOD in TGF-β-induced fibrogenesis [176]. In summary, TGF-β stimulates ROS production and suppresses antioxidant defense, leading to redox imbalance or oxidative stress (Fig. 1).
Fig. 1.

TGF-β induces redox imbalance by increasing ROS production and suppressing antioxidant defense. TGF-β1 increases ROS production by disrupting mitochondrial function and inducing ROS-generating enzymes NADPH oxidases (Noxs). TGF-β1 also suppresses the expression of the enzymes involved in the antioxidant defense, including superoxide dismutase (SOD), catalase, glutaredoxin (Grx), and glutamate cysteine ligase, which leads to a decrease in GSH concentration.
4. Redox regulation of TGF-β1 activity and expression
TGF-β is synthesized and secreted into the extracellular space as a large latent complex containing mature dimeric TGF-β bound to latency-associated protein (LAP) and latent TGF-β-binding protein (LTBP) [50], [177], [178]. Release of TGF-β from LAP, a process called latent TGF-β activation, is required for the binding of TGF-β to its receptors. Multiple mechanisms have been proposed for the activation of latent TGF-β, including conformation changes induced by thrombospondin-1 [179], [180], binding to integrins ανβ6 and ανβ8 [181], [182], proteolytic cleavage of LAP by plasmin and matrix metalloproteinase [183], [184], [185], [186], and oxidative modification of LAP or activation of MMPs, which then cleave LAP to release active TGF-β [89], [187], [188], [189], [190], [191], [192]. LAP is sensitive to oxidation and oxidized LAP loses its TGF-β binding capacity, leading to TGF-β activation. Oxidants have been shown to activate latent TGF-β directly through oxidation of LAP and indirectly through activation of MMPs such as MMP-2 and MMP-9, which in turn cleave LAP to release active TGF-β [89], [188], [192], [193]. In vivo redox regulation of TGF-β activity was also demonstrated in a recent study [176]. Cui et al. reported that administration of AdTGF-β1 suppressed the expression of ECSOD, which was associated with an increase in oxidative stress and TGF-β activation in rat lung tissue [176]. Concomitant administration of ECSOD expressing adenovirus AdECSOD, on the other hand, significantly reduced oxidative stress level and the amount of active TGF-β and attenuated AdTGF-β1-induced lung fibrosis [176]. The results strongly suggest that TGF-β activity and expression is regulated by redox status in vivo. Interestingly, it has been reported that redox-mediated activation was restricted to the TGF-β1 isoform [193]. Using site-specific mutation, a methionine residue at amino acid position 253 unique to LAP-TGFβ1 was identified to be critical for ROS-mediated activation of latent TGF-β1 [193].
Besides directly activating latent TGF-β, numerous studies have shown that ROS/RNS also upregulate TGF-β gene expression in various types of cells [194], [195], [196], [197], [198], [199], [200]. In cultured human alveolar epithelial cells, xanthine/xanthine oxidase derived ROS increased TGF-β1 production through a transcriptional mechanism whereas S-nitroso-N-acetyl-penicilamine generated RNS induced TGF-β1 through translational mechanisms [201]. Exogenous H2O2 has been shown to induce the mRNA and protein expression of both TGF-β1 and TGF-β2 in human umbilical vein endothelial cells (HUVECs) [199]. ROS generated endogenously in mitochondria or by NADPH oxidases have also been shown to induce TGF-β [196]. Shvedova et al. reported that mice deficient with gp91phox, the prototype of NADPH oxidases, had reduced levels of TGF-β, compared to wild type mice, indicating that Nox-derived ROS are the major source of ROS for the induction of TGF-β [196]. A recent study further showed that mitochondria-originated ROS were responsible for TGF-β induction in an allergic asthma model induced by ovalbumin as mitochondria-targeted antioxidant (mitoTEMPO) significantly reduced ovalbumin-induced mitochondrial ROS and TGF-β expression [198]. The signaling pathway mediating TGF-β induction by ROS remains to be determined. Using small molecule inhibitor and siRNA technique, Lin et al. reported that ROS mediated hepatitis C virus-induced TGF-β1 expression through activating p38/JNK/ERK and NF-kβ pathways in human hepatocellular carcinoma cells [197]. In summary, although numerous studies have demonstrated that ROS induce TGF-β gene expression the signaling pathway regulating ROS induction of TGF-β1, however, remains poorly understood. In summary, redox imbalance due to increased ROS production and/or decreased antioxidant defense, in turn, can activate latent TGF-β1 and also induce TGF-β1 expression (Fig. 2), forming a vicious cycle.
Fig. 2.
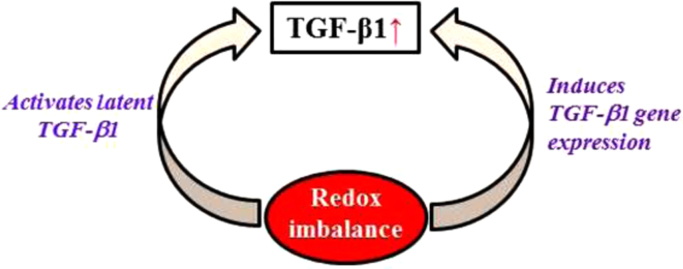
Redox imbalance increases TGF-β1 activity. Redox imbalance due to increased ROS and/or decreased antioxidants, in turn, activates latent TGF-β1 and induces TGF-β1 gene expression, leading to an increase in TGF-β1 activity.
5. ROS mediate TGF-β-induced fibrotic responses
TGF-β promotes fibrosis through diverse mechanisms, including activation of resident fibroblasts, stimulation of apoptosis in epithelial and endothelial cells, induction of epithelial– or endothelial–mesenchymal transition, production of ECM matrix proteins, and suppressing ECM degradation. ROS have been shown to mediate many of TGF-β-induced profibrotic effects. In this section, we will mainly focus on the role of ROS in TGF-β-induced fibroblast activation/myofibroblast differentiation, epithelial apoptosis, epithelial–mesenchymal transition (EMT/EnMT), and the expression of profibrogenic mediator, plasminogen activator inhibitor 1 (PAI-1) as elucidated in Fig. 3.
Fig. 3.

ROS mediate many of TGF-β1's profibrogenic effects. ROS mediate TGF-β1-induced myofibroblast differentiation, epithelial apoptosis, epithelial–mesenchymal transition (EMT), and the expression of profibrogenic mediators including PAI-1, which suppresses ECM degradation. By inducing myofibroblast differentiation and thereby ECM production and by suppressing ECM degradation through inducing PAI-1, TGF-β1 promotes ECM accumulation, fibrosis.
5.1. TGF-β, ROS, and fibroblast activation/myofibroblast differentiation
Myofibroblasts are the major producers of extracellular matrix (ECM) and therefore are key players in fibrogenesis. Fibroblast activation/differentiation to myofibroblasts is a key in the initiation and progression of fibrosis and TGF-β plays a pivotal role in myofibroblast differentiation. A large body of literature show that ROS, generated in mitochondria or by NADPH oxidases, are essential for TGF-β-mediated myofibroblast differentiation under different pathological conditions [85], [86], [92], [96], [111], [112], [117], [202]. Hecker et al. reported that knockdown of Nox4 with Nox4siRNA suppressed TGF-β1-induced production of H2O2 and expression of α-SMA, collagen, and fibronectin, the markers of myofibroblasts, in human fetal lung as well as IPF lung mesenchymal cells (hFLMCs) [111]. Most importantly, they showed that administration of diphenyleneiodonium (DPI), a Nox inhibitor, or Nox4 siRNA abrogated lung fibrosis in two murine lung fibrosis models. Their data support the notion that Nox4 induction is required for myofibroblast differentiation and the development of lung fibrosis [111]. Increased Nox4 expression has also been shown to mediate TGF-β-induced activation of fibroblasts from kidney, nasal polyss, liver, and heart [92], [96], [118], [202], [203], [204], although Nox4 was found to function upstream of Smad in cardiac fibroblasts [96] but downstream of Smad in human lung mesenchymal cells and nasal polys [92], [111]. Mitochondria-derived ROS are also involved in TGF-β-induced myofibroblast differentiation [85]. Interestingly, it was reported that mitochondria derived ROS mediated TGF-β-induced myofibroblast differentiation in human lung fibroblasts through inducing Nox4 as inhibition of mitochondrial ROS by mitochondrial targeted antioxidant suppressed TGF-β-induced Nox4 expression and myofibroblast differentiation [85]. Nonetheless, although it has been well documented that ROS mediate TGF-β-induced myofibroblast differentiation, the molecular mechanisms or signaling pathways whereby ROS mediate the phenotype transition remain unclear.
Increased resistance of myofibroblasts to apoptosis is evident in IPF [117], [205], [206] and in experimental lung fibrosis models [117], [207]. It is therefore believed that increased apoptosis resistance underlies the sustained myofibroblast activation observed in fibrotic tissues. The mechanism underlying increased resistance of myofibroblasts to apoptosis is unknown. Hecker et al. have reported that aged mice have impaired resolution of fibrosis upon bleomycin challenge, which is associated with the acquisition of senescent and apoptosis-resistant phenotype of fibroblasts [117]. They further show that Nox4 expression is increased in IPF lung myofibroblasts whereas Nrf2-mediated antioxidant response declined in lung fibroblasts isolated from aged mice, associated with senescence and apoptosis resistance phenotype [117]. Most importantly, they show that knockdown of Nox4 with siRNA reverses the aging-related senescence and apoptosis resistance phenotype of fibroblasts [117]. Their results suggest that redox imbalance resulting from elevated expression of Nox4 and an impaired Nrf2 antioxidant response underlies the acquisition of senescence and apoptosis resistance phenotypes in myofibroblasts from old mouse lung and IPF patient [117]. Nonetheless, although increasing evidence indicates that redox imbalance renders myofibroblasts resistance to apoptosis, the underlying mechanisms mediating the ROS effect is unknown.
5.2. TGF-β, ROS, and epithelial apoptosis
TGF-β affects diverse cellular processes including cell proliferation, differentiation, senescence, and apoptosis, depending on cell types. Epithelium apoptosis is juxtaposed with fibrosis and is believed to contribute importantly to fibrogenesis induced by different stimuli in different organs. Apoptosis of type II alveolar epithelial cells is evident in fibrotic lung diseases including IPF and in experimental fibrosis model [117], [205], [206], [208], [209], [210], [211], [212], [213], [214]. Studies have demonstrated that epithelial apoptosis precedes and is required for TGF-b-induced lung fibrosis [20]. Lee et al. reported that overexpression of TGF-β1 in the lung of the inducible TGF-β triple transgenic mice led to a transient wave of epithelial apoptosis that was followed by inflammation and lung fibrosis [20]. Knockout of the early growth response gene (Egr-1) or administration of a caspase inhibitor (Z-VAD-fmk) significantly reduced TGF-β-induced epithelial apoptosis as well as lung fibrosis [20]. Their studies suggest strongly that epithelial apoptosis precede and is required for TGF-β-induced lung fibrosis. Studies from other labs further show that TGF-β induces senescence [81], [82], [83], [85], [87] and apoptosis [76], [77], [78], [79], [80], [91] of epithelial cells in vitro; blocking ROS production from mitochondria or NADPH oxidases attenuated TGF-β-induced cell senescence or apoptosis, suggesting an important role of ROS in TGF-β mediated epithelial senescence or apoptosis. In a previous study, we showed that intranasal instillation of TGF-β expressing adenovirus (AdTGF-β223/225) suppressed the expression of GCL and reduced lung GSH, which was associated with increased lipid peroxidation and epithelial apoptosis as well as massive lung fibrosis in mice [25]. These data further support the notion that TGF-β induces redox imbalance in vivo, which underlies TGF-β-induced epithelial apoptosis and lung fibrosis.
5.3. ROS mediate TGF-induced epithelial–mesenchymal transition
Epithelial–mesenchymal transition (EMT/EnMT), a process characterized by the loss of epithelial characteristics and the acquisition of mesenchymal phenotype, is important in normal embryonic development and is also co-opted in the pathogenesis of diseases including cancer and fibrosis. EMT is believed to be an important source of myofibroblasts during development of fibrosis in different organs [215], [216], [217], [218], [219], [220], [221], [222], [223], [224], [225], [226]. TGF-β is the most potent inducer of EMT and can induce EMT in epithelial cells from different organs in vitro and in vivo [84], [90], [101], [218], [219], [227], [228], [229], [230]. Signaling pathways including Smad, MAPK, and PI3K pathways are involved in the induction of EMT by TGF-β [231], [232]. Importantly, ROS generated from different sources, including mitochondria and NOXs, have been shown to mediate TGF-β-induced EMT in the settings of cancer and fibrosis [84], [90], [101], [229], [233]. Hirage et al. reported that TGF-β induces Nox4 whereas Nox4 inhibitor DPI or siRNA blocked TGF-β-induced EMT in pancreatic cancer cells [233]. They further show that protein tyrosine phosphatase 1B (PTP1B) serves as a redox sensor in TGF-β-induced Nox4/ROS-mediated EMT in these tumor cells [233]. Nox4 has also been shown to mediate TGF-β-induced EMT in human breast epithelial cells [101]. Kim and Cho showed, on the other hand, that TGF-β induced Nox2, which was responsible for the increase in ROS and the induction of epithelial–mesenchymal transition in Hela cells [90]. They further identified a new EMT regulator YB-1 as a downstream target in ROS-induced EMT [90]. Mitochondria derived ROS have also been shown to mediate TGF-β-induced EMT in mouse mammary epithelial cells through a mitochondrial thioredoxin sensitive mechanism [84]. Evidence linking ROS to TGF-β-induced EMT in the context of fibrosis is also bounty [228], [234], [235], [236], [237]. Rhyu et al. showed that TGF-β increased ROS production and induced EMT in renal tubular epithelial cells whereas inhibitors of NOX (DPI and apocynin), mitochondria, and ERK MAPK blocked TGF-β-induced ROS production and/or EMT. Their data suggest that ROS play an important role in TGF-β1-induced EMT primarily through activation of MAPK in proximal tubular epithelial cells [228]. Felton et al. reported that TGF-β1 decreases intracellular GSH, increases ROS production, and induces EMT in rat alveolar type II cells (RLE-6TN and primary); N-acetylcysteine (NAC), GSH monoethyl ester reversed these effects induced by TGF-β1 [234]. They concluded that NAC prevents TGF-β-induced EMT in vitro at least in part through replenishment of intracellular GSH stores and suppression of TGF-β-induced intracellular ROS generation [234]. Nox4 derived ROS have also been shown to mediate TGF-β-induced EMT in renal tubular epithelial cells [236], [237]. Most interestingly, Lee et al. showed that metformin, an AMP-activated protein kinase (AMPK) activator that has been used in clinics for the treatment of diabetes, and 5-aminomidazole-4-carboxamide-1β riboside (AICAR), another AMPK activator suppressed TGF-β-induced EMT through inhibition of ROS production, suggesting the potential therapeutic value of metformin for fibrosis by inhibition of ROS-mediated pathogenesis [236].
5.4. TGF-β-mediated redox regulation of plasminogen activator inhibitor 1 gene expression
ROS have been shown to mediate the induction of many matrix structural proteins and pro-fibrotic mediators by TGF-β1. In this section, we will focus on one of these proteins, PAI-1. PAI-1 is a primary inhibitor of urokinase-type and tissue-type plasminogen activators (uPA and tPA, respectively). The primary function of PAI-1 is inhibition of fibrinolysis (hemostasis) through inhibition of tPA and uPA and thereby activation of the zymogen plasminogen. Besides breaking down fibrin, plasmin is also involved in the degradation of extracellular matrix (ECM) proteins directly or indirectly through activation of matrix metalloproteinases (MMPs). Therefore, PAI-1 is believed to play a role in the development of fibrosis through inhibition of plasminogen activation and thus ECM degradation. PAI-1 expression is increased in many fibrotic diseases including IPF [238], [239], [240], [241], [242], [243] and in experimental fibrosis models [244], [245]. Knockout of the PAI-1 gene or administration of PAI-1 siRNA attenuates, whereas overexpression of PAI-1 protein enhances, fibrotic responses induced by different stimuli [26], [53], [54], [168], [207], [242], [246], [247], [248]. In a previous study, we showed that inhibition of PAI-1 with a small molecule PAI-1 inhibitor attenuated TGF-β1-induced lung fibrosis in mice and the fibrotic responses in cultured lung fibroblasts [26]. In a recent study, we further showed that PAI-1 expression is increased with age in mouse lung tissue and lung fibroblasts, which was associated with increased apoptosis resistance of lung fibroblasts and increased sensitivity of old mice to bleomycin-induced lung fibrosis [207]. Inhibition of PAI-1 activity with a small molecule PAI-1 inhibitor or knockdown of PAI-1 protein with PAI-1 siRNA restored the sensitivity of lung fibroblasts from old mice to apoptosis [207]. Together, the data suggest that PAI-1 plays a critical role in the development of lung fibrosis, although the underlying mechanism is under debating.
Many cytokines and chemokines as well as growth factors induce PAI-1 and TGF-β is considered to be the most potent inducer. TGF-β induces PAI-1 gene expression in various types of cells in vitro and in vivo and an elevated PAI-1 level is also associated with increased TGF-β expression and ECM deposition in diverse pathological conditions, indicating a critical role of PAI-1 in TGF-β fibrogenesis [26], [53], [54], [100], [249], [250], [251], [252], [253], [254]. Both Smad dependent and independent pathways are involved in TGF-β induction of PAI-1 [255], [256], [257]. Ιmportantly, ROS have been shown to function as signaling molecules mediating TGF-β-induced PAI-1 expression in many of cell types [53], [54], [100], [254], [257], [258], [259]. GSH is the most abundant intracellular free thiol and an important antioxidant. Depletion of intracellular GSH leads to increased basal as well as TGF-β1 or H2O2-induced PAI-1 expression whereas treatment with N-acetylcysteine (NAC), a GSH precursor, suppresses TGF-β-induced PAI-1 expression in kidney mesangial cells [258]. Our previous studies have also shown that treatment with exogenous GSH or GSH ester suppresses TGF-β1-induced PAI-1 promoter activity, PAI-1 mRNA and protein expression in murine embryo fibroblasts (NIH3T3 cells) through inhibition of JNK and p38, but not Smad, pathways [53], [54]. GSH treatment also reduces ROS level and restores tPA activity as well as collagen degradation capacity in TGF-β treated NIH3T3 cells [54]. Tubulointerstitial fibrosis is a major feature of several chronic renal diseases and TGF-β is the key inducer of tubulointerstitial fibrosis. A critical role of PAI-1 in the development of kidney fibrosis has been well demonstrated by using mutant non-inhibitory PAI-1 protein [260], [261], [262]. Importantly, studies have shown that ROS are essential for TGF-β-mediated induction of PAI-1 in renal tubular epithelial cells or mesangial cells through activation of ERK and p38 MAPK pathways [258], [263].
Although it has been well documented that ROS mediate TGF-β-induced PAI-1 expression through activation of MAPK pathways, the redox sensitive target(s) remain(s) unclear. Samarakoon et al. reported that TGF-β induces PAI-1 in vascular smooth muscle cells (VSMCs) through two distinct pathways [264]. One of the pathways involves phosphorylation of epithelial growth factor receptor (EGFR) on Y845, a Src kinase target residue, which leads to activation of ERK1/2 and then induction PAI-1 [264]. Another pathway is through activation of RhoA-ROCK, which in turn phosphorylates Smad2 and thus induces PAI-1 [264]. MAPKs are activated by phosphorylation of threonine and tyrosine residues by MAPK kinases, which, in turn, are phosphorylated and activated by MAPK kinase kinases. Activated MAPKs, on the other hand, can be inactivated by serine/threonine specific MAPK phosphatase (SS-MKPs), tyrosine-specific MAPK phosphatases (TS-MKPs), or dual-specific MAPK phosphatases (DS-MKPs). Many of MAPK phosphatases (MKPs), especially DS-MKPs, contain a redox sensitive cysteine motif in their active sites and are sensitive to oxidative inactivation by ROS/RNS. Our previous study showed that TGF-β1 increased the expression and activity of Nox4 in the nucleus of mouse and human fibroblasts, which was associated with increased nuclear ROS production [100]. Most interestingly, we showed that increased nuclear ROS led to thiol modifications and inhibition of a nuclear DS-MKP, MKP-1 [100]. Knockdown of MKP-1 expression with MKP-1 siRNA enhanced TGF-β1-induced JNK/p38 phosphorylation and PAI-1 expression whereas knockdown of Nox4 expression reduced TGF-β1-stimulated nuclear ROS production, p38 phosphorylation, and PAI-1 expression. These data reveal a novel mechanism whereby ROS modulate TGF-β signaling and PAI-1 expression [100].
6. In summary
TGF-β, the most potent profibrogenic cytokine, increases ROS production and suppresses antioxidant system. ROS in turn activate/induce TGF-β and mediate many of TGF-β's profibrogenic effects, including fibroblast activation, epithelial/endothelial apoptosis, EMT, and synthesis of profibrogenic mediator such as PAI-1. Therapeutic targeting of TGF-β-mediated ROS production may provide beneficial effect for the treatment of fibrotic diseases.
Acknowledgments
The work was supported by Grants from National Institute of Aging (AG046701) and National Heart, Lung, and Blood Institute (P01 HL114470, Animal and Therapeutic Core) to Rui-Ming Liu.
References
- 1.Manoury B., Nenan S., Leclerc O., Guenon I., Boichot E., Planquois J.M., Bertrand C.P., Lagente V. The absence of reactive oxygen species production protects mice against bleomycin-induced pulmonary fibrosis. Respir. Res. 2005;6:11. doi: 10.1186/1465-9921-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lenz A.G., Costabel U., Maier K.L. Oxidized BAL fluid proteins in patients with interstitial lung diseases. Eur. Respir. J. 1996;9:307–312. doi: 10.1183/09031936.96.09020307. [DOI] [PubMed] [Google Scholar]
- 3.Rottoli P., Magi B., Cianti R., Bargagli E., Vagaggini C., Nikiforakis N., Pallini V., Bini L. Carbonylated proteins in bronchoalveolar lavage of patients with sarcoidosis, pulmonary fibrosis associated with systemic sclerosis and idiopathic pulmonary fibrosis. Proteomics. 2005;5:2612–2618. doi: 10.1002/pmic.200401206. [DOI] [PubMed] [Google Scholar]
- 4.Psathakis K., Mermigkis D., Papatheodorou G., Loukides S., Panagou P., Polychronopoulos V., Siafakas N.M., Bouros D. Exhaled markers of oxidative stress in idiopathic pulmonary fibrosis. Eur. J. Clin. Investig. 2006;36:362–367. doi: 10.1111/j.1365-2362.2006.01636.x. [DOI] [PubMed] [Google Scholar]
- 5.Liu R.M., Gaston Pravia K.A. Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free. Radic. Biol. Med. 2010;48:1–15. doi: 10.1016/j.freeradbiomed.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broekelmann T.J., Limper A.H., Colby T.V., McDonald J.A. Transforming growth factor beta 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc. Natl. Acad. Sci. USA. 1991;88:6642–6646. doi: 10.1073/pnas.88.15.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bergeron A., Soler P., Kambouchner M., Loiseau P., Milleron B., Valeyre D., Hance A.J., Tazi A. Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-beta and IL-10. Eur. Respir. J. 2003;22:69–76. doi: 10.1183/09031936.03.00014703. [DOI] [PubMed] [Google Scholar]
- 8.Bataller R., Brenner D.A. Liver fibrosis. J. Clin. Investig. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tzortzaki E.G., Antoniou K.M., Zervou M.I., Lambiri I., Koutsopoulos A., Tzanakis N., Plataki M., Maltezakis G., Bouros D., Siafakas N.M. Effects of antifibrotic agents on TGF-[beta]1, CTGF and IFN-[gamma] expression in patients with idiopathic pulmonary fibrosis. Respir. Med. 2007;101:1821–1829. doi: 10.1016/j.rmed.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Brosius F. New insights into the mechanisms of fibrosis and sclerosis in diabetic nephropathy. Rev. Endocr. Metab. Disord. 2008;9:245–254. doi: 10.1007/s11154-008-9100-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fahy R.J., Lichtenberger F., McKeegan C.B., Nuovo G.J., Marsh C.B., Wewers M.D. The acute respiratory distress syndrome: a role for transforming growth factor-beta 1. Am. J. Respir. Cell. Mol. Biol. 2003;28:499–503. doi: 10.1165/rcmb.2002-0092OC. [DOI] [PubMed] [Google Scholar]
- 12.Williams A.O., Flanders K.C., Saffiotti U. Immunohistochemical localization of transforming growth factor-beta 1 in rats with experimental silicosis, alveolar type II hyperplasia, and lung cancer. Am. J. Pathol. 1993;142:1831–1840. [PMC free article] [PubMed] [Google Scholar]
- 13.Coker R.K., Laurent G.J., Shahzeidi S., Lympany P.A., du Bois R.M., Jeffery P.K., McAnulty R.J. Transforming growth factors-beta 1, -beta 2, and -beta 3 stimulate fibroblast procollagen production in vitro but are differentially expressed during bleomycin-induced lung fibrosis. Am. J. Pathol. 1997;150:981–991. [PMC free article] [PubMed] [Google Scholar]
- 14.Rube C.E., Uthe D., Schmid K.W., Richter K.D., Wessel J., Schuck A., Willich N., Rube C. Dose-dependent induction of transforming growth factor beta (TGF-beta) in the lung tissue of fibrosis-prone mice after thoracic irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2000;47:1033–1042. doi: 10.1016/s0360-3016(00)00482-x. [DOI] [PubMed] [Google Scholar]
- 15.Kumar R.K., Herbert C., Foster P.S. Expression of growth factors by airway epithelial cells in a model of chronic asthma: regulation and relationship to subepithelial fibrosis. Clin. Exp. Allergy. 2004;34:567–575. doi: 10.1111/j.1365-2222.2004.1917.x. [DOI] [PubMed] [Google Scholar]
- 16.Ellmers L.J., Scott N.J.A., Medicherla S., Pilbrow A.P., Bridgman P.G., Yandle T.G., Richards A.M., Protter A.A., Cameron V.A. Transforming growth factor-{beta} blockade down-regulates the renin-angiotensin system and modifies cardiac remodeling after myocardial infarction. Endocrinology. 2008;149:5828–5834. doi: 10.1210/en.2008-0165. [DOI] [PubMed] [Google Scholar]
- 17.Churg A., Zhou S., Preobrazhenska O., Tai H., Wang R., Wright J.L. Expression of profibrotic mediators in small airways versus parenchyma after cigarette smoke exposure. Am. J. Respir. Cell. Mol. Biol. 2009;40:268–276. doi: 10.1165/rcmb.2007-0367OC. [DOI] [PubMed] [Google Scholar]
- 18.Ng Y.Y., Chen Y.M., Tsai T.J., Lan X.R., Yang W.C., Lan H.Y. Pentoxifylline inhibits transforming growth factor-beta signaling and renal fibrosis in experimental crescentic glomerulonephritis in rats. Am. J. Nephrol. 2009;29:43–53. doi: 10.1159/000150600. [DOI] [PubMed] [Google Scholar]
- 19.Hardie W.D., Le Cras T.D., Jiang K., Tichelaar J.W., Azhar M., Korfhagen T.R. Conditional expression of transforming growth factor-alpha in adult mouse lung causes pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004;286:L741–L749. doi: 10.1152/ajplung.00208.2003. [DOI] [PubMed] [Google Scholar]
- 20.Lee C.G., Cho S.J., Kang M.J., Chapoval S.P., Lee P.J., Noble P.W., Yehualaeshet T., Lu B., Flavell R.A., Milbrandt J., Homer R.J., Elias J.A. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J. Exp. Med. 2004;200:377–389. doi: 10.1084/jem.20040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vicencio A.G., Lee C.G., Cho S.J., Eickelberg O., Chuu Y., Haddad G.G., Elias J.A. Conditional overexpression of bioactive transforming growth factor-beta1 in neonatal mouse lung: a new model for bronchopulmonary dysplasia? Am. J. Respir. Cell Mol. Biol. 2004;31:650–656. doi: 10.1165/rcmb.2004-0092OC. [DOI] [PubMed] [Google Scholar]
- 22.Sime P.J., Xing Z., Graham F.L., Csaky K.G., Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J. Clin. Investig. 1997;100:768–776. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kolb M., Bonniaud P., Galt T., Sime P.J., Kelly M.M., Margetts P.J., Gauldie J. Differences in the fibrogenic response after transfer of active transforming growth factor-beta1 gene to lungs of "fibrosis-prone" and "fibrosis-resistant" mouse strains. Am. J. Respir. Cell Mol. Biol. 2002;27:141–150. doi: 10.1165/ajrcmb.27.2.4674. [DOI] [PubMed] [Google Scholar]
- 24.Bonniaud P., Margetts P.J., Kolb M., Schroeder J.A., Kapoun A.M., Damm D., Murphy A., Chakravarty S., Dugar S., Higgins L., Protter A.A., Gauldie J. Progressive transforming growth factor {beta}1-induced lung fibrosis is blocked by an orally active ALK5 kinase inhibitor. Am. J. Respir. Crit. Care Med. 2005;171:889–898. doi: 10.1164/rccm.200405-612OC. [DOI] [PubMed] [Google Scholar]
- 25.Liu R.M., Vayalil P.K., Ballinger C., Dickinson D.A., Huang W.T., Wang S., Kavanagh T.J., Matthews Q.L., Postlethwait E.M. Transforming growth factor beta suppresses glutamate-cysteine ligase gene expression and induces oxidative stress in a lung fibrosis model. Free. Radic. Biol. Med. 2012;53:554–563. doi: 10.1016/j.freeradbiomed.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang W.T., Vayalil P.K., Miyata T., Hagood J., Liu R.M. Therapeutic value of small molecule inhibitor to plasminogen activator inhibitor-1 for lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2012;46:87–95. doi: 10.1165/rcmb.2011-0139OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horowitz J.C., Rogers D.S., Simon R.H., Sisson T.H., Thannickal V.J. Plasminogen activation induced pericellular fibronectin proteolysis promotes fibroblast apoptosis. Am. J. Respir. Cell Mol. Biol. 2008;38:78–87. doi: 10.1165/rcmb.2007-0174OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mackinnon A.C., Gibbons M.A., Farnworth S.L., Leffler H., Nilsson U.J., Delaine T., Simpson A.J., Forbes S.J., Hirani N., Gauldie J., Sethi T. Regulation of transforming growth factor-beta1-driven lung fibrosis by galectin-3. Am. J. Respir. Crit. Care Med. 2012;185:537–546. doi: 10.1164/rccm.201106-0965OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reilkoff R.A., Peng H., Murray L.A., Peng X., Russell T., Montgomery R., Feghali-Bostwick C., Shaw A., Homer R.J., Gulati M., Mathur A., Elias J.A., Herzog E.L. Semaphorin 7a+ regulatory T cells are associated with progressive idiopathic pulmonary fibrosis and are implicated in transforming growth factor-beta1-induced pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013;187:180–188. doi: 10.1164/rccm.201206-1109OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giri S.N., Hyde D.M., Hollinger M.A. Effect of antibody to transforming growth factor beta on bleomycin induced accumulation of lung collagen in mice. Thorax. 1993;48:959–966. doi: 10.1136/thx.48.10.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Higashiyama H., Yoshimoto D., Kaise T., Matsubara S., Fujiwara M., Kikkawa H., Asano S., Kinoshita M. Inhibition of activin receptor-like kinase 5 attenuates bleomycin-induced pulmonary fibrosis. Exp. Mol. Pathol. 2007;83:39–46. doi: 10.1016/j.yexmp.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 32.Wang Q., Wang Y., Hyde D.M., Gotwals P.J., Koteliansky V.E., Ryan S.T., Giri S.N. Reduction of bleomycin induced lung fibrosis by transforming growth factor beta soluble receptor in hamsters. Thorax. 1999;54:805–812. doi: 10.1136/thx.54.9.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolb M., Margetts P.J., Galt T., Sime P.J., Xing Z., Schmidt M., Gauldie J. Transient transgene expression of decorin in the lung reduces the fibrotic response to bleomycin. Am. J. Respir. Crit. Care Med. 2001;163:770–777. doi: 10.1164/ajrccm.163.3.2006084. [DOI] [PubMed] [Google Scholar]
- 34.Li M., Krishnaveni M.S., Li C., Zhou B., Xing Y., Banfalvi A., Li A., Lombardi V., Akbari O., Borok Z., Minoo P. Epithelium-specific deletion of TGF-beta receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J. Clin. Investig. 2011;121:277–287. doi: 10.1172/JCI42090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ask K., Bonniaud P., Maass K., Eickelberg O., Margetts P.J., Warburton D., Groffen J., Gauldie J., Kolb M. Progressive pulmonary fibrosis is mediated by TGF-beta isoform 1 but not TGF-beta3. Int. J. Biochem. Cell Biol. 2008;40:484–495. doi: 10.1016/j.biocel.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Massague J., Seoane J., Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 37.Rahimi R.A., Leof E.B. TGF-beta signaling: a tale of two responses. J. Cell. Biochem. 2007;102:593–608. doi: 10.1002/jcb.21501. [DOI] [PubMed] [Google Scholar]
- 38.Derynck R., Zhang Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 39.Lan H.Y. Smad7 as a therapeutic agent for chronic kidney diseases. Front. Biosci. 2008;13:4984–4992. doi: 10.2741/3057. [DOI] [PubMed] [Google Scholar]
- 40.Corcoran J.B., McCarthy S., Griffin B., Gaffney A., Bhreathnach U., Borgeson E., Hickey F.B., Docherty N.G., Higgins D.F., Furlong F., Martin F., Godson C., Murphy M. IHG-1 must be localised to mitochondria to decrease Smad7 expression and amplify TGF-beta1-induced fibrotic responses. Biochim. Biophys. Acta. 2013;1833:1969–1978. doi: 10.1016/j.bbamcr.2013.03.027. [DOI] [PubMed] [Google Scholar]
- 41.Dooley S., Delvoux B., Streckert M., Bonzel L., Stopa M., ten Dijke P., Gressner A.M. Transforming growth factor beta signal transduction in hepatic stellate cells via Smad2/3 phosphorylation, a pathway that is abrogated during in vitro progression to myofibroblasts. TGFbeta signal transduction during transdifferentiation of hepatic stellate cells. FEBS Lett. 2001;502:4–10. doi: 10.1016/s0014-5793(01)02656-4. [DOI] [PubMed] [Google Scholar]
- 42.Schnabl B., Kweon Y.O., Frederick J.P., Wang X.F., Rippe R.A., Brenner D.A. The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology. 2001;34:89–100. doi: 10.1053/jhep.2001.25349. [DOI] [PubMed] [Google Scholar]
- 43.Lakos G., Takagawa S., Chen S.J., Ferreira A.M., Han G., Masuda K., Wang X.J., DiPietro L.A., Varga J. Targeted disruption of TGF-beta/Smad3 signaling modulates skin fibrosis in a mouse model of scleroderma. Am. J. Pathol. 2004;165:203–217. doi: 10.1016/s0002-9440(10)63289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan D.L., Zhao W.J., Wang Y.X., Han S.Y., Guo S. Oxymatrine inhibits collagen synthesis in keloid fibroblasts via inhibition of transforming growth factor-beta1/Smad signaling pathway. Int. J. Dermatol. 2012;51:463–472. doi: 10.1111/j.1365-4632.2011.05234.x. [DOI] [PubMed] [Google Scholar]
- 45.Kanasaki K., Koya D., Sugimoto T., Isono M., Kashiwagi A., Haneda M. N-Acetyl-seryl-aspartyl-lysyl-proline inhibits TGF-beta-mediated plasminogen activator inhibitor-1 expression via inhibition of Smad pathway in human mesangial cells. J. Am. Soc. Nephrol. 2003;14:863–872. doi: 10.1097/01.asn.0000057544.95569.ec. [DOI] [PubMed] [Google Scholar]
- 46.Zhan M., Kanwar Y.S. Hierarchy of molecules in TGF-beta1 signaling relevant to myofibroblast activation and renal fibrosis. Am. J. Physiol. Ren. Physiol. 2014;307:F385–F387. doi: 10.1152/ajprenal.00338.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fernandez I.E., Eickelberg O. The impact of TGF-beta on lung fibrosis: from targeting to biomarkers. Proc. Am. Thorac. Soc. 2012;9:111–116. doi: 10.1513/pats.201203-023AW. [DOI] [PubMed] [Google Scholar]
- 48.Dooley S., Hamzavi J., Breitkopf K., Wiercinska E., Said H.M., Lorenzen J., Ten Dijke P., Gressner A.M. Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology. 2003;125:178–191. doi: 10.1016/s0016-5085(03)00666-8. [DOI] [PubMed] [Google Scholar]
- 49.Fukasawa H., Yamamoto T., Togawa A., Ohashi N., Fujigaki Y., Oda T., Uchida C., Kitagawa K., Hattori T., Suzuki S., Kitagawa M., Hishida A. Down-regulation of Smad7 expression by ubiquitin-dependent degradation contributes to renal fibrosis in obstructive nephropathy in mice. Proc. Natl. Acad. Sci. USA. 2004;101:8687–8692. doi: 10.1073/pnas.0400035101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Biernacka A., Dobaczewski M., Frangogiannis N.G. TGF-beta signaling in fibrosis. Growth Factors. 2011;29:196–202. doi: 10.3109/08977194.2011.595714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu L., Hebert M.C., Zhang Y.E. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002;21:3749–3759. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stratton R., Rajkumar V., Ponticos M., Nichols B., Shiwen X., Black C.M., Abraham D.J., Leask A. Prostacyclin derivatives prevent the fibrotic response to TGF-beta by inhibiting the Ras/MEK/ERK pathway. FASEB J. 2002;16:1949–1951. doi: 10.1096/fj.02-0204fje. [DOI] [PubMed] [Google Scholar]
- 53.Vayalil P.K., Iles K.E., Choi J., Yi A.K., Postlethwait E.M., Liu R.M. Glutathione suppresses TGF-beta-induced PAI-1 expression by inhibiting p38 and JNK MAPK and the binding of AP-1, SP-1, and Smad to the PAI-1 promoter. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;293:L1281–L1292. doi: 10.1152/ajplung.00128.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vayalil P.K., Olman M., Murphy-Ullrich J.E., Postlethwait E.M., Liu R.M. Glutathione restores collagen degradation in TGF-beta-treated fibroblasts by blocking plasminogen activator inhibitor-1 expression and activating plasminogen. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005;289:L937–L945. doi: 10.1152/ajplung.00150.2005. [DOI] [PubMed] [Google Scholar]
- 55.Pannu J., Nakerakanti S., Smith E., ten Dijke P., Trojanowska M. Transforming growth factor-beta receptor type I-dependent fibrogenic gene program is mediated via activation of Smad1 and ERK1/2 pathways. J. Biol. Chem. 2007;282:10405–10413. doi: 10.1074/jbc.M611742200. [DOI] [PubMed] [Google Scholar]
- 56.Yuan Q., Wang R., Peng Y., Fu X., Wang W., Wang L., Zhang F., Peng Z., Ning W., Hu G., Wang Z., Tao L. Fluorofenidone attenuates tubulointerstitial fibrosis by inhibiting TGF-beta(1)-induced fibroblast activation. Am. J. Nephrol. 2011;34:181–194. doi: 10.1159/000329080. [DOI] [PubMed] [Google Scholar]
- 57.Lan R., Geng H., Polichnowski A.J., Singha P.K., Saikumar P., McEwen D.G., Griffin K.A., Koesters R., Weinberg J.M., Bidani A.K., Kriz W., Venkatachalam M.A. PTEN loss defines a TGF-beta-induced tubule phenotype of failed differentiation and JNK signaling during renal fibrosis. Am. J. Physiol. Ren. Physiol. 2012;302:F1210–F1223. doi: 10.1152/ajprenal.00660.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Samarakoon R., Overstreet J.M., Higgins P.J. TGF-beta signaling in tissue fibrosis: redox controls, target genes and therapeutic opportunities. Cell. Signal. 2013;25:264–268. doi: 10.1016/j.cellsig.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang F., Liu G.S., Dusting G.J., Chan E.C. NADPH oxidase-dependent redox signaling in TGF-beta-mediated fibrotic responses. Redox Biol. 2014;2:267–272. doi: 10.1016/j.redox.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sampson N., Berger P., Zenzmaier C. Redox signaling as a therapeutic target to inhibit myofibroblast activation in degenerative fibrotic disease. Biomed. Res. Int. 2014;2014:131737. doi: 10.1155/2014/131737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Manickam N., Patel M., Griendling K.K., Gorin Y., Barnes J.L. RhoA/Rho kinase mediates TGF-beta1-induced kidney myofibroblast activation through Poldip2/Nox4-derived reactive oxygen species. Am. J. Physiol. Ren. Physiol. 2014;307:F159–F171. doi: 10.1152/ajprenal.00546.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harvey K.A., Paranavitana C.N., Zaloga G.P., Siddiqui R.A. Diverse signaling pathways regulate fibroblast differentiation and transformation through Rho kinase activation. J. Cell. Physiol. 2007;211:353–363. doi: 10.1002/jcp.20939. [DOI] [PubMed] [Google Scholar]
- 63.Heasman S.J., Ridley A.J. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- 64.Ridley A.J. Rho family proteins: coordinating cell responses. Trends Cell. Biol. 2001;11:471–477. doi: 10.1016/s0962-8924(01)02153-5. [DOI] [PubMed] [Google Scholar]
- 65.Tsou P.S., Haak A.J., Khanna D., Neubig R.R. Cellular mechanisms of tissue fibrosis. 8. Current and future drug targets in fibrosis: focus on Rho GTPase-regulated gene transcription. Am. J. Physiol. Cell Physiol. 2014;307:C2–13. doi: 10.1152/ajpcell.00060.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou Y., Huang X., Hecker L., Kurundkar D., Kurundkar A., Liu H., Jin T.H., Desai L., Bernard K., Thannickal V.J. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J. Clin. Investig. 2013;123:1096–1108. doi: 10.1172/JCI66700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson L.A., Rodansky E.S., Haak A.J., Larsen S.D., Neubig R.R., Higgins P.D. Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-beta-induced fibrogenesis in human colonic myofibroblasts. Inflamm. Bowel Dis. 2014;20:154–165. doi: 10.1097/01.MIB.0000437615.98881.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jin L., Ying Z., Webb R.C. Activation of Rho/Rho kinase signaling pathway by reactive oxygen species in rat aorta. Am. J. Physiol. Heart Circ. Physiol. 2004;287:H1495–H1500. doi: 10.1152/ajpheart.01006.2003. [DOI] [PubMed] [Google Scholar]
- 69.Bailey S.R., Mitra S., Flavahan S., Flavahan N.A. Reactive oxygen species from smooth muscle mitochondria initiate cold-induced constriction of cutaneous arteries. Am. J. Physiol. Heart Circ. Physiol. 2005;289:H243–H250. doi: 10.1152/ajpheart.01305.2004. [DOI] [PubMed] [Google Scholar]
- 70.Heo J., Campbell S.L. Mechanism of redox-mediated guanine nucleotide exchange on redox-active Rho GTPases. J. Biol. Chem. 2005;280:31003–31010. doi: 10.1074/jbc.M504768200. [DOI] [PubMed] [Google Scholar]
- 71.Knock G.A., Snetkov V.A., Shaifta Y., Connolly M., Drndarski S., Noah A., Pourmahram G.E., Becker S., Aaronson P.I., Ward J.P. Superoxide constricts rat pulmonary arteries via Rho-kinase-mediated Ca(2+) sensitization. Free Radic. Biol. Med. 2009;46:633–642. doi: 10.1016/j.freeradbiomed.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aghajanian A., Wittchen E.S., Campbell S.L., Burridge K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PloS One. 2009;4:e8045. doi: 10.1371/journal.pone.0008045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Soliman H., Gador A., Lu Y.H., Lin G., Bankar G., MacLeod K.M. Diabetes-induced increased oxidative stress in cardiomyocytes is sustained by a positive feedback loop involving Rho kinase and PKCbeta2. Am. J. Physiol. Heart Circ. Physiol. 2012;303:H989–H1000. doi: 10.1152/ajpheart.00416.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pernomian L., Santos Gomes M., Baraldi Araujo Restini C., Naira Zambelli Ramalho L., Renato Tirapelli C., Maria de Oliveira A. The role of reactive oxygen species in the modulation of the contraction induced by angiotensin II in carotid artery from diabetic rat. Eur. J. Pharmacol. 2012;678:15–25. doi: 10.1016/j.ejphar.2011.12.036. [DOI] [PubMed] [Google Scholar]
- 75.Weidinger A., Kozlov A.V. Biological activities of reactive oxygen and nitrogen species: oxidative stress versus signal transduction. Biomolecules. 2015;5:472–484. doi: 10.3390/biom5020472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Herrera B., Alvarez A.M., Sanchez A., Fernandez M., Roncero C., Benito M., Fabregat I. Reactive oxygen species (ROS) mediates the mitochondrial-dependent apoptosis induced by transforming growth factor (beta) in fetal hepatocytes. FASEB J. 2001;15:741–751. doi: 10.1096/fj.00-0267com. [DOI] [PubMed] [Google Scholar]
- 77.Albright C.D., Salganik R.I., Craciunescu C.N., Mar M.H., Zeisel S.H. Mitochondrial and microsomal derived reactive oxygen species mediate apoptosis induced by transforming growth factor-beta1 in immortalized rat hepatocytes. J. Cell. Biochem. 2003;89:254–261. doi: 10.1002/jcb.10498. [DOI] [PubMed] [Google Scholar]
- 78.Herrera B., Murillo M.M., Alvarez-Barrientos A., Beltran J., Fernandez M., Fabregat I. Source of early reactive oxygen species in the apoptosis induced by transforming growth factor-[beta] in fetal rat hepatocytes. Free Radic. Biol. Med. 2004;36:16–26. doi: 10.1016/j.freeradbiomed.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 79.Black D., Lyman S., Qian T., Lemasters J.J., Rippe R.A., Nitta T., Kim J.S., Behrns K.E. Transforming growth factor beta mediates hepatocyte apoptosis through Smad3 generation of reactive oxygen species. Biochimie. 2007;89:1464–1473. doi: 10.1016/j.biochi.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ortiz C., Caja L., Bertran E., Gonzalez-Rodriguez A., Valverde A.M., Fabregat I., Sancho P. Protein-tyrosine phosphatase 1B (PTP1B) deficiency confers resistance to transforming growth factor-beta (TGF-beta)-induced suppressor effects in hepatocytes. J. Biol. Chem. 2012;287:15263–15274. doi: 10.1074/jbc.M111.303958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yoon Y.S., Lee J.H., Hwang S.C., Choi K.S., Yoon G. TGF beta1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene. 2005;24:1895–1903. doi: 10.1038/sj.onc.1208262. [DOI] [PubMed] [Google Scholar]
- 82.Byun H.O., Jung H.J., Seo Y.H., Lee Y.K., Hwang S.C., Hwang E.S., Yoon G. GSK3 inactivation is involved in mitochondrial complex IV defect in transforming growth factor (TGF) beta1-induced senescence. Exp. Cell Res. 2012;318:1808–1819. doi: 10.1016/j.yexcr.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 83.Wu J., Niu J., Li X., Wang X., Guo Z., Zhang F. TGF-beta1 induces senescence of bone marrow mesenchymal stem cells via increase of mitochondrial ROS production. BMC Dev. Biol. 2014;14:21. doi: 10.1186/1471-213X-14-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ishikawa F., Kaneko E., Sugimoto T., Ishijima T., Wakamatsu M., Yuasa A., Sampei R., Mori K., Nose K., Shibanuma M. A mitochondrial thioredoxin-sensitive mechanism regulates TGF-beta-mediated gene expression associated with epithelial–mesenchymal transition. Biochem. Biophys. Res. Commun. 2014;443:821–827. doi: 10.1016/j.bbrc.2013.12.050. [DOI] [PubMed] [Google Scholar]
- 85.Jain M., Rivera S., Monclus E.A., Synenki L., Zirk A., Eisenbart J., Feghali-Bostwick C., Mutlu G.M., Budinger G.R., Chandel N.S. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J. Biol. Chem. 2013;288:770–777. doi: 10.1074/jbc.M112.431973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bracey N.A., Gershkovich B., Chun J., Vilaysane A., Meijndert H.C., Wright J.R., Jr, Fedak P.W., Beck P.L., Muruve D.A., Duff H.J. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J. Biol. Chem. 2014;289:19571–19584. doi: 10.1074/jbc.M114.550624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Abe Y., Sakairi T., Beeson C., Kopp J.B. TGF-beta1 stimulates mitochondrial oxidative phosphorylation and generation of reactive oxygen species in cultured mouse podocytes, mediated in part by the mTOR pathway. Am. J. Physiol. Ren. Physiol. 2013;305:F1477–F1490. doi: 10.1152/ajprenal.00182.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hagler M.A., Hadley T.M., Zhang H., Mehra K., Roos C.M., Schaff H.V., Suri R.M., Miller J.D. TGF-beta signalling and reactive oxygen species drive fibrosis and matrix remodelling in myxomatous mitral valves. Cardiovasc. Res. 2013;99:175–184. doi: 10.1093/cvr/cvt083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pociask D.A., Sime P.J., Brody A.R. Asbestos-derived reactive oxygen species activate TGF-beta1. Lab. Investig. 2004;84:1013–1023. doi: 10.1038/labinvest.3700109. [DOI] [PubMed] [Google Scholar]
- 90.Kim Y.M., Cho M. Activation of NADPH oxidase subunit NCF4 induces ROS-mediated EMT signaling in HeLa cells. Cell. Signal. 2014;26:784–796. doi: 10.1016/j.cellsig.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 91.Sancho P., Bertran E., Caja L., Carmona-Cuenca I., Murillo M.M., Fabregat I. The inhibition of the epidermal growth factor (EGF) pathway enhances TGF-beta-induced apoptosis in rat hepatoma cells through inducing oxidative stress coincident with a change in the expression pattern of the NADPH oxidases (NOX) isoforms. Biochim. Biophys. Acta. 2009;1793:253–263. doi: 10.1016/j.bbamcr.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 92.Bondi C.D., Manickam N., Lee D.Y., Block K., Gorin Y., Abboud H.E., Barnes J.L. NAD(P)H oxidase mediates TGF-{beta}1-induced activation of kidney myofibroblasts. J. Am. Soc. Nephrol. 2010;21:93–102. doi: 10.1681/ASN.2009020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sturrock A., Cahill B., Norman K., Huecksteadt T.P., Hill K., Sanders K., Karwande S.V., Stringham J.C., Bull D.A., Gleich M., Kennedy T.P., Hoidal J.R. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006;290:L661–L673. doi: 10.1152/ajplung.00269.2005. [DOI] [PubMed] [Google Scholar]
- 94.Sturrock A., Huecksteadt T.P., Norman K., Sanders K., Murphy T.M., Chitano P., Wilson K., Hoidal J.R., Kennedy T.P. Nox4 mediates TGF-beta1-induced retinoblastoma protein phosphorylation, proliferation, and hypertrophy in human airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;292:L1543–L1555. doi: 10.1152/ajplung.00430.2006. [DOI] [PubMed] [Google Scholar]
- 95.Ismail S., Sturrock A., Wu P., Cahill B., Norman K., Huecksteadt T., Sanders K., Kennedy T., Hoidal J. NOX4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: the role of autocrine production of transforming growth factor-{beta}1 and insulin-like growth factor binding protein-3. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009;296:L489–L499. doi: 10.1152/ajplung.90488.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cucoranu I., Clempus R., Dikalova A., Phelan P.J., Ariyan S., Dikalov S., Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 97.Murillo M.M., Carmona-Cuenca I., Del Castillo G., Ortiz C., Roncero C., Sanchez A., Fernandez M., Fabregat I. Activation of NADPH oxidase by transforming growth factor-beta in hepatocytes mediates up-regulation of epidermal growth factor receptor ligands through a nuclear factor-kappaB-dependent mechanism. Biochem. J. 2007;405:251–259. doi: 10.1042/BJ20061846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Carmona-Cuenca I., Roncero C., Sancho P., Caja L., Fausto N., Fernandez M., Fabregat I. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J. Hepatol. 2008;49:965–976. doi: 10.1016/j.jhep.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 99.Hu T., RamachandraRao S.P., Siva S., Valancius C., Zhu Y., Mahadev K., Toh I., Goldstein B.J., Woolkalis M., Sharma K. Reactive oxygen species production via NADPH oxidase mediates TGF-{beta}-induced cytoskeletal alterations in endothelial cells. Am. J. Physiol. Ren. Physiol. 2005;289:F816–F825. doi: 10.1152/ajprenal.00024.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu R.M., Choi J., Wu J.H., Gaston Pravia K.A., Lewis K.M., Brand J.D., Mochel N.S., Krzywanski D.M., Lambeth J.D., Hagood J.S., Forman H.J., Thannickal V.J., Postlethwait E.M. Oxidative modification of nuclear mitogen-activated protein kinase phosphatase 1 is involved in transforming growth factor beta1-induced expression of plasminogen activator inhibitor 1 in fibroblasts. J. Biol. Chem. 2010;285:16239–16247. doi: 10.1074/jbc.M110.111732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Boudreau H.E., Casterline B.W., Rada B., Korzeniowska A., Leto T.L. Nox4 involvement in TGF-beta and SMAD3-driven induction of the epithelial-to-mesenchymal transition and migration of breast epithelial cells. Free Radic. Biol. Med. 2012;53:1489–1499. doi: 10.1016/j.freeradbiomed.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Buul J.D.V., Fernandez-Borja M., Anthony E.C., Hordijk P.L. Expression and localization of NOX2 and NOX4 in primary human endothelial cells. Antioxid. Redox Signal. 2005;7:308. doi: 10.1089/ars.2005.7.308. [DOI] [PubMed] [Google Scholar]
- 103.Chen K., Kirber M.T., Xiao H., Yang Y., Keaney J.F., Jr. Regulation of ROS signal transduction by NADPH oxidase 4 localization. J. Cell Biol. 2008;181:1129–1139. doi: 10.1083/jcb.200709049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Helmcke I., Heumuller S., Tikkanen R., Schroder K., Brandes R.P. Identification of structural elements in Nox1 and Nox4 controlling localization and activity. Antioxid. Redox Signal. 2009;11:1279–1287. doi: 10.1089/ars.2008.2383. [DOI] [PubMed] [Google Scholar]
- 105.Mittal M., Roth M., Konig P., Hofmann S., Dony E., Goyal P., Selbitz A.-C., Schermuly R.T., Ghofrani H.A., Kwapiszewska G., Kummer W., Klepetko W., Hoda M.A.R., Fink L., Hanze J., Seeger W., Grimminger F., Schmidt H.H.H.W., Weissmann N. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 2007;101:258–267. doi: 10.1161/CIRCRESAHA.107.148015. [DOI] [PubMed] [Google Scholar]
- 106.Kuroda Junya, Nakagawa K., Yamasaki Tomoko, Nakamura Kei-ichiro, Takeya Ryu, Kuribayashi Futoshi, Imajoh-Ohmi Shinobu, Igarashi Kazuhiko, Shibata Yosaburo, Sueishi Katsuo, Sumimoto Hideki. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells. 2005;10:1139–1151. doi: 10.1111/j.1365-2443.2005.00907.x. [DOI] [PubMed] [Google Scholar]
- 107.Case A.J., Li S., Basu U., Tian J., Zimmerman M.C. Mitochondrial-localized NADPH oxidase 4 is a source of superoxide in angiotensin II-stimulated neurons. Am. J. Physiol. Heart Circ. Physiol. 2013;305:H19–H28. doi: 10.1152/ajpheart.00974.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Block K., Gorin Y., Abboud H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA. 2009;106:14385–14390. doi: 10.1073/pnas.0906805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kim S.M., Kim Y.G., Jeong K.H., Lee S.H., Lee T.W., Ihm C.G., Moon J.Y. Angiotensin II-induced mitochondrial Nox4 is a major endogenous source of oxidative stress in kidney tubular cells. PloS One. 2012;7:e39739. doi: 10.1371/journal.pone.0039739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Casalena G., Daehn I., Bottinger E. Transforming growth factor-beta, bioenergetics, and mitochondria in renal disease. Semin. Nephrol. 2012;32:295–303. doi: 10.1016/j.semnephrol.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hecker L., Vittal R., Jones T., Jagirdar R., Luckhardt T.R., Horowitz J.C., Pennathur S., Martinez F.J., Thannickal V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009;15:1077–1081. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tobar N., Toyos M., Urra C., Mendez N., Arancibia R., Smith P.C., Martinez J. c-Jun N terminal kinase modulates NOX-4 derived ROS production and myofibroblasts differentiation in human breast stromal cells. BMC Cancer. 2014;14:640. doi: 10.1186/1471-2407-14-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yan F., Wang Y., Wu X., Peshavariya H.M., Dusting G.J., Zhang M., Jiang F. Nox4 and redox signaling mediate TGF-beta-induced endothelial cell apoptosis and phenotypic switch. Cell Death Dis. 2014;5:e1010. doi: 10.1038/cddis.2013.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Caja L., Sancho P., Bertran E., Iglesias-Serret D., Gil J., Fabregat I. Overactivation of the MEK/ERK pathway in liver tumor cells confers resistance to TGF-{beta}-induced cell death through impairing up-regulation of the NADPH oxidase NOX4. Cancer Res. 2009;69:7595–7602. doi: 10.1158/0008-5472.CAN-09-1482. [DOI] [PubMed] [Google Scholar]
- 115.Chan E.C., Peshavariya H.M., Liu G.S., Jiang F., Lim S.Y., Dusting G.J. Nox4 modulates collagen production stimulated by transforming growth factor beta1 in vivo and in vitro. Biochem. Biophys. Res. Commun. 2013;430:918–925. doi: 10.1016/j.bbrc.2012.11.138. [DOI] [PubMed] [Google Scholar]
- 116.Amara N., Goven D., Prost F., Muloway R., Crestani B., Boczkowski J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax. 2010;65:733–738. doi: 10.1136/thx.2009.113456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hecker L., Logsdon N.J., Kurundkar D., Kurundkar A., Bernard K., Hock T., Meldrum E., Sanders Y.Y., Thannickal V.J. Reversal of persistent fibrosis in aging by targeting nox4-nrf2 redox imbalance. Sci. Transl. Med. 2014;6:231ra247. doi: 10.1126/scitranslmed.3008182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jiang J.X., Chen X., Serizawa N., Szyndralewiez C., Page P., Schroder K., Brandes R.P., Devaraj S., Torok N.J. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic. Biol. Med. 2012;53:289–296. doi: 10.1016/j.freeradbiomed.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Michaeloudes C., Sukkar M.B., Khorasani N.M., Bhavsar P.K., Chung K.F. TGF-beta regulates Nox4, MnSOD and catalase expression, and IL-6 release in airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011;300:L295–L304. doi: 10.1152/ajplung.00134.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yeh Y.H., Kuo C.T., Chang G.J., Qi X.Y., Nattel S., Chen W.J. Nicotinamide adenine dinucleotide phosphate oxidase 4 mediates the differential responsiveness of atrial versus ventricular fibroblasts to transforming growth factor-beta. Circ. Arrhythm. Electrophysiol. 2013;6:790–798. doi: 10.1161/CIRCEP.113.000338. [DOI] [PubMed] [Google Scholar]
- 121.Rathore R., Zheng Y.M., Niu C.F., Liu Q.H., Korde A., Ho Y.S., Wang Y.X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2008;45:1223–1231. doi: 10.1016/j.freeradbiomed.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wosniak J., Jr, Santos C.X., Kowaltowski A.J., Laurindo F.R. Cross-talk between mitochondria and NADPH oxidase: effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid. Redox Signal. 2009;11:1265–1278. doi: 10.1089/ars.2009.2392. [DOI] [PubMed] [Google Scholar]
- 123.Daiber A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta. 2010;1797:897–906. doi: 10.1016/j.bbabio.2010.01.032. [DOI] [PubMed] [Google Scholar]
- 124.Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011;51:1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vendrov A.E., Vendrov K.C., Smith A., Yuan J., Sumida A., Robidoux J., Runge M.S., Madamanchi N.R. NOX4 NADPH oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxid. Redox Signal. 2015 doi: 10.1089/ars.2014.6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Roum J.H., Buhl R., McElvany N.G., Borok Z., Crystal R.G. Systemic deficiency of glutathione in cystic fibrosis. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1993;75:2419–2424. doi: 10.1152/jappl.1993.75.6.2419. [DOI] [PubMed] [Google Scholar]
- 127.Tirouvanziam R., Conrad C.K., Bottiglieri T., Herzenberg L.A., Moss R.B. High-dose oral N-acetylcysteine, a glutathione prodrug, modulates inflammation in cystic fibrosis. Proc. Natl. Acad. Sci. USA. 2006;103:4628–4633. doi: 10.1073/pnas.0511304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rahman I., Skwarska E., Henry M., Davis M., O'Connor C.M., FitzGerald M.X., Greening A., MacNee W. Systemic and pulmonary oxidative stress in idiopathic pulmonary fibrosis. Free Radic. Biol. Med. 1999;27:60–68. doi: 10.1016/s0891-5849(99)00035-0. [DOI] [PubMed] [Google Scholar]
- 129.Pacht E.R., Timerman A.P., Lykens M.G., Merola A.J. Deficiency of alveolar fluid glutathione in patients with sepsis and the adult respiratory distress syndrome. Chest. 1991;100:1397–1403. doi: 10.1378/chest.100.5.1397. [DOI] [PubMed] [Google Scholar]
- 130.Bunnell E., Pacht E.R. Oxidized glutathione is increased in the alveolar fluid of patients with the adult respiratory distress syndrome. Am. Rev. Respir. Dis. 1993;148:1174–1178. doi: 10.1164/ajrccm/148.5.1174. [DOI] [PubMed] [Google Scholar]
- 131.Laurent T., Markert M., Feihl F., Schaller M.D., Perret C. Oxidant-antioxidant balance in granulocytes during ARDS. Effect of N-acetylcysteine. Chest. 1996;109:163–166. doi: 10.1378/chest.109.1.163. [DOI] [PubMed] [Google Scholar]
- 132.Bernard G.R., Wheeler A.P., Arons M.M., Morris P.E., Paz H.L., Russell J.A., Wright P.E. A trial of antioxidants N-acetylcysteine and procysteine in ARDS. The Antioxidant in ARDS Study Group. Chest. 1997;112:164–172. doi: 10.1378/chest.112.1.164. [DOI] [PubMed] [Google Scholar]
- 133.Soltan-Sharifi M.S., Mojtahedzadeh M., Najafi A., Reza Khajavi M., Reza Rouini M., Moradi M., Mohammadirad A., Abdollahi M. Improvement by N-acetylcysteine of acute respiratory distress syndrome through increasing intracellular glutathione, and extracellular thiol molecules and anti-oxidant power: evidence for underlying toxicological mechanisms. Hum. Exp. Toxicol. 2007;26:697–703. doi: 10.1177/0960327107083452. [DOI] [PubMed] [Google Scholar]
- 134.Borok Z., Grimes G.J., Buhl R., Bokser A., Hubbard R.C., Holroyd K., Roum J.H., Czerski D.B., Cantin A.M., Crystal R.G. Effect of glutathione aerosol on oxidant-antioxidant imbalance in idiopathic pulmonary fibrosis. Lancet. 1991;338:215–216. doi: 10.1016/0140-6736(91)90350-x. [DOI] [PubMed] [Google Scholar]
- 135.Meyer A., Buhl R., Magnussen H. The effect of oral N-acetylcysteine on lung glutathione levels in idiopathic pulmonary fibrosis. Eur. Respir. J. 1994;7:431–436. doi: 10.1183/09031936.94.07030431. [DOI] [PubMed] [Google Scholar]
- 136.Behr J., Maier K., Degenkolb B., Krombach F., Vogelmeier C. Antioxidative and clinical effects of high-dose N-acetylcysteine in fibrosing alveolitis. Adjunctive therapy to maintenance immunosuppression. Am. J. Respir. Crit. Care Med. 1997;156:1897–1901. doi: 10.1164/ajrccm.156.6.9706065. [DOI] [PubMed] [Google Scholar]
- 137.Behr J., Degenkolb B., Krombach F., Vogelmeier C. Intracellular glutathione and bronchoalveolar cells in fibrosing alveolitis: effects of N-acetylcysteine. Eur. Respir. J. 2002;19:906–911. doi: 10.1183/09031936.02.00204902. [DOI] [PubMed] [Google Scholar]
- 138.Montaldo C., Cannas E., Ledda M., Rosetti L., Congiu L., Atzori L. Bronchoalveolar glutathione and nitrite/nitrate in idiopathic pulmonary fibrosis and sarcoidosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2002;19:54–58. [PubMed] [Google Scholar]
- 139.Beeh K.M., Beier J., Haas I.C., Kornmann O., Micke P., Buhl R. Glutathione deficiency of the lower respiratory tract in patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 2002;19:1119–1123. doi: 10.1183/09031936.02.00262402. [DOI] [PubMed] [Google Scholar]
- 140.Tiitto L.H., Peltoniemi M.J., Kaarteenaho-Wiik R.L., Soini Y.M., Paakko P.K., Sormunen R.T., Kinnula V.L. Cell-specific regulation of gamma-glutamylcysteine synthetase in human interstitial lung diseases. Hum. Pathol. 2004;35:832–839. doi: 10.1016/j.humpath.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 141.Boots A.W., Drent M., Swennen E.L.R., Moonen H.J.J., Bast A., Haenen G.R.M.M. Antioxidant status associated with inflammation in sarcoidosis: a potential role for antioxidants. Respir. Med. 2009;103:364–372. doi: 10.1016/j.rmed.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 142.Bianchi G., Bugianesi E., Ronchi M., Fabbri A., Zoli M., Marchesini G. Glutathione kinetics in normal man and in patients with liver cirrhosis. J. Hepatol. 1997;26:606–613. doi: 10.1016/s0168-8278(97)80426-6. [DOI] [PubMed] [Google Scholar]
- 143.Aboutwerat A., Pemberton P.W., Smith A., Burrows P.C., McMahon R.F.T., Jain S.K., Warnes T.W. Oxidant stress is a significant feature of primary biliary cirrhosis. Biochim. Et. Biophys. Acta – Mol. Basis Dis. 1637;142–150:2003. doi: 10.1016/s0925-4439(02)00225-9. [DOI] [PubMed] [Google Scholar]
- 144.Loguercio C., Federico A. Oxidative stress in viral and alcoholic hepatitis. Free Radic. Biol. Med. 2003;34:1–10. doi: 10.1016/s0891-5849(02)01167-x. [DOI] [PubMed] [Google Scholar]
- 145.Czeczot H., Scibior D., Skrzycki M., Podsiad M. Glutathione and GSH-dependent enzymes in patients with liver cirrhosis and hepatocellular carcinoma. Acta Biochim. Pol. 2006;53:237–242. [PubMed] [Google Scholar]
- 146.Priyanka Bandara J.G., Geoffrey Mc.Caughan, Daya Naidoo, Ora Lux, Chris Salonikas, Kench James, Byth Karen, Geoffrey C. Farrell. Antioxidant levels in peripheral blood, disease activity and fibrotic stage in chronic hepatitis C. Liver Int. 2005;25:518–526. doi: 10.1111/j.1478-3231.2005.01049.x. [DOI] [PubMed] [Google Scholar]
- 147.Marotta F., Yoshida C., Barreto R., Naito Y., Packer L. Oxidative-inflammatory damage in cirrhosis: effect of vitamin E and a fermented papaya preparation. J. Gastroenterol. Hepatol. 2007;22:697–703. doi: 10.1111/j.1440-1746.2007.04937.x. [DOI] [PubMed] [Google Scholar]
- 148.Osman H., Gabr O., Lotfy S., Gabr S. Serum levels of bcl-2 and cellular oxidative stress in patients with viral hepatitis. Indian J. Med. Microbiol. 2007;25:323–329. doi: 10.4103/0255-0857.37333. [DOI] [PubMed] [Google Scholar]
- 149.Ma J.Y., Barger M.W., Hubbs A.F., Castranova V., Weber S.L., Ma J.K. Use of tetrandrine to differentiate between mechanisms involved in silica-versus bleomycin-induced fibrosis. J. Toxicol. Environ. Healh A. 1999;57:247–266. doi: 10.1080/009841099157692. [DOI] [PubMed] [Google Scholar]
- 150.Candan F., Alagozlu H. Captopril inhibits the pulmonary toxicity of paraquat in rats. Hum. Exp. Toxicol. 2001;20:637–641. doi: 10.1191/096032701718890540. [DOI] [PubMed] [Google Scholar]
- 151.Arslan S.O., Zerin M., Vural H., Coskun A. The effect of melatonin on bleomycin-induced pulmonary fibrosis in rats. J. Pineal Res. 2002;32:21–25. doi: 10.1034/j.1600-079x.2002.10796.x. [DOI] [PubMed] [Google Scholar]
- 152.Nieto N., Cederbaum A.I. S-adenosylmethionine blocks collagen I production by preventing transforming growth factor-{beta} induction of the COL1A2 promoter. J. Biol. Chem. 2005;280:30963–30974. doi: 10.1074/jbc.M503569200. [DOI] [PubMed] [Google Scholar]
- 153.Iyer S.S., Ramirez A.M., Ritzenthaler J.D., Torres-Gonzalez E., Roser-Page S., Mora A.L., Brigham K.L., Jones D.P., Roman J., Rojas M. Oxidation of extracellular cysteine/cystine redox state in bleomycin-induced lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009;296:L37–L45. doi: 10.1152/ajplung.90401.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Atzori L., Chua F., Dunsmore S.E., Willis D., Barbarisi M., McAnulty R.J., Laurent G.J. Attenuation of bleomycin induced pulmonary fibrosis in mice using the heme oxygenase inhibitor Zn-deuteroporphyrin IX-2,4-bisethylene glycol. Thorax. 2004;59:217–223. doi: 10.1136/thx.2003.008979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.El-Medany A., Hagar H.H., Moursi M., At Muhammed R., El-Rakhawy F.I., El-Medany G. Attenuation of bleomycin-induced lung fibrosis in rats by mesna. Eur. J. Pharmacol. 2005;509:61–70. doi: 10.1016/j.ejphar.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 156.Boyacı H., Maral H., Turan G., Başyiğit İ., Dillioğlugil M., Yıldız F., Tugay M., Pala A., Erçin C. Effects of erdosteine on bleomycin-induced lung fibrosis in rats. Mol. Cell. Biochem. 2006;281:129–137. doi: 10.1007/s11010-006-0640-3. [DOI] [PubMed] [Google Scholar]
- 157.Deger Yeter, Yur F., Ertekin Ali, Mert Nihat, Dede Semiha, Mert Handan. Protective effect of alpha-tocopherol on oxidative stress in experimental pulmonary fibrosis in rats. Cell Biochem. Funct. 2007;25:633–637. doi: 10.1002/cbf.1362. [DOI] [PubMed] [Google Scholar]
- 158.Arafa H.M.M., Abdel-Wahab M.H., El-Shafeey M.F., Badary O.A., Hamada F.M.A. Anti-fibrotic effect of meloxicam in a murine lung fibrosis model. Eur. J. Pharmacol. 2007;564:181–189. doi: 10.1016/j.ejphar.2007.02.065. [DOI] [PubMed] [Google Scholar]
- 159.White A.C., Das S.K., Fanburg B.L. Reduction of glutathione is associated with growth restriction and enlargement of bovine pulmonary artery endothelial cells produced by transforming growth factor-beta 1. Am. J. Respir. Cell Mol. Biol. 1992;6:364–368. doi: 10.1165/ajrcmb/6.4.364. [DOI] [PubMed] [Google Scholar]
- 160.Sanchez A., Alvarez A.M., Benito M., Fabregat I. Apoptosis induced by transforming growth factor-beta in fetal hepatocyte primary cultures: involvement of reactive oxygen intermediates. J. Biol. Chem. 1996;271:7416–7422. doi: 10.1074/jbc.271.13.7416. [DOI] [PubMed] [Google Scholar]
- 161.Sanchez A., Alvarez A.M., Benito M., Fabregat I. Cycloheximide prevents apoptosis, reactive oxygen species production, and glutathione depletion induced by transforming growth factor beta in fetal rat hepatocytes in primary culture. Hepatology. 1997;26:935–943. doi: 10.1002/hep.510260420. [DOI] [PubMed] [Google Scholar]
- 162.Arsalane K., Dubois C.M., Muanza T., Begin R., Boudreau F., Asselin C., Cantin A.M. Transforming growth factor-beta1 is a potent inhibitor of glutathione synthesis in the lung epithelial cell line A549: transcriptional effect on the GSH rate-limiting enzyme gamma-glutamylcysteine synthetase. Am. J. Respir. Cell Mol. Biol. 1997;17:599–607. doi: 10.1165/ajrcmb.17.5.2833. [DOI] [PubMed] [Google Scholar]
- 163.Pittet J.F., Griffiths M.J., Geiser T., Kaminski N., Dalton S.L., Huang X., Brown L.A., Gotwals P.J., Koteliansky V.E., Matthay M.A., Sheppard D. TGF-beta is a critical mediator of acute lung injury. J. Clin. Invest. 2001;107:1537–1544. doi: 10.1172/JCI11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Jardine H., MacNee W., Donaldson K., Rahman I. Molecular mechanism of transforming growth factor (TGF)-beta1-induced glutathione depletion in alveolar epithelial cells. Involvement of AP-1/ARE and Fra-1. J. Biol. Chem. 2002;277:21158–21166. doi: 10.1074/jbc.M112145200. [DOI] [PubMed] [Google Scholar]
- 165.Franklin C.C., Rosenfeld-Franklin M.E., White C., Kavanagh T.J., Fausto N. TGFbeta1-induced suppression of glutathione antioxidant defenses in hepatocytes: caspase-dependent post-translational and caspase-independent transcriptional regulatory mechanisms. FASEB J. 2003;17:1535–1537. doi: 10.1096/fj.02-0867fje. [DOI] [PubMed] [Google Scholar]
- 166.Herrera B., Murillo M.M., Alvarez-Barrientos A., Beltran J., Fernandez M., Fabregat I. Source of early reactive oxygen species in the apoptosis induced by transforming growth factor-beta in fetal rat hepatocytes. Free Radic. Biol. Med. 2004;36:16–26. doi: 10.1016/j.freeradbiomed.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 167.Bakin A.V., Stourman N.V., Sekhar K.R., Rinehart C., Yan X., Meredith M.J., Arteaga C.L., Freeman M.L. Smad3-ATF3 signaling mediates TGF-[beta] suppression of genes encoding Phase II detoxifying proteins. Free Radic. Biol. Med. 2005;38:375–387. doi: 10.1016/j.freeradbiomed.2004.10.033. [DOI] [PubMed] [Google Scholar]
- 168.Liu R.-M., Liu Y., Forman H.J., Olman M., Tarpey M.M. Glutathione regulates transforming growth factor-{beta}-stimulated collagen production in fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004;286:L121–L128. doi: 10.1152/ajplung.00231.2003. [DOI] [PubMed] [Google Scholar]
- 169.McKone E.F., Shao J., Frangolias D.D., Keener C.L., Shephard C.A., Farin F.M., Tonelli M.R., Pare P.D., Sandford A.J., Aitken M.L., Kavanagh T.J. Variants in the glutamate-cysteine-ligase gene are associated with cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2006;174:415–419. doi: 10.1164/rccm.200508-1281OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Schafer F.Q., Buettner G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 171.Dayer R., Fischer B.B., Eggen R.I., Lemaire S.D. The peroxiredoxin and glutathione peroxidase families in Chlamydomonas reinhardtii. Genetics. 2008;179:41–57. doi: 10.1534/genetics.107.086041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kayanoki Y., Fujii J., Suzuki K., Kawata S., Matsuzawa Y., Taniguchi N. Suppression of antioxidative enzyme expression by transforming growth factor-beta 1 in rat hepatocytes. J. Biol. Chem. 1994;269:15488–15492. [PubMed] [Google Scholar]
- 173.Islam K.N., Kayanoki Y., Kaneto H., Suzuki K., Asahi M., Fujii J., Taniguchi N. TGF-beta1 triggers oxidative modifications and enhances apoptosis in HIT cells through accumulation of reactive oxygen species by suppression of catalase and glutathione peroxidase. Free Radic. Biol. Med. 1997;22:1007–1017. doi: 10.1016/s0891-5849(96)00493-5. [DOI] [PubMed] [Google Scholar]
- 174.Peltoniemi M., Kaarteenaho-Wiik R., Saily M., Sormunen R., Paakko P., Holmgren A., Soini Y., Kinnula V.L. Expression of glutaredoxin is highly cell specific in human lung and is decreased by transforming growth factor-beta in vitro and in interstitial lung diseases in vivo. Hum. Pathol. 2004;35:1000–1007. doi: 10.1016/j.humpath.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 175.Gao F., Kinnula V.L., Myllarniemi M., Oury T.D. Extracellular superoxide dismutase in pulmonary fibrosis. Antioxid. Redox Signal. 2008;10:343–354. doi: 10.1089/ars.2007.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Cui Y., Robertson J., Maharaj S., Waldhauser L., Niu J., Wang J., Farkas L., Kolb M., Gauldie J. Oxidative stress contributes to the induction and persistence of TGF-beta1 induced pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2011;43:1122–1133. doi: 10.1016/j.biocel.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 177.Saharinen J., Taipale J., Monni O., Keski-Oja J. Identification and characterization of a new latent transforming growth factor-beta -binding protein, LTBP-4. J. Biol. Chem. 1998;273:18459–18469. doi: 10.1074/jbc.273.29.18459. [DOI] [PubMed] [Google Scholar]
- 178.Todorovic V., Jurukovski V., Chen Y., Fontana L., Dabovic B., Rifkin D.B. Latent TGF-[beta] binding proteins. Int. J. Biochem. Cell Biol. 2005;37:38–41. doi: 10.1016/j.biocel.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 179.Schultz-Cherry S., Murphy-Ullrich J.E. Thrombospondin causes activation of latent transforming growth factor-beta secreted by endothelial cells by a novel mechanism. J. Cell Biol. 1993;122:923–932. doi: 10.1083/jcb.122.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Murphy-Ullrich J.E., Poczatek M. Activation of latent TGF-beta by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev. 2000;11:59–69. doi: 10.1016/s1359-6101(99)00029-5. [DOI] [PubMed] [Google Scholar]
- 181.Munger J.S., Huang X., Kawakatsu H., Griffiths M.J., Dalton S.L., Wu J., Pittet J.F., Kaminski N., Garat C., Matthay M.A., Rifkin D.B., Sheppard D. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 182.Mu D., Cambier S., Fjellbirkeland L., Baron J.L., Munger J.S., Kawakatsu H., Sheppard D., Broaddus V.C., Nishimura S.L. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J. Cell Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Lyons R.M., Keski-Oja J., Moses H.L. Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J. Cell Biol. 1988;106:1659–1665. doi: 10.1083/jcb.106.5.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Taipale J., Lohi J., Saarinen J., Kovanen P.T., Keski-Oja J. Human mast cell chymase and leukocyte elastase release latent transforming growth factor-beta 1 from the extracellular matrix of cultured human epithelial and endothelial cells. J. Biol. Chem. 1995;270:4689–4696. doi: 10.1074/jbc.270.9.4689. [DOI] [PubMed] [Google Scholar]
- 185.Taipale J., Koli K., Keski-Oja J. Release of transforming growth factor-beta 1 from the pericellular matrix of cultured fibroblasts and fibrosarcoma cells by plasmin and thrombin. J. Biol. Chem. 1992;267:25378–25384. [PubMed] [Google Scholar]
- 186.Yu Q., Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 187.Barcellos-Hoff M., Dix T. Redox-mediated activation of latent transforming growth factor-beta 1. Mol. Endocrinol. 1996;10:1077–1083. doi: 10.1210/mend.10.9.8885242. [DOI] [PubMed] [Google Scholar]
- 188.Vodovotz Y., Chesler L., Chong H., Kim S.J., Simpson J.T., DeGraff W., Cox G.W., Roberts A.B., Wink D.A., Barcellos-Hoff M.H. Regulation of transforming growth factor beta1 by nitric oxide. Cancer Res. 1999;59:2142–2149. [PubMed] [Google Scholar]
- 189.Wang L., Clutter S., Benincosa J., Fortney J., Gibson L.F. Activation of TGF-{beta}1/p38/Smad3 signaling in stromal cells requires ROS-mediated MMP-2 activity during bone marrow damage. Stem Cells. 2005;23:1122–1134. doi: 10.1634/stemcells.2004-0354. [DOI] [PubMed] [Google Scholar]
- 190.Amarnath S., Dong L., Li J., Wu Y., Chen W. Endogenous TGF-beta activation by reactive oxygen species is key to Foxp3 induction in TCR-stimulated and HIV-1-infected human CD4+CD25− T cells. Retrovirology. 2007;4:57. doi: 10.1186/1742-4690-4-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Koli K., Myllarniemi M., Keski-Oja J., Kinnula V.L. Transforming growth factor-beta activation in the lung: focus on fibrosis and reactive oxygen species. Antioxid. Redox Signal. 2008;10:333–342. doi: 10.1089/ars.2007.1914. [DOI] [PubMed] [Google Scholar]
- 192.Sullivan D.E., Ferris M., Pociask D., Brody A.R. The latent form of TGFbeta(1) is induced by TNFalpha through an ERK specific pathway and is activated by asbestos-derived reactive oxygen species in vitro and in vivo. J. Immunotoxicol. 2008;5:145–149. doi: 10.1080/15476910802085822. [DOI] [PubMed] [Google Scholar]
- 193.Jobling M.F., Mott J.D., Finnegan M.T., Jurukovski V., Erickson A.C., Walian P.J., Taylor S.E., Ledbetter S., Lawrence C.M., Rifkin D.B., Barcellos-Hoff M.H. Isoform-specific activation of latent transforming growth factor beta (LTGF-beta) by reactive oxygen species. Radiat. Res. 2006;166:839–848. doi: 10.1667/RR0695.1. [DOI] [PubMed] [Google Scholar]
- 194.Saito K., Ishizaka N., Aizawa T., Sata M., Iso-o N., Noiri E., Mori I., Ohno M., Nagai R. Iron chelation and a free radical scavenger suppress angiotensin II-induced upregulation of TGF-beta1 in the heart. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H1836–H1843. doi: 10.1152/ajpheart.00679.2004. [DOI] [PubMed] [Google Scholar]
- 195.Zhao W., Zhao T., Chen Y., Ahokas R.A., Sun Y. Oxidative stress mediates cardiac fibrosis by enhancing transforming growth factor-beta1 in hypertensive rats. Mol. Cell. Biochem. 2008;317:43–50. doi: 10.1007/s11010-008-9803-8. [DOI] [PubMed] [Google Scholar]
- 196.Shvedova A.A., Kisin E.R., Murray A.R., Kommineni C., Castranova V., Fadeel B., Kagan V.E. Increased accumulation of neutrophils and decreased fibrosis in the lung of NADPH oxidase-deficient C57BL/6 mice exposed to carbon nanotubes. Toxicol. Appl. Pharmacol. 2008;231:235–240. doi: 10.1016/j.taap.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 197.Lin W., Tsai W.L., Shao R.X., Wu G., Peng L.F., Barlow L.L., Chung W.J., Zhang L., Zhao H., Jang J.Y., Chung R.T. Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology. 2010;138:2509–2518. doi: 10.1053/j.gastro.2010.03.008. 2518. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Jaffer O.A., Carter A.B., Sanders P.N., Dibbern M.E., Winters C.J., Murthy S., Ryan A.J., Rokita A.G., Prasad A.M., Zabner J., Kline J.N., Grumbach I.M., Anderson M.E. Mitochondrial-targeted antioxidant therapy decreases transforming growth factor-beta-mediated collagen production in a murine asthma model. Am. J. Respir. Cell Mol. Biol. 2015;52:106–115. doi: 10.1165/rcmb.2013-0519OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Montorfano I., Becerra A., Cerro R., Echeverria C., Saez E., Morales M.G., Fernandez R., Cabello-Verrugio C., Simon F. Oxidative stress mediates the conversion of endothelial cells into myofibroblasts via a TGF-beta1 and TGF-beta2-dependent pathway. Lab. Invest. 2014;94:1068–1082. doi: 10.1038/labinvest.2014.100. [DOI] [PubMed] [Google Scholar]
- 200.Oruqaj G., Karnati S., Vijayan V., Kotarkonda L.K., Boateng E., Zhang W., Ruppert C., Gunther A., Shi W., Baumgart-Vogt E. Compromised peroxisomes in idiopathic pulmonary fibrosis, a vicious cycle inducing a higher fibrotic response via TGF-beta signaling. Proc. Natl. Acad. Sci. USA. 2015;112:E2048–E2057. doi: 10.1073/pnas.1415111112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bellocq A., Azoulay E., Marullo S., Flahault A., Fouqueray B., Philippe C., Cadranel J., Baud L. Reactive oxygen and nitrogen intermediates increase transforming growth factor-beta1 release from human epithelial alveolar cells through two different mechanisms. Am. J. Respir. Cell Mol. Biol. 1999;21:128–136. doi: 10.1165/ajrcmb.21.1.3379. [DOI] [PubMed] [Google Scholar]
- 202.Moon Y.M., Kang H.J., Cho J.S., Park I.H., Lee H.M. Nox4 mediates hypoxia-stimulated myofibroblast differentiation in nasal polyp-derived fibroblasts. Int. Arch. Allergy Immunol. 2012;159:399–409. doi: 10.1159/000337658. [DOI] [PubMed] [Google Scholar]
- 203.Sancho P., Mainez J., Crosas-Molist E., Roncero C., Fernandez-Rodriguez C.M., Pinedo F., Huber H., Eferl R., Mikulits W., Fabregat I. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PloS One. 2012;7:e45285. doi: 10.1371/journal.pone.0045285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Aoyama T., Paik Y.H., Watanabe S., Laleu B., Gaggini F., Fioraso-Cartier L., Molango S., Heitz F., Merlot C., Szyndralewiez C., Page P., Brenner D.A. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology. 2012;56:2316–2327. doi: 10.1002/hep.25938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Maher T.M., Evans I.C., Bottoms S.E., Mercer P.F., Thorley A.J., Nicholson A.G., Laurent G.J., Tetley T.D., Chambers R.C., McAnulty R.J. Diminished prostaglandin E2 contributes to the apoptosis paradox in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010;182:73–82. doi: 10.1164/rccm.200905-0674OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Buhling F., Wille A., Rocken C., Wiesner O., Baier A., Meinecke I., Welte T., Pap T. Altered expression of membrane-bound and soluble CD95/Fas contributes to the resistance of fibrotic lung fibroblasts to FasL induced apoptosis. Respir. Res. 2005;6:37. doi: 10.1186/1465-9921-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Huang W.T., Akhter H., Jiang C., MacEwen M., Ding Q., Antony V., Thannickal V.J., Liu R.M. Plasminogen activator inhibitor 1, fibroblast apoptosis resistance, and aging-related susceptibility to lung fibrosis. Exp. Gerontol. 2015;61:62–75. doi: 10.1016/j.exger.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Barbas-Filho J.V., Ferreira M.A., Sesso A., Kairalla R.A., Carvalho C.R., Capelozzi V.L. Evidence of type II pneumocyte apoptosis in the pathogenesis of idiopathic pulmonary fibrosis (IFP)/usual interstitial pneumonia (UIP) J. Clin. Pathol. 2001;54:132–138. doi: 10.1136/jcp.54.2.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Plataki M., Koutsopoulos A.V., Darivianaki K., Delides G., Siafakas N.M., Bouros D. Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest. 2005;127:266–274. doi: 10.1378/chest.127.1.266. [DOI] [PubMed] [Google Scholar]
- 210.Gunther A., Korfei M., Mahavadi P., von der Beck D., Ruppert C., Markart P. Unravelling the progressive pathophysiology of idiopathic pulmonary fibrosis. Eur. Respir. Rev.: Off. J. Eur. Respir. Soc. 2012;21:152–160. doi: 10.1183/09059180.00001012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Moodley Y.P., Caterina P., Scaffidi A.K., Misso N.L., Papadimitriou J.M., McAnulty R.J., Laurent G.J., Thompson P.J., Knight D.A. Comparison of the morphological and biochemical changes in normal human lung fibroblasts and fibroblasts derived from lungs of patients with idiopathic pulmonary fibrosis during FasL-induced apoptosis. J. Pathol. 2004;202:486–495. doi: 10.1002/path.1531. [DOI] [PubMed] [Google Scholar]
- 212.Hagimoto N., Kuwano K., Nomoto Y., Kunitake R., Hara N. Apoptosis and expression of Fas/Fas ligand mRNA in bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Cell. Mol. Biol. 1997;16:91–101. doi: 10.1165/ajrcmb.16.1.8998084. [DOI] [PubMed] [Google Scholar]
- 213.Budinger G.R., Mutlu G.M., Eisenbart J., Fuller A.C., Bellmeyer A.A., Baker C.M., Wilson M., Ridge K., Barrett T.A., Lee V.Y., Chandel N.S. Proapoptotic Bid is required for pulmonary fibrosis. Proc. Natl. Acad. Sci. USA. 2006;103:4604–4609. doi: 10.1073/pnas.0507604103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Degryse A.L., Tanjore H., Xu X.C., Polosukhin V.V., Jones B.R., McMahon F.B., Gleaves L.A., Blackwell T.S., Lawson W.E. Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010;299:L442–L452. doi: 10.1152/ajplung.00026.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Iwano M., Plieth D., Danoff T.M., Xue C., Okada H., Neilson E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Slattery C., Campbell E., McMorrow T., Ryan M.P. Cyclosporine A-induced renal fibrosis: a role for epithelial–mesenchymal transition. Am. J. Pathol. 2005;167:395–407. doi: 10.1016/S0002-9440(10)62984-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Saika S., Kono-Saika S., Ohnishi Y., Sato M., Muragaki Y., Ooshima A., Flanders K.C., Yoo J., Anzano M., Liu C.Y., Kao W.W., Roberts A.B. Smad3 signaling is required for epithelial–mesenchymal transition of lens epithelium after injury. Am. J. Pathol. 2004;164:651–663. doi: 10.1016/S0002-9440(10)63153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Margetts P.J., Bonniaud P., Liu L., Hoff C.M., Holmes C.J., West-Mays J.A., Kelly M.M. Transient overexpression of TGF-{beta}1 induces epithelial mesenchymal transition in the rodent peritoneum. J. Am. Soc. Nephrol. 2005;16:425–436. doi: 10.1681/ASN.2004060436. [DOI] [PubMed] [Google Scholar]
- 219.Kim K.K., Kugler M.C., Wolters P.J., Robillard L., Galvez M.G., Brumwell A.N., Sheppard D., Chapman H.A. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. USA. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wu Z., Yang L., Cai L., Zhang M., Cheng X., Yang X., Xu J. Detection of epithelial to mesenchymal transition in airways of a bleomycin induced pulmonary fibrosis model derived from an alpha-smooth muscle actin-Cre transgenic mouse. Respir. Res. 2007;8:1. doi: 10.1186/1465-9921-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Thiery J.P., Acloque H., Huang R.Y., Nieto M.A. Epithelial–mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 222.Chapman H.A. Epithelial–mesenchymal interactions in pulmonary fibrosis. Annu. Rev. Physiol. 2011;73:413–435. doi: 10.1146/annurev-physiol-012110-142225. [DOI] [PubMed] [Google Scholar]
- 223.Marmai C., Sutherland R.E., Kim K.K., Dolganov G.M., Fang X., Kim S.S., Jiang S., Golden J.A., Hoopes C.W., Matthay M.A., Chapman H.A., Wolters P.J. Alveolar epithelial cells express mesenchymal proteins in patients with idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2011;301:L71–L78. doi: 10.1152/ajplung.00212.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Flier S.N., Tanjore H., Kokkotou E.G., Sugimoto H., Zeisberg M., Kalluri R. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J. Biol. Chem. 2010;285:20202–20212. doi: 10.1074/jbc.M110.102012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Tanjore H., Xu X.C., Polosukhin V.V., Degryse A.L., Li B., Han W., Sherrill T.P., Plieth D., Neilson E.G., Blackwell T.S., Lawson W.E. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 2009;180:657–665. doi: 10.1164/rccm.200903-0322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.LeBleu V.S., Taduri G., O'Connell J., Teng Y., Cooke V.G., Woda C., Sugimoto H., Kalluri R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013;19:1047–1053. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Jain R., Shaul P.W., Borok Z., Willis B.C. Endothelin-1 induces alveolar epithelial–mesenchymal transition through endothelin type A receptor-mediated production of TGF-beta1. Am. J. Respir. Cell Mol. Biol. 2007;37:38–47. doi: 10.1165/rcmb.2006-0353OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Rhyu D.Y., Yang Y., Ha H., Lee G.T., Song J.S., Uh S.T., Lee H.B. Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial–mesenchymal transition in renal tubular epithelial cells. J. Am. Soc. Nephrol. 2005;16:667–675. doi: 10.1681/ASN.2004050425. [DOI] [PubMed] [Google Scholar]
- 229.Lee E.K., Jeon W.K., Chae M.Y., Hong H.Y., Lee Y.S., Kim J.H., Kwon J.Y., Kim B.C., Park S.H. Decreased expression of glutaredoxin 1 is required for transforming growth factor-beta1-mediated epithelial–mesenchymal transition of EpRas mammary epithelial cells. Biochem. Biophys. Res. Commun. 2010;391:1021–1027. doi: 10.1016/j.bbrc.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 230.Zhou Q., Yang M., Lan H., Yu X. miR-30a negatively regulates TGF-beta1-induced epithelial–mesenchymal transition and peritoneal fibrosis by targeting Snai1. Am. J. Pathol. 2013;183:808–819. doi: 10.1016/j.ajpath.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 231.Derynck R., Muthusamy B.P., Saeteurn K.Y. Signaling pathway cooperation in TGF-beta-induced epithelial–mesenchymal transition. Curr. Opin. Cell Biol. 2014;31:56–66. doi: 10.1016/j.ceb.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Lamouille S., Xu J., Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell. Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Hiraga R., Kato M., Miyagawa S., Kamata T., Nox4-derived R.O.S. signaling contributes to TGF-beta-induced epithelial–mesenchymal transition in pancreatic cancer cells. Anticancer Res. 2013;33:4431–4438. [PubMed] [Google Scholar]
- 234.Felton V.M., Borok Z., Willis B.C. N-acetylcysteine inhibits alveolar epithelial–mesenchymal transition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009;297:L805–L812. doi: 10.1152/ajplung.00009.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Zhou G., Dada L.A., Wu M., Kelly A., Trejo H., Zhou Q., Varga J., Sznajder J.I. Hypoxia-induced alveolar epithelial–mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009;297:L1120–L1130. doi: 10.1152/ajplung.00007.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Lee J.H., Kim J.H., Kim J.S., Chang J.W., Kim S.B., Park J.S., Lee S.K. AMP-activated protein kinase inhibits TGF-beta-, angiotensin II-, aldosterone-, high glucose-, and albumin-induced epithelial–mesenchymal transition. Am. J. Physiol. Ren. Physiol. 2013;304:F686–F697. doi: 10.1152/ajprenal.00148.2012. [DOI] [PubMed] [Google Scholar]
- 237.Liu X.X., Zhou H.J., Cai L., Zhang W., Ma J.L., Tao X.J., Yu J.N. NADPH oxidase-dependent formation of reactive oxygen species contributes to transforming growth factor beta1-induced epithelial–mesenchymal transition in rat peritoneal mesothelial cells, and the role of astragalus intervention. Chin. J. Integr. Med. 2014;20:667–674. doi: 10.1007/s11655-012-1176-x. [DOI] [PubMed] [Google Scholar]
- 238.Lardot C., Heusterpreute M., Mertens P., Philippe M., Lison D. Expression of plasminogen activator inhibitors type-1 and type-2 in the mouse lung after administration of crystalline silica. Eur. Respir. J. 1998;11:912–921. doi: 10.1183/09031936.98.11040912. [DOI] [PubMed] [Google Scholar]
- 239.El Solh A.A., Bhora M., Pineda L., Aquilina A., Abbetessa L., Berbary E. Alveolar plasminogen activator inhibitor-1 predicts ARDS in aspiration pneumonitis. Intensive Care Med. 2006;32:110–115. doi: 10.1007/s00134-005-2847-2. [DOI] [PubMed] [Google Scholar]
- 240.Idell S., Koenig K.B., Fair D.S., Martin T.R., McLarty J., Maunder R.J. Serial abnormalities of fibrin turnover in evolving adult respiratory distress syndrome. Am. J. Physiol. Lung Cell. Mol. Physiol. 1991;261:L240–L248. doi: 10.1152/ajplung.1991.261.4.L240. [DOI] [PubMed] [Google Scholar]
- 241.Kotani I., Sato A., Hayakawa H., Urano T., Takada Y., Takada A. Increased procoagulant and antifibrinolytic activities in the lungs with idiopathic pulmonary fibrosis. Thromb. Res. 1995;77:493–504. doi: 10.1016/0049-3848(95)00025-9. [DOI] [PubMed] [Google Scholar]
- 242.Senoo T., Hattori N., Tanimoto T., Furonaka M., Ishikawa N., Fujitaka K., Haruta Y., Murai H., Yokoyama A., Kohno N. Suppression of plasminogen activator inhibitor-1 by RNA interference attenuates pulmonary fibrosis. Thorax. 2010;65:334–340. doi: 10.1136/thx.2009.119974. [DOI] [PubMed] [Google Scholar]
- 243.Chang W., Wei K., Jacobs S.S., Upadhyay D., Weill D., Rosen G.D. SPARC suppresses apoptosis of idiopathic pulmonary fibrosis fibroblasts through constitutive activation of beta-catenin. J. Biol. Chem. 2010;285:8196–8206. doi: 10.1074/jbc.M109.025684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Olman M.A., Mackman N., Gladson C.L., Moser K.M., Loskutoff D.J. Changes in procoagulant and fibrinolytic gene expression during bleomycin-induced lung injury in the mouse. J. Clin. Investig. 1995;96:1621–1630. doi: 10.1172/JCI118201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Zhang Y., Ma J. Changes of coagulation and fibrinolysis systen in bronchoalveolar lavage fluid in lung fibrosis. Beijing Da Xue Xue Bao. 2005;37:516–519. [PubMed] [Google Scholar]
- 246.Eitzman D.T., McCoy R.D., Zheng X., Fay W.P., Shen T., Ginsburg D., Simon R.H. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J. Clin. Investig. 1996;97:232–237. doi: 10.1172/JCI118396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Hattori N., Degen J.L., Sisson T.H., Liu H., Moore B.B., Pandrangi R.G., Simon R.H., Drew A.F. Bleomycin-induced pulmonary fibrosis in fibrinogen-null mice. J. Clin. Invest. 2000;106:1341–1350. doi: 10.1172/JCI10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Chuang-Tsai S., Sisson T.H., Hattori N., Tsai C.G., Subbotina N.M., Hanson K.E., Simon R.H. Reduction in fibrotic tissue formation in mice genetically deficient in plasminogen activator inhibitor-1. Am. J. Pathol. 2003;163:445–452. doi: 10.1016/S0002-9440(10)63674-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Samarakoon R., Higgins C.E., Higgins S.P., Kutz S.M., Higgins P.J. Plasminogen activator inhibitor type-1 gene expression and induced migration in TGF-beta1-stimulated smooth muscle cells is pp60(c-src)/MEK-dependent. J. Cell. Physiol. 2005;204:236–246. doi: 10.1002/jcp.20279. [DOI] [PubMed] [Google Scholar]
- 250.Guo B., Inoki K., Isono M., Mori H., Kanasaki K., Sugimoto T., Akiba S., Sato T., Yang B., Kikkawa R., Kashiwagi A., Haneda M., Koya D. MAPK//AP-1-dependent regulation of PAI-1 gene expression by TGF-[beta] in rat mesangial cells. Kidney Int. 2005;68:972–984. doi: 10.1111/j.1523-1755.2005.00491.x. [DOI] [PubMed] [Google Scholar]
- 251.Yang C., Patel K., Harding P., Sorokin A., Glass W.F., II Regulation of TGF-[beta]1/MAPK-mediated PAI-1 gene expression by the actin cytoskeleton in human mesangial cells. Exp. Cell Res. 2007;313:1240–1250. doi: 10.1016/j.yexcr.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Grandaliano G., Di Paolo S., Monno R., Stallone G., Ranieri E., Pontrelli P., Gesualdo L., Schena F.P. Protease-activated receptor 1 and plasminogen activator inhibitor 1 expression in chronic allograft nephropathy: the role of coagulation and fibrinolysis in renal graft fibrosis. Transplantation. 2001;72:1437–1443. doi: 10.1097/00007890-200110270-00018. [DOI] [PubMed] [Google Scholar]
- 253.de Gouville A.-C., Boullay V., Krysa G., Pilot J., Brusq J.-M., Loriolle F., Gauthier J.-M., Papworth S.A., Laroze A., Gellibert F., Huet S. Inhibition of TGF-[beta] signaling by an ALK5 inhibitor protects rats from dimethylnitrosamine-induced liver fibrosis. Br. J. Pharmacol. 2005;145:166–177. doi: 10.1038/sj.bjp.0706172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Liu R.M. Oxidative stress, plasminogen activator inhibitor 1, and lung fibrosis. Antioxid. Redox Signal. 2008;10:303–319. doi: 10.1089/ars.2007.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Jarman E.R., Khambata V.S., Cope C., Jones P., Roger J., Ye L.Y., Duggan N., Head D., Pearce A., Press N.J., Bellenie B., Sohal B., Jarai G. An inhibitor of NADPH oxidase-4 attenuates established pulmonary fibrosis in a rodent disease model. Am. J. Respir. Cell Mol. Biol. 2014;50:158–169. doi: 10.1165/rcmb.2013-0174OC. [DOI] [PubMed] [Google Scholar]
- 256.Mayoral R., Valverde A.M., Llorente Izquierdo C., Gonzalez-Rodriguez A., Bosca L., Martin-Sanz P. Impairment of transforming growth factor beta signaling in caveolin-1-deficient hepatocytes: role in liver regeneration. J. Biol. Chem. 2010;285:3633–3642. doi: 10.1074/jbc.M109.072900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Samarakoon R., Chitnis S.S., Higgins S.P., Higgins C.E., Krepinsky J.C., Higgins P.J. Redox-induced Src kinase and caveolin-1 signaling in TGF-beta1-initiated SMAD2/3 activation and PAI-1 expression. PloS One. 2011;6:e22896. doi: 10.1371/journal.pone.0022896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Jiang Z., Seo J.Y., Ha H., Lee E.A., Kim Y.S., Han D.C., Uh S.T., Park C.S., Lee H.B. Reactive oxygen species mediate TGF-beta1-induced plasminogen activator inhibitor-1 upregulation in mesangial cells. Biochem. Biophys. Res. Commun. 2003;309:961–966. doi: 10.1016/j.bbrc.2003.08.102. [DOI] [PubMed] [Google Scholar]
- 259.Nakayama N., Nakamura T., Okada H., Iwaki S., Sobel B.E., Fujii S. Modulators of induction of plasminogen activator inhibitor type-1 in HepG2 cells by transforming growth factor-beta. Coron. Artery Dis. 2011;22:468–478. doi: 10.1097/MCA.0b013e32834a3817. [DOI] [PubMed] [Google Scholar]
- 260.Huang Y., Haraguchi M., Lawrence D.A., Border W.A., Yu L., Noble N.A. A mutant, noninhibitory plasminogen activator inhibitor type 1 decreases matrix accumulation in experimental glomerulonephritis. J. Clin. Investig. 2003;112:379–388. doi: 10.1172/JCI18038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Huang Y., Border W.A., Lawrence D.A., Noble N.A. Mechanisms underlying the antifibrotic properties of noninhibitory PAI-1 (PAI-1R) in experimental nephritis. Am. J. Physiol. Ren. Physiol. 2009;297:F1045–F1054. doi: 10.1152/ajprenal.00024.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Huang Y., Border W.A., Lawrence D.A., Noble N.A. Noninhibitory PAI-1 enhances plasmin-mediated matrix degradation both in vitro and in experimental nephritis. Kidney Int. 2006;70:515–522. doi: 10.1038/sj.ki.5000353. [DOI] [PubMed] [Google Scholar]
- 263.Rhyu D.Y., Park J., Sharma B.R., Ha H. Role of reactive oxygen species in transforming growth factor-beta1-induced extracellular matrix accumulation in renal tubular epithelial cells. Transplant. Proc. 2012;44:625–628. doi: 10.1016/j.transproceed.2011.12.054. [DOI] [PubMed] [Google Scholar]
- 264.Samarakoon R., Higgins S.P., Higgins C.E., Higgins P.J. TGF-beta1-induced plasminogen activator inhibitor-1 expression in vascular smooth muscle cells requires pp60(c-src)/EGFR(Y845) and Rho/ROCK signaling. J. Mol. Cell. Cardiol. 2008;44:527–538. doi: 10.1016/j.yjmcc.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]