

Summary
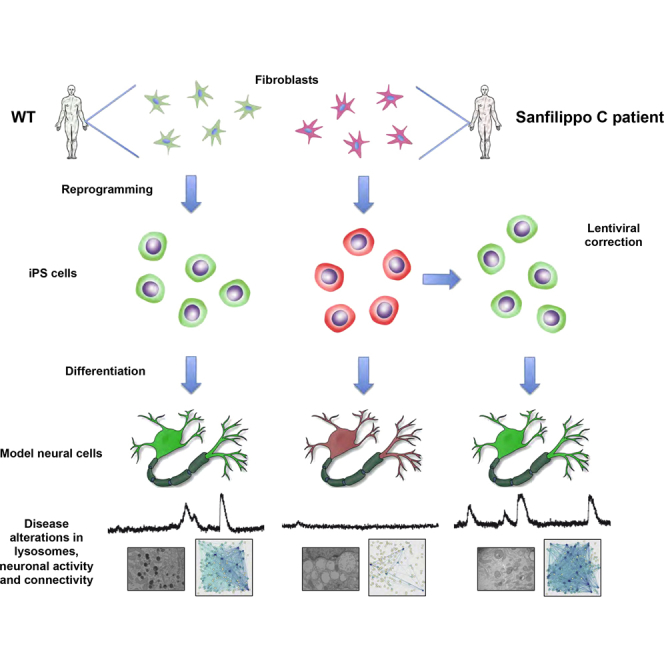
Induced pluripotent stem cell (iPSC) technology has been successfully used to recapitulate phenotypic traits of several human diseases in vitro. Patient-specific iPSC-based disease models are also expected to reveal early functional phenotypes, although this remains to be proved. Here, we generated iPSC lines from two patients with Sanfilippo type C syndrome, a lysosomal storage disorder with inheritable progressive neurodegeneration. Mature neurons obtained from patient-specific iPSC lines recapitulated the main known phenotypes of the disease, not present in genetically corrected patient-specific iPSC-derived cultures. Moreover, neuronal networks organized in vitro from mature patient-derived neurons showed early defects in neuronal activity, network-wide degradation, and altered effective connectivity. Our findings establish the importance of iPSC-based technology to identify early functional phenotypes, which can in turn shed light on the pathological mechanisms occurring in Sanfilippo syndrome. This technology also has the potential to provide valuable readouts to screen compounds, which can prevent the onset of neurodegeneration.
Graphical Abstract
Highlights
-
•
Fibroblasts from two Sanfilippo C patients were reprogrammed to obtain iPSCs
-
•
iPSCs were successfully differentiated to neural cells that mimic the disease
-
•
Networks of patients’ neurons show altered activity and connectivity
-
•
Early functional phenotypes are prevented in gene-corrected patients’ neurons
iPSC technology has been used to model dozens of diseases. In this article, Raya, Grinberg, and colleagues take a step forward and show that this technology can also be used to model pre-symptomatic stages of human diseases. In particular, they used it to generate neurons from Sanfilippo C patients and found functional alterations in how these neurons interacted with one another, before the neurons themselves had any obvious alteration.
Introduction
Sanfilippo syndrome, also known as mucopolysaccharidosis type III (MPS III), is a lysosomal storage disorder (LSD) with an autosomal recessive inheritance pattern. Four different subtypes have been described (type A, OMIM 252900; type B, OMIM 252920; type C, OMIM 252930; and type D, OMIM 252940), which share clinical characteristics, including severe early onset CNS degeneration that typically results in death within the second or third decade of life (Valstar et al., 2008). Each subtype is caused by mutations in a different gene encoding for enzymes involved in the degradation pathway of the glycosaminoglycan (GAG) heparan sulfate (Neufeld and Muenzer, 2001). The lack of activity of any of these enzymes leads to the accumulation of partially degraded heparan sulfate chains within the lysosomes. Subtype C (MPS IIIC) is caused by mutations in the HGSNAT gene, encoding acetyl-CoA α-glucosaminide N-acetyltransferase (EC 2.3.1.78), a lysosomal membrane enzyme. The prevalence of MPS IIIC ranges between 0.07 and 0.42 per 100,000 births, depending on the population (Poupetová et al., 2010). The HGSNAT gene was identified by two independent groups in 2006 (Fan et al., 2006, Hřebíček et al., 2006), and 64 different mutations have been identified since then (Human Gene Mutation Database Professional 2014.3). A mouse model has been very recently developed (Martins et al., 2015), but a cellular model for Sanfilippo type C has yet to be developed.
The ability to reprogram somatic cells back to a pluripotent state (Takahashi and Yamanaka, 2006, Takahashi et al., 2007) has created new opportunities for generating in vitro models of disease-relevant cells differentiated from patient-specific induced pluripotent stem cell (iPSC) lines (recently reviewed by Cherry and Daley, 2013, Inoue et al., 2014, Trounson et al., 2012). This approach has been shown to be particularly useful in the case of congenital or early-onset monogenic diseases. In particular, iPSC-based models of various LSD have been established, including Gaucher disease (Mazzulli et al., 2011, Panicker et al., 2012, Park et al., 2008, Schöndorf et al., 2014, Tiscornia et al., 2013), Hurler syndrome (Tolar et al., 2011), Pompe disease (Higuchi et al., 2014, Huang et al., 2011), Sanfilippo B syndrome (Lemonnier et al., 2011), and Niemann-Pick type C1 (Maetzel et al., 2014, Trilck et al., 2013). In all these cases, disease-relevant cell types derived from patient-specific iPSCs not only displayed morphologic, biochemical, and/or functional hallmarks of the disease but also have the capacity of being used as a drug-screening platform to find therapies that are capable of reverting LSD-related phenotypes.
In this study, we set out to test whether patient-specific iPSC-derived cells could be used to investigate the existence of early functional alterations prior to the appearance of disease-related phenotypes identified in patients. For this purpose, we generated iPSCs from fibroblasts of Sanfilippo C syndrome patients (SFC-iPSCs) and differentiated them into neurons, which recapitulate the pathological phenotypes observed in vivo, such as low acetyl-CoA α-glucosaminide N-acetyltransferase activity, accumulation of GAGs, and an increase in lysosome size and number. Moreover, we found that neural networks organized in vitro from control iPSC-derived neurons grew in complexity over time, as quantified in terms of neuronal activity, network activity, and effective connectivity, a measure of neuronal network function as defined by information theory and analyzed through generalized transfer entropy (GTE) methods. In contrast, networks of SFC-iPSC-derived neurons showed early defects in neuronal activity and alterations in effective connectivity and network organization over time.
The identification of early functional phenotypes in SFC-iPSC-derived neurons attests to the validity of iPSC-based technology to model pre-symptomatic stages of human diseases, thus widening the spectrum of potential applications of somatic cell reprogramming for biomedical research.
Results
Generation and Characterization of Patient-Specific iPSCs
Fibroblasts from two unrelated Spanish patients with Sanfilippo type C syndrome (SFC6 and SFC7) and two healthy individuals (WT1 and WT2) were collected. Patient SFC6 was a compound heterozygote carrying a splicing mutation (c.633+1G > A) and a missense mutation (c.1334T > C; p.L445P), both of which were found only in this patient (Canals et al., 2011). This patient was also carrying the Robertsonian translocation der(13;14)(q10;q10), which is the most common chromosome rearrangement in humans and is usually phenotypically silent (Engels et al., 2008). Patient SFC7 was a homozygote for the most prevalent mutation in Spanish patients, accounting for 50% of the mutated alleles (Canals et al., 2011), affecting another splicing site (c.372-2A > G), which results in residual enzyme activity (Matos et al., 2014) and a typically less severe clinical progression. The effects of these mutations on the splicing process and the transferase protein have been previously described (Canals et al., 2011).
Fibroblasts were reprogrammed at early passages (5–7) through the retroviral delivery of SOX2, KLF4, and OCT4 (3F) or SOX2, KLF4, OCT4, and c-MYC (4F) to generate up to 15 independent iPSC lines for each individual. We selected clones displaying embryonic stem cell-like morphology and positive alkaline phosphatase staining. Four clones representing each individual were chosen to be thoroughly characterized and shown to be fully reprogrammed, as judged by the silencing of the reprogramming transgenes, activation of endogenous pluripotency-associated factors, expression of pluripotency-associated transcription factors and surface markers, demethylation of OCT4 and NANOG promoters, pluripotent differentiation ability in vitro and/or in vivo, and karyotype stability after more than 15 passages (Figures 1A–1G and S1A–S1F; Table S1).
Figure 1.

Generation and Characterization of Control and SFC-iPSC Lines Using 3 Reprogramming Factors
A total of 17 independent iPSC lines were obtained after reprogramming control (WT1 and WT2) and SFC (SFC6 and SFC7) fibroblasts with retroviruses expressing OCT4, SOX2, and KLF4. Two lines per fibroblast sample were selected for complete characterization: WT1-iPS#3.1, WT1-iPS#3.6, WT2-iPS#3.1, WT2-iPS#3.2, SFC6-iPS#3.1, SFC6-iPS#3.2, SFC7-iPS#3.1, and SFC7-iPS#3.2. Shown are representative images of the characterization of WT (WT1-iPS#3.6), SFC6 (SFC6-iPS#3.1), and SFC7 (SFC7-iPS#3.1) iPSC lines.
(A) Left: SFC fibroblasts before being transduced with retroviruses carrying reprogramming factors. The scale bar represents 50 μm. Center: Typical human embryonic stem cell (hESC)-like colonies obtained after SFC fibroblast reprogramming. The scale bar represents 400 μm. Right: Positive alkaline phosphatase staining of the hESC-like SFC-iPSC colonies. The scale bar represents 400 μm.
(B) Bisulfite genomic sequencing of the OCT4 and NANOG promoters showing demethylation pattern in WT-, SFC6-, and SFC7-iPSCs.
(C) RT-qPCR analyses of the expression levels of the indicated retroviral-derived reprogramming factors (TRANS-) and endogenous (ENDO-) genes in WT-, SFC6- and SFC7-iPSC.
(D) Karyotype of WT-, SFC6-, and SFC7-iPSCs, which are identical to that of parent fibroblasts (including the known balanced Robertsonian translocation der[13;14][q10;q10] of SFC6).(E) Representative colonies of WT-, SFC6-, and SFC7-iPSCs stained positive for the pluripotency markers OCT4, SOX2, and NANOG (green) and SSEA3, SSEA4, and TRA-1-81 (red). The scale bar represents 100 μm.
(F) Immunofluorescence analyses with specific markers on WT-, SFC6-, and SFC7-iPSCs differentiated in vitro to generate cell derivatives of all three primary germ layers. Endoderm, α-fetoprotein (green), FOX2A (red); mesoderm, GATA4 (green), α-SMA (red); and ectoderm, TUJ1 fetoprotein (green), GFAP (red). The scale bar represents 100 μm.
(G) Immunofluorescence analyses with specific markers on sections from the same teratoma induced after injecting WT-, SFC6-, or SFC7-iPSCs, showing cell derivatives of the three embryo germ layers. Endoderm, α-fetoprotein (green), FOX2A (red); mesoderm, SOX9 (green), CS (red); and ectoderm, TUJ1 fetoprotein (green), GFAP (red). The scale bar represents 100 μm.
Patient-Specific Neurons Recapitulate Known Sanfilippo C Phenotypes
Wild-type (WT)- and SFC-iPSCs were then differentiated to pure masses of neural precursors using a previously described protocol (Cho et al., 2008) that involves the formation of embryoid bodies and the culture of neural precursor cells to form spherical neural masses (SNMs), which can be expanded and subsequently differentiated to mature neurons after culturing them for several weeks in neuronal induction medium (Figure 2A; see Supplemental Experimental Procedures for further details). SNMs derived from WT- and SFC-iPSC lines homogeneously expressed neural progenitor markers such as PAX6, NESTIN, and SOX2, as well as proliferation markers such as Ki67 (Figure S2). Furthermore, when iPSC-derived SNMs were cultured in neuronal induction media supplemented with N2 and B27, differentiation into mature and synaptically active neurons was evident within 3 to 5 weeks (Figures 2B and 2C). After about 3 weeks in neuronal medium, the cultures formed dense neuronal networks and stained for dendritic marker MAP2 and synaptic marker SYNAPSIN. Under these conditions, SNMs mainly generated MAP2-positive mature neurons (63% ± 3% of differentiated cells, mean ± SEM; n = 37), but also GFAP-positive cells (10% ± 1% of differentiated cells, mean ± SEM; n = 37), confirming their neurogenic capacity (Figure 2B). MAP2-positive neurons showed expression of SYNAPSIN, indicating their capability to form synapses (Figure 2C).
Figure 2.

Neural Differentiation of Control and SFC-Specific iPSC Lines
(A) Scheme illustrating the differentiation protocol from iPSC to mature neurons through the establishment of SNMs enriched in neural progenitors (NPs).
(B) Left images show immunofluorescence analyses on representative neural differentiation cultures from SNMs derived from WT2-iPSC#3.1 (WT) and SFC7-iPSC#4.8 (SFC) iPSC lines, stained for typical markers of mature neurons (MAP2 in green) and astrocytes (GFAP in red). Central and right images display the green and red channels in white, for clarity. Similar results were obtained from neural differentiation cultures of SNMs derived from WT1-iPSC#3.1, WT1-iPSC#3.6, WT1-iPSC#4.10, WT1-iPSC#4.12, WT2-iPSC#3.2, WT2-iPSC#4.2, WT2-iPSC#4.5, SFC6-iPSC#3.1, SFC6-iPSC#3.2, SFC6-iPSC#4.6, SFC6-iPSC#4.7, SFC7-iPSC#3.1, SFC7-iPSC#3.2, and SFC7-iPSC#4.9 iPSC lines. The scale bar represents 50 μm.
(C) Representative image of the immunofluorescence analysis of a neuron differentiated from SNMs derived from the SFC7-iPSC#4.8 iPSC line, stained with MAP2 (green), a mature neuronal marker, and SYNAPSIN (red), a marker for synapses (upper image). Magnification of a neuron prolongation with specific staining for SYNAPSIN (lower image). Similar stainings were obtained from neural differentiation cultures of SNMs derived from WT1-iPSC#3.1, WT1-iPSC#3.6, WT1-iPSC#4.10, WT1-iPSC#4.12, WT2-iPSC#3.1, WT2-iPSC#3.2, WT2-iPSC#4.2, WT2-iPSC#4.5, SFC6-iPSC#3.1, SFC6-iPSC#3.2, SFC6-iPSC#4.6, SFC6-iPSC#4.7, SFC7-iPSC#3.1, SFC7-iPSC#3.2, and SFC7-iPSC#4.9 iPSC lines. The scale bar represents 5 μm.
See also Figure S2.
Mutation analysis confirmed that SFC-iPSCs bore the mutations present in the patients’ fibroblasts, resulting in the expected splicing defects (data not shown). SFC-iPSCs showed no detectable acetyl-CoA α-glucosaminide N-acetyltransferase activity, consistent with the low enzyme activity levels found in patients’ fibroblasts (1.78% and 3.02% of that of WT fibroblasts for SFC6 and SFC7, respectively; Figure 3A). Similarly, SNMs and neural cultures derived from SFC-iPSCs showed low enzyme activity levels, representing less than 1% of those found in control cells (Figures 3C and 3D). Total GAG content in patients’ fibroblasts approximately doubled that of control cells (Figure 3E). In contrast, SFC-iPSC-derived neural cultures accumulated GAG over time, reaching statistically significant differences compared with controls only after 9 weeks of culture (Figure 3F).
Figure 3.

HGSNAT Activity and GAG Storage in Control and SCF-Specific Cell Types
(A–D) Analyses of the HGSNAT enzyme activity expressed in nmol × h−1 × mg−1 protein for the WT-, SFC6-, and SFC7-fibroblasts (A), iPSCs (B), SNMs (C), and neurons (D). The data show mean ± SD of three independent experiments performed in triplicate (WT1, SFC6, and SFC7 fibroblasts and iPSCs, SNMs, and neurons derived from the WT1-iPSC#3.6, SFC6-iPSC#3.1, and SFC7-iPSC#3.1 iPSC lines), and three independent experiments performed in duplicate (WT2 fibroblasts and iPSC, SNMs, and neurons derived from the WT1-iPSC#3.1, WT1-iPSC#4.10, WT2-iPSC#3.1, WT2-iPSC#3.2, WT2-iPSC#4.2, WT2-iPSC#4.5, SFC6-iPSC#3.2, SFC6-iPSC#4.6, SFC6-iPSC#4.7, SFC7-iPSC#3.2, SFC7-iPSC#4.8, and SFC7-iPSC#4.9 iPSC lines). ∗∗p < 0.01 (WT versus patients), ∗∗∗p < 0.001 (WT versus patients).
(E) Analyses of the GAG levels expressed as percentage of WT levels, in WT1-, SFC6-, and SFC7-fibroblasts (E) and neurons differentiated for 3, 6, and 9 weeks from SNMs derived from the SFC6-iPSC#3.1 and SFC7-iPSC#3.1 iPSC lines or after 9 weeks from gene-complemented (treated) SNMs derived from the SFC6-iPSC#3.1 and SFC7-iPSC#3.1 iPSC lines, relative to neurons differentiated at the same time points from SNMs derived from the WT1-iPSC#3.6 iPSC line (F). The data show mean ± SD of three independent experiments performed in duplicate. ∗∗p < 0.01 (WT versus patients).
See also Figure S3.
Gene-corrected controls were generated by lentiviral (LV) complementation of SNMs with WT cDNA for the HGSNAT gene under a cytomegalovirus (CMV) promoter (LV.CMV.HGSNAT.ires.GFP; Figure S3A). The vector also expressed GFP downstream of an internal ribosome entry site element. Neural cells differentiated from gene-corrected SNMs showed high activity of the enzyme, between 50- and 150-fold higher than those differentiated from WT-SNMs transduced with a control LV.CMV.GFP vector (Figures S3B–S3E), demonstrating long-term sustained transgene expression. Gene complementation of SFC-derived neural cultures prevented the statistically significant accumulation of GAG observed in GFP-transduced SCF cells after 9 weeks of differentiation, when compared with control cells (Figure 3F).
Analysis by transmission electron microscopy (TEM) revealed marked differences in lysosomes between controls and patients, which increased along time. Whereas lysosomes for control cultures showed typical morphology and size, we found only large vacuoles with an empty-like appearance in patients’ samples (Figure 4A), similar to those described in a mouse model of Sanfilippo B (Vitry et al., 2010). These vacuoles were derived from lysosomes, as judged by positive immuno-gold staining for LAMP1 (Figure 4B). The differences in lysosome size increased with culture time and reached 80% for both patients compared with controls at 9 weeks (Figure 4C). In addition, immunostaining for LAMP2 in isolated neurons of these cultures analyzed 9 weeks after differentiation showed significant differences between controls and patients (Figures 4D and 4E), consistent with the 80% detected by the TEM analysis, and illustrating that these differences can be detected specifically in iPSC-derived neurons. After transduction with the HGSNAT cDNA, some cells presented lysosomes with a morphology between the affected type and those of WT, suggesting a partial correction (Figure 4A).
Figure 4.

Lysosome Alterations in SFC-iPSC-Derived Neurons
(A) TEM micrograph representative of WT-, SFC6-, SFC7-, and gene-complemented SFC6-iPSC-derived neurons after 9 weeks of differentiation, showing lysosomal-derived accumulation vesicles (dark in the WT, empty-like in both patients, and gray in the gene-complemented sample). Neural cultures for these experiments were derived from SNMs established from the WT1-iPSC#3.6, SFC6-iPSC#3.1, and SFC7-iPSC#3.1 iPSC lines. The scale bar represents 2 μm.
(B) Immuno-gold analysis with anti-LAMP1 antibodies of SFC6-iPSC#3.1-derived neurons after 9 weeks of differentiation demonstrating the lysosomal origin of accumulation vesicles (gold particles indicated by black arrows). The scale bar represents 0.2 μm.
(C) Comparative analysis of lysosome size in WT, SFC6, and SFC7 samples, measured in TEM micrographs of fibroblasts and iPSC-derived neurons at the indicated times of differentiation. Neurons were the most abundant cell type in these preparations and were readily identified thanks to their round-shaped nuclei with weakly compacted chromatin and prominent nucleolus, scarce electrodense cytoplasm with numerous organelles, and synaptic contacts. The values shown indicate the size of SFC6 and SFC7 lysosomes in each cell type relative to the size of WT lysosomes in equivalent samples. The data show means ± SE of at least 200 lysosomes for each sample, obtained in 3 independent experiments. ∗∗∗p < 0.001 (patients versus WT).
(D and E) Representative images of the immunoanalysis of LAMP2 staining per neuronal area in neurons (MAP2-positive cells) differentiated from WT (line WT2-iPSC#3.1, left image) and SFC (line SFC7-iPSC#4.8, right image) iPSC lines (D) and comparative analysis between control and patients at 9 weeks of differentiation (E). The values indicate the percentage of MAP2 stained area also stained for LAMP2. The data show mean ± SE of at least 15 neurons for each sample, obtained in 3 independent experiments. ∗∗p < 0.01 (patients versus controls).
See also Figure S4.
Network-Broad Activity Alterations in Sanfilippo C Neuronal Cultures
Next, we allowed control- and SFC-iPSC-derived neurons to form networks and used a calcium fluorescence imaging assay to monitor functional neuronal activity after 3, 6, and 9 weeks. Typical recordings of spontaneous activity at week 9 for the different conditions are shown in Movies S1, S2, and S3. WT-iPSC-derived neurons showed repeated firing episodes of large amplitude (“bursts”) that were not present in SFC6-iPSC neurons. SFC7-iPSC neurons showed a slightly richer activity than SFC6-iPSC neurons, but the amplitude of the bursts was still low compared with WT-iPSC neurons (Figure 5A). Gene complementation with HGSNAT significantly changed the activity of the patient-specific iPSC-derived neurons, and both SCF6- and SCF7-iPSC-derived neurons exhibited activity patterns similar to those of WT.
Figure 5.

Altered Neuronal Activity in SFC-Derived Neuronal Networks
(A) Representative traces of spontaneous neuronal activity at week 9 of differentiation analyzed by calcium fluorescence imaging. Top traces correspond to control (GFP-transduced) neuronal networks; bottom traces correspond to gene complemented (HGSNAT-transduced) networks. Sharp increases in the fluorescence signal denote bursting episodes. Firings events of low amplitude (arrowheads) correspond to single spikes.
(B) Neuronal activity (total number of firings per neuron monitored along 30 min recording) of three independent untransduced cultures, at 3, 6, and 9 weeks after differentiation for each cell line and at each time point.
(C) Relative change in activity of control (GFP-transduced) or gene complemented (HGSNAT-transduced) SFC6 and SFC7 neuronal networks with respect to their equivalent WT networks. Three independent cultures were analyzed for each cell line at each time point.
(D) Fraction of active neurons at week 9 in control (GFP-transduced) or gene complemented (HGSNAT-transduced) WT, SFC6, and SFC7 neuronal networks. Three independent cultures were analyzed for each cell line at each time point.
(E) Neuronal activity of control (GFP-transduced) or gene complemented (HGSNAT-transduced) WT, SFC6, or SFC7 neuronal networks at week 9 of differentiation. In (B) through (E), error bars are root-mean-square error. Three independent cultures were analyzed for each line at each time point. ∗p < 0.05 (patients versus WT), ∗∗p < 0.01 (patients versus WT), #p < 0.05 (GFP- versus HGSNAT-transduced), ##p < 0.01 (GFP- versus HGSNAT-transduced).
The overall network performance was first quantified by means of two standard descriptors: (1) neuronal activity, which was defined as the average number of bursting episodes per neuron during the 30 min recording time, and (2) fraction of active neurons, which was defined as the ratio between those cells that showed at least one bursting episode and the total population monitored (see detailed definition and measurement in Experimental Procedures). We first considered the scenario of cultures that were not transduced to test the reliability of our analysis. The network activity for WT cultures was close to 1, indicating that most of the neurons in the network were active (Figure 5B). Activity was maintained within experimental error at weeks 6 and 9. This stability of WT measurements allowed us to associate possible changes in activity due solely to the disorder. Indeed, neuronal activity in the patients’ neurons showed a gradual decrease, although the loss of activity was more evident in networks of SFC6-iPSC-derived neurons (Figure 5B).
We next considered the cultures that were transduced by LV.CMV.GFP and LV.CMV.HGSNAT.ires.GFP and carried out identical measurements (Figure 5C). For clarity, we compared the relative change in activity of the cultures from patients with respect to their WT counterparts (namely, WT solely transduced with GFP or WT transduced with both GFP and HGSNAT). We verified that WT GFP-transduced neurons showed the same trend as the untransduced ones within statistical error. SFC6 and SFC7 neuronal networks gradually decayed in activity, and at week 9, activity loss reached about 70% and 45%, respectively, compared with controls. The reproducibility of this trend (evidenced by the small error bars and their similar trends in Figures 5B and 5C) indicates that the transduction protocol itself did not influence the behavior of the neurons and the development of the network. On the other hand, HGSNAT-transduced cultures showed a significant increase in activity over time, reaching activity levels similar to controls at 9 weeks (Figure 5C).
The analysis of the fraction of active neurons in the network allowed further characterization of the differences between the HGSNAT-treated and untreated cultures. As shown in Figure 5D, GFP-only SFC6 and SFC7 exhibited at week 9 about 70% and 50% less active neurons, respectively, compared with WT controls. However, after transduction with HGSNAT, both SFC6 and SFC7 maintained a fraction of active neurons comparable with the WTs. We also include in Figure 5E the values of network activity at week 9 (corresponding to the time point of Figure 5C). GFP-transduced SFC6 and SFC7 cultures showed a significant loss of network activity, while the corresponding HGSNAT-transduced counterparts reached activity levels indistinguishable from the control condition.
Effective Connectivity Degradation in Patient-Specific Neuronal Networks
Effective network connectivity was determined by identifying causal influences among neurons through GTE, an information theory method that allows drawing a functional map of neuronal interactions in the network (Orlandi et al., 2014, Stetter et al., 2012). A total of 30 cultures were analyzed, extending from week 3 to 9, and including all the conditions (WT, SFC, and gene-corrected SFC). Figure 6 provides the connectivity maps of three representative neuronal cultures at weeks 3 and 9, comparing the WT and SFC7 case with and without HGSNAT transduction. The WT and SFC7-HGSNAT-transduced cultures displayed comparable network structures, where most of the neurons established a similar pattern of connections with other neurons including a uniform increase in connectivity. In contrast, in the GFP-only SFC7 cultures, strong connections were formed only within a subset of neurons, leaving most of the remaining neurons weakly connected or disconnected, particularly at week 9. In general, from the total of 30 cultures analyzed for network effective connectivity, this extreme feature was almost exclusive to SFC6 and SFC7 cultures at week 9, with 4 of 6 cultures showing such a trait. All 9 WT cultures at any week showed uniform connectivity characteristics, and of the 15 HGSNAT-transduced cultures analyzed, only 1 SFC6-transduced culture at week 9 exhibited this extreme trait.
Figure 6.

Alterations in Effective Connectivity in SFC-Derived Neuronal Networks
Structure of representative WT and control (GFP-transduced) or gene complemented (HGSNAT-transduced) SFC7-iPSC-derived neuronal networks at 3 (top) and 9 (bottom) weeks of differentiation, as reconstructed through GTE methods. In all the depicted networks, circles show the actual position of the neurons in the culture and are color-coded according to their relative connectivity. For easier visualization, the number of neurons in each network is limited to 150, which are randomly chosen from the original set, and only those connections with p values < 0.002 are represented.
As a complementary measure of network connectivity, we also analyzed the cultures for the existence of assortativity (Newman, 2002). A network is said to be assortative when nodes with many connections preferentially connect to one another. In turn, nodes with few connections also tend to connect to one another. Conversely, a network is said to be disassortative if nodes with many connections tend to connect with nodes with few connections. In our experiments, of a total of 30 analyzed cultures, 24 were assortative and 6 disassortative. Interestingly, 4 of the disassortative networks corresponded to the SFC6 and SFC7 cultures at week 9 (see, e.g., Figure 6, bottom central panel).
Each of the above analyses (i.e., the presence of unconnected neurons and disassortativity traits) is not significant when observed independently. However, their concurrent presence is highly indicative of network-wide changes in SFC6 and SFC7 untreated cultures compared with WT and HGSNAT-transduced ones. Indeed, a combined Fisher’s permutation test yields an achieved significance level (ASL) of 0.02 between the SFC and WT cultures and an ASL of 0.03 between the SFC and SFC-HGSNAT transduced cultures, whereas the ASL between SFC-HGSNAT and WT is 0.46, showing no statistically significant differences. The reasonable significance of the former group supports the idea that the disorder causes important network topology changes, ultimately driving the affected networks toward a state of high fragility.
Discussion
In this work, we have generated a neuronal model of Sanfilippo type C by reprogramming fibroblasts from two patients using the iPSC technology. The generation of a neuronal model is relevant because the main features of the disease cannot be studied in fibroblasts. The fact that we used samples from two patients validates the results and allows the detection of slight inter-individual differences, although the study of additional Sanfilippo type C patient-specific iPSCs would be necessary to generalize our conclusions.
iPSC technology has been widely used to model different types of diseases, including those affecting the CNS (Durnaoglu et al., 2011, Okano and Yamanaka, 2014). Some other LSDs have been modeled using iPSC technology, which were later differentiated to the human cellular type of interest for each case. For Pompe’s disease, cardiomyocytes exhibit the highest accumulation of glycogen, impaired autophagy, vacuolation, mitochondrial aberrances, and shorter survival times, features that were reverted after the overexpression of the normal gene (Huang et al., 2011). In the case of Hurler disease, hematopoietic and non-hematopoietic cells showed GAG accumulation and could be rescued by introducing the normal copy of the gene (Tolar et al., 2011). For Sanfilippo B syndrome, patient-derived neurons presented storage vesicles and Golgi disorganization (Lemonnier et al., 2011). In the case of Gaucher disease, iPSC-derived macrophages showed impaired lysosomal function and red blood cell clearance, recapitulating the hallmarks of the disease in this cell type, which could be reverted after administration of the recombinant enzyme (Panicker et al., 2012). Moreover, Gaucher disease-specific macrophages and neurons displayed low enzyme activity that could be partially rescued using small compounds with chaperone activity (Tiscornia et al., 2013); and dopaminergic neurons accumulated glucosylceramide and α-synuclein and showed autophagy and lysosomal defects and dysregulation of calcium homeostasis, all of which could be reverted after gene correction (Schöndorf et al., 2014). Finally, for Niemann-Pick type C1, iPSC-derived neurons exhibited spontaneous action potentials, confirming their maturation and accumulated cholesterol (Trilck et al., 2013). In another work regarding this disease, hepatic and neuronal cells presented lower cell viability, cholesterol storage, and impaired autophagy, features that could be reverted after gene correction (Maetzel et al., 2014). In many of these studies, the phenotypes observed could not be analyzed in fibroblasts, highlighting the importance of developing iPSC-derived models. Gene complementation provides an important experimental control that allows the assurance that the phenotypes detected are due to the genetic defect in the patient, rather than reprogramming artifacts.
In the present study, enzyme activity was dispensable for reprogramming and iPSC maintenance. This is in contrast with Fanconi anemia (Raya et al., 2009, Navarro et al., 2014), and also with Sanfilippo type B (Lemonnier et al., 2011) and Pompe disease (Higuchi et al., 2014), in all of which gene complementation was needed to achieve reprogramming. We hypothesize that the dispensability of enzyme activity for iPSC generation and maintenance might be related to overall low lysosomal activity in these cells. Thus, all the iPSCs generated in the present study (from either controls or SFC patients) showed relatively small numbers of lysosomes in comparison with fibroblasts, as judged by immunostaining with LAMP2 (Figure S4). Moreover, enzyme activity was very low in control iPSCs compared with fibroblasts (Figures 3A and 3B), as were the activities of other lysosomal enzymes (β-hexosaminidase and β-glucocerebrosidase; data not shown), in agreement with previous results (Tiscornia et al., 2013). Taken together, all these data suggest that iPSCs present a small number of lysosomes in comparison with other cell types.
To reduce the variability associated with neural differentiation of iPSCs (Falk et al., 2012), we established iPSC-derived SNMs consisting of neural progenitor cells that can be expanded and subsequently differentiated to neurons and glia (Cho et al., 2008). Mature human neurons that exhibit the principal features of the disease have been successfully generated after culturing iPSC-derived neural precursors cells for 3 to 9 weeks in neuronal differentiation medium. iPSC differentiation, including mature and functional neurons as the main cellular type, was proved through the expression of mature neuronal markers as well as synaptic proteins. Importantly, neuronal cultures derived from SFC patients showed lack of enzyme activity and an accumulation of GAGs and alteration of lysosomes. Apart from the lack of enzyme activity, which is always lacking in SFC cultures, other alterations appeared to be progressive. GAGs accumulated over time, but in our experiments, these differences did not reach statistical significance versus controls until 9 weeks (around 50% respect to control), which could indicate a lower rate of synthesis and storage in this cell type compared to fibroblasts. Lysosomal alterations displayed through TEM/LAMP1 and immunostaining with LAMP2 were first noticed at 3 to 6 weeks and became dramatic at 9 weeks (almost doubled in size). This timeline of appearance of alterations is in concordance with the progressive nature of the disease and highlights current difficulties in predicting the extent of neurological decline, because the lack of enzyme activity is not predictive, and the analysis of GAG storage and lysosome size and number requires invasive techniques.
The fact that our SFC-iPSC-derived neural cultures developed alterations that recapitulated those seen in patients prompted us to investigate whether we could detect early functional alterations predating known pathological signs of the disease. For this purpose, we used calcium imaging to analyze neuronal function in patients’ cells. An important decrease in spontaneous activity of SFC neurons compared with WT controls was already detected at 3 weeks of differentiation, particularly for the most severe case (SFC6). The different behavior in SFC6 and SFC7 could be due to the particular features of their mutations. In this regard, we have recently showed that the c.372-2G > A mutation, borne by SFC7, gives rise to a protein lacking 4 amino acids that has some residual activity (Matos et al., 2014). These data suggest that the decline of neuronal activity correlates with the severity of the neurological phenotype observed in patients.
Moreover, we combined a direct quantification of neuronal activity with advanced functional connectivity analyses framed in the context of transfer entropy (TE) (Stetter et al., 2012). The latter provided evidences for broad changes in network structure, unraveling extensive disconnection of neurons, the emergence of a subset of highly connected cells, and the evolution of the network toward a disassortative structure. Although the presence of unconnected neurons is a clear indicator of a dysfunctional network, the existence of assortative or disassortative traits is not. Indeed, several naturally occurring networks may fall into one kind or another (Honey et al., 2007, Pan et al., 2010). Theoretical studies (Schmeltzer et al., 2014) and experiments in vitro (Teller et al., 2014) have shown that assortative networks are resilient to attack since the highly connected nodes shape a core that preserves the functionality of network. In contrast, disassortative networks are highly vulnerable, since the targeted loss of the few highly connected nodes may cause network-wide failure (Newman, 2002). These ideas, translated to our study, suggest that the affected, disassortative cultures could completely collapse upon such targeted damage, making them highly vulnerable networks.
Our results and analyses suggest that the disorder notably disrupts the topology of the network. In the context of the patients, these alterations could significantly affect the normal operation of the brain. Such an aspect is important in the framework of studies that highlight the relation between altered network topology upon disease and the degradation of brain’s operability and cognitive tasks (Bassett and Bullmore, 2009, van den Heuvel and Sporns, 2013).
The spontaneous activity of SFC-derived neurons was recovered after transduction, in the SNM stage, with lentiviruses carrying the WT HGSNAT cDNA, and subsequent differentiation. The lack of recovery at 3 weeks and the partial recovery at 6 weeks contrast with the total recovery at 9 weeks of differentiation. We believe these differences are due to the fact that LV transduction was around 60% efficient, which could initially slow down the development of the network compared with the WT case. At later stages, we hypothesize that the large fraction of healthy neurons suffices to foster broad circuit connectivity and, ultimately, high neuronal activity. We note that the high expression levels of the transduced cells do not seem to alter their individual activity. Indeed, the WT-derived cultures overexpressing the HGSNAT cDNA did not show any significant alteration in these properties. However, our results indicate that the transduced cells do play a role in maintaining or restoring broad network activity and connectivity. Thus, we conclude that neuronal network activity and development could be reestablished with a therapeutic approach that rescues only a fraction (though sufficiently high) of the total neurons. However, GAG storage or lysosome appearance by TEM showed only partial reversion after 9 weeks of differentiation. Whether longer times are needed for complete reversion of these features, or whether this is not at all possible, will require further investigation. The availability of the human cellular model described here provides an excellent tool to investigate this and other issues.
In summary, the cellular model introduced here reproduces the major features of the Sanfilippo type C syndrome, especially specific neuronal traits. We have demonstrated that most of the phenotypic features of this neuronal model can be reversed after gene complementation, using lentiviruses overexpressing the cDNA of the HGSNAT gene. Moreover, our neuronal model could be used as a tool to test different possible therapeutic strategies. This is particularly relevant because no cellular model was available for Sanfilippo type C syndrome, and a mouse model has only very recently been developed (Martins et al., 2015). Our findings prove the usefulness of iPSC-derived neuronal models to detect early functional phenotypes that can shed light onto the molecular and cellular processes that lead to the brain dysfunction in these patients, as well as providing valuable readouts for screening of potential therapeutic compounds that prevent, rather than revert, the onset of neurodegeneration. Moreover, the neuronal activity and effective connectivity analyses could be applicable to other neurodegenerative diseases in which iPSC-based models are available, such as Parkinson’s or Alzheimer’s disease, autism, and others. Further studies are needed to establish whether this technique would be able to detect differences in the neuronal activity or the network structure before the onset of disease. Such an approach could foster the development of in vivo analyses for early diagnosis of patients affected by neurological diseases, as well as their monitoring during potential treatments.
Experimental Procedures
A complete description of experimental procedures can be found in Supplemental Experimental Procedures.
Network Reconstruction
Network reconstruction was carried out by identifying causal influences between neurons through TE (Schreiber, 2000, Stetter et al., 2012, Vicente et al., 2011). TE is an information-theoretic measure that identifies the flow of information between two time traces. The measure is model free (i.e., it does not require previous knowledge of the dynamics of the system) and is able to detect linear and non-linear interactions between any pair of traces. In a more formal description of TE, one considers two signals X and Y (any two neuronal fluorescence traces in our case), with the goal to assess the influence of X on Y (X → Y). TE measures the amount of uncertainty reduced in predicting the future of Y by taking into account both the pasts of Y and X, rather than the past of Y alone. Mathematically, this operation can be written as
where denotes the value of Y at time t and the past k-th values of Y. P(·) is the probability of observing that particular sequence, whereas P(·|·) is the conditional probability. The sum is performed over all possible values of .
There are different variations of the TE measure depending on the data under analysis, for example, fMRI (Honey et al., 2007), magnetoencephalography (Wibral et al., 2011), spike trains (Ito et al., 2011), or fluorescence data, as in our case. Here we use a version named GTE (Orlandi et al., 2014, Stetter et al., 2012) that was specifically developed for neuronal fluorescence signals (see Stetter et al., 2012, for details).
The analysis of the recorded spontaneous activity traces within the context of GTE was as follows. We initially computed the first derivative of the fluorescence trace and the resulting values binned with n = 2 intervals for TE computation. TE was then applied to all pairs of neurons in the network. TE assigns a score for every neuronal pair, but only those pairs with scores above a given significance level were considered putative connections. Significance was established by comparing the raw TE scores and the bootstrapped versions (to account for bias) and those obtained by shuffling the data from only the presynaptic neuron. Bootstrapped versions were obtained by generating surrogates of the fluorescence data for every neuronal pair while preserving temporal correlations between the pairs. For the representative networks, a paired Z test was performed with the bias-corrected and shuffled scores, and only those values above the 97.5th percentile (p < 0.002) were considered putative connections. For the connectivity and assortativity analysis, only the connections with the top 10% bias-corrected score were considered.
Statistical Analysis
Differences between conditions respect to controls were evaluated using a Mann-Whitney non-parametric U test, and statistical significance was set at p < 0.05, except for the network reconstruction experiment (see above).
Acknowledgments
The authors would like to thank M. Cozar of the Genetics Department (UB) and the Institut de Bioquímica Clínica, Barcelona, for helpful insights and A. di Domenico for critical reading of the manuscript. We are also grateful for the permanent support, including financial aid, from Jonah’s Just Begun – Foundation to Cure Sanfilippo, Association Sanfilippo Sud, Fundación Stop Sanfilippo, and Asociación MPS España. This study was partially funded by grants from MINECO (SAF2011-25431, SAF2012-33526, BFU2010-21823), the Catalan Government (2014SGR932, 2009SGR971, and 2009SGR14), ISCIII (Red de Terapia Celular – TerCel RD12/0019/0019, FIS2010-21924-C02-02), and the ERC-2013-StG grant of the European Research Council to A.C. I.C. was partially supported by a grant from the University of Barcelona (APIF).
Published: September 24, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, four figures, three tables, and three movies and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2015.08.016.
Contributor Information
Daniel Grinberg, Email: dgrinberg@ub.edu.
Angel Raya, Email: araya@cmrb.eu.
Supplemental Information
The field of view covers an area of 1.4 × 1 mm2 (width × height). The real duration of the recording is 30 min. Video is accelerated 150× to show the rich spontaneous activity of the network along the measurement.
{kind=link}
The network shows no activity. Video settings are as in Movie S1.
{kind=link}
Network activity is similar to WT conditions. Video settings are as in Movie S1.
{kind=link}
References
- Bassett D.S., Bullmore E.T. Human brain networks in health and disease. Curr. Opin. Neurol. 2009;22:340–347. doi: 10.1097/WCO.0b013e32832d93dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals I., Elalaoui S.C., Pineda M., Delgadillo V., Szlago M., Jaouad I.C., Sefiani A., Chabás A., Coll M.J., Grinberg D., Vilageliu L. Molecular analysis of Sanfilippo syndrome type C in Spain: seven novel HGSNAT mutations and characterization of the mutant alleles. Clin. Genet. 2011;80:367–374. doi: 10.1111/j.1399-0004.2010.01525.x. [DOI] [PubMed] [Google Scholar]
- Cherry A.B.C., Daley G.Q. Reprogrammed cells for disease modeling and regenerative medicine. Annu. Rev. Med. 2013;64:277–290. doi: 10.1146/annurev-med-050311-163324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho M.-S., Hwang D.-Y., Kim D.-W. Efficient derivation of functional dopaminergic neurons from human embryonic stem cells on a large scale. Nat. Protoc. 2008;3:1888–1894. doi: 10.1038/nprot.2008.188. [DOI] [PubMed] [Google Scholar]
- Durnaoglu S., Genc S., Genc K. Patient-specific pluripotent stem cells in neurological diseases. Stem Cells Int. 2011;2011:212487. doi: 10.4061/2011/212487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engels H., Eggermann T., Caliebe A., Jelska A., Schubert R., Schüler H.M., Panasiuk B., Zaremba J., Latos-Bieleńska A., Jakubowski L. Genetic counseling in Robertsonian translocations der(13;14): frequencies of reproductive outcomes and infertility in 101 pedigrees. Am. J. Med. Genet. A. 2008;146A:2611–2616. doi: 10.1002/ajmg.a.32500. [DOI] [PubMed] [Google Scholar]
- Falk A., Koch P., Kesavan J., Takashima Y., Ladewig J., Alexander M., Wiskow O., Tailor J., Trotter M., Pollard S. Capture of neuroepithelial-like stem cells from pluripotent stem cells provides a versatile system for in vitro production of human neurons. PLoS ONE. 2012;7:e29597. doi: 10.1371/journal.pone.0029597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X., Zhang H., Zhang S., Bagshaw R.D., Tropak M.B., Callahan J.W., Mahuran D.J. Identification of the gene encoding the enzyme deficient in mucopolysaccharidosis IIIC (Sanfilippo disease type C) Am. J. Hum. Genet. 2006;79:738–744. doi: 10.1086/508068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi T., Kawagoe S., Otsu M., Shimada Y., Kobayashi H., Hirayama R., Eto K., Ida H., Ohashi T., Nakauchi H., Eto Y. The generation of induced pluripotent stem cells (iPSCs) from patients with infantile and late-onset types of Pompe disease and the effects of treatment with acid-α-glucosidase in Pompe’s iPSCs. Mol. Genet. Metab. 2014;112:44–48. doi: 10.1016/j.ymgme.2014.02.012. [DOI] [PubMed] [Google Scholar]
- Honey C.J., Kötter R., Breakspear M., Sporns O. Network structure of cerebral cortex shapes functional connectivity on multiple time scales. Proc. Natl. Acad. Sci. U S A. 2007;104:10240–10245. doi: 10.1073/pnas.0701519104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hřebíček M., Mrázová L., Seyrantepe V., Durand S., Roslin N.M., Nosková L., Hartmannová H., Ivánek R., Cízkova A., Poupetová H. Mutations in TMEM76∗ cause mucopolysaccharidosis IIIC (Sanfilippo C syndrome) Am. J. Hum. Genet. 2006;79:807–819. doi: 10.1086/508294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.-P., Chen P.-H., Hwu W.-L., Chuang C.-Y., Chien Y.-H., Stone L., Chien C.-L., Li L.-T., Chiang S.-C., Chen H.-F. Human Pompe disease-induced pluripotent stem cells for pathogenesis modeling, drug testing and disease marker identification. Hum. Mol. Genet. 2011;20:4851–4864. doi: 10.1093/hmg/ddr424. [DOI] [PubMed] [Google Scholar]
- Inoue H., Nagata N., Kurokawa H., Yamanaka S. iPS cells: a game changer for future medicine. EMBO J. 2014;33:409–417. doi: 10.1002/embj.201387098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S., Hansen M.E., Heiland R., Lumsdaine A., Litke A.M., Beggs J.M. Extending transfer entropy improves identification of effective connectivity in a spiking cortical network model. PLoS ONE. 2011;6:e27431. doi: 10.1371/journal.pone.0027431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemonnier T., Blanchard S., Toli D., Roy E., Bigou S., Froissart R., Rouvet I., Vitry S., Heard J.M., Bohl D. Modeling neuronal defects associated with a lysosomal disorder using patient-derived induced pluripotent stem cells. Hum. Mol. Genet. 2011;20:3653–3666. doi: 10.1093/hmg/ddr285. [DOI] [PubMed] [Google Scholar]
- Maetzel D., Sarkar S., Wang H., Abi-Mosleh L., Xu P., Cheng A.W., Gao Q., Mitalipova M., Jaenisch R. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem Cell Reports. 2014;2:866–880. doi: 10.1016/j.stemcr.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins C., Hůlková H., Dridi L., Dormoy-Raclet V., Grigoryeva L., Choi Y., Langford-Smith A., Wilkinson F.L., Ohmi K., DiCristo G. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain. 2015;138:336–355. doi: 10.1093/brain/awu355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos L., Canals I., Dridi L., Choi Y., Prata M.J., Jordan P., Desviat L.R., Pérez B., Pshezhetsky A.V., Grinberg D. Therapeutic strategies based on modified U1 snRNAs and chaperones for Sanfilippo C splicing mutations. Orphanet J. Rare Dis. 2014;9:180. doi: 10.1186/s13023-014-0180-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli J.R., Xu Y.-H., Sun Y., Knight A.L., McLean P.J., Caldwell G.A., Sidransky E., Grabowski G.A., Krainc D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro S., Moleiro V., Molina-Estevez F.J., Lozano M.L., Chinchon R., Almarza E., Quintana-Bustamante O., Mostoslavsky G., Maetzig T., Galla M. Generation of iPSCs from genetically corrected Brca2 hypomorphic cells: implications in cell reprogramming and stem cell therapy. Stem Cells. 2014;32:436–446. doi: 10.1002/stem.1586. [DOI] [PubMed] [Google Scholar]
- Neufeld E.F., Muenzer J. The mucopolysaccharidoses. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 3421–3452. [Google Scholar]
- Newman M.E. Assortative mixing in networks. Phys. Rev. Lett. 2002;89:208701. doi: 10.1103/PhysRevLett.89.208701. [DOI] [PubMed] [Google Scholar]
- Okano H., Yamanaka S. iPS cell technologies: significance and applications to CNS regeneration and disease. Mol. Brain. 2014;7:22. doi: 10.1186/1756-6606-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlandi J.G., Stetter O., Soriano J., Geisel T., Battaglia D. Transfer entropy reconstruction and labeling of neuronal connections from simulated calcium imaging. PLoS ONE. 2014;9:e98842. doi: 10.1371/journal.pone.0098842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan R.K., Chatterjee N., Sinha S. Mesoscopic organization reveals the constraints governing Caenorhabditis elegans nervous system. PLoS ONE. 2010;5:e9240. doi: 10.1371/journal.pone.0009240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panicker L.M., Miller D., Park T.S., Patel B., Azevedo J.L., Awad O., Masood M.A., Veenstra T.D., Goldin E., Stubblefield B.K. Induced pluripotent stem cell model recapitulates pathologic hallmarks of Gaucher disease. Proc. Natl. Acad. Sci. U S A. 2012;109:18054–18059. doi: 10.1073/pnas.1207889109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park I.H., Arora N., Huo H., Maherali N., Ahfeldt T., Shimamura A., Lensch M.W., Cowan C., Hochedlinger K., Daley G.Q. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poupetová H., Ledvinová J., Berná L., Dvoráková L., Kozich V., Elleder M. The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J. Inherit. Metab. Dis. 2010;33:387–396. doi: 10.1007/s10545-010-9093-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raya A., Rodríguez-Pizà I., Guenechea G., Vassena R., Navarro S., Barrero M.J., Consiglio A., Castellà M., Río P., Sleep E. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature. 2009;460:53–59. doi: 10.1038/nature08129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeltzer C., Soriano J., Sokolov I.M., Rüdiger S. Percolation of spatially constrained Erdős-Rényi networks with degree correlations. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2014;89:012116. doi: 10.1103/PhysRevE.89.012116. [DOI] [PubMed] [Google Scholar]
- Schöndorf D.C., Aureli M., McAllister F.E., Hindley C.J., Mayer F., Schmid B., Sardi S.P., Valsecchi M., Hoffmann S., Schwarz L.K. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014;5:4028. doi: 10.1038/ncomms5028. [DOI] [PubMed] [Google Scholar]
- Schreiber T. Measuring information transfer. Phys. Rev. Lett. 2000;85:461–464. doi: 10.1103/PhysRevLett.85.461. [DOI] [PubMed] [Google Scholar]
- Stetter O., Battaglia D., Soriano J., Geisel T. Model-free reconstruction of excitatory neuronal connectivity from calcium imaging signals. PLoS Comput. Biol. 2012;8:e1002653. doi: 10.1371/journal.pcbi.1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Teller S., Granell C., De Domenico M., Soriano J., Gómez S., Arenas A. Emergence of assortative mixing between clusters of cultured neurons. PLoS Comput. Biol. 2014;10:e1003796. doi: 10.1371/journal.pcbi.1003796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiscornia G., Vivas E.L., Matalonga L., Berniakovich I., Barragán Monasterio M., Eguizábal C., Gort L., González F., Ortiz Mellet C., García Fernández J.M. Neuronopathic Gaucher’s disease: induced pluripotent stem cells for disease modelling and testing chaperone activity of small compounds. Hum. Mol. Genet. 2013;22:633–645. doi: 10.1093/hmg/dds471. [DOI] [PubMed] [Google Scholar]
- Tolar J., Park I.H., Xia L., Lees C.J., Peacock B., Webber B., McElmurry R.T., Eide C.R., Orchard P.J., Kyba M. Hematopoietic differentiation of induced pluripotent stem cells from patients with mucopolysaccharidosis type I (Hurler syndrome) Blood. 2011;117:839–847. doi: 10.1182/blood-2010-05-287607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trilck M., Hübner R., Seibler P., Klein C., Rolfs A., Frech M.J. Niemann-Pick type C1 patient-specific induced pluripotent stem cells display disease specific hallmarks. Orphanet J. Rare Dis. 2013;8:144. doi: 10.1186/1750-1172-8-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trounson A., Shepard K.A., DeWitt N.D. Human disease modeling with induced pluripotent stem cells. Curr. Opin. Genet. Dev. 2012;22:509–516. doi: 10.1016/j.gde.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Valstar M.J., Ruijter G.J.G., van Diggelen O.P., Poorthuis B.J., Wijburg F.A. Sanfilippo syndrome: a mini-review. J. Inherit. Metab. Dis. 2008;31:240–252. doi: 10.1007/s10545-008-0838-5. [DOI] [PubMed] [Google Scholar]
- van den Heuvel M.P., Sporns O. Network hubs in the human brain. Trends Cogn. Sci. 2013;17:683–696. doi: 10.1016/j.tics.2013.09.012. [DOI] [PubMed] [Google Scholar]
- Vicente R., Wibral M., Lindner M., Pipa G. Transfer entropy—a model-free measure of effective connectivity for the neurosciences. J. Comput. Neurosci. 2011;30:45–67. doi: 10.1007/s10827-010-0262-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitry S., Bruyère J., Hocquemiller M., Bigou S., Ausseil J., Colle M.-A., Prévost M.-C., Heard J.M. Storage vesicles in neurons are related to Golgi complex alterations in mucopolysaccharidosis IIIB. Am. J. Pathol. 2010;177:2984–2999. doi: 10.2353/ajpath.2010.100447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wibral M., Rahm B., Rieder M., Lindner M., Vicente R., Kaiser J. Transfer entropy in magnetoencephalographic data: quantifying information flow in cortical and cerebellar networks. Prog. Biophys. Mol. Biol. 2011;105:80–97. doi: 10.1016/j.pbiomolbio.2010.11.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The field of view covers an area of 1.4 × 1 mm2 (width × height). The real duration of the recording is 30 min. Video is accelerated 150× to show the rich spontaneous activity of the network along the measurement.
The network shows no activity. Video settings are as in Movie S1.
Network activity is similar to WT conditions. Video settings are as in Movie S1.