

Abstract
G protein-coupled receptors (GPCRs) constitute the largest and the most physiologically important membrane protein family that recognizes a variety of environmental stimuli, and are drug targets in the treatment of numerous diseases. Recent progress on GPCR structural studies shed light on molecular mechanisms of GPCR ligand recognition, activation and allosteric modulation, as well as structural basis of GPCR dimerization. In this review, we will discuss the structural features of GPCRs and structural insights of different aspects of GPCR biological functions.
Keywords: activation, allosteric modulation, GPCR, ligand recognition, structure
INTRODUCTION
GPCRs represent a large family of integral membrane proteins that are encoded by more than 800 genes in the human genome (Zhang et al., 2006). Based on amino acid sequence similarity within the seven-transmembrane (7TM) helical segments, GPCRs are divided into five families: the rhodopsin family (class A, 701 members), the secretin family (class B, 15 members), the adhesion family (24 members), the glutamate family (class C, 15 members), and the frizzled/taste family (class F, 24 members) (Fredriksson et al., 2003). GPCRs recognize a variety of extracellular stimuli, including photons, ions, organic odorants, purines, amines, neurotransmitters, chemokines, hormones, and lipids etc. Upon ligand binding, GPCRs undergo conformational changes, coupling to G protein, activating downstream signaling networks and initiating a broad range of physiological and pathological processes (Gether, 2000). GPCRs are important and highly attractive drug targets for numerous human diseases, such as central nervous system disorders, inflammatory diseases, metabolic imbalances, cardiac diseases, monogenic diseases, cancer, and many more (Thomsen et al., 2005). Indeed, 40–50 % of the total sales of marketed drugs and ∼25 % of the top selling drugs target on GPCRs (Schlyer and Horuk, 2006). GPCR structures are key for not only understanding the molecular mechanisms of GPCR signal transduction, but also developing new therapeutics for the treatment of human diseases.
Structural studies of GPCR remain challenging and intractable due to the lack of a naturally abundant source of protein, the conformational heterogeneity in solutions, and the relatively small structured polar surface available for forming crystal lattice contacts. The first GPCR structure is the crystal structure of bovine rhodopsin in complex with 11-cis-retinal solved by Palczewski and colleagues in 2000 (Palczewski et al., 2000). After seven-year extensive research and technology development, the high-resolution structure of the human β2-adrenergic receptor (β2AR) – the first structure of a GPCR bound to a diffusible ligand was obtained (Cherezov et al., 2007). Then, the first active-state GPCR structure of ligand free rhodopsin (opsin) was followed in 2008 (Scheerer et al., 2008). In the past few years, with many technology breakthroughs in membrane protein engineering and crystallography (Caffrey, 2011; Chae et al., 2010; Rosenbaum et al., 2007), GPCR structure determination has experienced an exponential growth trend. To date, over 100 structures of 28 different class A GPCRs and 5 structures of the transmembrane domain of receptors from classes B, C and F have been determined in complex with ligands of varied pharmacology. These structures shed unprecedented light on structural similarity and diversity of the GPCR super-family, and molecular basis of GPCR ligand recognition, activation, allosteric modulation and dimerization. In this review, we will focus on how the recent structural and biophysical insights contribute to our understanding of GPCR structure-function relationship. We also outline open challenges in GPCR structural studies and discuss new exciting directions for GPCR research to get a comprehensive understanding of biological and therapeutic mechanisms of the GPCR superfamily.
SOLVED GPCR STRUCTURES
So far, a total of 127 GPCR structures (Table 1) have been solved for the following class A GPCRs: rhodopsin (bovine rhodopsin and squid rhodopsin) (Caffrey, 2011; Kang et al., 2015; Palczewski et al., 2000; Park et al., 2008; Scheerer et al., 2008; Szczepek et al., 2014); eight aminergic receptors, including β2-adrenergic receptor (β2AR) (Cherezov et al., 2007; Rasmussen et al., 2007; 2011; Rosenbaum et al., 2007; 2011), β1-adrenergic receptor (β1AR) (Huang et al., 2013; Warne et al., 2012), H1 histamine receptor (H1R) (Shimamura et al., 2011), D3 dopamine receptor (D3R) (Chien et al., 2010), 5-hydroxytryptamine receptors (5-HT1B and 5-HT2B) (Wacker et al., 2013; Wang et al., 2013a) and muscarinic acetylcholine receptors (M2R and M3R) (Haga et al., 2012; Kruse et al., 2012); three nucleoside receptors, including A2A adenosine receptor (A2AAR) (Jaakola et al., 2008; Lebon et al., 2011; Xu et al., 2011), P2Y1 purinergic receptor (P2Y1R) (Zhang et al., 2015a) and P2Y12 purinergic receptor (P2Y12R) (Zhang et al., 2014a; 2014b); ten peptide receptors, including chemokine receptors CXCR4 and CCR5 (Tan et al., 2013; Wu et al., 2010), κ-opioid receptor (κ-OR) (Wu et al., 2012), μ-opioid receptor (μ-OR) (Manglik et al., 2012), δ-opioid receptor (δ-OR) (Granier et al., 2012), nociceptin/orphanin FQ peptide receptor (NOP) (Thompson et al., 2012), neurotensin receptor 1 (NTSR1) (White et al., 2012), angiotensin II type 1 receptor (AT1R) (Zhang et al., 2015b), orexin type 2 receptor (OX2R) (Yin et al., 2015), and protease-activated receptor 1 (PAR1) (Zhang et al., 2012); and three lipid-activated GPCRs, including sphingosine-1-phosphate receptor 1 (S1P1) (Hanson et al., 2012), free fatty acid receptor 1 (FFAR1) (Srivastava et al., 2014) and lysophosphatidic acid receptor 1 (LPA1) (Chrencik et al., 2015). Five structures of 7TM domain of the members from classes B, C and F GPCRs, including corticotropin releasing factor receptor 1 (CRF1R) (Hollenstein et al., 2013), glucagon receptor (GCGR) (Siu et al., 2013), metabotropic glutamate type 1 receptor (mGluR1) (Wu et al., 2014), metabotropic glutamate type 5 receptor (mGluR5) (Dore et al., 2014), and smoothened receptor (SMO) (Wang et al., 2013b).
Table 1.
Crystal structures of GPCRs.
| Receptor | Number of structures | PDB ID |
|---|---|---|
| Rhodopsin | 27 | 1F88, 1HZX, 1L9H, 1GZM, 1U19, 2HPY, 2G87, 2I35, 2I37, 2J4Y, 2PED, 3CAP, 3C9L, 3C9M, 3DQB, 3PQR, 2X72, 2ZIY, 2Z73, 3AYN, 3OAX, 4BEY, 4BEZ, 4A4M, 4J4Q, 4PXF, 4ZWJ |
| β2AR | 15 | 2RH1, 2R4R, 3D4S, 3NY8, 3NY9, 3NYA, 3PDS, 3P0G, 3SN6, 3KJ6, 4GBR, 4LDE, 4LDL, 4LDO, 4QKX |
| β1AR | 16 | 2VT4, 2Y00, 2Y02, 2Y03, 2Y04, 2Y01, 2YCW, 2YCX, 2YCZ, 2YCY, 4AMI, 4AMJ, 4GPO, 3ZPR, 3ZPQ, 4BVN |
| A2AAR | 13 | 3EML, 3QAK, 2YDO, 2YDV, 3PWH, 3UZA, 3UZC, 3VG9, 3VGA, 3REY, 3RFM, 4UHR,4EIY |
| CXCR4 | 6 | 3ODU, 3OE0, 3OE6, 3OE8, 3OE9, 4RWS |
| CCR5 | 1 | 4MBS |
| D3R | 1 | 3PBL |
| H1R | 1 | 3RZE |
| M2R | 3 | 3UON, 4MQS, 4MQT |
| M3R | 4 | 4DAJ, 4U14, 4U15, 4U16 |
| κ-OR | 1 | 4DJH |
| μ-OR | 2 | 4DKL, 5C1M |
| δ-OR | 3 | 4EJ4, 4RWA, 4N6H |
| NOP | 1 | 4EA3 |
| PAR1 | 1 | 3VW7 |
| NTSR1 | 5 | 4GRV, 3ZEV, 4BUO, 4BV0, 4BWB |
| 5-HT1B | 2 | 4IAR, 4IAQ |
| 5-HT2B | 2 | 4IB4, 4NC3 |
| S1P1 | 2 | 3V2W, 3V2Y |
| CRF1R | 1 | 4K5Y |
| GCGR | 1 | 4L6R |
| SMO | 5 | 4JKV, 4N4W, 4O9R, 4QIM,4QIN |
| mGluR1 | 1 | 4OR2 |
| mGluR5 | 3 | 5CGD, 5CGC, 4OO9 |
| P2Y12R | 3 | 4NTJ, 4PXZ, 4PY0 |
| P2Y1R | 2 | 4XNV, 4XNW |
| FFAR1 | 1 | 4PHU |
| AT1R | 1 | 4YAY |
| OX2R | 1 | 4S0V |
| LPA1 | 3 | 4Z34, 4Z35, 4Z36 |
STRUCTURAL CHARACTERISTICS OF GPCRS
GPCRs share a common structural architecture of 7TM helical bundle connected by three extracellular loops (ECL1-3) and three intracellular loops (ICL1-3). The N-terminus of GPCR is at the extracellular side, and the C-terminus locates intracellularly. The N-terminus and the extracellular loops are responsible for recognizing a vast variety of ligands and modulating ligand access. The 7TM bundle region forms the structural core, binding ligands and transducing extracellular signals to the intracellular region through conformational changes. The intracellular (IC) parts interact with cytosolic G-proteins, arrestins, G protein-coupled receptor kinases (GRKs) and other downstream signaling effectors.
The solved GPCR structures reveal some common structural features among the receptors. One of the most conserved motifs is the D[E]R3.50Y [superscript indicates residue numbering using the Ballesteros-Weinstein nomenclature (Ballesteros and Weinstein, 1995)] motif in helix III, which often forms a so-called “ionic lock” by making a salt bridge with D/E6.30 in helix VI, for example, in the rhodopsin structure (Palczewski et al., 2000). The ionic lock was suggested as a characteristic of the inactive conformation of GPCRs, blocking the G-protein binding at the cytoplasmic region (discussed in the following part). The W6.48xP motif in helix VI is also considered as one of the micro-switches that have substantially different conformations in the active state versus the inactive state of the receptor. Another conserved motif is the NP7.50 xxY motif in helix VII, which also plays an important role in GPCR activation (discussed in the following part). In addition to the above conserved motifs in the transmembrane domain, the extracellular loop regions in the GPCR structures also show some similarities. A highly conserved disulfide bond between the residue Cys3.25 at the extra-cellular tip of helix III and a cysteine residue in ECL2 was observed in most of the GPCR structures, except for the structure of P2Y12R bound to the antagonist AZD1283 (Zhang et al., 2014b). This disulfide bond plays an important role in stabilizing the conformation of the extracellular region and shapes the entrance to the ligand-binding pocket. The conformation of the intracellular region of GPCRs is relatively conserved, which may correlate with the limited types of downstream binding partners. In most of the solved GPCR structures, the C-terminus contains a 3-4 turn α-helix, helix VIII, that runs parallel to the membrane and is characterized by a common [F(RK)xx(FL)xxx] amphiphilic motif.
Although all the known GPCR structures are characterized by a similar 7TM topology, remarkable structural diversity was observed, especially in the dynamic extracellular loop region. This region presents a diverse repertoire of secondary structures in different GPCR structures. The largest extracellular loop, ECL2, structurally differs between receptors, but its conformation is likely to be conserved in a subfamily-specific manner. For example, ECL2 exhibits an α-helical structure in adrenergic receptor structures, and a hairpin structure in all the peptide receptor structures. In contrast to ECL2, ECL1 and ECL3 are relatively short and tend to lack distinct secondary structural elements. Besides the structural differences in the extracellular region, striking diversities were also observed in the 7TM helical bundle. The seven α-helices display different tilts and rotations in various GPCR structures, which make the ligand-binding pockets of the receptors differ dramatically in size and shape (discussed in the following part).
LIGAND-BINDING MODES OF GPCRS
In the known GPCR structures, ligands usually bind to the canonical 7TM helical bundle. However, size, shape and electro-static properties of the ligand-binding pockets are dramatically distinct between different receptors (Fig. 1). In the structure of the class C GPCR, mGluR5, the negative allosteric modulator mavoglurant penetrates into the 7TM bundle of the receptor, locating much deeper compared to the ligands in the other GPCR structures. The ligand-binding sites in the CRF1R and mGluR1 structures are closer to the extracellular surface than that in mGluR5, but deeper than in the class A GPCRs and SMO. In the SMO structure, the ligand LY294068 occupies a long and narrow binding pocket, which is connected to the extracellular aqueous environment though a small opening formed by ECL2 and ECL3 (Wang et al., 2014). Remarkable differences were also found in the ligand-binding pockets of class A GPCRs. The ligands of peptide receptors, such as chemokine receptors and opioid receptors, bind to cavities that are shallower and more open compared to the ligand-binding pockets of the receptors with endogenous small-molecule ligands, such as aminergic receptors. This may correlate with the fact that peptides are much larger molecules that need more binding space than the small molecules and can not enter deeply into the 7TM bundle like the small-molecule ligands.
Fig. 1.
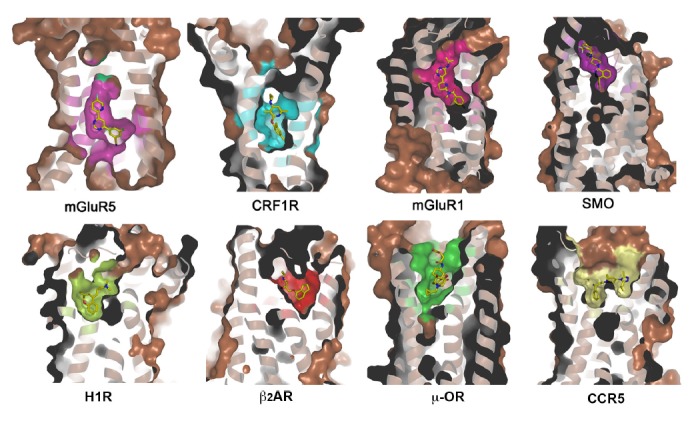
Ligand-binding pockets of mGluR5, CRF1R, mGluR1, SMO, H1R, β2AR, μ-OR and CCR5. Receptors are shown in cartoon and surface representations. Ligands are shown as yellow sticks.
Despite of the high overall structural similarity, ligand-binding modes can be distinct between receptors in the same sub-family (Fig. 2). The structures of purinergic receptors P2Y1R and P2Y12R reveal very different features in binding their nucleotide-like ligands, although these two receptors recognize the same endogenous ligand ADP. The ligand-binding sites for the nucleotide-like antagonist MRS2500 in P2Y1R and the nucleotide agonist 2MeSADP in P2Y12R are spatially distinct with only a minor overlap. In the P2Y1R structure, the adenine group of MRS2500 is closed to helices VI and VII, whereas the adenine ring of 2MeSADP reaches deep into the binding pocket and interacts with helices III and IV in the P2Y12R structure (Zhang et al., 2014b; 2015a). In the structures of chemokine receptors CXCR4 and CCR5, the ligand-binding modes are distinct as well. The antagonist IT1t in the CXCR4 structure occupies part of the binding pocket formed by helices I, II, III and VII and ECL2, while in the CCR5 structure, the ligand maraviroc locates in a deeper binding site and occupies a larger area of the pocket defined by residues from helices I, II, III, V, VI and VII, making no contacts with the extracellular loops (Tan et al., 2013; Wu et al., 2010). These findings highlight the diversity of GPCR ligand-binding mode.
Fig. 2.

Comparison of the ligand-binding pockets in the structures of P2Y1R-MRS2500, P2Y12R-2MeSADP, CXCR4-IT1t and CCR5-maraviroc complexes. The receptors are shown in cartoon and surface representations, and are colored in magenta, green, yellow and cyan, respectively. The ligands MRS2500, 2MeSADP, IT1t and maraviroc are shown as green, magenta, cyan and yellow sticks, respectively.
Additionally, there are differences on the approach routes that GPCR ligands enter the binding pockets. In most of the GPCR structures, there is a large extracellular vestibule as part of an extended hydrophilic channel to the orthosteric binding pocket where the ligands gain access into the binding pockets. However, in the lipid-activated GPCRs, such as S1P1 and FFAR1, receptor’s N-terminus or extracellular loops form a cap-like structure over the ligand-binding pocket, blocking the entrance to the ligand-binding cavity through the extracellular region of the receptor (Hanson et al., 2012; Srivastava et al., 2014). The ligands of these receptors are highly lipophilic and bind to the receptors mainly through hydrophobic interactions. This ligand-binding mode and the hydrophobic nature of the ligands suggest that these ligands most likely gain access into the binding pockets through the lipid bilayer. A similar approach route of ligand was proposed in the structure of P2Y1R bound to the non-nucleotide antagonist BPTU. In this structure, BPTU binds to a shallow and highly hydrophobic pocket on the external receptor interface with the lipid bilayer. This is the first structurally characterized selective GPCR ligand located entirely outside of the 7TM helical bundle (Zhang et al., 2015a).
GPCR ACTIVATION
It is widely believed that there is an equilibrium between two conformations of GPCR, the inactive state (R) and the active state (R*). In the absence of ligands, the receptor activity is in basal level. The efficacy of ligands reflects their ability to alter the equilibrium between the inactive state and the active state. Full agonists preferentially bind to and stabilize the active conformation, and shift the equilibrium to R*, while inverse agonists stabilize the inactive conformation, and shift the equilibrium to R. Partial agonists have some affinity for both R and R*, and are therefore less effective in shifting the equilibrium towards R* (Kobilka and Deupi, 2007).
To date, the structures of several different GPCRs, rhodopsin, β1AR, β2AR, A2AAR, M2R, P2Y12R and μ-OR, have been determined in both antagonist-bound and agonist-bound states (Haga et al., 2012; Huang et al., 2013; 2015; Jaakola et al., 2008; Kang et al., 2015; Lebon et al., 2011; 2012; Manglik et al., 2012; Palczewski et al., 2000; Park et al., 2008; Rasmussen et al., 2011; Rosenbaum et al., 2007; Xu et al., 2011; Zhang et al., 2014a; 2014b). These structures provide insights into the molecular mechanisms about how the ligands bind at the extracellular side of the receptors and trigger conformational changes in the receptors’ 7TM domains, where G-protein and other effectors bind and initiate a series of downstream signaling pathways. In spite of the remarkable diversity of ligands and ligand-binding sites of GPCRs, the receptors share similar activation mechanisms. Some conserved motifs in GPCRs are defined as molecular micro-switches, which are believed to be important in the regulation of receptor activation and signaling, acting as switches between inactive states and active states (Nygaard et al., 2009). In the rhodopsin and A2AAR structures, R3.50 in the D[E]R3.50 Y motif interacts with E6.30 in helix VI to form the ionic lock, which is broken in the active structures (Scheerer et al., 2008; Xu et al., 2011). W6.48 within the CW6.48 xP motif is classified as a rotamer toggle switch. As seen in the rhodopsin and A2AAR structures, a conformational change of the side chain of W6.48 is induced by interacting with agonists, leading to a movement of helix VI, which is considered as a major ligand-dependent trigger in GPCR activation (Park et al., 2008; Xu et al., 2011). Additionally, the NP7.50xxY motif was also proposed as one of the important micro-switches. In the structure of ligand-free opsin, the Y7.53 residue rotates to face into the helical bundle, blocking helix VI from moving back towards helix III to adopt the inactive conformation (Park et al., 2008).
Multiple biochemical and biophysical studies indicate that receptor activation is associated with a relatively large-scale rearrangement of the transmembrane helices, leading to different receptor conformations and differential effects on downstream signaling proteins (Kobilka, 2007; Nygaard et al., 2009). Such conformational changes have been observed in the structures of several different GPCRs, such as rhodopsin, A2AAR, β2AR and β1AR (Huang et al., 2013; Park et al., 2008; Rasmussen et al., 2011; Xu et al., 2011). The helical rearrangements include an outward movement of helices V and VI and an inward motion of helix VII, leading to an opening of the helical bundle at the cytoplasmic side, which facilitates the G-protein coupling and subsequent activation (Fig. 3A). However, in the P2Y12R structures, remarkable conformational changes occur in the extracellular part. Compared with the antagonist-bound P2Y12R structure, the extracellular tips of helices VI and VII in the agonist-bound structure shift 5–10 Å towards the central axis of the 7TM helical bundle (Fig. 3B). This finding suggests that not only the intracellular parts but also the extracellular parts of GPCRs play critical roles in receptor activation.
Fig. 3.
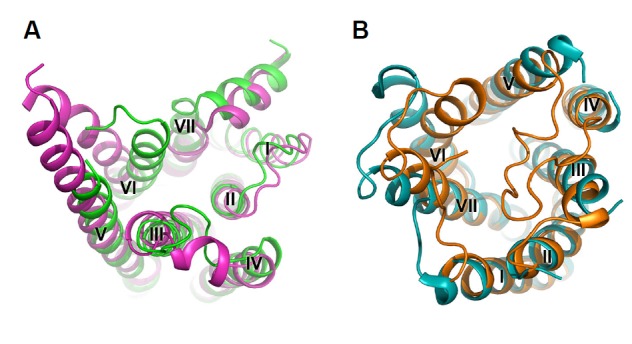
Comparison between active structures and inactive structures of β2AR and P2Y12R. A, Intracellular side of the active structure (magenta) and the inactive structure (green) of β2AR. B, Extracellular side of the active structure (orange) and the inactive structure (cyan) of P2Y12R. The receptors are shown in cartoon representations.
Upon activation, GPCRs couple to G protein, changing GDP that binds to G protein to GTP. The active GPCRs can also interact with GRKs, which phosphorylate GPCRs to trigger a high affinity binding to arrestins. This blocks the interaction between the receptors and G protein, turning off G-protein signaling, and leads to GPCR desensitization (Nygaard et al., 2009). The complex structures of β2AR bound to Gs protein (β2AR-Gs) (Rasmussen et al., 2011), bovine rhodopsin in complex with a G-protein peptide (rhodopsin-GαCT) (Scheerer et al., 2008) and human rhodopsin bound to a pre-activated mouse visual arrestin (Kang et al., 2015) provide molecular basis of GPCR signaling. In the structures of β2AR-Gs and rhodopsin-GαCT, the C-terminus of Gα inserts into the intracellular side of the receptor’s 7TM helical bundle, leading to the rearrangement of helices V, VI and VII. The rhodopsin-arrestin structure shows a large rhodopsin-arrestin interface formed by ICL1, N-terminus of helix VIII and the C-terminal region of helix VII. These structures provide insights into the interactions between GPCRs and G-proteins, as well as arrestins. However, more complex structures between different GPCRs and various G proteins, arrestins, as well as GRKs are desperately needed for better understanding the molecular mechanisms of GPCR signaling transduction.
ALLOSTERIC MODULATION
GPCRs can not only recognize orthosteric ligands but also are affected by a variety of endogenous allosteric modulators, including lipids, ions, waters and sterols (Chini and Parenti, 2009). These modulators regulate receptor function by binding to alternative regions in the receptors, allosteric binding sites, instead of the conventional orthosteric binding sites. In recent years, there has been increasing therapeutic interest in ligands that bind to the allosteric sites of GPCRs. Allosteric ligands can potentially be more selective than the orthosteric ligands because of their little conserved inherent activity, making them highly beneficial therapeutic candidates with less adverse effects from overdose. Allosteric ligands have been identified for many GPCRs, such as adenosine receptors, histamine receptors, dopamine receptors, serotonin receptors, muscarinic receptors, chemokine receptors and class C GPCRs. The previously solved GPCR structures reveal various allosteric binding sites within the 7TM helical bundle (in CCR5, mGluR1 and mGluR5) (Dore et al., 2014; Tan et al., 2013; Wu et al., 2014), in the extracellular region of the receptor (in M2R) (Haga et al., 2012), or on the external lipidic interface of the transmembrane domain (in P2Y1R) (Zhang et al., 2015a).
The GPCR structures deepen our understanding about GPCR allosteric modulation, and open the door to high selective and novel therapeutic agents for the treatment of multiple human disorders. The A2AAR structure suggests that sodium ion may act as an allosteric modulator to alter the dynamics and activation profiles of GPCRs (Gutierrez-de-Teran et al., 2013). In the β2AR structure, the specific cholesterol binding modulates receptor thermostability and binding affinity for inverse agonists (Cherezov et al., 2007). The CCR5 structure demonstrates that the allosteric inverse agonist maraviroc inhibits receptor function by stabilizing the receptor in an inactive conformation (Tan et al., 2013). The unique allosteric binding site of the P2Y1R antagonist BPTU on the external receptor surface broaden the new scope of future GPCR ligand discovery outside of the canonical orthosteric ligand-binding pocket (Zhang et al., 2015a).
GPCR DIMERIZATION
A growing body of biochemical and biophysical evidence indicates that many GPCRs can form homo- or hetero-dimers or larger oligomers (Lee et al., 2003). These dimers and oligomers have been proposed to be important for modulation of GPCR signal transduction and mediation of crosstalk between GPCR pathways (Lohse, 2010; Milligan, 2009; Pin et al., 2004).
Parallel dimers with substantial receptor-receptor interfaces have been observed in the structures of some GPCRs, including β2AR, CXCR4, κ-OR, μ-OR and mGluR1 (Manglik et al., 2012; Rasmussen et al., 2007; Wu et al., 2010; 2012; 2014). In the rhodopsin and κ-OR structures, a parallel receptor dimer is formed via helices I, II and VIII. In the CXCR4 and μ-OR structures, a parallel, symmetric dimer is formed via helices V and VI. A receptor dimer mediated by interactions between receptor’s helix I and cholesterol molecules was observed in the 7TM domain structure of mGluR1. In the β2AR structure, the dimer is mediated by ordered lipids consisting of two cholesterol molecules and two palmitic acid molecules in the C-terminal region of the receptor. Although the above structural information provides insights into the GPCR dimerization, whether the dimerization is necessary for signal transduction and how do the dimers function in physiological state are still unclear. More structural evidence of GPCR dimerization is needed to elucidate what is likely to be a common underlying mechanism for GPCR dimerization.
CONCLUSION
Recent tremendous breakthroughs in GPCR structural studies provide us opportunities to learn the structural features of GPCRs and the molecular mechanisms of various GPCR physiological behaviors, such as ligand recognition, receptor activation, allosteric modulation and dimerization, etc. However, the current structural information is far away from enough to understand all the biological functions of GPCRs. More structures from different receptor sub-families, such as adhesion receptors, olfactory receptors and many orphan receptors, are essential to fully understand the ligand-binding landscape of the GPCR superfamily. Although several trans-membrane domain structures of classes B, C and F receptors have been solved, the full-length structures of non-rhodopsin family GPCRs are necessitous to reveal the mechanisms of receptor activation for these specific families. Determining the complex structures of GPCRs coupling to the intracellular signaling effectors is still considered as one of the major tasks of the GPCR structural studies, but remains extremely challenging. Additionally, the structures of GPCR homo- or heterodimers are key for understanding how GPCRs regulate the cell signaling by changing their stoichiometry. The medicinal importance of GPCRs is beyond doubt. Obtaining GPCR structures will immensely facilitate structure-based drug discovery and certainly open up further therapeutic opportunities for many severe diseases.
Acknowledgments
This work was supported by CAS Strategic Priority Research Program XDB08020300, and the National Science Foundation of China grants 31422017 and 31370729.
REFERENCES
- Ballesteros J., Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- Caffrey M. Crystallizing membrane proteins for structure-function studies using lipidic mesophases. Biochem. Soc. Trans. 2011;39:725–732. doi: 10.1042/BST0390725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae P.S., Rasmussen S.G., Rana R.R., Gotfryd K., Chandra R., Goren M.A., Kruse A.C., Nurva S., Loland C.J., Pierre Y., et al. Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nat. Methods. 2010;7:1003–1008. doi: 10.1038/nmeth.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V., Rosenbaum D.M., Hanson M.A., Rasmussen S.G., Thian F.S., Kobilka T.S., Choi H.J., Kuhn P., Weis W.I., Kobilka B.K., et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien E.Y., Liu W., Zhao Q., Katritch V., Han G.W., Hanson M.A., Shi L., Newman A.H., Javitch J.A., Cherezov V., et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini B., Parenti M. G-protein-coupled receptors, cholesterol and palmitoylation: facts about fats. J. Mol. Endocrinol. 2009;42:371–379. doi: 10.1677/JME-08-0114. [DOI] [PubMed] [Google Scholar]
- Chrencik J.E., Roth C.B., Terakado M., Kurata H., Omi R., Kihara Y., Warshaviak D., Nakade S., Asmar-Rovira G., Mileni M., et al. Crystal structure of antagonist bound human lysophosphatidic Acid Receptor 1. Cell. 2015;161:1633–1643. doi: 10.1016/j.cell.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore A.S., Okrasa K., Patel J.C., Serrano-Vega M., Bennett K., Cooke R.M., Errey J.C., Jazayeri A., Khan S., Tehan B., et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature. 2014;511:557–562. doi: 10.1038/nature13396. [DOI] [PubMed] [Google Scholar]
- Fredriksson R., Lagerstrom M.C., Lundin L.G., Schioth H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Gether U. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr. Rev. 2000;21:90–113. doi: 10.1210/edrv.21.1.0390. [DOI] [PubMed] [Google Scholar]
- Granier S., Manglik A., Kruse A.C., Kobilka T.S., Thian F.S., Weis W.I., Kobilka B.K. Structure of the delta-opioid receptor bound to naltrindole. Nature. 2012;485:400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-de-Teran H., Massink A., Rodriguez D., Liu W., Han G.W., Joseph J.S., Katritch I., Heitman L.H., Xia L., Ijzerman A.P., et al. The role of a sodium ion binding site in the allosteric modulation of the A(2A) adenosine G protein-coupled receptor. Structure. 2013;21:2175–2185. doi: 10.1016/j.str.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga K., Kruse A.C., Asada H., Yurugi-Kobayashi T., Shiroishi M., Zhang C., Weis W.I., Okada T., Kobilka B.K., Haga T., et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature. 2012;482:547–551. doi: 10.1038/nature10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson M.A., Roth C.B., Jo E., Griffith M.T., Scott F.L., Reinhart G., Desale H., Clemons B., Cahalan S.M., Schuerer S.C., et al. Crystal structure of a lipid G protein-coupled receptor. Science. 2012;335:851–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenstein K., Kean J., Bortolato A., Cheng R.K., Dore A.S., Jazayeri A., Cooke R.M., Weir M., Marshall F.H. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature. 2013;499:438–443. doi: 10.1038/nature12357. [DOI] [PubMed] [Google Scholar]
- Huang J., Chen S., Zhang J.J., Huang X.Y. Crystal structure of oligomeric beta1-adrenergic G protein-coupled receptors in ligand-free basal state. Nat. Struct. Mol. Biol. 2013;20:419–425. doi: 10.1038/nsmb.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.J., Manglik A., Venkatakrishnan A.J., Laeremans T., Feinberg E.N., Sanborn A.L., Kato H.E., Livingston K.E., Thorsen T.S., Kling R.C., et al. Structural insights into mu-opioid receptor activation. Nature. 2015;524:315–321. doi: 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakola V.P., Griffith M.T., Hanson M.A., Cherezov V., Chien E.Y., Lane J.R., Ijzerman A.P., Stevens R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y., Zhou X.E., Gao X., He Y., Liu W., Ishchenko A., Barty A., White T.A., Yefanov O., Han G.W., et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature. 2015;523:561–567. doi: 10.1038/nature14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka B.K. G protein coupled receptor structure and activation. Biochim. Biophys. Acta. 2007;1768:794–807. doi: 10.1016/j.bbamem.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka B.K., Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol. Sci. 2007;28:397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Kruse A.C., Hu J., Pan A.C., Arlow D.H., Rosenbaum D.M., Rosemond E., Green H.F., Liu T., Chae P.S., Dror R.O., et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature. 2012;482:552–556. doi: 10.1038/nature10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G., Warne T., Edwards P.C., Bennett K., Langmead C.J., Leslie A.G., Tate C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G., Warne T., Tate C.G. Agonist-bound structures of G protein-coupled receptors. Curr Opin Struct Biol. 2012;22:482–490. doi: 10.1016/j.sbi.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Lee S.P., O’Dowd B.F., George S.R. Homo- and hetero-oligomerization of G protein-coupled receptors. Life Sci. 2003;74:173–180. doi: 10.1016/j.lfs.2003.09.028. [DOI] [PubMed] [Google Scholar]
- Lohse M.J. Dimerization in GPCR mobility and signaling. Curr. Opin. Pharmacol. 2010;10:53–58. doi: 10.1016/j.coph.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Manglik A., Kruse A.C., Kobilka T.S., Thian F.S., Mathiesen J.M., Sunahara R.K., Pardo L., Weis W.I., Kobilka B.K., Granier S. Crystal structure of the mu-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G. G protein-coupled receptor hetero-dimerization: contribution to pharmacology and function. Br J. Pharmacol. 2009;158:5–14. doi: 10.1111/j.1476-5381.2009.00169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard R., Frimurer T.M., Holst B., Rosenkilde M.M., Schwartz T.W. Ligand binding and micro-switches in 7TM receptor structures. Trends Pharmacol. Sci. 2009;30:249–259. doi: 10.1016/j.tips.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Palczewski K., Kumasaka T., Hori T., Behnke C.A., Motoshima H., Fox B.A., Le Trong I., Teller D.C., Okada T., Stenkamp R.E., et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Park J.H., Scheerer P., Hofmann K.P., Choe H.-W., Ernst O.P. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- Pin J.P., Kniazeff J., Binet V., Liu J., Maurel D., Galvez T., Duthey B., Havlickova M., Blahos J., Prezeau L., et al. Activation mechanism of the heterodimeric GABA(B) receptor. Biochem. Pharmacol. 2004;68:1565–1572. doi: 10.1016/j.bcp.2004.06.035. [DOI] [PubMed] [Google Scholar]
- Rasmussen S.G., Choi H.J., Rosenbaum D.M., Kobilka T.S., Thian F.S., Edwards P.C., Burghammer M., Ratnala V.R., Sanishvili R., Fischetti R.F., et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- Rasmussen S.G., DeVree B.T., Zou Y., Kruse A.C., Chung K.Y., Kobilka T.S., Thian F.S., Chae P.S., Pardon E., Calinski D., et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum D.M., Cherezov V., Hanson M.A., Rasmussen S.G., Thian F.S., Kobilka T.S., Choi H.J., Yao X.J., Weis W.I., Stevens R.C., et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- Rosenbaum D.M., Zhang C., Lyons J.A., Holl R., Aragao D., Arlow D.H., Rasmussen S.G.F., Choi H.-J., DeVree B.T., Sunahara R.K., et al. Structure and function of an irreversible agonist-β2 adrenoceptor complex. Nature. 2011;469:236–240. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheerer P., Park J.H., Hildebrand P.W., Kim Y.J., Krauss N., Choe H.W., Hofmann K.P., Ernst O.P. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- Schlyer S., Horuk R. I want a new drug: G-protein-coupled receptors in drug development. Drug Discov. Today. 2006;11:481–493. doi: 10.1016/j.drudis.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Shimamura T., Shiroishi M., Weyand S., Tsujimoto H., Winter G., Katritch V., Abagyan R., Cherezov V., Liu W., Han G.W., et al. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011;475:65–70. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu F.Y., He M., de Graaf C., Han G.W., Yang D., Zhang Z., Zhou C., Xu Q., Wacker D., Joseph J.S., et al. Structure of the human glucagon class B G-protein-coupled receptor. Nature. 2013;499:444–449. doi: 10.1038/nature12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A., Yano J., Hirozane Y., Kefala G., Gruswitz F., Snell G., Lane W., Ivetac A., Aertgeerts K., Nguyen J., et al. High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature. 2014;513:124–127. doi: 10.1038/nature13494. [DOI] [PubMed] [Google Scholar]
- Szczepek M., Beyriere F., Hofmann K.P., Elgeti M., Kazmin R., Rose A., Bartl F.J., von Stetten D., Heck M., Sommer M.E., et al. Crystal structure of a common GPCR-binding interface for G protein and arrestin. Nat. Commun. 2014;5:4801. doi: 10.1038/ncomms5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q., Zhu Y., Li J., Chen Z., Han G.W., Kufareva I., Li T., Ma L., Fenalti G., Li J., et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science. 2013;341:1387–1390. doi: 10.1126/science.1241475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A.A., Liu W., Chun E., Katritch V., Wu H., Vardy E., Huang X.P., Trapella C., Guerrini R., Calo G., et al. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature. 2012;485:395–399. doi: 10.1038/nature11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen W., Frazer J., Unett D. Functional assays for screening GPCR targets. Curr. Opin. Biotechnol. 2005;16:655–665. doi: 10.1016/j.copbio.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Wacker D., Wang C., Katritch V., Han G.W., Huang X.P., Vardy E., McCorvy J.D., Jiang Y., Chu M.H., Siu F.Y., et al. Structural features for functional selectivity at serotonin receptors. Science. 2013;340:615–619. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Jiang Y., Ma J., Wu H., Wacker D., Katritch V., Han G.W., Liu W., Huang X.P., Vardy E., et al. Structural basis for molecular recognition at serotonin receptors. Science. 2013a;340:610–614. doi: 10.1126/science.1232807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Wu H., Katritch V., Han G.W., Huang X.P., Liu W., Siu F.Y., Roth B.L., Cherezov V., Stevens R.C. Structure of the human smoothened receptor bound to an antitumour agent. Nature. 2013b;497:338–343. doi: 10.1038/nature12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Wu H., Evron T., Vardy E., Han G.W., Huang X.P., Hufeisen S.J., Mangano T.J., Urban D.J., Katritch V., et al. Structural basis for Smoothened receptor modulation and chemoresistance to anticancer drugs. Nat. Commun. 2014;5:4355. doi: 10.1038/ncomms5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T., Edwards P.C., Leslie A.G., Tate C.G. Crystal structures of a stabilized beta1-adrenoceptor bound to the biased agonists bucindolol and carvedilol. Structure. 2012;20:841–849. doi: 10.1016/j.str.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J.F., Noinaj N., Shibata Y., Love J., Kloss B., Xu F., Gvozdenovic-Jeremic J., Shah P., Shiloach J., Tate C.G., et al. Structure of the agonist-bound neurotensin receptor. Nature. 2012;490:508–513. doi: 10.1038/nature11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B., Chien E.Y., Mol C.D., Fenalti G., Liu W., Katritch V., Abagyan R., Brooun A., Wells P., Bi F.C., et al. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Wacker D., Mileni M., Katritch V., Han G.W., Vardy E., Liu W., Thompson A.A., Huang X.P., Carroll F.I., et al. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Wang C., Gregory K.J., Han G.W., Cho H.P., Xia Y., Niswender C.M., Katritch V., Meiler J., Cherezov V., et al. Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science. 2014;344:58–64. doi: 10.1126/science.1249489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F., Wu H., Katritch V., Han G.W., Jacobson K.A., Gao Z.G., Cherezov V., Stevens R.C. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J., Mobarec J.C., Kolb P., Rosenbaum D.M. Crystal structure of the human OX2 orexin receptor bound to the insomnia drug suvorexant. Nature. 2015;519:247–250. doi: 10.1038/nature14035. [DOI] [PubMed] [Google Scholar]
- Zhang Y., DeVries M.E., Skolnick J. Structure modeling of all identified G protein-coupled receptors in the human genome. Plos Comput. Biol. 2006;2:88–99. doi: 10.1371/journal.pcbi.0020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Srinivasan Y., Arlow D.H., Fung J.J., Palmer D., Zheng Y., Green H.F., Pandey A., Dror R.O., Shaw D.E., et al. High-resolution crystal structure of human protease-activated receptor 1. Nature. 2012;492:387–392. doi: 10.1038/nature11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Zhang K., Gao Z.G., Paoletta S., Zhang D., Han G.W., Li T., Ma L., Zhang W., Muller C.E., et al. Agonist-bound structure of the human P2Y12 receptor. Nature. 2014a;509:119–122. doi: 10.1038/nature13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Zhang J., Gao Z.G., Zhang D., Zhu L., Han G.W., Moss S.M., Paoletta S., Kiselev E., Lu W., et al. Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature. 2014b;509:115–118. doi: 10.1038/nature13083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D., Gao Z.G., Zhang K., Kiselev E., Crane S., Wang J., Paoletta S., Yi C., Ma L., Zhang W., et al. Two disparate ligand-binding sites in the human P2Y1 receptor. Nature. 2015a;520:317–321. doi: 10.1038/nature14287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Unal H., Gati C., Han G.W., Liu W., Zatsepin N.A., James D., Wang D., Nelson G., Weierstall U., et al. Structure of the Angiotensin receptor revealed by serial femtosecond crystallography. Cell. 2015b;161:833–844. doi: 10.1016/j.cell.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]