

Abstract
During T cell activation, mitochondrial content increases to meet the high energy demand of rapid cell proliferation. With this increase, the level of reactive oxygen species (ROS) also increases and causes the rapid apoptotic death of activated cells, thereby facilitating T cell homeostasis. Nicotinamide (NAM) has previously been shown to enhance mitochondria quality and extend the replicative life span of human fibroblasts. In this study, we examined the effect of NAM on CD8+ T cell activation. NAM treatment attenuated the increase of mitochondrial content and ROS in T cells activated by CD3/CD28 antibodies. This was accompanied by an accelerated and higher-level clonal expansion resulting from attenuated apoptotic death but not increased division of the activated cells. Attenuation of ROS-triggered pro-apoptotic events and upregulation of Bcl-2 expression appeared to be involved. Although cells activated in the presence of NAM exhibited compromised cytokine gene expression, our results suggest a means to augment the size of T cell expansion during activation without consuming their limited replicative potential.
Keywords: mitochondria, nicotinamide, ROS, T cell activation
INTRODUCTION
Upon activation by foreign antigens presented by antigen-presenting cells, naïve T cells undergo clonal expansion and differentiation to become effector cells. After antigen clearance, the clonally expanded T cells are rapidly removed through apoptosis (Brenner et al., 2008). During T cell activation, mitochondrial biogenesis and respiration increase (D’Souza et al., 2007) in order to supply enough ATP for an increase in cell size, followed by rapid proliferation and differentiation during the expansion period (Kaminski et al., 2007). Failure to generate enough energy would lead to infertile T cell activation.
Increased mitochondrial respiration is also important in another aspect of T cell activation: the rapid increase in mitochondrial production of reactive oxygen species (ROS) (Grayson et al., 2003). High-levels of ROS affect both the expansion and removal of activated cells (Hildeman et al., 1999). ROS modulate the activities of transcription factors such as NF-κB (Hoyos et al., 1989) and AP-1 (Beiqing et al., 1996), which promote the expression of IL-2, a major autocrine factor for T cell proliferation (Meuer et al., 1984). Meanwhile, activated T cells are removed via the activation-induced cell death (AICD) mechanism, which is predominantly triggered by Fas-L. Fas-L induction is known to be enhanced by ROS (Stranges et al., 2007). The rapid downregulation of T cell numbers during the contraction phase is also mediated by apoptosis, known as activation-caused autonomous death (ACAD) (Hildeman et al., 2002), which is dependent on the intrinsic death pathway that involves pro- and anti-apoptotic members of Bcl-2 family proteins (Hildeman et al., 2003a). ROS have been suggested to be responsible for Bcl-2 downregulation in activated T cells (Hildeman et al., 2003b). Furthermore, the increase of mitochondrial content itself may also contribute to apoptosis. Accumulated cytochrome c in these mitochondria is released via Ca+2-dependent mitochondrial permeability transition, which is also triggered by ROS (Crompton, 1999). Therefore, alteration in the status of mitochondria and ROS would be expected to change the level and the kinetics of T cell activation and thereby substantially affect the adaptive immune response and T cell homeostasis.
Nicotinamide (NAM), a vitamin B3 derivative, is converted to NAD+ through a salvage pathway (Liu et al., 1982). When administered at high doses, it exerts positive effects on the proliferation, survival, and longevity of cells (Kang et al., 2006; Maiese et al., 2009), possibly by increasing the cellular level of NAD+. For this reason, NAM is actively investigated for therapeutic applications to human diseases, although underlying mechanisms of its actions are not fully understood (Maiese et al., 2009). In our previous studies, NAM accelerated autophagy-mediated mitochondrial turnover and induced a substantial decrease in ATP and ROS content (Jang et al., 2012; Kang and Hwang, 2009). Furthermore, NAM treatment caused an increase in the replicative lifespan of human fibroblasts and keratinocytes. This was suggested to be mediated by an increase in mitochondrial quality, defined by increased mitochondrial membrane potential and decreased levels of mitochondrial content and mitochondrial ROS production and, thereby, a decrease in ROS generation (Jang et al., 2012; Kang and Hwang, 2009; Kang et al., 2006).
In this study, CD8+ cell activation was used as a model to investigate whether NAM treatment alters the outcome of cellular activity in which mitochondria and ROS play important roles. We observed that NAM treatment attenuated the increase in mitochondrial content in CD8+ cells during their activation. Unexpectedly, this was accompanied by an increase in the size of population expansion. Decreased apoptotic cell death, likely caused by attenuated ROS production, underlies the increase in the expansion size of the activated cells. Our results suggest that NAM can affect the physiology of T cells and that the extent of population growth during T cell activation can be manipulated through modulation of levels of mitochondria and ROS.
MATERIALS AND METHODS
CD8+ cell isolation and activation
Four healthy males (three in their twenties and one in his fifties) and one female (in her twenties) donated 10 cc of blood in compliance with the protocol approved by the IRB of Sookmyung Women’s University (SM-IRB-08-0225). CD8+ T cells were purified using the Dynal CD8+ Isolation Kit (Invitrogen, USA) and activated by the treatment of Dynabeads conjugated with anti-CD3 and anti-CD28 antibodies (Invitrogen). The Roswell Park Memorial Institute (RPMI) medium was replaced every two days with fresh supplements of human IL-2 (60 UI/ml; Sigma-Aldrich, USA) and IL-15 (5 ng/ml; ProSpec, USA). At the start of the activation, 5 mM NAM was added. The individual donors provided written informed consent to publish these case details.
Determination of cell division number
The method developed by Lyons and Parish (1994) was used. After incubation with 0.5 μM carboxyfluorescein succinimidyl ester (CFSE; Sigma-Aldrich), cells were divided into two plates and activated in the presence or absence of NAM. After three or seven days, cells were analyzed by flow cytometry by using a BD FACSCanto (BD Biosciences, USA).
Flow cytometry
To analyze cell cycle or annexin V-positivity, cells were stained with either 10 μg/ml propidium iodide (PI), 0.2 μg/ml annexin V-FITC (BD Biosciences), or both PI + annexin V-FITC. In order to determine mitochondrial content, the levels of mitochondrial superoxide or hydroxyl radicals, or cytosolic Ca+2 concentration ([Ca+2]cyt), cells were stained with either 30 nM MitoTracker Green, 0.1 μM MitoSox or DHR123, or 5 μM Fluo-3 (all from Invitrogen), respectively. Cells were then analyzed by flow cytometry by using a BD FACSCanto. Raw data were analyzed using the CellQuest 3.2 software (BD Biosciences).
Quantification of tumor necrosis factor α (TNF-α) secretion
The amount of TNF-α in 24 h-conditioned medium was determined with a TNF-α ELISA Kit (Invitrogen) following the manufacturer’s protocol and by reading the chromogen density in an ELx808 microplate reader (BioTek, USA).
Western analysis
Cells were lyzed in radioimmunoprecipitation assay (RIPA) buffer [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS]. Equal quantities of proteins were subjected to SDS-PAGE and western analysis using antibodies against electron transport chain proteins (Total OXPHOS Human Antibody Cocktail; MitoSciences, USA), p62/SQSTM1 (Santa Cruz Biotechnology, Inc., USA), and LC3, Bcl-2, caspase 3, and Erk (Cell Signaling Technology, USA).
Real time PCR analysis
Total RNA was isolated using TRizol reagent (Invitrogen), and 5 μg was converted to cDNA by MuLV reverse transcriptase (Promega, USA). For quantitative PCR, an equal volume of the cDNA reactions was mixed with TOPreal™ qPCR 2X PreMIX (SYBR green; Enzynomics, Korea) and primers with following sequences: 5′-AAG AAT CCC AAA CTC ACC AGG AT-3′ and 5′-TAG ACA CTG AAG ATG TTT CAG TTC TG-3′ for interleukin-2 (IL-2); 5′-TGA CCA GAG CAT CCA AAA GA-3′ and 5′-CTC TTC GAC CTC GAA ACA GC-3′ for interferon-γ (IFN-γ); 5′-CAA GGG ATT GGA ATT GAG GA-3′ and 5′-TGG AAG AAA AAT GGG CTT TG-3′ for Fas; and 5′-TGG GGA TGT TTC AGC TCT TC-3′ and 5′-CAG AGG CAT GGA CCT TGA GT-3′ for Fas-L.
Statistical analyses
For most analyses, the mean ± standard error of the mean from two or three different samples was plotted. For changes in mitochondrial content and ROS levels, the differences between the naïve and activated groups were considered significant at p < 0.02. For changes in the annexin V-positive population, the differences between the control and NAM-treated groups were considered significant at p < 0.1.
RESULTS
NAM attenuates the increase of mitochondrial and ROS content during T cell activation
In our previous studies, the cellular content of mitochondria was reduced following treatment with 5 mM NAM in human fibroblasts (Jang et al., 2012; Kang and Hwang, 2009). To determine whether NAM treatment exerts a suppressive effect against the increase in mitochondria during T cell activation, we examined isolated primary CD8+ T cells in vitro. Upon activation with a pulse of antibodies to CD3 and CD28, the population rapidly expanded, and the mitochondrial content increased during the expansion period (Figs. 1A–1C). However, this increase was dramatically suppressed in cells activated in the presence of NAM (Fig. 1A). Furthermore, the continuous increase of mitochondrial ROS, both superoxide and hydroxyl radials, during activation was substantially attenuated following NAM treatment (Fig. 1B and 1C). The suppressive effect of NAM was reproduced in T cells from other donors, with differences in the extent of the effect observed (Supplementary Fig. 1). These results suggest that NAM treatment effectively suppresses mitochondria and ROS levels even during T cell activation where they are actively produced.
Fig. 1.

Effect of NAM on mitochondria and ROS contents, and autophagy during T cell activation. A million naïve CD8+ T cells were activated in a medium supplemented with (━•━) or without (--○--) 5mM NAM. On every second day, one-tenth million cells were stained with MTG (A), MitoSox (B), or DHR123(C) and applied to flow cytometry. The mean of the numbers from three cultures is plotted. (*P < 0.02 compared to the value of naïve cells) (Box attached to (A); Western analysis on mitochondrial proteins (ATP synthase subunit alpha, cytochrome c oxidase subunit 1, Succinate dehydrogenase) and Erk from the cells activated for 8 days. (D) Either naïve cells or those activated for 2 days in the absence (−) or presence (+NAM) of 5mM NAM were probed for LC3 to visualize autophagosomes (green) and nuclei (blue). Typical fields were photographed. (E) Cells were activated in the absence (lane 2; lanes 5 and 7) or presence (lane 3; lanes 6 and 8) of NAM for 2 (left) or 3 days (right). In the latter cases, the effect of bafilomycin A1 was further checked for both the cultures (lanes 7 and 8). Equal amount of proteins was applied to western analysis for p62, LC3 or Erk.
Autophagy takes place in T cells during activation and has been reported to either promote or inhibit cell death (Bell et al., 2008; Kovacs et al., 2012). Meanwhile, NAM has been suggested to induce the autophagy-mediated removal of mitochondria in human fibroblasts and carcinoma cells (Jang et al., 2012). In this study, the effect of NAM on autophagy during T cell activation was determined. T cell activation resulted in an increase in the number of LC3-positive puncta, indicating higher-level autophagosome formation. NAM treatment caused a further increase in their number and size (Fig. 1D), suggesting a greater level of autophagy activation. The level of LC3-type II, a marker of autophagy activation (Klionsky et al., 2008), increased, while the level of p62/SQSTM1 (p62), whose degradation is a marker of active autophagy flux (Bjørkøy et al., 2005), decreased in cells activated in the presence of NAM (Fig. 1E). These results suggest an activated autophagy.
We also observed an increase in the levels of p62 and LC3-type II upon treatment with bafilomycin A1, an inhibitor of autolysis (Bjørkøy et al., 2005), indicating an actively ongoing autophagy flux in the NAM-treated cells (Fig. 1E). In addition, this augmented autophagy was accompanied by a fragmental change in mitochondria structure (Supplemental Fig. 2). Mitochondrial fragmentation through active fission has been proposed to be essential to their autophagic turn-over (Gomes and Scorrano, 2003). These results together demonstrate that NAM treatment effectively induces mitochondrial turnover and, thereby, attenuates the increase of mitochondrial content even during T cell activation.
NAM induces greater expansion of the activated T cell population
Next, we investigated how NAM treatment affects T cell expansion. As expected, the populations of CD8+ cells isolated from five different donors all expanded similarly upon activation (Fig. 2A and Supplementary Fig. 3). Different cells exhibited variation in the magnitude and kinetics of the activation-induced increase in cell number, possibly because of differences in body conditions of the donors. We found no correlation between the age or gender of the donors and the activation kinetics. When activated in the presence of NAM, the cell populations grew, but cell numbers increased faster and to greater extent than expected. In most cases, NAM treatment resulted in an almost two-fold increase in cell number (Fig. 2A). This occurred in the absence of changes to the length of the expansion phase: the onset of the contraction phase was unaffected. Higher-level population expansion was also demonstrated by the increase of T cells positive for CD44, a cell-surface marker of active/memory T cells (Russell, 1995) (Fig. 2B).
Fig. 2.
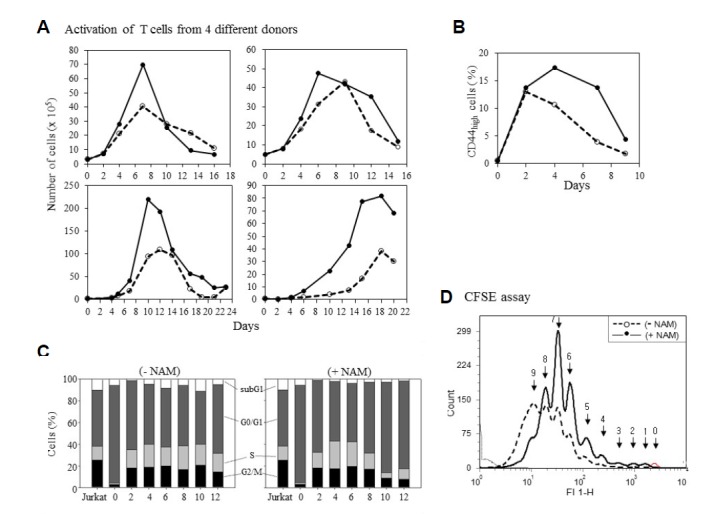
Effect of NAM on T cell population expansion. (A) One or 5×105 CD8+ T cells isolated from four healthy donors were activated in a medium supplemented with (━•━) or without (--○--) 5mM NAM. On every second or third day, cells were fed with fresh media containing NAM while viable cells were counted. (The cells of the upper two graphs were used in the rest of the experiments). Or, cells (of the upper left donor) were probed with CD44 antibody and applied to flow cytometry. % CD44-positive cells is plotted (B). (C) At every two days, cells from control (− NAM) or NAM-treated (+NAM) cultures were stained with PI and applied to flow cytometry. The size of sub-G1, G0G1, S, and G2/M fractions is plotted as bars. Jurkat cells were used as an actively proliferating cell control. (D) Naïve T cells were stained with CFSE, divided into two plates, and activated in the absence or presence of NAM. Cell distribution profiles from CFSE-fluorescence flow cytometry were determined at day 7. The cells not activated (no-division) produced the small right-most peak (arrow 0). The arrows point to the individual peaks that represent the cumulative numbers of division.
The greater population expansion might be caused by an increase in cell proliferation. This possibility was investigated by comparing cell cycle profiles and the number of cell divisions during activation. Upon activation, the length of the G0/G1 phase was reduced, while that of both the S and G2/M phases increased (Fig. 2C). The expanded S and G2/M phases were maintained throughout the 12-day activation in the absence of NAM (Fig. 2C). However, in the presence of NAM, both phases became substantially shorter after day eight, and inversely, the length of the G0/G1 phase increased (Fig. 2C). This suggests that cells stopped dividing earlier when activated in the presence of NAM.
This possibility is supported by the results of the cell division assay using CFSE (Fig. 2D). CFSE-treated naïve cells were divided into two groups and activated in the presence or absence of NAM. At day seven, the cells activated in the absence of NAM were distributed among 10 different peaks, indicating up to nine divisions. Meanwhile, in the NAM-treated population, the last (9th) peak was almost negligible, while the 7th peak was prominent. This indicates that the NAM-treated population underwent fewer cell divisions. Together, these results demonstrate that NAM treatment induces a greater level of population expansion and that this is not attributed to the higher-level division of activated T cells.
NAM causes downregulation of apoptosis during T cell activation
During activation, antigen-stimulated T cells undergo apoptosis through either AICD or ACAD. Both of these processes are affected by high levels of ROS (Brenner et al., 2008; D’Souza et al., 2007; Hildeman et al., 2002; 2003a; Meuer et al., 1984; Stranges et al., 2007). The cell cycle profiles in Fig. 2C reveal another difference induced by NAM; the attenuated increase of the sub-G1 fraction. The increase in the rate and the extent of population growth upon NAM treatment was found to be associated with a decrease in apoptotic death. First, the fraction of the cells positive for annexin V gradually increased during activation, but this increase was substantially attenuated by NAM treatment (Fig. 3A). The increase in the expression of Fas and Fas-L mRNA was largely abolished (Fig. 3B), indicating an attenuation of the extrinsic signaling of apoptosis, the major route of T cell death during activation (Jambrina et al., 2003). The pattern of caspase 3 cleavage, a marker of apoptosis execution, also suggests attenuated apoptosis (Fig. 3C). Caspase 3 cleavage increased until day six and then decreased in this particular T cell sample. NAM treatment attenuated and delayed the cleavage until day six.
Fig. 3.
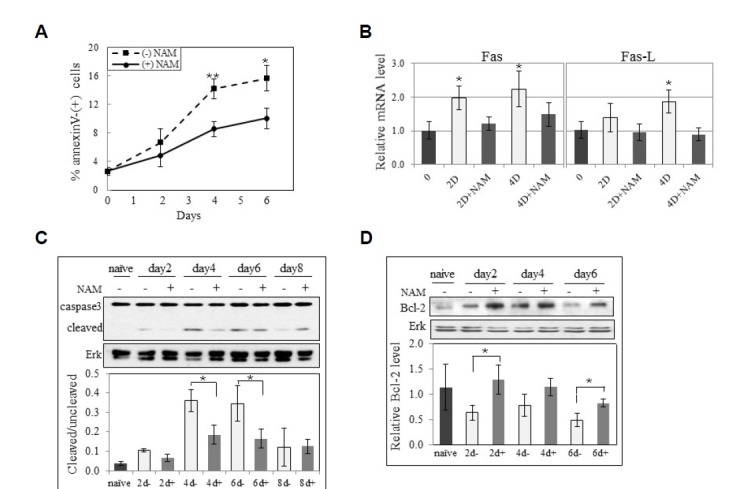
Effect of NAM on apoptosis during activation. (A) T cells activated in the presence (━•━) or absence (--▪--) of NAM were collected at days 2, 4, and 6, and probed with PI or FITC-annexin V, and applied to flow cytometry to determine % total annexin V-positive cells. The averaged numbers of two independent cultures are plotted. (*P < 0.1 and **P < 0.05 between the cells activated with or without NAM) (B) mRNA from T cells naïve or activated for 2 days (−/+NAM) were applied to qPCR for Fas or Fas ligand. Actin mRNA served a control. Mean values from 3 measurements of a biological sample are plotted. ((*P < 0.1 compared to naïve cells) (C, D) Proteins from cells either naïve or activated for 2, 4, 6, 8 days or 2, 4, 6 days in the absence or presence of NAM were applied to western analysis for caspase 3 and Erk (C), or Bcl-2 and Erk (D). Bands in two blots from one biological sample were quantified through densitometry, and mean values relative to that from naïve cells are plotted. (*P < 0.1 between the cells activated with or without NAM)
In previous studies, the cell-beneficiary effects of NAM could not be reproduced following simple treatment with anti-oxidants (Kang et al., 2006; Kwak et al., 2015). Therefore, NAM was expected to activate certain pathways that mediate cell survival. We assessed whether NAM potentiates Bcl-2 expression, which promotes cell survival against apoptotic stimuli and is involved in T cell activation (Brennan et al., 1997; Marte and Downward, 1997). The activation triggered decline of Bcl-2 levels (Hildeman et al., 2003b), but this was less steep in the presence of NAM, suggesting a reduced suppression of the Bcl-2 anti-apoptotic pathway (Fig. 3D). Therefore, NAM appears to potentiate T cell population expansion possibly through the augmentation of Akt-Bcl-2 survival pathway. However, it cannot be determined whether this is induced indirectly by ROS attenuation or directly by another unknown effect of NAM.
NAM attenuates the increase of [Ca+2]cyt and cytokine expression during activation
Upon T cell receptor activation, [Ca+2]cyt increases, and the extra Ca+2 is transported to the mitochondria, resulting in the release of cytochrome c (Hogan et al., 2003). Increase of cytosolic [Ca+2]cyt triggers the intrinsic pathway of apoptosis. This also appears to be attenuated by NAM treatment (a representative result is presented in Fig. 4A), suggesting that the increased T cell expansion may be attributed in part to the attenuation [Ca+2]cyt, although how this is caused by NAM remains unknown.
Fig. 4.
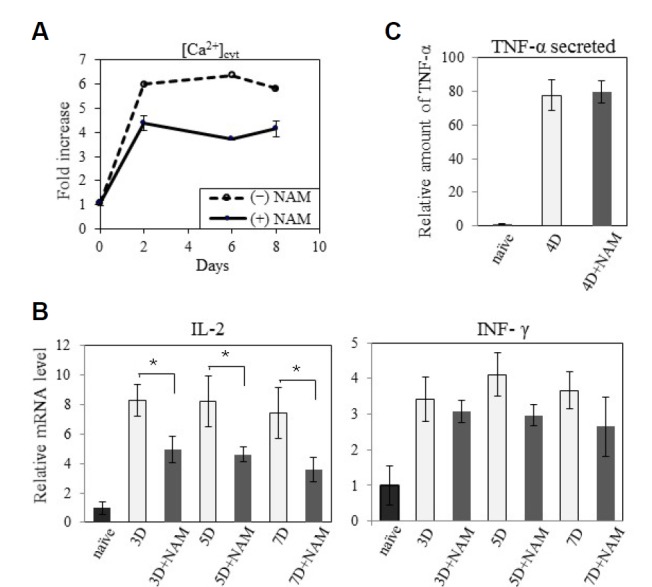
Effect of NAM on the levels of [Ca2+]cyt and cytokine expression. (A) At day 2, 6, or 8 of activation in the presence (━•━) or absence (--○--) of NAM, cells were stained with Fluo-3 and applied to [Ca2+]cyt quantification through flow cytometry. Average of two values normalized by that of naïve cells is plotted. (B) mRNAs from T cells naïve or activated for 3, 5, or 7 days were applied to qPCR for IL-2 or INF-γ using actin mRNA as control. Average of three measurements normalized by that of naïve cells is plotted. (P < 0.1 between the cells activated with or without NAM) (C) At day 4 post activation, 1 × 106 cells were suspended in 1 ml of fresh media and further incubated for 24 h (during this, cell numbers increase to 1.22 and 1.59 × 106 respectively). The amount of TNF-α in the conditioned medium was determined in TNF-α ELISA kit. Measurements from three different wells were averaged and fold change compared to the values from naïve cells was plotted.
High [Ca+2]cyt also activates calcineurin (Giannakou and Partridge, 2004), which functions in the activation of nuclear factor of activated T-cells (NFAT) responsible for the expression of cytokine genes necessary for effector function (Giannakou and Partridge, 2004). Therefore, expression of certain cytokine genes is expected to be altered in NAM-treated cells. Indeed, the increase in the levels of IL-2 and IFN-γ mRNA was reduced in NAM-treated cells (Fig. 4B). Meanwhile, the amount of TNF-α secreted in the medium increased 80-fold upon activation, irrespective of NAM presence (Fig. 4C). However, considering the greater number of cells in the culture, this suggests a substantially lower level of TNF-α secretion from individual cells activated in the presence of NAM. These results demonstrate that the cytokines, IL-2, IFN-γ, and TNF-α, are produced at lower levels concurrently with the decrease in [Ca+2]cyt. Therefore, effector function represented by cytokine secretion is compromised by the NAM-mediated acceleration of the population expansion.
DISCUSSION
This study demonstrates that NAM treatment increases the size of the population expansion during CD8+ T cell activation through a decrease in apoptotic cell death. This attenuated cell death is likely partially attributed to the suppression of the activation-induced increase of mitochondrial ROS generation. However, we also observed that greater population expansions are accompanied by compromised expression of key cytokines and, therefore, compromised effector function.
NAM has previously been shown to reduce ROS levels and inhibit ROS-induced apoptosis in many types of adherent cells (Maiese et al., 2009). The mechanisms underlying these antioxidative and anti-apoptotic effects have not been explored in detail. An association with mitochondria quality enhancement has been proposed by our studies, but the mechanisms by which this occurs have not yet been determined. It is likely mediated by the activation of certain key cellular factors. Inside cells, NAM is readily converted to NAD+ (Liu et al., 1982), which modulates the activity of several proteins, including the sirtuin family proteins (Shore, 2000). In particular, SIRT1 positively affects cell survival, either by preventing apoptosis (Lee et al., 2008) or activating autophagy (Effros et al., 2005; Ma et al., 2015).
Our previous studies suggested the possible involvement of SIRT1 in NAM-induced autophagy activation and mitochondria/ROS suppression (Jang et al., 2012; Kang Hwang, 2009). Combined with these previous findings, the lower level of mitochondria accumulation and higher level of autophagy activation in T cells activated in the presence of NAM suggest that the attenuated ROS accumulation may be due to SIRT1-induced mitophagy enhancement. However, SRT1720, a potent activator of SIRT1, was observed to exert a marginal effect on the level of T cell expansion (HJ Choi, unpublished result), suggesting that SIRT1 activation itself may not be sufficient to achieve the observed level of T cell expansion.
SIRT3, a mitochondrial member of the sirtuin family, plays a potent antioxidative role by activating SOD2 and reducing glutathione (Qiu et al., 2010; Someya et al., 2010). Furthermore, SIRT3 promotes efficient mitochondrial ATP production (Ahn et al., 2008; Hirschey et al., 2010), which is required to occur in the NAM-treated cells. Whether SIRT3 activation promotes T cell expansion remains to be determined. Our results also suggest that events other than ROS suppression may be attributed to Bcl-2 upregulation. Enhanced Akt activation is a good candidate. Akt activation has been proposed to be induced by NAM (Chong et al., 2005).
One important message from our study is that the increase in the T cell population is achieved in the absence of additional cell division. This may have practical considerations. Potentiation of T cell activation, which is accompanied by an increase in functional effector cells, would help the body eliminate infected cells at faster rates and allow quicker infection clearance. Moreover, immunosenescence, a major cause of mortality in the elderly, is triggered by the limited proliferation capacity of T cells, which, in turn, is caused by replicative senescence imposed by telomere shortening (Effros et al., 2005). Therefore, augmenting T cell expansion without extra cell divisions may be a useful strategy in alleviating declining immune functions.
However, the reduced cytokine expression suggests that the cell population expanded in the presence of NAM may suffer from a reduced potency in effector function. Therefore, NAM alone may not directly be used to augment CD8+ T cell responses in infection-induced immunopathology. Because of complicated interactions between different immune cells and because of nutrient and cytokine differences, the in vitro response may differ from that in vitro. Still, the results validly suggest that a higher level T cell expansion without extra cell divisions can be achieved by altering the cellular level of NAD+.
Acknowledgments
This study was supported by a grant of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Korea (A121601). The authors declare that their experiments complied with the current laws of the Republic of Korea, inclusive of ethics approval.
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Ahn B.-H., Kim H.-S., Song S., Lee I.H., Liu J., Vassilopoulos A., Deng C.-X., Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA. 2008;105:14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beiqing L., Chen M., Whisler R.L. Sublethal levels of oxidative stress stimulate transcriptional activation of c-jun and suppress IL-2 promoter activation in Jurkat T cells. J. Immunol. 1996;157:160–169. [PubMed] [Google Scholar]
- Bell B.D., Leverrier S., Weist B.M., Newton R.H., Arechiga A.F., Luhrs K.A., Morrissette N.S., Walsh C.M. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc. Natl. Acad. Sci. USA. 2008;105:16677–16682. doi: 10.1073/pnas.0808597105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørkøy G., Lamark T., Brech A., Outzen H., Perander M., øvervatn A., Stenmark H., Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan P., Babbage J.W., Burgering B.M.T., Groner B., Reif K., Cantrell D.A. Phosphatidylinositol 3-kinase couples the interleukin-2 receptor to the cell cycle regulator E2F. Immunity. 1997;7:679–689. doi: 10.1016/s1074-7613(00)80388-x. [DOI] [PubMed] [Google Scholar]
- Brenner D., Krammer P.H., Arnold R. Concepts of activated T cell death. Crit. Rev. Oncol. Hematol. 2008;66:52–64. doi: 10.1016/j.critrevonc.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Chong Z.-Z., Lin S.-H., Li F., Maiese K. The sirtuin inhibitor nicotinamide enhances neuronal cell survival during acute anoxic injury through AKT, BAD, PARP, and mitochondrial associated “anti-apoptotic” pathways. Curr. Neurovasc. Res. 2005;2:271–285. doi: 10.2174/156720205774322584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- D’Souza A.D., Parikh N., Kaech S.M., Shadel G.S. Convergence of multiple signaling pathways is required to coordinately up-regulate mtDNA and mitochondrial biogenesis during T cell activation. Mitochondrion. 2007;7:374–385. doi: 10.1016/j.mito.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effros R.B., Dagarag M., Spaulding C., Man J. The role of CD8 T-cell replicative senescence in human aging. Immunol. Rev. 2005;205:147–157. doi: 10.1111/j.0105-2896.2005.00259.x. [DOI] [PubMed] [Google Scholar]
- Giannakou M.E., Partridge L. The interaction between FOXO and SIRT1: Tipping the balance towards survival. Trends Cell Biol. 2004;14:408–412. doi: 10.1016/j.tcb.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Gomes L.C., Scorrano L. Mitochondrial morphology in mitophagy and macroautophagy. Biochim. Biophys. Acta. 2013;1833:205–212. doi: 10.1016/j.bbamcr.2012.02.012. [DOI] [PubMed] [Google Scholar]
- Grayson J.M., Laniewski N.G., Lanier J.G., Ahmed R. Mitochondrial potential and reactive oxygen intermediates in antigen-specific CD8+ T cells during viral infection. J. Immunol. 2003;170:4745–4751. doi: 10.4049/jimmunol.170.9.4745. [DOI] [PubMed] [Google Scholar]
- Hildeman D.A., Mitchell T., Teague T.K., Henson P., Day B.J., Kappler J., Marrack P.C. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity. 1999;10:735–744. doi: 10.1016/s1074-7613(00)80072-2. [DOI] [PubMed] [Google Scholar]
- Hildeman D.A., Zhu Y., Mitchell T.C., Bouillet P., Strasser A., Kappler J., Marrack P. Activated T cell death in vivo mediated by proapoptotic Bcl-2 family member Bim. Immunity. 2002;16:759–767. doi: 10.1016/s1074-7613(02)00322-9. [DOI] [PubMed] [Google Scholar]
- Hildeman D.A., Mitchell T., Aronow B., Wojciechowski S., Kappler J., Marrack P. Control of Bcl-2 expression by reactive oxygen species. Proc. Natl. Acad. Sci. USA. 2003a;100:15035–15040. doi: 10.1073/pnas.1936213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildeman D.A., Mitchell T., Kappler J., Marrack P. T cell apoptosis and reactive oxygen species. J. Clin. Invest. 2003b;111:575–581. doi: 10.1172/JCI18007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey M.D., Shimazu T., Goetzman E., Jing E., Schwer B., Lombard D.B., Grueter C.A., Harris C., Biddinger S., Ilkayeva O.R., et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan P.G., Hogan P.G., Chen L., Chen L. Transcriptional regulation by calcium, calcineurin. NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- Hoyos B., Ballard D.W., Bohnlein E., Siekevitz M., Greene W.C. Kappa B-specific DNA binding proteins: role in the regulation of human interleukin-2 gene expression. Science. 1989;244:457–460. doi: 10.1126/science.2497518. [DOI] [PubMed] [Google Scholar]
- Jambrina E., Alonso R., Alcalde M., Rodríguez M. del C., Serrano A., Martínez-A. C., García-Sancho J., Izquierdo M. Calcium influx through receptor-operated channel induces mitochondria-triggered paraptotic cell death. J. Biol. Chem. 2003;278:14134–14145. doi: 10.1074/jbc.M211388200. [DOI] [PubMed] [Google Scholar]
- Jang S.Y., Kang H.T., Hwang E.S. Nicotinamide-induced mitophagy: Event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J. Biol. Chem. 2012;287:19304–19314. doi: 10.1074/jbc.M112.363747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski M., Kiessling M., Süss D., Krammer P.H., Gülow K. Novel role for mitochondria: protein kinase Ctheta-dependent oxidative signaling organelles in activation-induced T-cell death. Mol. Cell. Biol. 2007;27:3625–3639. doi: 10.1128/MCB.02295-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H.T., Hwang E.S. Nicotinamide enhances mitochondria quality through autophagy activation in human cells. Aging Cell. 2009;8:426–438. doi: 10.1111/j.1474-9726.2009.00487.x. [DOI] [PubMed] [Google Scholar]
- Kang H.T., Lee H.I., Hwang E.S. Nicotinamide extends replicative lifespan of human cells. Aging Cell. 2006;5:423–436. doi: 10.1111/j.1474-9726.2006.00234.x. [DOI] [PubMed] [Google Scholar]
- Klionsky D.J., Elazar Z., Seglen P.O., Rubinsztein D.C. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy. 2008;4:849–850. doi: 10.4161/auto.6845. [DOI] [PubMed] [Google Scholar]
- Kovacs J.R., Li C., Yang Q., Li G., Garcia I.G., Ju S., Roodman D.G., Windle J.J., Zhang X., Lu B. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ. 2012;19:144–152. doi: 10.1038/cdd.2011.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak J.Y., Ham H.J., Kim C.M., Hwang E.S. Nicotinamide exerts antioxidative effects on senescent cells. Mol. Cells. 2015;38:229–235. doi: 10.14348/molcells.2015.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I.H., Cao L., Mostoslavsky R., Lombard D.B., Liu J., Bruns N.E., Tsokos M., Alt F.W., Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G., Foster J., Manlapaz Ramos P., Olivera B.M. Nucleoside salvage pathway for NAD biosynthesis in Salmonella typhimurium. J. Bacteriol. 1982;152:1111–1116. doi: 10.1128/jb.152.3.1111-1116.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons A.B., Parish C.R. Determination of lymphocyte division by flow cytometry. J. Immunol. Methods. 1994;171:131–137. doi: 10.1016/0022-1759(94)90236-4. [DOI] [PubMed] [Google Scholar]
- Ma Y., Nie H., Chen H., Li J., Hong Y., Wang B., Wang C., Zhang J., Cao W., Zhang M., et al. NAD+/NADH metabolism and NAD+-dependent enzymes in cell death and ischemic brain injury: current advances and therapeutic implications. Curr. Med. Chem. 2015;22:1239–1247. doi: 10.2174/0929867322666150209154420. [DOI] [PubMed] [Google Scholar]
- Maiese K., Chong Z.Z., Hou J., Shang C. The vitamin nicotinamide: Translating nutrition into clinical care. Molecules. 2009;14:3446–3485. doi: 10.3390/molecules14093446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marte B.M., Downward J. PKB/Akt: Connecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biochem. Sci. 1997;22:355–358. doi: 10.1016/s0968-0004(97)01097-9. [DOI] [PubMed] [Google Scholar]
- Meuer S.C., Hussey R.E., Cantrell D.A., Hodgdon J.C., Schlossman S.F., Smith K.A., Reinherz E.L. Triggering of the T3-Ti antigen-receptor complex results in clonal T-cell proliferation through an interleukin 2-dependent autocrine pathway. Proc. Natl. Acad. Sci. USA. 1984;81:1509–1513. doi: 10.1073/pnas.81.5.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X., Brown K., Hirschey M.D., Verdin E., Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Russell J.H. Activation-induced death of mature T cells in the regulation of immune responses. Curr. Opin. Immunol. 1995;7:382–388. doi: 10.1016/0952-7915(95)80114-6. [DOI] [PubMed] [Google Scholar]
- Shore D. The Sir2 protein family: A novel deacetylase for gene silencing and more. Proc. Natl. Acad. Sci. USA. 2000;97:14030–14032. doi: 10.1073/pnas.011506198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya S., Yu W., Hallows W.C., Xu J., Vann J.M., Leeuwenburgh C., Tanokura M., Denu J.M., Prolla T.A. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under Caloric Restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranges P.B., Watson J., Cooper C.J., Choisy-Rossi C.M., Stonebraker A.C., Beighton R.A., Hartig H., Sundberg J.P., Servick S., Kaufmann G., et al. Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity. 2007;26:629–641. doi: 10.1016/j.immuni.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.