

Abstract
The direct interaction of drugs with the cell membrane is often neglected when drug effects are studied. Systematic investigations are hindered by the complexity of the natural membrane and model membrane systems can offer a useful alternative. Here some examples are reviewed of how model membrane architectures including vesicles, Langmuir monolayers and solid supported membranes can be used to investigate the effects of drug molecules on the membrane structure, and how these interactions can translate into effects on embedded membrane proteins.
Keywords: Model membranes, Drugs, Drug–membrane interaction, Review
1. Introduction
The majority of drugs are designed to target membrane proteins, since most diseases are related to the malfunction of these proteins (Yildirim et al., 2007). For example, drugs have been designed to block channel activities or inhibit protein binding (Cohen, 2002). Whilst drug–protein interactions have been systematically studied, the interactions of drugs with the membrane surrounding the proteins are often neglected. Membrane proteins require an adequate membrane as a surrounding support to ensure their structural and functional integrity. The natural membrane is a complex system, composed of a wide variety of different constituents such as lipids, carbohydrates and proteins. The exact membrane composition varies from organism to organism, even though there are some common characteristics for membranes from similar organisms (Escriba et al., 2008, Dowhan, 1997).
Membrane proteins themselves are in most cases rather fragile and unstable and typically denature once extracted from a membrane. Therefore, to fully understand the functional properties of a membrane protein, it has to be studied being embedded in a lipid bilayer membrane. The natural cell membrane, however, is a very complex and highly diverse system, comprising a large variety of different lipids, sterols and carbohydrates, yet the composition of the membrane and its structure play an important role in the functioning of the embedded membrane proteins. For example, changes in the membrane curvature can lead to an opening or closing of mechanosensitive membrane channels (Perozo et al., 2002).
Despite the importance of the membrane, the influence of drugs on its structure and function is often neglected in drug related studies. Similarly, how drug-induced changes of the membrane properties influence the function of embedded membrane proteins is rarely investigated. This is partially due to the high complexity of the membrane, which renders systematic investigations very challenging. Additionally, experiments using whole cells or natural cell membrane patches are often time- and cost-intensive and mostly not suitable for routine screening. Finally, non-specific drug–membrane interactions, where the drug binds to the membrane, effectively reduce the available free drug, and thus make the treatment potentially less efficient (McLure et al., 2000, Nagar and Korzekwa, 2012, Smith et al., 2010). Thus it is clear that probing the membrane role in drug interactions is critical to a complete understanding.
Biomimetic model membrane systems offer an alternative platform to the natural membrane and enable the study of membrane–drug interactions under very defined and controlled conditions. The underlying structure of any membrane is a lipid bilayer. Different model systems have been developed to mimic the fundamental structural and functional properties of this bilayer. Very prominent examples for model membrane systems are vesicles or liposomes, Langmuir monolayers, solid supported bilayers and tethered bilayer lipid membranes. All of these systems offer certain advantages and disadvantages for the study of drug–membrane interactions. However, all of these architectures have been extensively characterised and analysed using a variety of biophysical techniques. The intrinsic properties of these model systems are well studied. No model system will mimic all properties of a natural membrane, however, it has been shown that specific characteristics of a membrane can be simulated very accurately using model systems. This offers the possibility for systematic investigation into membrane-related processes. At the same time it is important that the results obtained from model systems are correlated and validated with findings in natural systems.
Here, some model membrane systems will be introduced with the specific emphasis on their use in drug–membrane interaction studies (see Fig. 1).
Figure 1.
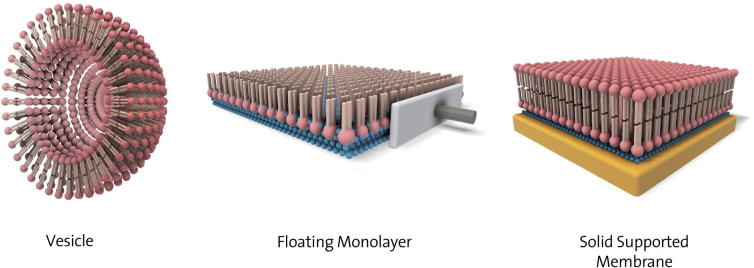
Schematic of different model membrane architectures: Vesicles, Langmuir monolayer and solid supported membranes. Vesicles are spherical assemblies of lipids (headgroups shown as balls, tails as sticks). Floating or Langmuir monolayers are assemblies of lipids at the air/water (blue balls) interface, whilst solid supported membranes are lipids bilayers at a solid support, often separated by a thin water layer.
2. Model membrane systems
Liposomes or vesicles are spherical phospholipid bilayers that can be formed by extrusion of an aqueous lipid dispersion through a membrane with pores of defined size or by sonication of lipid dispersion. Liposomes can be relatively easily prepared as unilamellar or multilamellar structures. The composition of the bilayer can be varied including a wide variety of different lipids and other membrane components (Peetla et al., 2009, Chan and Boxer, 2007, Olson et al., 1979).
Whilst vesicles are easily accessible, the number of techniques that can be used limits studies using vesicles. In principle, two different types of experiments can be performed. Changes in the shape and size of the vesicles due to an external stimulus, e.g. the interaction with a drug, can be monitored using scattering techniques, such as light scattering, small angle X-ray or neutron scattering. These experiments, however, do not give any insight into changes in the functionality of the membrane. Functional properties of the membrane such as the transport of molecules across the bilayer using vesicles can be done by fluorescence studies (Domenech et al., 2009). In such an experiment, a liposome is typically loaded with a fluorescent dye and for example pore formation in the bilayer would lead to the efflux of the dye and thus a change in the measured fluorescence.
Multilamellar vesicles were used to investigate the interaction of the antibacterial compound Rifabutin with membranes of various compositions (Pinheiro et al., 2013). Typically, bacterial membranes contain a higher amount of phosphatidylglycerol headgroups, whereas mammalian cell membranes are dominated by phosphatidylcholine and phosphatidylethanolamine headgroups. In model systems, bacterial membranes are thus often mimicked by using for examples dipalmitoylphosphatidylglycerol (DPPG) lipids, whilst mammalian membranes are represented by dipalmitoylphosphatidylcholine (DPPC).
Rifabutin is a wide spectrum antibiotic, which has an intracellular target and thus has to cross the membrane in order to be active. Analysis of structural changes in the membrane induced by the presence of the drug can lead to indications about the mechanism of action. Small and wide angle X-ray diffraction techniques (SAXS and WAXS) were employed by Pinheiro et al. to show a preferential interaction of Rifabutin with membranes in the gel phase of a mammalian model system, which explains the non-toxic effect of the drug. In contrast, the drug induced pronounced structural changes in a bacterial model system even though the membrane was in a fluid phase, in good agreement with in vivo results (Pinheiro et al., 2013).
In another example of the use of liposomes, the effect of oritavancin, an antibiotic on membrane permeability and lipid organisation was studied (Domenech et al., 2009). Oritavancin is a lipoglycopeptide, a new class of antibiotics derived from glycopeptides that have been synthetically modified to contain a lipophilic side chain. Lipoglycopeptides have shown improved activity against multiple bacterial strains that have become resistant to glycopeptides. The lipophilic side chain is thought to induce a novel mechanism of action accounting for its increased activity possibly via membrane destabilisation.
Liposomes composed of cardiolipin (CL) and POPG (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol), both common in bacterial cell membranes but not in mammalian cellular membranes were studied. Calcein leakage from liposomes was used to measure membrane permeabilisation caused by oritavancin. It was shown that the level of permeabilisation was dependent on the lipid composition and hence the physical properties of the bilayers, specifically the surface charge, lipid packing, propensity to induce negative curvature and fluidity. Liposomes containing CL showed the highest rate and amount of calcein release, followed by POPC (1-Palmitoyl-2-oleoylphosphatidylcholine), POPG and then DPPG containing liposomes.
Liposomes can also be studied by nuclear magnetic resonance (NMR) techniques and circular dichroism. For example, the interaction of a synthetic antimicrobial peptide, P5, with liposomes was studied using these two techniques (Fernandez et al., 2011). P5 was designed based on the structure of two naturally occurring antimicrobial peptides, cecropin A and magainin 2, with additional leucine and lysine substitutions to increase both the hydrophobicity and net charge. This peptide has been shown to be active against a wide range of Gram-positive and Gram-negative bacteria as well as to show anti-fungal and anti-tumour activity whilst not being haemolytic. Both zwitterionic (DMPC, 1,2-dimyristoyl-sn-glycero-3-phosphocholine) and anionic (DMPG, 1,2-dimyristoyl-sn-glycero-3-phosphoglycerol) liposomes were used to mimic the charge of mammalian and bacterial cellular membranes, respectively. P5 was shown to be unstructured in solution as well as in the presence of DMPC liposomes using CD. In the presence of DMPC/DMPG (2:1) liposomes, however, P5 underwent a conformational change to about 40% α-helical structure.
The order and range of motion of the lipid hydrocarbon chains were also investigated using 2H NMR. A small increase in disorder of the hydrocarbon chains close to the head groups was observed upon addition of P5 to the DMPC liposomes. It was suggested that P5 perturbed the periphery of the DMPC liposomes whilst retaining the overall membrane integrity. A larger disorder of the entire hydrocarbon chain region was observed upon addition of P5 to the DMPC/DMPG liposomes indicating the peptide penetrated further into the centre of the bilayer. 31P NMR also confirmed a significant perturbation of the DMPG containing liposomes after P5 exposure, causing an increase in the dynamics of the head groups. The selective interaction of P5 with membranes containing anionic lipids confirmed the selective activity of P5 against bacterial cells.
Another class of frequently studied drugs are cationic amphiphilic drugs (CADs). For example, the effect of two CADs, haloperidol and spiperone on membranes has been investigated by studying liposomes using various techniques (Baciu et al., 1847). Small angle X-ray scattering (SAXS), solid state NMR and fluorescence microscopy (FM) showed rapid partitioning of both CADs into the bilayer, due to their amphiphilic nature, planarity and relatively small size. Disappearance of the fluid lamellar phase was observed by SAXS and NMR after incorporation of the CADs whilst fluorescence and transmission microscopy showed degradation of giant unilamellar vesicles (GUVs). Lipid hydrolysis was thought to occur via the tertiary amine on the CADs (protonated at physiological pH), which could protonate the ester carbonyl moiety of 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) and catalyse ester hydrolysis. The products of DOPC ester hydrolysis, a single chain phospholipid and a fatty acid were observed using magic angle spinning NMR. Small fragments of the CAD containing GUVs were seen using FM following GUV degradation. The authors have postulated that these are micelles formed from the single chain phospholipids, which retain and transport the CAD to a neighbouring cellular membrane on either side of the degraded membrane. The formation of an inverse hexagonal phase resulting from the oleic acid enriched membrane was observed after 3 weeks using SAXS and NMR.
Other common model systems are supported bilayer membranes, where a lipid bilayer is placed on a solid support (Castellana and Cremer, 2006). Such architectures have the advantage that the solid support offers stability to the bilayer, making more time-consuming analytical approaches possible. Also, the solid supports allows for the use of a wide variety of surface analytical techniques. Typical supports are ‘flat’ substrates such as mica or silicon, (Sackmann, 1996) and gold electrodes, (Naumann et al., 2003, Vogel et al., 2012) which allow for additional electrochemical characterisation of the membrane and possible binding processes at the membrane interface.
Supported bilayers allow, for example, analysis of the binding kinetics of a drug using surface plasmon resonance techniques. This information can give deeper insight into the interaction mechanism and can lead to the targeted development of new drugs with optimised properties. The use of model systems in general allow more systematic investigations of the drug–membrane interaction or the screening of certain parameters, whereas in natural systems, such investigations are often complicated by a wide variety of components.
Mica and silicon substrates allow for high-resolution imaging of the membrane itself and associated proteins, for example by using atomic force microscopy (AFM). This has been used to investigate changes in the nanomechanical properties of the membrane upon exposure to a drug. For example, it has been shown that ethanol significantly changes membrane properties such as thickness, Young’s modulus and bending stiffness (Stetter and Hugel, 2013).
The AFM was also used to probe the influence of chlorpromazine hydrochloride (CPZ) with supported phospholipid bilayers (Nussio et al., 2008). Addition of the drug to a pre-formed DMPC bilayer on a solid support led to changes in the molecular organisation of the membrane, indicating that the drug modifies the melting temperature of the phospholipid as well as the phase behaviour. These results were in good agreement with SPR data, which also indicated a modification of the melting temperature due to membrane–drug interactions (Nussio et al., 2007).
Solid supported membranes can also be deposited on conductive substrates, making electrical characterisation of the membrane possible allowing determination of the resistive and capacitive characteristics of the membrane. Changes in these parameters due to the influence of a drug can give indications about the type of interaction occurring. For example, electrochemical impedance spectroscopy has been used to determine how different antimicrobial peptides interact with a lipid bilayer and allowed for a differentiation between a pore-forming and a non pore-forming interaction mechanism (Chang et al., 2008).
Langmuir monolayers are another useful biophysical model system to study certain aspects of membranes (Junghans et al., 2010). They are formed by spreading a lipid on an air/water interface; the packing density of the lipids can then be varied and the surface pressure is measured. The surface pressure gives a direct measure of the status of the lipid layer and interactions of drug molecules with this layer can be detected as changes in the surface pressure. Additionally, visualisation of the layer is possible, either by optical, fluorescence or Brewster angle microscopy. Although these monolayers represent only one half of a bilayer, they can be used to study biophysical interactions between lipids and drugs (Peetla et al., 2010, Peetla and Labhasetwar, 2008, Peetla and Labhasetwar, 2009).
Langmuir monolayers have the advantage over other model systems, in that it is comparably much easier to control the composition and density of the lipid layer. For example, different amounts of cholesterol or lipids with different head groups can be assembled into monolayers. Also, multi-component mixtures are relatively easy to achieve. Screening experiments that probe the influence of different lipids in the membrane on the effect a drug molecule has on the membrane structure are therefore easily feasible.
Peetla et al. showed that lipid extracts from drug resistant and drug sensitive cells showed distinctively different pressure-area isotherms, indicating different biophysical properties of the membranes. Lipids extracted from membranes resistant to the anti-cancer drug doxorubicin not only showed much more rigid phase behaviour than lipids extracted from drug sensitive cells but they also showed much more pronounced interactions with the drug, indicating that the resistance mechanism is most probably due to the drug interacting with the membrane. In vivo, this would lead to the drug being trapped inside the lipid bilayer, thus effectively reducing the concentration of the free drug. Similar results have been obtained for the drug moxifloxacin, which shows stronger hydrophobic interactions with Langmuir monolayers than the similar drug ciprofloxacin. In vivo, this corresponds to a higher cellular accumulation of moxifloxacin (Michot et al., 2005).
Ambike et al. used Langmuir monolayers to probe the influence of the membrane composition on drug–membrane interactions (Ambike et al., 2011). The addition of cholesterol to a lipid layer enhanced the interactions with the anti-cancer drug Gemcitabine, which in vivo shows preferential binding to cholesterol-rich membranes.
The power of model membrane systems can be shown, when multiple model architectures are used to study drug–membrane interactions of the same drug. In a comprehensive study, Nunes et al. have used liposomes, Langmuir monolayers and solid supported membranes to investigate the influence of nonsteroidal anti-inflammatory drugs on the structure of a DPPC membrane (Nunes et al., 2011). Again, the lipid was chosen to represent the most abundant headgroups in natural membranes. Using the model systems offers the advantage of being able to control the pH of the membrane surrounding medium, thus simulating en environment representing physiological conditions (pH 7.4) and inflamed tissue (pH 5). By measuring changes in the phase transition temperature in liposomes due to the presence of a drug, the authors found indications of a preferential headgroup interaction at pH 7.4 in contrast to interactions with the hydrophobic membrane core at pH 5. Results from these rather indirect measurements have been correlated with changes in Langmuir isotherms of the same lipid due to the presence of the drugs and similar effects have been observed. Additionally, Langmuir monolayers can also be characterised using complementary techniques such as fluorescence microscopy, Brewster angle microscopy or infrared reflection-adsorption spectroscopy. The latter technique has been used to confirm the preferential drug–headgroup interactions at pH 7.4 and the larger change in the membrane inner structure at pH 5. Finally, changes to the membrane structure have been visualised in solid supported membranes using the Atomic Force Microscope. The stronger effect at pH 5 could also be correlated to the tendency of gastritic toxicity of some of these anti-inflammatory drugs.
A special class of membrane proteins are ion channels. They allow for the selective passage of ions across the lipid bilayer membrane and malfunction of these proteins is often the cause for a disease. Functional studies of these proteins are however difficult, since they require sensitive electronic equipment, such as a patch-clamp amplifier when investigations are to be made at a single molecule level. Patch-clamp experiments using whole cell are also labour intensive and complicated. Again, model membrane systems can provide a useful alternative. Ensemble measurements of protein function can be performed for example using supported membranes or tethered bilayer membranes (Köper, 2007, Braunagel et al., 2011). Additionally, planar lipid bilayer systems or black lipid membranes (BLM) allow for single molecule measurements in a controlled environment. Such a BLM system has been used to investigate the effect of tubulin-binding chemotherapeutic drugs on the lipid membrane (Ashrafuzzaman et al., 2012). By combining experimental and theoretical studies, it has been shown that thiocolchicoside and taxol can induce pore formation in lipid membranes, which can add to the cytotoxic effects of these drugs.
3. Summary
The effect of drugs on the structure and function of cell membranes is an important part of the overall effectiveness of a drug. These effects can be studied systematically using model membrane systems. Vesicles, Langmuir monolayer or solid supported membranes provide the advantage of a system with reduced complexity, access to a wide variety of characterisation techniques and control over the individual constituents. Whilst model membranes will never be able to entirely replace studies using whole cells, they can provide a useful first screening platform for the investigation of drug–membrane interactions.
Footnotes
Peer review under responsibility of King Saud University.
References
- Ambike A. Interaction of self-assembled squalenoyl gemcitabine nanoparticles with phospholipid-cholesterol monolayers mimicking a biomembrane. Langmuir. 2011;27(8):4891–4899. doi: 10.1021/la200002d. [DOI] [PubMed] [Google Scholar]
- Ashrafuzzaman M. Chemotherapy drugs form ion pores in membranes due to physical interactions with lipids. Chem. Biol. Drug. Des. 2012;80(6):992–1002. doi: 10.1111/cbdd.12060. [DOI] [PubMed] [Google Scholar]
- Baciu M. Degradative transport of cationic amphiphilic drugs across phospholipid bilayers. Philos. Trans. R. Soc. A: Math. Phys. Eng. Sci. 1847;2006(364):2597–2614. doi: 10.1098/rsta.2006.1842. [DOI] [PubMed] [Google Scholar]
- Braunagel J., Junghans A., Köper I. Membrane-based sensing approaches. Aust. J. Chem. 2011;64(1):54–61. [Google Scholar]
- Castellana E.T., Cremer P.S. Solid supported lipid bilayers: from biophysical studies to sensor design. Surf. Sci. Rep. 2006;61(10):429–444. doi: 10.1016/j.surfrep.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan Y.H.M., Boxer S.G. Model membrane systems and their applications. Curr. Opin. Chem. Biol. 2007;11:581–587. doi: 10.1016/j.cbpa.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W.K. Characterization of antimicrobial peptide activity by electrochemical impedance spectroscopy. Biochim. Biophys. Acta. 2008;1778(10):2430–2436. doi: 10.1016/j.bbamem.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P. Protein kinases – the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002;1(4):309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- Domenech O. Interactions of oritavancin, a new lipoglycopeptide derived from vancomycin, with phospholipid bilayers: effect on membrane permeability and nanoscale lipid membrane organization. Biochim. Biophys. Acta. 2009;1788(9):1832–1840. doi: 10.1016/j.bbamem.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Dowhan W. Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu. Rev. Biochem. 1997;66:199–232. doi: 10.1146/annurev.biochem.66.1.199. [DOI] [PubMed] [Google Scholar]
- Escriba P.V. Membranes: a meeting point for lipids, proteins and therapies. J. Cell. Mol. Med. 2008;12(3):829–875. doi: 10.1111/j.1582-4934.2008.00281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez D.I. Interactions of a synthetic Leu-Lys-rich antimicrobial peptide with phospholipid bilayers. Eur. Biophys. J. 2011;40(4):471–480. doi: 10.1007/s00249-010-0660-5. [DOI] [PubMed] [Google Scholar]
- Junghans A. Protein–lipid interactions at the air–water interface. Langmuir. 2010;26(14):12049–12053. doi: 10.1021/la100036v. [DOI] [PubMed] [Google Scholar]
- Köper I. Insulating tethered bilayer lipid membranes to study membrane proteins. Mol. Biosyst. 2007:651–657. doi: 10.1039/b707168j. [DOI] [PubMed] [Google Scholar]
- McLure J.A., Miners J.O., Birkett D.J. Nonspecific binding of drugs to human liver microsomes. Br. J. Clin. Pharmacol. 2000;49(5):453–461. doi: 10.1046/j.1365-2125.2000.00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michot J.M. Influence of efflux transporters on the accumulation and efflux of four quinolones (ciprofloxacin, levofloxacin, garenoxacin, and moxifloxacin) in J774 macrophages. Antimicrob. Agents Chemother. 2005;49(6):2429–2437. doi: 10.1128/AAC.49.6.2429-2437.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagar S., Korzekwa K. Commentary: nonspecific protein binding versus membrane partitioning: it is not just semantics. Drug Metab. Dispos. 2012;40(9):1649–1652. doi: 10.1124/dmd.112.046599. [DOI] [PubMed] [Google Scholar]
- Naumann R. Tethered lipid bilayers on ultraflat gold surfaces. Langmuir. 2003;19:5435–5443. [Google Scholar]
- Nunes C. NSAIDs interactions with membranes: a biophysical approach. Langmuir. 2011;27(17):10847–10858. doi: 10.1021/la201600y. [DOI] [PubMed] [Google Scholar]
- Nussio M.R. Characterisation of the binding of cationic amphiphilic drugs to phospholipid bilayers using surface plasmon resonance. ChemMedChem. 2007;2(3):366–373. doi: 10.1002/cmdc.200600252. [DOI] [PubMed] [Google Scholar]
- Nussio M.R. Kinetics membrane disruption due to drug interactions of chlorpromazine hydrochloride. Langmuir. 2008;25(2):1086–1090. doi: 10.1021/la803288s. [DOI] [PubMed] [Google Scholar]
- Olson F. Preparation of liposomes of defined size distribution by extrusion through polycarbonate membranes. Biochim. Biophys. Acta. 1979;557(1):9–23. doi: 10.1016/0005-2736(79)90085-3. [DOI] [PubMed] [Google Scholar]
- Peetla C., Labhasetwar V. Biophysical characterization of nanoparticle−endothelial model cell membrane interactions. Mol. Pharmaceutics. 2008;5(3):418–429. doi: 10.1021/mp700140a. [DOI] [PubMed] [Google Scholar]
- Peetla C., Labhasetwar V. Effect of molecular structure of cationic surfactants on biophysical interactions of surfactant-modified nanoparticles with a model membrane and cellular uptake. Langmuir. 2009;25(4):2369–2377. doi: 10.1021/la803361y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peetla C., Stine A., Labhasetwar V. Biophysical interactions with model lipid membranes: applications in drug discovery and drug delivery. Mol. Pharmaceutics. 2009;6(5):1264–1276. doi: 10.1021/mp9000662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peetla C. Drug resistance in breast cancer cells: biophysical characterization of and doxorubicin interactions with membrane lipids. Mol. Pharmaceutics. 2010;7(6):2334–2348. doi: 10.1021/mp100308n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo E. Physical principles underlying the transduction of bilayer deformation forces during mechanosensitive channel gating. Nat. Struct. Biol. 2002;9(9):696–703. doi: 10.1038/nsb827. [DOI] [PubMed] [Google Scholar]
- Pinheiro M. The influence of Rifabutin on human and bacterial membrane models: implications for its mechanism of action. J. Phys. Chem. B. 2013;117(20):6187–6193. doi: 10.1021/jp403073v. [DOI] [PubMed] [Google Scholar]
- Sackmann E. Supported membranes: scientific and practical applications. Science. 1996;271:43–48. doi: 10.1126/science.271.5245.43. [DOI] [PubMed] [Google Scholar]
- Smith D.A., Di L., Kerns E.H. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat. Rev. Drug Discov. 2010;9(12):929–939. doi: 10.1038/nrd3287. [DOI] [PubMed] [Google Scholar]
- Stetter F.W., Hugel T. The nanomechanical properties of lipid membranes are significantly influenced by the presence of ethanol. Biophys. J. 2013;104(5):1049–1055. doi: 10.1016/j.bpj.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel N., Zieleniecki J., Koper I. As flat as it gets: ultrasmooth surfaces from template-stripping procedures. Nanoscale. 2012;4(13):3820–3832. doi: 10.1039/c2nr30434a. [DOI] [PubMed] [Google Scholar]
- Yildirim M.A. Drug–target network. Nat. Biotechnol. 2007;25(10):1119–1126. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]