

Traumatic spinal cord injury (SCI) causes motor paralysis, sensory anesthesia and autonomic dysfunction below the lesion site and additionally some SCI patients refer neuropathic pain together with these signs and symptoms. Clinical and experimental studies have revealed the main pathological changes of injured spinal cord implicated in all these signs and symptoms, including neuropathic pain. After few hours of traumatic SCI, it is usual to observe broken blood brain barrier with plasma and blood cells extravasation, cell necrosis, disruption of ascending and descending spinal cord pathways and increased potassium and glutamate. Glutamate contributes to excitotoxicity of neurons whereas potassium facilitates ectopic depolarization of survival neurons and activation of resident microglia. Reactive microglia cells are able to secrete several pro-inflammatory cytokines (e.g., tumor necrosis factor-alpha (TNF-alpha), interleukin-1 (IL-1), IL-6) and chemokines (C-C motif) ligand 2 (CCL2) or monocyte chemoattractant protein 1 (MCP1) that contribute to the reactivation and migration of more microglial cells located far to lesion site, and also astrocytes that contribute to the secretion of more pro-inflammatory agents. Chemokine attracts blood cells, including neutrophils, lymphocytes and monocytes that infiltrate on injured spinal cord parenchyma, and contribute to eliminate the cellular debris, but also secrete more pro-inflammatory agents. All these cellular and biochemical changes were observed during the first weeks post-injury. Finally, reactive astrocytes and microglial cells form the glial scar around the lesion site, and astrocytes secrete several proteoglycan that inhibit the re-growth of regenerated central axons across the lesion site. Apoptosis of oligodendrocytes, and wallerian degeneration of nude axons also were seen. The associated myelin proteins (e.g., NOGO, OMpG, MAG, LINGO) that appeared in the injured spinal cord parenchyma also contribute to inhibit the regeneration of central axons. In summary, disruption of spinal cord pathways, persistent pro-inflammatory environment, necrosis and apoptosis of neurons, glia and endothelial cells, and inhibitory environment to axonal regeneration are the main changes observed in injured spinal cord (Silva et al., 2014) (Figure 1A and B).
Figure 1.
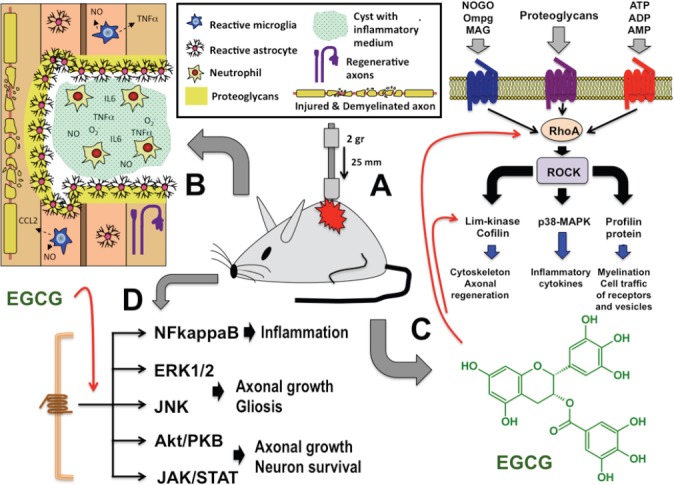
Effect of epigallocatechin-3-gallate (EGCG) after spinal cord injury (SCI).
Spinal cord contusion by means of weight drop technique (A) causes an insult that produces disruption of ascending and descending spinal cord pathways, demyelination of these spinal pathways, and a core of injured tissue surrounded by a glial scare that includes reactive astrocytes and microglial cells and several inhibitory proteoglycans. This core is formed by infiltrated neutrophils that secreted several reactive oxygen species and pro-inflammatory mediators. In addition, reactive glial cells also secrete these pro-inflammatory factors (B). Some of these factors that appeared in injured spinal cord are able to stimulate RhoA-ROCK intracellular pathway, which in turn, activates downstream effectors implicated in axonal regeneration, synthesis and secretion of inflammatory cytokines and modulation of cell traffic vesicles and neurotransmission receptors (C). EGCG treatment is able to reduce the expression of RhoA in the injured spinal cord, but also modulates other intracellular pathways including NFkappaB, ERK1/2, JNK, Akt/PKB and JAK/STAT (D). All these cell signallings are implicated in the promotion of neuroprotection and functional recovery of injured nervous system (for details see text). NFkappaB: Transcription factor nuclear factor-κB; ERK1/2: extracellular signal-related kinases 1 and 2; JNKs: JUN amino-terminal kinases; Akt/PKB: serine/threonine kinase Akt/protein kinase B; JAK/STAT: Janus kinase/signal transducer and activator of transcription.
Of interest is that the majority of biochemical changes that appeared in injured spinal cord parenchyma also were implicated in the development of neuropathic pain. It is well reported that pro-inflammatory mediators (e.g., TNF-alpha, IL-1, IL-6) and glutamate are able to depolarize nociceptive dorsal horn neurons. Glutamate also causes central sensitization of these neurons. Neurotrophins (e.g., nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), glial cell derived neurotrophic factor (GDNF)) released by reactive glial cells contribute to the generation of neuropathic pain by increasing excitatory and reducing inhibitory transmission, as well as enhancing descending facilitation in the dorsal horn (Vranken 2012).
For promoting neuroprotection after SCI, a variety of promising drugs have been tested in animal models, but few have had potential application to SCI patients, including anti-apoptotic agents (e.g., erythropoietin, caspase inhibitors, inhibitor to the p38 mitogen activated protein kinase), nonsteroidal anti-inflammatory drugs (e.g., indomethacin, ibuprofen), antibodies against integrin (e.g., CD11/CD18) and CD95 ligand activity, microglia modulators (e.g., minocycline, glibenclamide), neurotransmitter-receptor antagonists (e.g., N-methyl-D-aspartate (NMDA), purinergic receptors), ion channel antagonists (e.g., nipodipine, riluzol), glucocorticoids (e.g., methylprednisolone), statins (e.g., atorvastatin), gangliosides (e.g., GM1), cyclooxygenase inhibitors, steroids, neuroimmunophilin ligand (e.g., FK506 or tacrolimus), anti-inflammatory cytokines (e.g., IL-10). Pharmacological treatment was also tested in animal models for promoting axonal regeneration such as antibodies against myelin inhibitors (e.g., NOGO, OmpG, MAG), neutralization of inhibitory proteoglycans (e.g., chondroitinase-ABC), biomaterials to improve axonal regeneration (e.g., collagen, neurogel, matrigel, fibronectin, carbon fibers, nitrocellulose), growth factors (e.g., NGF, neurotrophin 3 (NT3), BDNF, GDNF, insulin-like growth factor 1 (IGF-1), FGF-2), and Rho inhibitors (e.g., C3-transferase) (Tohda and Kuboyama 2011; Silva et al., 2014). In addition, some of these pro-regenerative and neuroprotective therapies were also used for alleviating neuropathic pain after SCI (Vranken 2012).
A promising pharmacological therapy for promoting neuroprotection, axonal regeneration and alleviation of neuropathic pain after SCI is the use of epigallocatechin-3-gallate (EGCG). Green tea (Camellia sinensis) is a complex mixture of compounds including polyphenols, flavonoids, flavonols, and other constituents such as amino acids, organic acids, lipids, vitamins, polysaccharides, and thiamine. Catechins are a type of polyphenol and are the main astringency component in green tea. The chief catechins are (−)-epicatechin (EC), (−)-epicatechin-3-gallate (ECG), (−)-epigallocatechin (EGC), and (−)-EGCG. Approximately 30–45% of the dry weight of green tea contains phenolic compounds and EGCG is one of the most abundant catechins that contain around 50–80% of the total catechins. In animal models of SCI, EGCG promote neuroprotection reducing lipid peroxidation, apoptosis and attenuation of pro-inflammatory cytokines production (Khalatbary et al., 2010; Khalatbary and Ahmadvand 2011). Intravenous infusion of EGCG in acute or chronic phase following SCI in rats promotes locomotor recovery and alleviates neuropathic pain. EGCG treatment of SCI rats results in a significant decrease in the lesion size and gliosis with an increase in the number of spared neurons and extensive arborization and axonal growth (Renno et al., 2014). After SCI, EGCG treatment also reduces thermal hyperalgesia and this effect may be attributable to a decrease of astro- and micro-gliosis, and a down-regulation of pro-inflammatory cytokines such as TNF-alpha, and also RhoA protein (Álvarez-Pérez et al., 2015).
A molecular target down regulated by EGCG treatment is the RhoA protein (Álvarez-Pérez et al., 2015). The family of Rho GTP-ases is intracellular signal transducers that link cell surface signals to multiple intracellular responses. There are 22 mammalian Rho GTPases, which are divided into 8 subclasses based on sequence homology. Rho has three kinds of isomers: RhoA, RhoB and RhoC, RhoA mainly in the nervous system. Rho normally exists in two forms: one is the non-activated form combined with GDP (Rho-GDP), and the other is the activated form combined with GTP (Rho-GTP). Rho signal transduction relies on the activation of downstream effector kinases, such as Rho-associated kinase (ROCK), a member of the AGC family of serine-threonine kinases. Rho-ROCK activation, in turn, activates downstream effectors, including (i) Lim kinase and cofilin, which are responsible for modifying the actin cytoskeleton; (ii) p38-MAPK which is responsible to regulate the expression of pro-inflammatory cytokines (TNF-alpha, IL-6, IL-1), cyclooxygenase-2 and inducible nitric oxide synthase; and (iii) profilin proteins that regulate myelination and cell membrane traffic of glutamate receptors and synaptic vesicles. It is well known that myelin-associated inhibitors, chondroitin sulfate proteoglycans, ATP and ADP are extracellular molecules that are able to activate the Rho-ROCK cascade (Birbach 2008; Forgione and Fehlings 2014), and consequently to cause growth cone collapse, up-expression of pro-inflammatory mediators, over-expression of glutamate receptors and reduction of myelination (Figure 1C). In summary, Rho-ROCK pathway activation after SCI interferes the axonal regeneration, the re-myelination of injured axons, and potentiate an inflammatory environment that enhances neuropathic pain. Of interest is that the use of Rho-ROCK antagonists and/or inhibitors (e.g., C3-transferase, Cethrin, Y-27632) reverses some of these events after injury (Forgione and Fehlings 2014). Consequently, EGCG that reduces the expression RhoA also can modulate the activation of Rho-ROCK pathway, promoting axonal regeneration and re-myelination of injured axons for reducing pro-inflammatory mediators. There are experimental evidences that after injury EGCG treatment blocks the anti-neuritogenic effect of NOGO, enhances the pro-neuritogenic effect of growth factors, accelerates the regeneration, reduces the expression of pro-inflammatory cytokines and oxidative stress, and inhibits pro-apoptotic pathways. All these cellular and biochemical changes are reflected in a higher degree of motor recovery and alleviation of neuropathic pain after injury.
Besides RhoA, other intracellular pathways also are modulated by EGCG such as nuclear factor-kappaB (NF-κB), ERK1/2, AkT, JAK/STAT and JNK (Figure 1D). The transcription factor NF-κB is a ubiquitously expressed dimeric molecule with post-translationally regulated activity. NF-κB is implicated in neuron survival against neuronal apoptosis, and plays an essential role in glial cell activation. Many stimuli trigger NF-κB activation both in astrocytes and microglia, resulting in the production of proinflammatory mediators such as chemokines, cytokines, prostaglandins, nitric oxide (NO) and reactive oxygen species (ROS). Activation of NF-κB in glia can be neuroprotective or promote neuronal death depending on the context, i.e. cell type, stimulus, duration and threshold levels of effectors. NF-κB activation also promotes myelination of oligodendrocytes and NF-κB activation in oligodendrocytes is important in response to stress and injury. On the other hand, the extracellular signal-related kinases 1 and 2 (ERK1/2) have been identified as critically important in mediating the effects of several growth factors that regulate oligodendroglial remyelination after injury. In addition, ERK1/2 cell signaling is also implicated in axonal growth and glia reactivation after injury. The serine/threonine kinase Akt, also known as protein kinase B (PKB), is a central node in cell signaling downstream of growth factors, cytokines, and other cellular stimuli. Akt-mediated phosphorylation of several intracellular proteins and Akt contributes to activation of the various cellular processes including survival, growth, proliferation, glucose uptake, metabolism, and angiogenesis. In the nervous system, Akt-mediated suppression of neuronal cell death occurs via multifarious mechanisms including alterations in gene expression, inhibition of caspase-9 and suppression of cytochrome c release by mitochondria. In addition, Akt also mediates astrocyte reactivity and the Akt-mTOR pathway regulates axonal growth after injury. JAK/STAT pathway is also implicated in the axon regeneration and survival of several neurons, such as dorsal root ganglion neurons, retinal neurons and spinal cord neurons. This cell signaling pathway plays a key role in regulating cytokine-dependent gene expression and cellular survival. Finally, JUN amino-terminal kinases (JNKs) are components of a classical mitogen activated protein kinase (MAPK) signaling cascade that serves to filter noise and allow signal amplification while maintaining upstream kinase complexity, enabling signaling diversity. The DLK/JNK pathway is related to axonal regeneration after injury, and JNK/c-Jun signaling pathway regulates gliosis.
According to the above information, EGCG treatment may modulate several cell signaling pathways implicated in neuronal survival, axonal regeneration and remyelination after injury, and in the modulation of astrocyte and microglia reactivity after central nervous system injuries, for promoting motor recovery and alleviating neuropathic pain after peripheral nerve injury (Xifró et al., 2015) and SCI (Renno et al., 2014; Álvarez-Pérez et al., 2015). Thus, these findings provide a rationale for the preclinical development of novel EGCG-derivatives with higher potency than EGCG for enhancing axonal regeneration, preservation of brain and/or spinal cord parenchyma, and reducing gliosis after peripheral and central nervous system injuries. In fact, our experience suggest that novel EGCG-derivatives showed better functional response for alleviating thermal hyperalgesia after chronic constriction injury (CCI) of sciatic nerve than the natural compound EGCG (Xifró et al., 2015). In addition, our experience also suggests that EGCG treatment alleviates thermal hyperalgesia after SCI (Álvarez-Pérez et al., 2015), and in both experimental models of neuropathic pain (CCI and SCI), EGCG treatment reduces the expression of pro-inflammatory cytokines (e.g., IL-1, IL-6, TNF-alpha) and modulates the expression of a transcription factor (NF-κB) and a small GTPase (RhoA) in the spinal cord (Álvarez-Pérez et al., 2015; Xifró et al., 2015).
As can be checked in “clinicaltrials.gov” database, several studies have been conducted to evaluate the effects of EGCG in the nervous system. Specifically, EGCG has been tested in subjects with multiple sclerosis (NCT01451723, NCT01417312) and Huntington's disease (NCT01357681). Furthermore, EGCG was also tested in healthy young adults for evaluating cerebral blood-flow and brain-electrical activity (NCT00981292). According to this information, the tested doses of EGCG were 200 mg per capsule twice daily (NCT01451723), 600 mg daily intake for 3 months (NCT01417312), and one dosage with 135 or 270 mg of EGCG administered on each of three separate study days (NCT00981292). As discussed in a previous paper (Álvarez-Pérez et al., 2015), in healthy volunteers single oral doses of EGCG up to 1,600 mg are safe and very well tolerated. On the other hand, the dose at which EGCG causes hepatotoxicity is controversial. Goodin et al. (2006) indicated hepatotoxicity at 50 mg/kg (Goodin et al., 2006), and Church et al. (2015) relate these changes at doses of 500–1,500 mg/kg in mice.
In the light of the above, EGCG treatment may be a potential drug to promote neuroprotection and functional recovery after nervous system injury. However, additional experimental research will be necessary to further explore the biological mechanisms of this polyphenol in order to become a suitable and safe therapeutic treatment.
The present work was funded by Accions Singulars de R+D (Sing 12/17) del Vicerectorat de Recerca de la Universitat de Girona, Girona, Spain.
References
- Álvarez-Pérez B, Homs J, Bosch-Mola M, Puig T, Reina F, Verdú E, Boadas-Vaello P. Epigallocatechin-3-gallate treatment reduces thermal hyperalgesia after spinal cord injury by down-regulating RhoA expression in mice. Eur J Pain. 2015 doi: 10.1002/ejp.722. doi: 10.1002/ejp.722. [DOI] [PubMed] [Google Scholar]
- Birbach A. Profilin, a multi-modal regulator of neuronal plasticity. Bioessays. 2008;30:994–1002. doi: 10.1002/bies.20822. [DOI] [PubMed] [Google Scholar]
- Church RJ, Gatti DM, Urban TJ, Long N, Yang X, Shi Q, Eaddy JS, Mosedale M, Ballard S, Churchill GA, Navarro V, Watkins PB Threadgill DW, Harrill AH. Sensitivity to hepatotoxicity due to epigallocatechin gallate is affected by genetic background in diversity outbred mice. Food Chem Toxicol. 2015;76:19–26. doi: 10.1016/j.fct.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgione N, Fehlings MG. Rho-ROCK inhibition in the treatment of spinal cord injury. World Neurosurg. 2014;82:e535–539. doi: 10.1016/j.wneu.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Goodin MG, Bray BJ, Rosengren RJ. Sex- and strain-dependent effects of epigallocatechin gallate (EGCG) and epicatechin gallate (ECG) in the mouse. Food Chem Toxicol. 2006;44:1496–1504. doi: 10.1016/j.fct.2006.04.012. [DOI] [PubMed] [Google Scholar]
- Khalatbary AR, Ahmadvand H. Anti-inflammatory effect of the epigallocatechin gallate following spinal cord trauma in rat. Iran Biomed J. 2011;15:31–37. [PMC free article] [PubMed] [Google Scholar]
- Khalatbary AR, Tiraihi T, Boroujeni MB, Ahmadvand H, Tavafi M, Tamjidipoor A. Effects of epigallocatechin gallate on tissue protection and functional recovery after contusive spinal cord injury in rats. Brain Res. 2010;1306:168–175. doi: 10.1016/j.brainres.2009.09.109. [DOI] [PubMed] [Google Scholar]
- Renno WM, Al-Khaledi G, Mousa A, Karam SM, Abul H, Asfar S. (-)-Epigallocatechin-3-gallate (EGCG) modulates neurological function when intravenously infused in acute and, chronically injured spinal cord of adult rats. Neuropharmacology. 2014;77:100–119. doi: 10.1016/j.neuropharm.2013.09.013. [DOI] [PubMed] [Google Scholar]
- Silva NA, Sousa N, Reis RL, Salgado AJ. From basics to clinical: a comprehensive review on spinal cord injury. Prog Neurobiol. 2014;114:25–57. doi: 10.1016/j.pneurobio.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Tohda C, Kuboyama T. Current and future therapeutic strategies for functional repair of spinal cord injury. Pharmacol Ther. 2011;132:57–71. doi: 10.1016/j.pharmthera.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Vranken JH. Elucidation of pathophysiology and treatment of neuropathic pain. Cent Nerv Syst Agents Med Chem. 2012;12:304–314. doi: 10.2174/187152412803760645. [DOI] [PubMed] [Google Scholar]
- Xifró X, Vidal-Sancho L, Boadas-Vaello P, Turrado C, Alberch J, Puig T, Verdú E. Novel epigallocatechin-3-gallate (EGCG) derivative as a new therapeutic strategy for reducing neuropathic pain after chronic constriction nerve injury in mice. PLoS One. 2015;10:e0123122. doi: 10.1371/journal.pone.0123122. [DOI] [PMC free article] [PubMed] [Google Scholar]