

Abstract
Cdx2 is a homeodomain transcription factor that regulates normal intestinal cell differentiation. Cdx2 is frequently lost during progression of colorectal cancer (CRC) and is widely viewed as a colorectal tumour suppressor. A previous study suggested that activation of protein kinase C (PKC) may be responsible for Cdx2 down-regulation in CRC cells. Here we show that activation of PKC does indeed promote down-regulation of Cdx2 at both the mRNA and protein levels. However, PKC-dependent loss of Cdx2 is dependent upon activation of the Raf–MEK–ERK1/2 pathway. Indeed, specific activation of the ERK1/2 pathway using the conditional kinase ΔRaf-1:ER is sufficient to inhibit Cdx2 transcription. The Raf–MEK–ERK1/2 pathway is hyper-activated in a large fraction of colorectal cancers due to mutations in K-Ras and we show that treatment of CRC cell lines with MEK inhibitors causes an increase in Cdx2 expression. Furthermore, activation of the ERK1/2 pathway promotes the phosphorylation and proteasome-dependent degradation of the Cdx2 protein. The inhibitory effect of ERK1/2 upon Cdx2 in CRC cells is in sharp contrast to its stimulatory effect upon Cdx2 expression in trophectoderm and trophoblast stem cells. These results provide important new insights into the regulation of the Cdx2 tumour suppressor by linking it to ERK1/2, a pathway which is frequently activated in CRC.
Keywords: Cdx2, Colorectal cancer, PKC, Raf-1, ERK, TS cells
1. Introduction
Cdx2 is a homeodomain transcription factor that is important in the differentiation of enterocytes in the adult gut [1], differentiation of trophectoderm in the pre-implantation embryo [2], and A–P patterning of the post-implantation embryo [3]. Different DNA regulatory elements around the Cdx2 gene are required for each of these three functions [4].
Cdx2 expression in the gut has attracted particular attention because many lines of evidence support the view that Cdx2 functions as a tumour suppressor in colorectal cancer (CRC). In CRC, Cdx2 expression is reduced relative to adjacent normal tissue [5,6] and differentiation is poor in tumours that lose Cdx2 [7,8]. Increasing Cdx2 expression in the undifferentiated intestinal cell line IEC-6 induces differentiation [1]. The RKO CRC cell line has normal APC (adenomatous polyposis coli) and β-catenin but an inactivating truncation in Cdx2 [9]. Heterozygous Cdx2+/− mice are hyper-sensitive to chemical mutagenesis, developing invasive intestinal adenocarcinomas [5], and an increased adenomatous polyp burden when crossed with mice heterozygous for an APC gene mutation [10]. However, some recent reports have questioned whether Cdx2 has a typical tumour suppressor role [11,12]. Thus, no reduction in Cdx2 was found in primary tumours relative to surrounding normal tissue except in advanced, undifferentiated tumours [12], and Cdx2 tumour promoter activity has also been reported [11]. It has been suggested that Cdx2 may function as a suppressor of metastasis rather than of tumourigenesis [11].
Despite the frequent down-regulation of Cdx2 in CRC, mutations in the Cdx2 locus are rare. Epigenetic silencing of the Cdx2 gene occurs in some CRC cell lines [13,14] but not in others, even though their Cdx2 expression is repressed [15,16]. Somatic cell hybrid fusions between CRC cells with intact Cdx2 expression and those lacking Cdx2 exhibit reduced Cdx2, suggesting a dominant repression mechanism, such as signalling pathway activation, as an additional factor in Cdx2 repression [15].
Mutational activation of K-Ras is a common and relatively early event in CRC. The K-Ras effector PI3K apparently plays no role in Cdx2 repression [15,17]. Nonetheless, Ras has been implicated in the down-regulation of Cdx2. Expression of mutant H-Ras in Caco-2 cells resulted in increased protein kinase C (PKC) and extracellular signal-regulated kinases (ERK) 1/2 activity and enhanced tumourigenicity [18,19] and subsequent studies have shown that H-Ras12V-induced down-regulation of Cdx2 in Caco-2 and HT29 CRC cells was dependent upon PKC but not ERK1/2 [17].
Several aspects of this study seem at odds with our current understanding of Ras-dependent signalling. For example, it is difficult to interpret the effects of expression of H-Ras12V when it is K-Ras and not H-Ras that is mutated in CRC [20], especially as the Ras proteins may differ in their coupling to various downstream effectors [21,22]. Furthermore, the suggestion that PKC acts downstream of Ras should not ignore the fact that PKC can also activate the ERK1/2 pathway in many cell types, either by activation of Raf or Ras [23,24]. Prompted by these issues we have re-examined the regulation of Cdx2 expression in CRC cells and find that PKC-dependent Cdx2 down-regulation does require the ERK1/2 pathway. Indeed, selective activation of the ERK1/2 pathway is sufficient to inhibit Cdx2 transcription whereas inhibition of the ERK1/2 pathway increases Cdx2 expression.
2. Materials and methods
2.1. Cell culture and transfection
HEK293, LS174T and SW48 cells were purchased from ATCC and maintained in Dulbecco’s modified Eagles medium (DMEM) supplemented with 2 mM glutamine, 100 mg/ml penicillin-streptomycin and 10% FBS. HEK293 cells expressing ΔRaf-1:ER* (HR1 cells; [25]), DKO4 [26] and DKO4 ΔRaf-1:ER cells [27] were maintained in phenol red-free DMEM.
Trophoblast stem cells (TS cells) were grown in standard conditions (20% FBS, 1 mM Na-pyruvate, 50 U/ml penicillin, 50 μg/ml streptomycin, 50 μM 2-mercaptoethanol, 25 ng/ml bFGF and 1 μg/ml heparin in RPMI1640 (Invitrogen), with 70% of the medium preconditioned on embryonic feeder cells [28]).
For luciferase reporter assays cells were transfected using Lipofectamine 2000 (Invitrogen) or Superfect (Qiagen) according to the manufacturers’ instructions. Following transfection cells were incubated for 4 h at 37 °C and 5% CO2 before the transfection mix was removed and replaced with complete medium containing drugs as indicated. Cells were then incubated overnight at 37 °C with 5% CO2 and cell extracts were prepared and processed for firefly and Renilla luciferase activity according to the Dual Luciferase Assay System manual (Promega).
2.2. Materials
The DUSP6 expression plasmid was provided by Prof Stephen Keyse, Biomedical Research Centre, Ninewells Hospital, Dundee. AZD6244 (ARRY-142886) was provided by Dr Paul Smith, AstraZeneca. PD184352 was obtained by custom synthesis. U0126 was purchased from Promega. All other chemicals were from Sigma (highest grade available) and used at the following final concentrations: 4-hydroxytamoxifen (4-HT): 100 nM; phorbol 12-myristate 13-acetate (PMA): 2.5 μM; MEK inhibitors U0126: 10 μM, PD184352: 1 μM, AZD6244: 1 μM; proteasome inhibitors MG132: 10 μM and lactacystin 1 μM; protein biosynthesis inhibitor emetine: 10 μM. Fetal bovine serum (FBS) and all other cell culture reagents were from Gibco Life Technologies. The following antibodies were used: total ERK1, phospho-ERK1/2, phospho-PKC (pan) (#9371), the phospho-PKC Antibody Sampler Kit (#9921), PKCα (#2056) and PKCδ (#2058) were from Cell Signaling Technology; anti-Cdx2 (CDX2-88) was from BioGenex; anti-Elf5 (ab13581) was from abcam. Horseradish peroxidase-conjugated secondary antibodies were from Bio-Rad, and detection was carried out with the enhanced chemiluminescence (ECL) system (Amersham).
2.3. Cloning of DNA constructs and site-directed mutagenesis
A Cdx2-luciferase construct was made by insertion of mouse Cdx2 upstream sequence (bases −1043 to −8, relative to the ATG start codon), prepared by PCR with HindIII and XhoI ends, into the multiple cloning site of pGL3-Basic vector. In a mutation of this construct, the mouse Cdx2 AP-1 site, 5′-TGTGTCAT-3′, identified by Lorentz et al. [17] at position commencing ~−330 relative to the ATG site, was changed to 5′-CCATGGGT-3′ with insertion of a NcoI site. Further deletions or mutations of the Cdx2 promoter sequence comprised the following regions (all relative to the ATG): Cdx2:Ets mut: like Cdx2:Luc WT but Ets recognition sequence changed from 5′-AAGGAGGAAGCT-3′ to 5′-AAGCCATGGGCT-3′ (at ~position −510); Cdx2:del1: −543 to −8; Cdx2:del2: −406 to −8; Cdx2:del3: −323 to −8; Cdx2:NFκB del: −406 to −247 and −156 to −8. Successful cloning of constructs was verified by DNA sequencing (Cogenics/Lark).
2.4. Preparation of cell lysates and Western blotting
Preparation of whole-cell lysates was done in TG lysis buffer and equal amounts of protein were used for Western blotting as described previously [29]. Densitometric analysis of Western blots was carried out using ImageJ software.
2.5. Real-time (Q)RT-PCR and statistical analysis
Total RNA was extracted using TRI-reagent (Sigma). 1.5 μg of total RNA was reverse transcribed into cDNA using the Reverse Transcription kit (Applied Biosystems). Real-time (Q)RT-PCR was performed according to the protocol supplied with the Taqman Reverse Transcription reagents (Applied Biosystems) in a Chromo-4 Thermal Cycler (Bio-Rad) or Stratagene Mx3005P lightcycler. Cdx2 mRNA levels were normalised to the control mRNAs HMBS or SDHA in DKO4 cells; these genes were identified as the most stable reference genes across different treatments using the geNorm software [30]. For mouse TS cells, Dynein and SDHA were used as reference genes which both yielded identical results; thus, the levels of Cdx2 and Elf5 mRNA depicted in Fig. 9 were normalised to Dynein expression levels. Oligonucleotide sequences for human genes were: Cdx2: 5′-AACCAGGACGAAAGACAAATATCGA-3′ (forward) and 5′-GCTAGCTCGGCTTTCCTCCG-3′ (reverse); HMBS: 5′-GGCAATGCGGCTGCAA-3′ (forward) and 5′-GGGTACCCACGCGAATCAC-3′ (reverse) [30]; SDHA 5′-TGGGAACAAGAGGGCATCTG-3′ (forward) and 5′-CCACCACTGCATCAAATTCATG-3′ (reverse) [30]. Oligonucleotide sequences for mouse specific analysis in TS cells were: Cdx2: 5′-AGTGAGCTGGCTGCCACACT-3′ (forward) and 5′-GCTGCTGCTGCTTCTTCTTGA-3′ (reverse); Elf5: 5′-TCTCCAGAACATTCGCTCGC-3′ (forward) and 5′-AATCAGCATAGTCTTTGATGCCAGTC-3′ (reverse); Dynein: 5′-GACCTCAGGCTCAGACGAAGAC-3′ (forward) and 5′-AAGACGCTCATGGCATCACA-3′ (reverse); SDHA: 5′-TGGTGAGAACAAGAAGGCATCA-3′ (forward) and 5′-CGCCTACAACCACAGCATCA-3′ (reverse). PCR with all oligos (spanning two exons) yielded a single PCR product of the predicted size and their identity was confirmed by DNA sequencing.
Fig. 9.
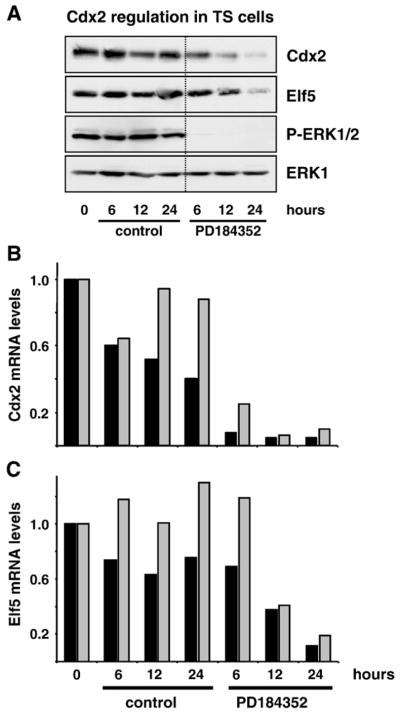
Both Cdx2 and Elf5 are down-regulated in response to inhibiting the ERK1/2 pathway in trophoblast stem cells (TS cells). (A) TS cells were grown in complete TS cell medium with or without the potent MEK inhibitor PD184352 for 6, 12 or 24 h; whole-cell extracts were then fractionated by SDS-PAGE and subjected to Western blotting with antibodies to Cdx2, Elf5, P-ERK1/2 and ERK1. Cdx2 and Elf5 protein levels decreased markedly in a time-dependent manner when ERK1/2 activity was inhibited by PD184352, and the down-regulation of Cdx2 was clearly preceding that of Elf5. Blots shown are from a single experiment which was performed twice yielding identical results. (B and C) Normalised real-time (Q)RT-PCR analyses of Cdx2 and Elf5 mRNA levels from two independent sets of experiments (black and gray bars) performed in parallel with the protein studies in (A). (B) When TS cells were maintained in complete TS cell medium, the expression levels of Cdx2 were not or only moderately affected over a time-course of 24 h. When ERK1/2 pathway activity was inhibited by PD184352, Cdx2 mRNA levels decreased drastically compared to their untreated controls at all time points (reduction ~90%). (C) Similarly, Elf5 mRNA levels remained constant in complete growth medium but decreased progressively upon PD184352 treatment. This down-regulation was lagging behind the decrease of Cdx2 levels, starting at 12 h and reaching a maximal reduction of ~85% only after 24 h.
For statistical analysis in Figs. 3 and 4 the amount of Cdx2 mRNA was normalised to the mean of the two reference genes HMBS and SDHA and the data of at least 4 independent experiments were analysed using the SPSS software package. As the data were standardised to the first values (serum-free control), a one-sample t-test analysis was performed if a condition was compared to the control. For comparing conditions with each other an independent sample t-test was used.
Fig. 3.
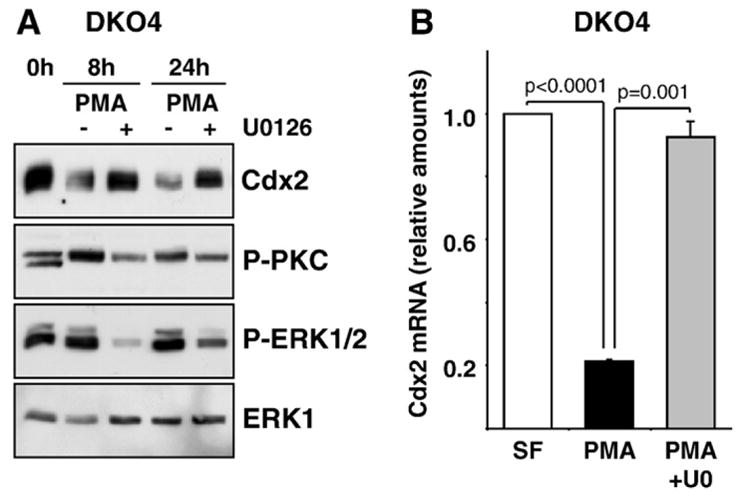
PMA stimulation represses Cdx2 expression via the ERK1/2 pathway in colorectal DKO4 cells. (A) DKO4 cells were stimulated with PMA for 8 or 24 h and whole-cell extracts were fractionated by SDS-PAGE and subjected to Western blotting with antibodies to pan-P-PKC, P-ERK1/2, ERK1 and Cdx2. Cells treated with PMA showed reduced levels of Cdx2 and the MEK inhibitor U0126 reversed this down-regulation. (B) DKO4 cells were stimulated with PMA or PMA plus U0126 for 4 h and Cdx2 mRNA levels were determined by real-time (Q)RT-PCR. Shown is the mean of four independent experiments where Cdx2 levels were normalised to the expression of the control genes HMBS and SDHA. SF: serum-free control.
Fig. 4.
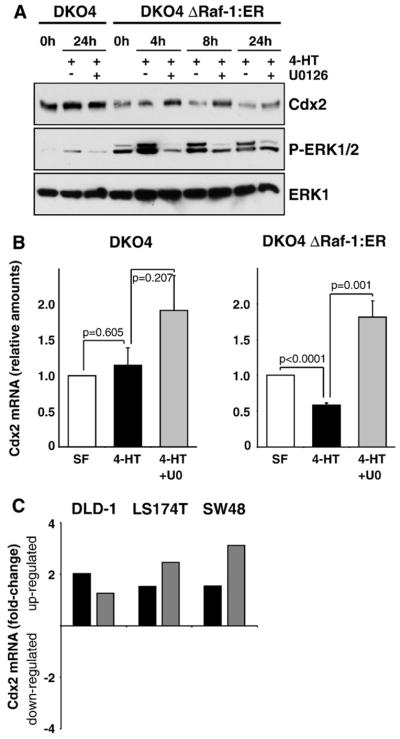
Activation of the Raf–MEK–ERK1/2 pathway is sufficient to down-regulate Cdx2. (A) DKO4 or DKO4 ΔRaf-1:ER cells were treated with 4-HT with or without U0126 for the indicated times; whole-cell extracts were then fractionated by SDS-PAGE and subjected to Western blotting with antibodies to P-ERK1/2, ERK1 and Cdx2. In DKO4 ΔRaf-1:ER cells, Cdx2 levels were reduced after specific activation of the ERK1/2 pathway by 4-HT and this was blocked by treatment with U0126. Parental DKO4 cells did not respond to 4-HT. (B and C) Normalised real-time (Q)RT-PCR analyses of Cdx2 mRNA levels of four (DKO4) or nine (DKO4 ΔRaf-1:ER) independent experiments. (B) DKO4 ΔRaf-1:ER were stimulated with 4-HT with or without U0126 for 4 h (right panel); this leads to down-regulation of Cdx2 mRNA levels by more than 40% which was reversed by U0126. Parental DKO4 cells were unaffected by 4-HT treatment (left panel). (C) The CRC cell lines DLD-1, LS174T and SW48 were incubated in complete medium with or without U0126 for 6 h. Inhibiting ERK1/2 activity with U0126 consistently increased Cdx2 mRNA levels in these cell lines by up to 3-fold. Shown here is the fold-change ratio of U0126-treated over control cells of two independently performed experiments (black and dark gray bars). UO: U0126.
3. Results
3.1. Crosstalk between the ERK1/2 and PKC signalling pathways
Activation of PKC has previously been demonstrated to reduce Cdx2 expression [17]. Since PKC can activate the ERK1/2 pathway in many cell types we first examined the relationship between these two. Initially we studied DKO4 cells, a derivative of the DLD-1 CRC cell line in which the activated K-Ras13D allele has been deleted by homologous recombination [26], and made comparisons with DKO4 ΔRaf-1:ER cells, a clonal derivative of DKO4 cells expressing the ΔRaf-1:ER fusion protein [31]. Stimulation of these cells with 4-hydroxytamoxifen (4-HT) induces a rapid activation of the Raf–MEK–ERK1/2 pathway [27].
To monitor PKC activation we used a phospho-specific pan-PKC antibody. This antibody was raised to phospho-Ser660 of PKCβII in the C-terminal hydrophobic site and also recognises PKCα, βI, δ, ε, η and θ when they are phosphorylated at the homologous residue. Stimulation of DKO4 cells with phorbol 12-myristate 13-acetate (PMA) activated PKC as judged by immunoblotting with the phospho-specific PKC antibody, which revealed both an increase in P-PKC levels and reduced mobility of the anti-P-PKC immune-reactive species (Fig. 1A). Under these conditions PMA caused only a modest increase in the basal level of ERK1/2 activity as judged by immunoblotting with a P-ERK1/2 antibody whereas U0126, a pan-MEK inhibitor, abolished ERK1/2 activity (Fig. 1A). Importantly, DKO4 cells, which do not express ΔRaf-1:ER, did not respond to 4-HT treatment. In DKO4 ΔRaf-1:ER cells PMA again caused a small increase in P-ERK1/2 which was abolished by U0126 (Fig. 1B). As expected, 4-HT treatment to activate ΔRaf-1:ER caused a robust activation of ERK1/2, which was again abolished by U0126.
Fig. 1.
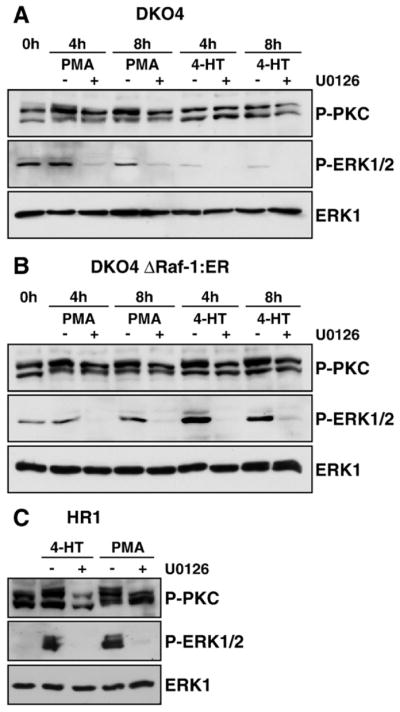
Crosstalk between the ERK1/2 and PKC signalling pathways. DKO4 (A) or DKO4 ΔRaf-1:ER cells (B) were treated with PMA or 4-HT with or without U0126 in serum-free medium for 4 or 8 h each. Whole-cell extracts were fractionated by SDS-PAGE and subjected to Western blotting with antibodies to pan-P-PKC, P-ERK1/2 and ERK1. In both cell lines treatment with PMA caused a stimulation of P-PKC as well as a slight increase of P-ERK; U0126 treatment abolished ERK activation and partially reverted P-PKC activation. Treatment with 4-HT leads to a U0126-reversible activation of P-ERK1/2 in DKO4 ΔRaf-1:ER only, and this in turn increased PKC phosphorylation. (C) HR1 cells were stimulated for 5 h with 4-HT or PMA with or without U0126. Activation of PKC with PMA promoted PKC and ERK1/2 phosphorylation, which was MEK-dependent. Selective activation of ERK1/2 also promoted PKC phosphorylation. PMA: phorbol 12-myristate 13-acetate, 4-HT: 4-hydroxytamoxifen. Results are shown from single experiments representative of three in each case.
Surprisingly we noted that 4-HT treatment also increased P-PKC levels and changed the mobility of the anti-P-PKC immune-reactive species (Fig. 1B). This was not an artefact of 4-HT treatment but was a result of activation of ΔRaf-1:ER and the ERK1/2 pathway since this effect was reversed by U0126 treatment at all time points (Fig. 1B). Indeed, U0126 also reduced the levels of phosphorylated PKC after stimulation with PMA as judged by blotting with the pan phospho-specific PKC antibody (Fig. 1A and B). These results were not confined to DKO4 cells or their derivatives. HR1 cells are a clone of HEK293 cells stably expressing ΔRaf-1:ER* [25]; these cells have a very low basal level of P-ERK1/2 (Fig. 1C). Treatment of HR1 cells with either 4-HT or PMA resulted in activation of ERK1/2 and an increase in P-PKC and in both cases these responses were inhibited by U0126 (Fig. 1C).
These results suggest that whilst activation of PMA-sensitive isoforms of PKC can lead to activation of ERK1/2, ERK1/2 activation in turn can promote phosphorylation of certain PKC isoforms at sites known to be associated with activation. To test this hypothesis we treated both HR1 and DKO4 ΔRaf-1:ER cells with 4-HT or PMA in the presence or absence of U0126 and performed Western blots with a panel of antibodies directed against different phospho-specific PKC isoforms. Whilst the phosphorylation of some of these isoforms appeared to be unchanged (not shown), the signals obtained with the phospho-PKC α/βII and phospho-PKC δ antibodies were increased in ERK1/2-dependent fashion (Fig. 2A). Intriguingly, the bands for phospho-PKC α/βII and phospho-PKC δ resolved at the same apparent molecular weight as the 4-HT-regulated upper and lower bands detected by the pan-P-PKC antibody, respectively. Densitometric analysis revealed that the total levels of PKC α remained constant suggesting that these results represented a change in the stoichiometry of phosphorylation for PKC α or βII (Fig. 2B). In contrast, the total amounts of PKC β appeared to be elevated with 4-HT or PMA compared to the respective U0126 treatments. Thus, the increased levels of phosphorylated PKC δ might be explained by a change in the abundance of this isoform which subsequently undergoes auto-phosphorylation.
Fig. 2.
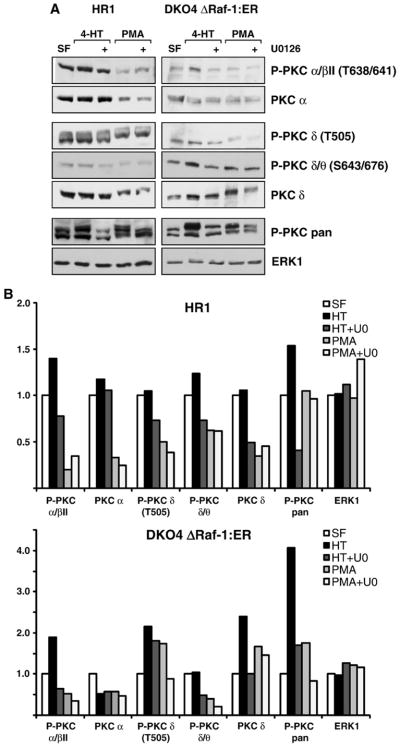
The crosstalk between ERK and PKC involves different PKC isoforms and mechanisms. (A) HR1 and DKO4 ΔRaf-1:ER cells were treated with PMA or 4-HT with or without U0126 in serum-free medium for 4 h. Whole-cell extracts were fractionated by SDS-PAGE and subjected to Western blotting with antibodies to P-PKC pan, to several different phospho-specific isoforms of PKC as indicated, to the non-phosphorylated isoforms PKC α and PKC δ, and to total ERK1. In both cell lines treatment with 4-HT or PMA caused a stimulation of P-PKC pan as seen before, and U0126 treatment partially reversed this P-PKC activation. Whereas the phospho-specific PKC isoform βII was regulated in a MEK/ERK-dependent manner the total levels of the PKC α subunit remained constant suggesting a change in stoichiometry of phosphorylation of PKC α or βII. Phospho-PKC δ also displayed MEK/ERK-dependent regulation, however, as the total levels of PKC δ were also affected, the differential PKC δ phosphorylation might be explained by auto-phosphorylation or changes in abundance. (B) Densitometric analysis of the Western blots shown in (A), the intensities are relative to the serum-free sample which was set to 1 in each case. Results are shown from single experiments representative of three in each case.
Having defined the key cell systems we moved on to examine the regulation of Cdx2 expression.
3.2. PKC-dependent down-regulation of Cdx2 requires the ERK1/2 signalling pathway
As a first step to examine a potential role for ERK1/2 in PKC-dependent Cdx2 regulation DKO4 cells were stimulated with PMA. This resulted in phosphorylation of PKC, a weak increase in P-ERK1/2 and a reduction in Cdx2 expression (Fig. 3A). The pan-MEK inhibitor U0126 abolished ERK1/2 activation but also reversed the effects of PMA on Cdx2, indicating that PMA-dependent down-regulation of Cdx2 requires ERK1/2 activation (Fig. 3A). Real-time (Q)RT-PCR analysis revealed that PMA treatment caused a 75% reduction in Cdx2 mRNA levels (Fig. 3B), which was completely reversed by U0126. These results indicate that the ERK1/2 pathway is required for PKC-dependent down-regulation of Cdx2 at both the RNA and protein levels.
3.3. Activation of the ERK1/2 signalling pathway is sufficient to down-regulate Cdx2 in CRC cells
Prompted by these results we set out to determine if ERK1/2 activation alone was sufficient to down-regulate Cdx2 expression. Stimulation of DKO4 ΔRaf-1:ER cells with 4-HT resulted in activation of ERK1/2 and a progressive down-regulation of Cdx2 protein levels (Fig. 4A). In contrast, DKO4 cells, which do not express ΔRaf-1:ER, were unaffected by 4-HT (Fig. 4A). We also observed that 4-HT treatment of DKO4 ΔRaf-1:ER cells resulted in a ~40% down-regulation of Cdx2 mRNA levels, which was reversed by U0126 (Fig. 4B, right panel), whereas DKO4 cells were again refractory to 4-HT (Fig. 4B, left panel). These results demonstrate that specific activation of the ERK1/2 pathway is sufficient to down-regulate Cdx2. A role for ERK1/2 in regulating Cdx2 expression was not restricted to cells containing the ΔRaf-1:ER construct; when we tested a panel of several different CRC cell lines DLD-1, LS174T and SW48 cells displayed up to three-fold increased Cdx2 mRNA levels in response to inhibition of the ERK1/2 pathway by U0126 (Fig. 4C). Taken together, these results demonstrate for the first time that the ERK1/2 pathway is sufficient to down-regulate Cdx2 mRNA and protein and is required for the previously described ability of PKC to reduce Cdx2 expression.
3.4. Activation of ERK1/2 inhibits transcription from the Cdx2 promoter
As Cdx2 mRNA levels were reduced after activation of the ERK1/2 pathway (Fig. 4B) we wanted to test whether Cdx2 was regulated at the level of transcription. We therefore constructed a luciferase reporter vector with the putative Cdx2 promoter region comprising 1 kb of sequence upstream of the transcriptional start site of the mouse Cdx2 gene, termed Cdx2:Luc (Fig. 5A); a similar construct was used before [17]. For these experiments we first used HR1 cells (HEK293 cells expressing ΔRaf-1:ER*). HR1 cells displayed a marked decrease in Cdx2:Luc reporter activity when stimulated with 4-HT, and this was reversed when the ERK1/2 pathway was inhibited with U0126 (Fig. 5B); in contrast, parental HEK293 cells did not respond to 4-HT treatment. In addition, when HR1 cells were transfected with DUSP6, a dual specificity phosphatase that dephosphorylates and thereby inactivates ERK1/2 directly, the 4-HT-dependent down-regulation of Cdx2:Luc was reversed (Fig. 5C). Activation of ERK1/2 was also sufficient to down-regulate Cdx2:Luc transcription in colorectal DKO4 ΔRaf-1:ER cells; in this case the 4-HT-induced decrease in Cdx2:Luc reporter activity was reversed by U0126, and also by the 2nd generation MEK1/2-specific inhibitors PD184352 or AZD6244 (ARRY-142886) (Fig. 5D). The fact that both pharmacological and molecular genetic targeting of the ERK1/2 pathway was able to block the down-regulation of Cdx2 reporter activity confirms that ERK1/2 activation can inhibit transcription from the Cdx2 promoter.
Fig. 5.

Activation of the ERK1/2 pathway inhibits Cdx2 transcription. (A) Schematic representation of the Cdx2:Luc luciferase reporter construct harbouring the putative Cdx2 promoter sequence. (B–D) Cdx2:Luc promoter reporter assays in different cell lines were carried out in triplicate and normalised for transfection efficiency. (B) HR1 or HEK293 cells were stimulated with 4-HT with or without U0126 for 20 h prior to assay of Luc activity. Activation of ΔRaf-1:ER* in HR1 cells reduced Cdx2 reporter activity by ~50% and this was reversed by U0126; parental HEK293 cells were unaffected by 4-HT. (C) Co-transfection of the ERK1/2 phosphatase DUSP-6 also reversed the 4-HT-dependent inhibition of the Cdx2: Luc reporter. (D) Activation of ΔRaf-1:ER in DKO4 ΔRaf-1:ER cells inhibited Cdx2:Luc reporter activity and this was reversed by three different MEK inhibitors. kb, kilobase pairs; PD: PD184352; AZD: AZD6244; D6: DUSP6 (dual specificity phosphatase 6).
Activation of PKC by PMA also caused a down-regulation of Cdx2: Luc activity in HR1 cells, albeit to a slightly lesser degree than that observed in response to activation of ΔRaf-1:ER* (Fig. 6A); this was reversed by the selective MEK1/2 inhibitor PD184352 indicating that it required signalling to ERK1/2. Thus ERK1/2 activation is required for PKC-dependent repression of the Cdx2 promoter. An AP-1 site within the promoter region of Cdx2 has previously been implicated in the PKC-dependent inhibition of Cdx2 transcription via changes in the balance of the transcription factors c-Jun and c-Fos [17]. To determine if the same site was required for ERK1/2-dependent inhibition of Cdx2 transcription we constructed a mutant version of the Cdx2:Luc reporter in which the AP-1 site was disrupted. However, in side-by-side analysis the reduction in luciferase expression by ΔRaf-1:ER* or PMA was essentially identical for both the Cdx2:Luc WT and Cdx2:Luc AP-1 mut promoter constructs and in both cases this was reversed by either U0126 or PD184352 (Fig. 6A). In an effort to define in greater detail region(s) mediating the effects of ERK1/2 we generated several mutations or truncations of the putative promoter of Cdx2 to delete transcription factor binding sites previously implicated in the regulation of Cdx2 activity, including Lef/Tcf, Ets, Oct-1 and NF-κB [17,32–34]. However, all of the promoter reporter constructs we tested were still inhibited by ERK activation to varying degrees, and this could be reversed with the MEK inhibitor U0126 (Fig. 6B).
Fig. 6.

The ERK1/2-mediated regulation of the Cdx2 promoter is independent of previously implicated transcription factor binding sites. (A) HR1 cells were transfected with the wild-type Cdx2 reporter (Cdx2:Luc WT) or a mutated version in which a putative AP-1 site was changed from 5′-TGTGTCAT-3′ to 5′-CCATGGGT-3′ (Cdx2:AP-1 mut). Cells were then stimulated with either 4-HT±U0126 or PMA±PD184352 prior to assay of luciferase activity. The mutated AP-1 construct displayed a pattern of MEK-dependent regulation identical to the wild-type reporter sequence in response to 4-HT or PMA. Averaged results of triplicates are shown from a single experiment representative of three giving similar results. (B) Shown on the left are schematic representations of Cdx2:Luc WT or various mutated or truncated Cdx2 promoter reporter constructs. Next to each construct a representative result of at least three independent transfection experiments into HR1 cells is shown. Even though the putative binding sites for several different transcription factors including AP-1, Ets, Wnt, Oct-1 and NF-κB were either mutated or truncated, the remaining Cdx2 promoter reporter fragment still showed MEK/ERK1/2-dependent regulation (4-HT or 4-HT+ U0126 treatment).
3.5. Activation of the ERK1/2 pathway also promotes Cdx2 protein turnover
In the course of our studies we noted that the Cdx2 protein was often not detected as a single distinct band at ~37 kDa but was rather present as at least two discrete forms of different molecular weight. In fact, there have been reports demonstrating phosphorylation of Cdx2 by various kinases [35–38]; a role for ERK has been shown in one study, where it phosphorylated Cdx2 at Ser60, thereby decreasing its transcriptional activity [38]. In addition, phosphorylation is often linked to changes in protein stability. To address this latter possibility DKO4 ΔRaf-1:ER cells were treated with emetine to stop new protein synthesis and stimulated with 4-HT to activate ERK1/2 in the presence or absence of the proteasome inhibitor MG132 (Fig. 7). Cdx2 was present as both slower and faster migrating forms at equal levels in cells treated with emetine alone, but activation of ERK1/2 (4-HT) shifted this ratio so that the slower running (upper) band was the predominant form; this was accompanied by a reduction in total Cdx2 protein consistent with increased turnover (Fig. 7). Inclusion of MG132 (a proteasome inhibitor) in addition to 4-HT treatment prevented the reduction in total Cdx2, effectively trapping the hyper-phosphorylated form of Cdx2. Taken together, these results suggest that activation of ERK1/2 results in the phosphorylation of Cdx2, thereby marking it for proteasomal degradation.
Fig. 7.
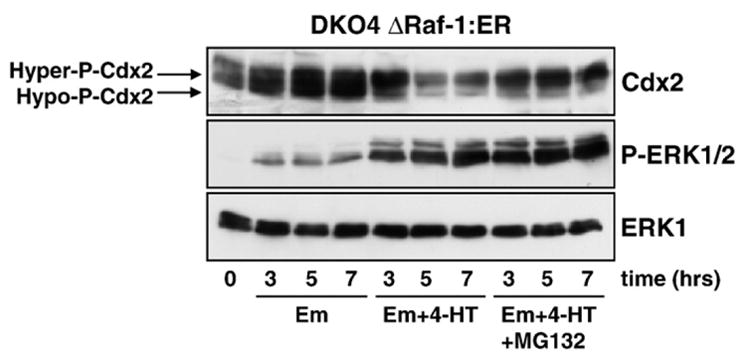
Cdx2 is phosphorylated and degraded in a proteasome-dependent manner in response to ERK1/2 activation. DKO4 ΔRaf-1:ER cells were incubated in serum-free medium containing emetine to inhibit new protein synthesis. Activation of ERK1/2 (4-HT) induced the down-regulation of Cdx2 which was accompanied by a shift in its mobility pattern on SDS-PAGE gels towards a slower migrating, hyper-phosphorylated form. Inhibition of the proteasome by MG132 (in the presence of 4-HT) reduced the degradation of Cdx2 and the remaining protein was predominantly in the hyper-phosphorylated form. MG132 did not prevent the activation of ERK1/2 by ΔRaf-1:ER. Em: emetine. Results are shown from a single experiment representative of three.
Attempts to demonstrate poly-ubiquitination of Cdx2 proved to be technically challenging. We were unable to demonstrate poly-Ub forms of endogenous Cdx2 in DKO4 ΔRaf-1:ER cells (data not shown). Similarly, when we co-expressed Cdx2 with flag-Ub we were unable to detect Cdx2-Ub conjugates (Fig. 8A). In both cases it is likely that this reflects the activity of de-ubiquitinating enzymes (DUBs) both pre- and post-lysis. However, as an alternative surrogate marker for ubiquitination we used Ub-binding domains (UBA domains) of the yeast protein Dsk2, which selectively bind K48-linked poly-Ub chains [39]. Cdx2 was expressed in HR1 cells, which were then left untreated or treated with 4-HT and lactacystin to activate ERK1/2 and inhibit the proteasome (Fig. 8B). Transfected cell extracts were then incubated with wild-type GST-Dsk2 beads (WT) or those containing two point mutations that abolish Ub binding (mut) in a pull-down assay. Following washing and Western blotting we found that wild-type UBA beads could precipitate Cdx2 and the amount recovered was increased upon activation of ERK1/2. As a specificity control much less Cdx2 was recovered with the mutant UBA beads and neither WT nor mutant beads were able to precipitate PKB/AKT from cell extracts. These results suggest that Cdx2 is modified by endogenous Ub, though the efficiency of recovery may again be hampered by DUB activities.
Fig. 8.
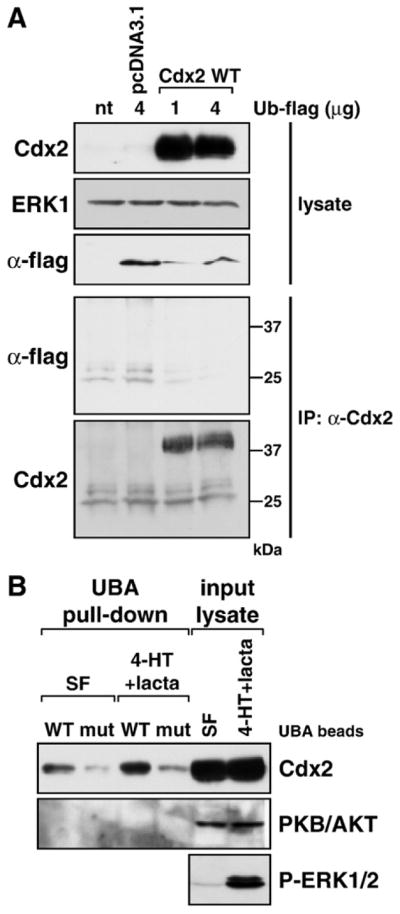
Ubiquitination of Cdx2 in response to ERK1/2 activation detected by binding to an immobilised UBA domain. (A) HR1 cells were transfected with wild-type Cdx2 or the control vector pcDNA3.1 in addition to a flag-tagged ubiquitin expression construct, and following overnight incubation were treated with the very potent proteasome inhibitor lactacystin (lacta) for 6 h. Expression of transfected Cdx2 and ubiquitin (α-flag) proteins was demonstrated in whole-cell lysates, ERK1 served as a loading control. Following immunoprecipitation of Cdx2 we were unable to detect extensive ubiquitination of Cdx2 in the pulled-down fraction (α-flag), even though Cdx2 was pulled down very nicely (α-Cdx2). (B) HR1 cells were transfected with wild-type Cdx2 and kept in either serum-free medium or in medium containing 4-HT plus lactacystin for 6 h to promote Cdx2 phosphorylation and ubiquitination as well as inhibit its proteasomal degradation. Western blots demonstrated that transfected Cdx2 was expressed well and that 4-HT+ lacta treatment activated the MEK/ERK1/2 pathway. Ubiquitinated proteins were pulled down using either wild-type or mutated UBA beads (designated WT or mut) and the pulled-down fraction was subjected to Western blotting against Cdx2 or protein kinase B (PKB/AKT). The amount of UBA-bound Cdx2 was increased upon 4-HT+lacta treatment compared to the serum-free sample. Endogenous PKB/AKT was not pulled down by UBA beads under any condition and served as a specificity control.
3.6. Contrasting effects of the ERK1/2 pathway on Cdx2 in trophoblast versus CRC cells
Whilst we have shown that the ERK1/2 pathway can repress Cdx2 in CRC cells, a recent report described completely the opposite effect in the developing trophectoderm (TE) where ERK activation is necessary for correct Cdx2 expression [40]. This same study also demonstrated that the activation of Ras-ERK in embryonic stem (ES) cells leads to their differentiation into trophoblast stem (TS) cells. In addition, another recent paper [41] showed that Cdx2 activates the transcription factor Elf5 that in turn also activates Cdx2 in a feed-back amplifying loop to potentiate Cdx2 expression in the trophoblast lineage. To address this apparent contradiction we examined Elf5 and Cdx2 expression in TS cells. We found that selective inhibition of the ERK1/2 pathway by PD184352 reduced expression of both the Cdx2 and Elf5 proteins (Fig. 9A) and also suppressed Cdx2 and Elf5 mRNA levels (Fig. 9B and C). Time course analysis revealed that the reduction in Cdx2 mRNA levels was almost complete within 6 h of PD184352 treatment (Fig. 9B) and clearly preceded the reduction in Elf5 mRNA (Fig. 9C). Similarly, the reduction in Cdx2 protein levels preceded that for Elf5 protein (Fig. 9A). These results are consistent with the view that in TS cells the ERK1/2 pathway activates Cdx2 which then, in turn, activates Elf5. In contrast to results for TS cells [41] we found that ectopic expression of Elf5 in CRC cells did not enhance Cdx2 expression (not shown). This is perhaps not surprising since we have failed to detect endogenous Elf5 in CRC cells in any case.
4. Discussion
The commonly observed down-regulation of Cdx2 expression in CRC may contribute to tumour progression [1,15] and/or to metastasis [16]. This down-regulation is attributable, at least in part, to derangement of signalling pathways [15]. The pathways that have been implicated include Ras and PKC [17], the p38 MAP kinase [37] and PI3K/PTEN [33]; notably, a role for the ERK1/2 pathway was ruled out [17].
4.1. ERK1/2 signalling represses Cdx2 expression in CRC cells
Lorentz et al. reported that Ras-dependent repression of Cdx2 was mediated by PKC signalling [17]. Since the MEK inhibitor PD98059 did not inhibit this effect they concluded that it was independent of ERK1/2. Whilst we could certainly demonstrate that Cdx2 protein and mRNA are down-regulated upon activation of PKC by PMA we found that this effect was reversed by MEK inhibitors, indicating that the effect of PKC is in large part dependent upon the ERK1/2 pathway. The reason for the disparity between our results and those of the earlier study is not clear but could reflect any of the following. Lorentz et al. used cell lines engineered to express H-Ras12V; however it is difficult to interpret these studies when it is K-Ras not H-Ras that is mutated in CRC. When Ras was studied in our experiments we used human CRC cell lines carrying a single activated K-Ras allele (DLD-1 or LS174T); use of such model systems, with the relevant Ras oncoprotein, is important since the Ras proteins may differ in their coupling to downstream effector pathways [21,22]. In addition, Lorentz et al. argued against a role for ERK1/2 based on the use of PD98059; this 1st generation MEK inhibitor has poor solubility and poor potency and the authors did not assay P-ERK1/2 levels to confirm that the inhibitor was effective. In our own studies we demonstrated inhibition of ERK1/2 by U0126 but also used two other distinct and potent 2nd generation MEK1/2 inhibitors, PD184352 and AZD6244.
Our results suggest that ERK1/2-dependent repression of Cdx2 expression in CRC cells can potentially be attributed to two distinct mechanisms (Fig. 10A). First, we found that ERK1/2 signalling repressed transcription from a 1 kb Cdx2 promoter, providing a likely explanation for the reduction in mRNA levels. Whilst we have not yet identified the promoter element responsible we could demonstrate that the effects of ERK1/2 and PKC were independent of the previously described AP-1, Ets, Oct-1 or NF-κB sites in the Cdx2 promoter [17,32,33]; definition of the ERK1/2-regulated element and the relevant binding proteins/transcriptional regulators should be a priority for future studies. In addition, we also found that activation of the ERK1/2 pathway could promote the phosphorylation and proteasome-dependent turnover of the Cdx2 protein. Furthermore, we were able to show that this correlated with the ability of Cdx2 to bind to the Dsk2 UBA domains in pull-down assays, a surrogate marker for K48-linked poly-ubiquitination. However the apparent abundance of poly-Ub forms was low and this may reflect DUB activity in cells pre- or post-lysis. In addition, we cannot rule out the possibility that Cdx2 is targeted for proteasomal degradation by other mechanisms. Analysis of the Cdx2 primary amino acid sequence with the FoldIndex© and IUPred algorithms predict extensive regions of disorder (data not shown), and disordered proteins can be degraded by the uncapped 20S proteasomes without the need for prior poly-Ub [42]. Future work should aim to address in detail the mechanism by which Cdx2 is degraded.
Fig. 10.

Models for ERK-dependent regulation of Cdx2 expression in CRC or TS cells. (A) In CRC cells activation of PKC can lead to the activation of the Ras-regulated Raf–MEK–ERK1/2 pathway, which in turn represses Cdx2 transcription and targets Cdx2 for proteasome-dependent turnover. In addition the activation of the ERK1/2 pathway leads to increased phosphorylation of PKC isoforms at a site associated with their activation, revealing a potential new link between ERK1/2 and the PKCs. In addition to such a feed-forward loop, which could serve to enhance Cdx2 repression still further, the activation of PKC might also repress Cdx2 transcription in an ERK1/2-independent pathway. (B) In TS cells activation of the Ras–Raf–MEK–ERK pathway increases the levels of Cdx2, a process which is required for correct trophoblast cell fate determination. Elf5 levels are also regulated by the Raf–MEK–ERK pathway, at least indirectly, via a co-stimulatory loop between Cdx2 and Elf5 which serves to reinforce the commitment to the trophoblast lineage.
The kinase responsible for phosphorylating Cdx2 and mediating these effects remains to be defined. ERK1/2 might phosphorylate Cdx2 and thereby lead to its degradation in a proteasome-dependent manner; indeed, ERK1/2 has been shown to phosphorylate Cdx2 at Ser60 to regulate transcriptional activity [38]. However, we cannot exclude the involvement of kinases downstream of ERK1/2 in this context. For example, CDK2 physically associates with and phosphorylates mouse Cdx2 at residue Serine281, leading to its poly-ubiquitination and proteolysis [35,36]. Thus activation of CDK2 several stages downstream of ERK1/2 activation might be responsible for turnover of Cdx2 following activation of ΔRaf-1:ER; however, our unpublished results suggest that the CDK2 inhibitor roscovitine fails to block Cdx2 turnover at doses at which it is clearly effective at inhibiting cell cycle progression and cyclin A expression (data not shown). Clearly identification of the kinase responsible for ERK1/2-driven Cdx2 turnover is a priority for future investigation.
4.2. Interplay between PKC and ERK1/2 signalling
At first glance the demonstration that PKC-dependent repression of Cdx2 requires ERK1/2 activity suggests a linear pathway in which PKC activates the ERK1/2 pathway, which is then responsible for the delivery of the signal to repress Cdx2. However, in many instances PKC activation caused only a modest increase in P-ERK1/2 levels. Whilst this may simply reflect the already high level of P-ERK1/2 in many CRC cells, we cannot rule out a model in which repression of Cdx2 requires the coincident activation of PKC and ERK1/2 acting independently, perhaps at different promoter elements (Fig. 10A). This notwithstanding, the ERK1/2 pathway was sufficient to repress the Cdx2 promoter and was required for the effects of PKC.
Our finding that the selective activation of ERK1/2 signalling can induce the phosphorylation of certain PKC isoforms takes the model of Cdx2 regulation one step further to include a potential feed-forward mechanism between ERK1/2 and PKC, causing the efficient down-regulation of Cdx2 upon receipt of a stimulatory signal (Fig. 10A). The existence of such a loop was initially indicated by analysing protein lysates with a pan phospho-specific PKC antibody, which recognises a phosphorylated serine residue in the C-terminal hydrophobic domain of many PKC isoforms. Indeed, an involvement of ERK1/2 in the stimulation of certain PKC isoforms has been suggested before [43,44]. Our preliminary analyses with a panel of phospho-specific antibodies directed against individual isoforms of PKC have corroborated these observations. Whilst we detected a higher level of the phosphorylated forms of PKC α/βII and δ, other isoforms such as P-PKC ζ/λ (T410/403), θ (T538) or PKD/PKC μ (S744/748) were not regulated. As we only studied a subset of different PKC isoforms this list is not comprehensive. However, our results suggest that the mechanisms involved in the ERK-dependent phosphorylation of PKCs are likely to be a combination of both direct changes in phosphorylation (PKC α/βII) and regulation of the abundance of certain isoforms followed by auto-phosphorylation events (e.g., PKC δ). It will be interesting to dissect the molecular mechanisms underlying the interplay between these pathways in greater detail in the future, in particular with respect to the regulation of Cdx2.
4.3. Contrasting effects of ERK1/2 signalling upon Cdx2 expression in CRC and trophoblast stem (TS) cells
Whilst we have shown that ERK1/2 signalling represses Cdx2 expression in CRC cells the opposite effect has recently been reported in the trophoblast compartment (Fig. 10B) [40]. In TS cells we confirmed that ERK1/2 is required to promote Cdx2 expression, and also showed that it promotes Elf5 expression. In TS cells Elf5 has previously been shown to be involved in a co-stimulatory loop with Cdx2, providing a possible mechanism for potentiation and maintenance of Cdx2 expression in the trophoblast compartment [41]. In contrast to TS cells Cdx2 expression in CRC cells was not activated by Elf5 and this is consistent with the lack of endogenous Elf5 in CRC cells. The contrasting responses of Cdx2 to ERK1/2 signalling in CRC and TS cells is most likely explained by the previous suggestion that separate DNA control elements are used to regulate Cdx2 expression in each of these two cell types [4]. Further work is needed to identify the DNA control elements and transcription factors that mediate the ERK1/2-dependent repression of Cdx2 expression in CRC cells.
Acknowledgments
We thank Dr Paul Smith (AstraZeneca) for the provision of AZD6244 (ARRY-142886) and Prof Stephen Keyse for the DUSP6 expression plasmid. We would like to thank Kathryn Hadfield-Moorhouse and Kathy Balmanno for technical assistance during the very early and late stages of the project. We are also grateful to Dr Anne Segonds-Pichon for advice and help concerning statistics. This work was funded by the BBSRC and a Synergy Grant from the Babraham Institute.
References
- 1.Suh E, Traber PG. Mol Cell Biol. 1996;16:619–625. doi: 10.1128/mcb.16.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Strumpf D, Mao CA, Yamanaka Y, Ralston A, Chawengsaksophak K, Beck F, Rossant J. Development. 2005;132:2093–2102. doi: 10.1242/dev.01801. [DOI] [PubMed] [Google Scholar]
- 3.Gaunt SJ, Drage D, Trubshaw RC. Development. 2008;135:2511–2520. doi: 10.1242/dev.015909. [DOI] [PubMed] [Google Scholar]
- 4.Benahmed F, Gross I, Gaunt SJ, Beck F, Jehan F, Domon-Dell C, Martin E, Kedinger M, Freund JN, Duluc I. Gastroenterology. 2008;135:1238–1247. doi: 10.1053/j.gastro.2008.06.045. [DOI] [PubMed] [Google Scholar]
- 5.Bonhomme C, Duluc I, Martin E, Chawengsaksophak K, Chenard MP, Kedinger M, Beck F, Freund JN, Domon-Dell C. Gut. 2003;52:1465–1471. doi: 10.1136/gut.52.10.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mallo GV, Rechreche H, Frigerio JM, Rocha D, Zweibaum A, Lacasa M, Jordan BR, Dusetti NJ, Dagorn JC, Iovanna JL. Int J Cancer. 1997;74:35–44. doi: 10.1002/(sici)1097-0215(19970220)74:1<35::aid-ijc7>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 7.Ee HC, Erler T, Bhathal PS, Young GP, James RJ. Am J Pathol. 1995;147:586–592. [PMC free article] [PubMed] [Google Scholar]
- 8.Hinoi T, Tani M, Lucas PC, Caca K, Dunn RL, Macri E, Loda M, Appelman HD, Cho KR, Fearon ER. Am J Pathol. 2001;159:2239–2248. doi: 10.1016/S0002-9440(10)63074-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.da Costa LT, He TC, Yu J, Sparks AB, Morin PJ, Polyak K, Laken S, Vogelstein B, Kinzler KW. Oncogene. 1999;18:5010–5014. doi: 10.1038/sj.onc.1202872. [DOI] [PubMed] [Google Scholar]
- 10.Aoki K, Tamai Y, Horiike S, Oshima M, Taketo MM. Nat Genet. 2003;35:323–330. doi: 10.1038/ng1265. [DOI] [PubMed] [Google Scholar]
- 11.Dang LH, Chen F, Ying C, Chun SY, Knock SA, Appelman HD, Dang DT. Oncogene. 2006;25:2264–2272. doi: 10.1038/sj.onc.1209247. [DOI] [PubMed] [Google Scholar]
- 12.Witek ME, Nielsen K, Walters R, Hyslop T, Palazzo J, Schulz S, Waldman SA. Clin Cancer Res. 2005;11:8549–8556. doi: 10.1158/1078-0432.CCR-05-1624. [DOI] [PubMed] [Google Scholar]
- 13.Kawai H, Tomii K, Toyooka S, Yano M, Murakami M, Tsukuda K, Shimizu N. Oncol Rep. 2005;13:547–551. [PubMed] [Google Scholar]
- 14.Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, Bergman Y, Simon I, Cedar H. Nat Genet. 2007;39:232–236. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- 15.Hinoi T, Loda M, Fearon ER. J Biol Chem. 2003;278:44608–44616. doi: 10.1074/jbc.M307435200. [DOI] [PubMed] [Google Scholar]
- 16.Lu X, Freund JN, Muller M, Ravey J, Nicolas JP, Gueant JL, Namour F. Biochimie. 2008;90:697–704. doi: 10.1016/j.biochi.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 17.Lorentz O, Cadoret A, Duluc I, Capeau J, Gespach C, Cherqui G, Freund JN. Oncogene. 1999;18:87–92. doi: 10.1038/sj.onc.1202280. [DOI] [PubMed] [Google Scholar]
- 18.Baron-Delage S, Capeau J, Barbu V, Chastre E, Levy P, Gespach C, Cherqui G. J Biol Chem. 1994;269:18686–18693. [PubMed] [Google Scholar]
- 19.Delage S, Chastre E, Empereur S, Wicek D, Veissiere D, Capeau J, Gespach C, Cherqui G. Cancer Res. 1993;53:2762–2770. [PubMed] [Google Scholar]
- 20.Bos JL. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 21.Voice JK, Klemke RL, Le A, Jackson JH. J Biol Chem. 1999;274:17164–17170. doi: 10.1074/jbc.274.24.17164. [DOI] [PubMed] [Google Scholar]
- 22.Yan J, Roy S, Apolloni A, Lane A, Hancock JF. J Biol Chem. 1998;273:24052–24056. doi: 10.1074/jbc.273.37.24052. [DOI] [PubMed] [Google Scholar]
- 23.Kolch W, Heidecker G, Kochs G, Hummel R, Vahidi H, Mischak H, Finkenzeller G, Marme D, Rapp UR. Nature. 1993;364:249–252. doi: 10.1038/364249a0. [DOI] [PubMed] [Google Scholar]
- 24.Marais R, Light Y, Mason C, Paterson H, Olson MF, Marshall CJ. Science. 1998;280:109–112. doi: 10.1126/science.280.5360.109. [DOI] [PubMed] [Google Scholar]
- 25.Boughan PK, Argent RH, Body-Malapel M, Park JH, Ewings KE, Bowie AG, Ong SJ, Cook SJ, Sorensen OE, Manzo BA, Inohara N, Klein NJ, Nunez G, Atherton JC, Bajaj-Elliott M. J Biol Chem. 2006;281:11637–11648. doi: 10.1074/jbc.M510275200. [DOI] [PubMed] [Google Scholar]
- 26.Shirasawa S, Furuse M, Yokoyama N, Sasazuki T. Science. 1993;260:85–88. doi: 10.1126/science.8465203. [DOI] [PubMed] [Google Scholar]
- 27.Ries S, Biederer C, Woods D, Shifman O, Shirasawa S, Sasazuki T, McMahon M, Oren M, McCormick F. Cell. 2000;103:321–330. doi: 10.1016/s0092-8674(00)00123-9. [DOI] [PubMed] [Google Scholar]
- 28.Tanaka S, Kunath T, Hadjantonakis AK, Nagy A, Rossant J. Science. 1998;282:2072–2075. doi: 10.1126/science.282.5396.2072. [DOI] [PubMed] [Google Scholar]
- 29.Balmanno K, Cook SJ. Oncogene. 1999;18:3085–3097. doi: 10.1038/sj.onc.1202647. [DOI] [PubMed] [Google Scholar]
- 30.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Genome Biol. 2002;3:Research0034.1–0034.11. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woods D, Parry D, Cherwinski H, Bosch E, Lees E, McMahon M. Mol Cell Biol. 1997;17:5598–5611. doi: 10.1128/mcb.17.9.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jin T, Li H. J Biol Chem. 2001;276:14752–14758. doi: 10.1074/jbc.M008277200. [DOI] [PubMed] [Google Scholar]
- 33.Kim S, Domon-Dell C, Wang Q, Chung DH, Di Cristofano A, Pandolfi PP, Freund JN, Evers BM. Gastroenterology. 2002;123:1163–1178. doi: 10.1053/gast.2002.36043. [DOI] [PubMed] [Google Scholar]
- 34.Pilon N, Oh K, Sylvestre JR, Bouchard N, Savory J, Lohnes D. Dev Biol. 2006;289:55–63. doi: 10.1016/j.ydbio.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 35.Boulanger J, Vezina A, Mongrain S, Boudreau F, Perreault N, Auclair BA, Laine J, Asselin C, Rivard N. J Biol Chem. 2005;280:18095–18107. doi: 10.1074/jbc.M502184200. [DOI] [PubMed] [Google Scholar]
- 36.Gross I, Lhermitte B, Domon-Dell C, Duluc I, Martin E, Gaiddon C, Kedinger M, Freund JN. Oncogene. 2005;24:7955–7963. doi: 10.1038/sj.onc.1208945. [DOI] [PubMed] [Google Scholar]
- 37.Houde M, Laprise P, Jean D, Blais M, Asselin C, Rivard N. J Biol Chem. 2001;276:21885–21894. doi: 10.1074/jbc.M100236200. [DOI] [PubMed] [Google Scholar]
- 38.Rings EH, Boudreau F, Taylor JK, Moffett J, Suh ER, Traber PG. Gastroenterology. 2001;121:1437–1450. doi: 10.1053/gast.2001.29618. [DOI] [PubMed] [Google Scholar]
- 39.Funakoshi M, Sasaki T, Nishimoto T, Kobayashi H. Proc Natl Acad Sci U S A. 2002;99:745–750. doi: 10.1073/pnas.012585199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu CW, Yabuuchi A, Chen L, Viswanathan S, Kim K, Daley GQ. Nat Genet. 2008;40:921–926. doi: 10.1038/ng.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ng RK, Dean W, Dawson C, Lucifero D, Madeja Z, Reik W, Hemberger M. Nat Cell Biol. 2008;10:1280–1290. doi: 10.1038/ncb1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsvetkov P, Asher G, Paz A, Reuven N, Sussman JL, Silman I, Shaul Y. Proteins. 2008;70:1357–1366. doi: 10.1002/prot.21614. [DOI] [PubMed] [Google Scholar]
- 43.Khundmiri SJ, Dean WL, McLeish KR, Lederer ED. J Biol Chem. 2005;280:8705–8713. doi: 10.1074/jbc.M408606200. [DOI] [PubMed] [Google Scholar]
- 44.Toma O, Weber NC, Wolter JI, Obal D, Preckel B, Schlack W. Anesthesiology. 2004;101:1372–1380. doi: 10.1097/00000542-200412000-00018. [DOI] [PubMed] [Google Scholar]