

Abstract
Purpose.
Retinal diseases such as macular degeneration and glaucoma are disorders that target specific retinal neurons that can ultimately lead to vision loss. Under these conditions and pathologies, retinal neurons can die via apoptosis that may be due to increased oxidative stress. The neuroprotective effects of 17β-estradiol (E2) and three synthetic nonfeminizing estrogen analogs (ZYC-26, ZYC-23, and ZYC-3) were investigated to examine their abilities to protect retinal neurons against glutamate toxicity.
Methods.
Using an in vitro model of glutamate-induced cell death in 661W cells, a mouse cone photoreceptor cell line, shown to express both estrogen receptors (ERs) via immunoblotting, was pretreated with E2 and its analogs and cell viability were assessed.
Results.
It was observed that E2 and estrogen analogs, ZYC-26 and ZYC-3, were protective against a 5 mM glutamate insult in 661W cells. The neuroprotective abilities of ZYC-26 and ZYC-3 were autonomous of estrogen receptor-α (ERα) and ERβ demonstrated by their ability to protect in the presence of ICI 182780, a pan-ER antagonist with a high affinity for the estrogen receptor. Treatment with PPT and DPN, ERα- and ERβ-specific agonists, respectively, did not protect the 661W cells from the glutamate insult. Studying the membrane ER (mER) or GPR30 did show that activation of the receptor by G1 protected the retinal neuron from insult, whereas G15, an antagonist of the mER was not able to antagonize the protection previously seen.
Conclusions.
These data demonstrate that nonfeminizing estrogens may emerge as useful compounds for neuroprotection of retinal cells.
In many retinal diseases involving elevated ROS, there are few therapies to prevent neuronal cell loss due to a lack of understanding of the pathologies themselves. Here we show that nonfeminizing estrogen analogs can serve as a prophylactic treatment in retinal cells exposed to a glutamate induces ROS insult.
Introduction
Glutamate is the predominant excitatory neurotransmitter in the mammalian central nervous system (CNS) and the vertical pathway of the mammalian retina, but at high concentrations, it has been shown to be neurotoxic through inhibition of the glutamate/cystine antiporter. This inhibition disrupts glutathione synthesis, which increases reactive oxygen species (ROS).1 The production of ROS is a normal consequence of metabolism, but in the retina, elevated levels of ROS has been associated with diseases such as age-related macular degeneration (AMD)2 and retinitis pigmentosa (RP).3 Although different enzyme systems within the cell can generate ROS, the mitochondrial electron transport chain is the predominant contributor.4–6
In retinal diseases such as RP and AMD there is an eventual loss of photoreceptor cells that can lead to partial or complete blindness. AMD alone has been shown to be the leading cause of vision loss in the elderly population in the western hemisphere.7,8 It has been predicted that within the next decade the number of AMD cases is expected to reach 3 million.9 As with many of these retinal diseases and other neurodegenerative diseases, there are very few therapies to combat the neuronal cell loss due to a lack of understanding of the pathologies themselves.
With AMD, the positioning of the retinal pigment epithelium (RPE) and its role create an idyllic atmosphere for the elevation and accumulation of ROS,10 which in turn can disrupt mitochondrial function within these cells. The development of mitochondrial dysfunction can lead to the death of the RPE11 via apoptosis, which as a consequence can result in death to the underlying photoreceptor cells.12,13 These factors can eventually lead to vision loss.
Epidemiologic studies have indicated a noticeable relationship between the development of AMD in women and the reduction of circulating estrogen as seen in menopause.14–17 These studies help to demonstrate the importance of estrogen. Estrogens have been shown to be neuroprotective in in vivo models, including transient and permanent middle cerebral artery occlusion,18–20 global forebrain ischemia,21 photothrombotic focal ischemia,22 and glutamate-induced focal cerebral ischemia,23 and in in vitro systems including H2O2,24–29 serum deprivation,30–33 oxygen-glucose deprivation,34,35 and excitotoxicity.34,36–39 The ability of estrogen to protect the mitochondrial respiratory chain from oxidative damage has been demonstrated in a plethora of tissues, including the brain, breast, myocardium, and lens epithelium.40 In particular, there has been over a decade of studies demonstrating convincingly the neuroprotective abilities of 17β-estradiol (E2).18,24,30,36,41,42 The mechanism by which it protects, however, is still inadequately understood.42,43 Estrogens possess a phenolic ring that provides antioxidant/redox cycling activity in neurons and other tissues.44–47
The discovery of a compound that can protect retinal neurons from insult or damage and reduce vision loss would be very beneficial. Although estrogens are viable candidates, their use as a neuroprotective agent is limited by their feminizing characteristics. We discovered nonfeminizing estrogens48 that can provide potent neuroprotection, but lack binding to estrogen receptors (ERs). In the present study, we demonstrate the neuroprotective effects of E2 and estrogen analogs in 661W cells exposed to oxidative stress initiated by a glutamate insult. Here we assessed the neuroprotective effects of the derivatives of estrone, 2-adamantyl-estra-1,3,5(10)-trien-3-ol-17-one (ZYC-3), 2-adamantyl, 4 methyl estrone (ZYC-26), and 2-adamantyl, 3–0-methyl estradiol (ZYC-23) in a neuronal cell culture model. The mechanisms by which estrogens are capable of protecting against oxidative stress are thought to occur via an ER-mediated mechanism49,50 or their antioxidant activity.46 Here we demonstrate that E2 and its analogs increase cell viability in 661W cells exposed to glutamate toxicity through a mechanism that is independent of the ER. The ability for estrogen and its analogs to protect within the 661W model was also dependent on the presence of a phenolic ring. These properties allow these compounds to function as neuroprotective agents that could provide protection from further damage to retinal neurons as well as provide prophylactic protection from future damage due to eye disease or even eye trauma.
Materials and Methods
Cell Culture
661W cells were generously donated by Muayyad Al-Ubaidi (University of Oklahoma Health Sciences Center, Oklahoma City, OK). The 661W cell line, a mouse retinal tumor cell line derived from cone photoreceptor cells, was maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% dialyzed fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were grown in a humidified atmosphere of 5% CO2 and 95% air at 37°C. Only the first seven cell line passages were used for experiments.
Chemicals and Reagents
The following materials were used for experiments in our investigations: 17β-estradiol (E2; Steraloids, Inc., Newport, RI); l-glutamic acid (Sigma-Aldrich, St. Louis, MO); G1 and G15, an agonist and antagonist, respectively, at the membrane ER G-protein–coupled receptor (GPR30) (Calbiochem, San Diego, CA); ICI 182780 (4,4′,4′-(4-propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol [PPT]) and diarylpropiolnitrile (DPN; Tocris Bioscience, Ellisville, MO); and 2-adamantyl, 4 methyl estrone (ZYC-26), 2-adamantyl-estra-1,3,5(10)-trien-3-ol-17-one (ZYC-3), and 2-adamantyl, 3–0-methyl estradiol (ZYC-23) (synthesized in the laboratories of Covey and colleagues51).
Treatment Paradigm
661W cells in culture were removed using 0.25% trypsin-EDTA (Invitrogen Corp., Carlsbad, CA) and plated at a density of 3000 cells/well in 96-well plates. Cell media consisted of DMEM with 10% dialyzed FBS, and 1% penicillin–streptomycin solution. After 24 hours, cells were treated with either dimethyl sulfoxide vehicle control (DMSO; Sigma-Aldrich) or 5 mM glutamate in the presence of 1 nM to 10 μM E2, DPN, PPT, ZYC-26, ZYC-3, ZYC-23, or G1. To study the specific involvement of the ERα and ERβ, cells were also treated with the ER antagonist ICI 182780 in the presence of the different ZYC compounds. We also assessed the involvement of the membrane ER receptor (GPR30) by studying its antagonist G15 in the presence of concentrations of estrogens that provide neuroprotection in 661W cells. The structures of estrogen-related compounds used are depicted in Figure 1.
Figure 1.
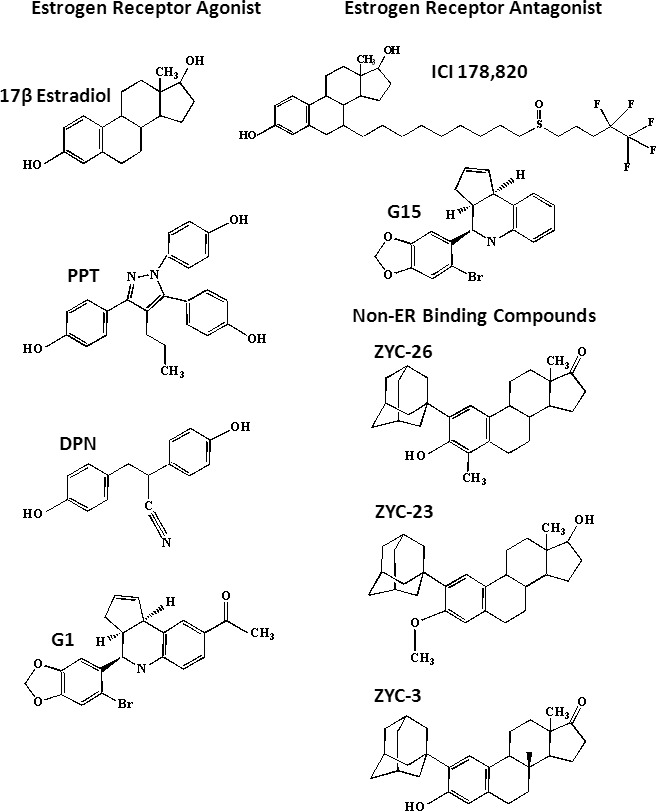
Structures of compounds used to evaluate neuroprotection against glutamate toxicity in 661W cells.
Calcein-AM Cell Imaging
Cell viability was assessed using the membrane-permeant calcein-AM dye (Molecular Probes, Eugene, OR), a fluorogenic esterase substrate that is capable of penetrating the membrane of live cells. Once inside the cell, calcein-AM undergoes hydrolysis by the intracellular esterases, causing the calcein to be released and to fluoresce strongly, thereby enabling the measurement of change in fluorescent units. Cells were plated at a density of 3000 cells/well in 96-well plates. After 24 hours of drug and glutamate treatment, the media was removed from each of the wells in the 96-well plates and the cells were rinsed once in phosphate buffer (pH 7.2) (PBS; Fisher Scientific, Pittsburgh, PA). Then to each well was added 100 μL of 1 μg/mL calcein-AM (Calbiochem) in PBS and the plate was then incubated at 37°C for 20 minutes. Fluorescence was determined using an excitation/emission filter set of 485/530 nm read with a commercial plate reader (Tecan Infinite M200; Tecan Systems, Inc., San Jose, CA).
Immunoblotting
For immunoblotting analysis, cells were harvested by scraping, washed in PBS, resuspended, then homogenized and sonicated in RIPA buffer (1× PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM sodium orthovanadate, 10 mg/mL aprotinin, and 100 mg/mL phenylmethyl sulfonyl fluoride [PMSF]). Lysates were then centrifuged at 12,000g for 10 minutes at 4°C, and supernatants were collected for analysis. The protein concentrations were measured using a commercial protein assay (Bio-Rad Laboratories, Hercules, CA) before Western blot analysis. Protein from 661W cells and MCF7 cells was mixed with loading buffer, boiled for 5 minutes, separated by SDS-PAGE, and then transferred to polyvinylidene difluoride (PVDF) membranes (Immunobilon-P; Millipore, Bedford, MA). Membranes were blocked with 5% dry milk in PBS. Proteins were probed with specific ERα or ERβ antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) at 1:1000 dilution and incubated overnight at 4°C. The blots were washed and applied with goat-anti-rabbit secondary antibodies (Santa Cruz Biotechnology) at 1:1000 dilution. After washing, the blots were developed with an enhanced chemiluminescent kit (Pierce, Rockford, IL).
Statistical Analysis
All data are presented as mean ±1 SD. These data were analyzed using the ANOVA with Tukey's post hoc test for statistical evaluation against an α-level of 0.05. All bar graphs and EC50 (half-maximal effective concentration) calculations were made using a commercial statistics and data analysis software program (GraphPad Prism 5; GraphPad Software Inc., La Jolla, CA). For all groups, n = 8 wells and experiments were repeated three times to ensure consistency.
Results
ERα Is Expressed in 661W Cells
To determine whether 661W cells expressed ERα or ERβ, we performed Western blot analysis for the presence of these intracellular proteins. It has previously been shown that within the retina, ERs are distributed within the outer plexiform layer, the inner nuclear layer, the ganglion cell layer, and the nerve fiber.52 Our Western blot analysis confirmed the protein expression of ERα in 661W cells (Fig. 2). MCF7 cells were used as a positive control since it has previously been shown to express both ERα and ERβ,53 and HEK 293T cells were used as a negative control since they do not express ERα or ERβ.
Figure 2.

ERα is expressed in the 661W cells.
Effects of Glutamate on Cell Viability in 661W Cells
To determine whether 661W cells were susceptible to glutamate toxicity, cells were cultured and incubated with concentrations of glutamate ranging from 1 to 10 mM for 24 hours. Exposure of 661W cells to increasing concentrations of glutamate resulted in a dose-dependent decrease in cell viability (Fig. 3). Viability was reduced from 100.0 ± 4.2% in the vehicle control to 97.9 ± 1.4, 77.2 ± 3.0, 60.0 ± 2.7, 42.9 ± 2.3, 34.3 ± 2.1, 21.0 ± 1.7, and 12.9 ± 1.4% in the presence of 1, 2, 3, 4, 5, 7.5, and 10 mM glutamate, respectively. At a concentration of 5 mM glutamate, cell viability was reduced by approximately 60–70%. We used the concentration of 5 mM glutamate to evaluate the neuroprotective properties of each of the compounds of interest.
Figure 3.

Effects of glutamate on cell viability of 661W cells. Depicted are mean ± SD (n = 8 per group). *P < 0.05 vs. vehicle control.
Effects of 17β-Estradiol on Cell Viability in Glutamate-Treated 661W Cells
E2 has been well described as a neuroprotective agent in a variety of in vivo18–23 and in vitro models.24–39 Here we assessed its effects in 661W cells against oxidative damage following glutamate exposure. We tested E2 concentrations ranging from 10 nM to 10 μM in 661W cells treated with 5 mM glutamate (Fig. 4A). E2 modestly increased cell viability (P < 0.05) at the 10 μM concentration only with an EC50 of 4.4 μM.
Figure 4.

Effects of E2 (A) and nonfeminizing estrogen analogs ZYC-26 (B), ZYC-3 (C), and ZYC-23 (D) on cell viability in glutamate-treated 661W cells. Depicted are mean ± SD (n = 8 per group). *P < 0.05 vs. glutamate alone–treated cells. ICI, ICI 182780.
Effects of Nonfeminizing Estrogens on Cell Viability in Glutamate-Treated 661W Cells
Treatment of the 661W cells with phenolic compounds ZYC-26 and ZYC-3 provided protection, whereas treatment with the nonphenolic compound ZYC-23 did not. ZYC-26, one of the nonfeminizing estrogen compounds, protected 661W cells against 5 mM glutamate-induced oxidative damage. We saw statistically significant neuroprotection at concentrations ranging from 100 nM to 10 μM (Fig. 4B). Viability increased from 79.2 ± 2.1% at 100 nM, to 85.0 ± 1.2% at 1 μM to 94.6 ± 1.5% at 10 μM, with an EC50 of 65.4 nM. Preincubation with 500 nM ICI 182780 with 1 μM ZYC-26 and 5 mM glutamate did not produce any significant changes in cell viability (Fig. 4B). Similarly, ZYC-3, also a nonfeminizing estrogen, was able to provide statistically significant protection in the 661W cells exposed to a 5 mM glutamate insult at concentrations ranging from 100 nM to 10 μM (Fig. 4C). ZYC-3 increased cell viability at 100 nM from 32.3 ± 0.9% to 88.3 ± 1.6% at 10 μM, with an EC50 of 444 nM. In contrast, ZYC-23 was not able to provide any protection from a 5 mM glutamate insult at the concentration range of 10 nM to 10 μM (Fig. 4D). ZYC-23 has an O-methyl group attached at the 3 position of the phenolic A ring (Fig. 1), abolishing the antioxidant characteristics of the compound. To determine whether the protection observed involved ERα or ERβ, we used 500 nM ICI 182780, a concentration 1500-fold greater than the IC50 (dose that causes half-maximal inhibition by an antagonist) of ICI 182780 in both ERα and ERβ.54 The addition of ICI 182780 at 500 nM during treatment with 1 μM ZYC-3 produced no significant attenuation of cell viability. These data support the concept that for E2 and nonfeminizing estrogens to be protective, it is necessary that they possess a phenol ring; this establishes the importance of the phenolic ring in the preservation of cell viability in 661W cells.
Effects of ERα- and ERβ-Specific Agonists on Cell Viability in Glutamate-Treated 661W Cells
The inability of the ER antagonist ICI 182780 to block the neuroprotection by nonfeminizing estrogens suggested that the ERs are not involved in the action in 661W cells. We then assessed the effects of specific ER agonists to determine whether they were protective in 661W cells. Cells treated with PPT, an ERα-specific agonist, were not protected from a 5 mM glutamate insult at concentrations ranging from 10 nM to 10 μM (Fig. 5A). Similarly, DPN, a relatively selective ERβ agonist had no protective effect on 661W cells against a 5 mM glutamate insult at concentrations ranging from 10 nM to 10 μM (Fig. 5B).
Figure 5.

Effects of ERα preferring agonist, PPT (A) and an ERβ-preferring agonist DPN (B) on cell viability in glutamate-treated 661W cells. Depicted are mean ± SD (n = 8 per group). *P < 0.05 vs. glutamate alone–treated cells.
Effects of the GPR30 Agonist and Antagonist on Cell Viability in Glutamate-Treated 661W Cells
The membrane ER (mER) or GPR30 is a membrane-associated Gq-protein–coupled ER. Since our data did not implicate ERα and ERβ in mediating the observed neuroprotection, we assessed another potential ER, the GPR30. To assess the role of this receptor, we utilized the GPR30-preferring ER agonist G1, which provided a dose-dependent, statistically significant neuroprotection against a 5 mM glutamate insult at concentrations 100 nM, 1 μM, and 10 μM (Fig. 6). Treatment with the GPR30 antagonist G15 did not markedly affect the neuroprotective activity of G1 (Fig. 7A). We assessed the ability of G15 to antagonize the neuroprotective effects of E2. G15 did not antagonize E2-induced protection from glutamate toxicity (Fig. 7B). G15 only modestly inhibited the protective effects of ZYC-26 and ZYC-3, again suggesting that their mechanism of protection was not via GPR30 (Figs. 7C, 7D).
Figure 6.

Effects of G1 on cell viability in glutamate-treated 661W cells. Depicted are mean ± SD (n = 8 per group). *P < 0.05 vs. glutamate alone–treated cells.
Figure 7.

Effects of the GPR30 antagonist, G15, on G1 (A), E2 (B), ZYC-26 (C), and ZYC-3 (D)–induced neuroprotection of cell viability in glutamate-treated 661W cells. Depicted are mean ± SD (n = 8 per group). *P < 0.05 vs. glutamate alone–treated cells.
Discussion
Age-related macular degeneration (AMD) is the leading cause of visual loss in patients older than 60 years of age in the United States and developing countries. The etiology of AMD remains unknown, but researchers have shown that a combination of genetic factors55–57 and oxidative stress factors in the retina may influence the pathology of the disease.58,59 Oxidative stress was shown to contribute to the development of AMD by the Age-Related Eye Disease Study.60 Other factors that have also been shown to influence AMD include environment, sex, smoking, diet, and nutrition. Several of these factors induce the generation of elevated reactive oxygen species (ROS) in the retina, which can eventually lead to increased oxidative damage.61,62
One of the earliest studies demonstrating the neuroprotective effects of estrogen focused on the ability of a synthetic estrogen analog to protect in an in vivo model of retinitis pigmentosa (RP). In that study, which used animals at postnatal day 9, the estrogen analog treatment resulted in an outer nuclear layer that was almost twice the thickness in untreated controls.63 An in vitro model of N-methyl-d-aspartate–induced excitotoxic glaucomatous retinal ganglion cell (RGC) death also treated with the estrogen analog showed that the primary RGCs were protected from the insult when treated.63 In another study, 17β-estradiol protected RGCs from changes in the synaptic connectivity associated with ischemia prior to apoptosis and ischemia-induced apoptosis.64 In a study that used RGCs, it was shown that RGC survival was greatly reduced in rats that had received oophorectomies, but in similar animals that received intravitreal 17β-estradiol injections, RGC loss was reduced.65
In our study, we demonstrated that the nonfeminizing estrogen analogs ZYC-26 and ZYC-3, as well as the GPR30 agonist G1, are able to protect 661W cells from glutamate-induced cell death. The neuroprotective abilities of these compounds were independent of ERα and ERβ in view of the observations that PPT and DPN failed to offer any protection within this model and ICI 182780 did not antagonize the observed neuroprotection. Interestingly, GPR30 may mediate some level of protection, based on our observation that G1 was potently neuroprotective. However, the putative membrane ER did not mediate the neuroprotection seen with the nonfeminizing estrogens, given that G15 was only minimally effective in antagonizing the neuroprotective effects of ZYC-26 and ZYC-3 (Figs. 7C, 7D).
The relative potency of the compounds tested for protection of 661W cells was ZYC-26 > G1 > ZYC-3 > E2 > > > ZYC-23, PPT, and DPN (the Table). The most potent of these compounds, ZYC-3, ZYC-26, and G1, were nonfeminizing51,66 compared with the inactive compounds: PPT, an ERα agonist, DPN, an ERβ agonist, and ZYC-23. The inverse relationship between potency in this 661W model and ER binding argues that neuroprotection and ER binding are clearly distinguishable processes. We have made similar observations concerning separation of ER binding and neuroprotection in hippocampal cell types and in animal models of cerebral ischemia.48
Table.
Potency of Estrogen-Related Compounds in Protecting 661W Cells from Glutamate-Induced Cell Death
|
Compounds |
ED50 (μM) |
| E2 | 4.4 |
| ZYC-3 | 0.44 |
| ZYC-26 | 0.065 |
| G1 | 0.12 |
| ZYC-23 | >10 |
| PPT | >10 |
| DPN | >10 |
Inasmuch as PPT and DPN both possess phenol rings (Fig. 1) and exert antioxidant activity in other toxicity models,67 we were surprised that neither compound provided protection of 661W cells against glutamate insult. Although these data clearly demonstrate the lack of ER mediation of the responses observed, they are at odds with the idea that phenolic rings are essential for neuroprotection of estrogen-like compounds. It is also worth noting that G1, which was potently neuroprotective in this assay, is not an effective antioxidant66,68 and lacks a phenolic ring (Fig. 1). Collectively, the observed neuroprotection against a known prooxidant stressor, high concentrations of glutamate, is not likely due to the pure antioxidant activities of the compounds tested.
The present study demonstrates that E2, nonfeminizing estrogens, and G1 are neuroprotective through a mechanism that does not require classical ERs or GPR30. These data support the non–ER-mediated action of E2 and nonfeminizing estrogens that have been seen in a number of studies both in vivo18–23 and in vitro.24–39 There are multiple lines of evidence that the effects observed are not mediated by ERs. First, ZYC-3 and ZYC-26 do not bind to either ERα or ERβ. Second, their effects are not antagonized by the pan-ER antagonist, ICI 182780. Third, the ER-selective agonists PPT and DPN did not show any evidence of neuroprotection in this model. Although G1 was active in this model, its effects were not antagonized by G15. Similarly, the effects of E2, ZYC-26, and ZYC-3 were not antagonized either by ICI 182780 or G15. Finally, the effective doses of compounds were much higher than the low nM ED50 for estrogens at ERα or ERβ. Collectively, these data provide strong support for the idea that neuroprotection in 661W cells is a non–ER-mediated mechanism.
In summary, the present observations suggest that the treatment of neurodegenerative eye diseases could be achieved with nonfeminizing estrogens and thereby could avoid the side effects of estrogens, even with chronic administration.
Acknowledgments
The authors thank Robert Luedtke for his help with ChemDraw software, Tim Richardson for his help with EC50 calculations, and Saumyendra Sarkar for his help with Western blot.
Footnotes
Supported in part by Department of Defense Grant W81XWH-10-2-0003.
Disclosure: E. Nixon, None; J.W. Simpkins, None
References
- 1. Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989; 2: 1547– 1558. [DOI] [PubMed] [Google Scholar]
- 2. Bailey TA, Kanuga N, Romero IA, Greenwood J, Luthert PJ, Cheetham ME. Oxidative stress affects the junctional integrity of retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2004;. 45: 675– 684. [DOI] [PubMed] [Google Scholar]
- 3. Usui S, Oveson BC, Lee SY, et al. NADPH oxidase plays a central role in cone cell death in retinitis pigmentosa. J Neurochem. 2009;. 110: 1028– 1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gutteridge JM, Halliwell B. Comments on review of Free Radicals in Biology and Medicine, second edition, by Halliwell Barry, Gutteridge John M. C. Free Radic Biol Med. 1992;. 12: 93– 95. [DOI] [PubMed] [Google Scholar]
- 5. Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;. 59: 527– 605. [DOI] [PubMed] [Google Scholar]
- 6. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005; 120: 483– 495. [DOI] [PubMed] [Google Scholar]
- 7. Augood CA, Vingerling JR, de Jong PT, et al. Prevalence of age-related maculopathy in older Europeans: the European Eye Study (EUREYE). Arch Ophthalmol. 2006;. 124: 529– 535. [DOI] [PubMed] [Google Scholar]
- 8. Javitt JC, Zhou Z, Maguire MG, Fine SL, Willke RJ. Incidence of exudative age-related macular degeneration among elderly americans. Ophthalmology. 2003; 110: 1534– 1539. [DOI] [PubMed] [Google Scholar]
- 9. Friedman DS, O'Colmain BJ, Munoz B, et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;. 122: 564– 572. [DOI] [PubMed] [Google Scholar]
- 10. Beatty S, Nolan J, Kavanagh H, O'Donovan O. Macular pigment optical density and its relationship with serum and dietary levels of lutein and zeaxanthin. Arch Biochem Biophys. 2004; 430: 70– 76. [DOI] [PubMed] [Google Scholar]
- 11. Liang FQ, Godley BF. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Exp Eye Res. 2003;. 76: 397– 403. [DOI] [PubMed] [Google Scholar]
- 12. Hinton DR, He S, Lopez PF. Apoptosis in surgically excised choroidal neovascular membranes in age-related macular degeneration. Arch Ophthalmol. 1998;. 116: 203– 209. [DOI] [PubMed] [Google Scholar]
- 13. Jin GF, Hurst JS, Godley BF. Hydrogen peroxide stimulates apoptosis in cultured human retinal pigment epithelial cells. Curr Eye Res. 2001;. 22: 165– 173. [DOI] [PubMed] [Google Scholar]
- 14. Feskanich D, Cho E, Schaumberg DA, Colditz GA, Hankinson SE. Menopausal and reproductive factors and risk of age-related macular degeneration. Arch Ophthalmol. 2008;. 126: 519– 524. [DOI] [PubMed] [Google Scholar]
- 15. Haan MN, Klein R, Klein BE, et al. Hormone therapy and age-related macular degeneration: the Women's Health Initiative Sight Exam Study. Arch Ophthalmol. 2006;. 124: 988– 992. [DOI] [PubMed] [Google Scholar]
- 16. Vingerling JR, Dielemans I, Witteman JC, Hofman A, Grobbee DE, de Jong PT. Macular degeneration and early menopause: a case-control study. BMJ. 1995;. 310: 1570– 1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Klein R, Klein BE, Jensen SC, Meuer SM. The five-year incidence and progression of age-related maculopathy: the Beaver Dam Eye Study. Ophthalmology. 1997;. 104: 7– 21. [DOI] [PubMed] [Google Scholar]
- 18. Simpkins JW, Rajakumar G, Zhang YQ, et al. Estrogens may reduce mortality and ischemic damage caused by middle cerebral artery occlusion in the female rat. J Neurosurg. 1997;. 87: 724– 730. [DOI] [PubMed] [Google Scholar]
- 19. Alkayed NJ, Harukuni I, Kimes AS, London ED, Traystman RJ, Hurn PD. Gender-linked brain injury in experimental stroke. Stroke. 1998;. 29: 159– 166. [DOI] [PubMed] [Google Scholar]
- 20. Dubal DB, Kashon ML, Pettigrew LC, et al. Estradiol protects against ischemic injury. J Cereb Blood Flow Metab. 1998;. 18: 1253– 1258. [DOI] [PubMed] [Google Scholar]
- 21. Sudo S, Wen TC, Desaki J, et al. Beta-estradiol protects hippocampal CA1 neurons against transient forebrain ischemia in gerbil. Neurosci Res. 1997;. 29: 345– 354. [DOI] [PubMed] [Google Scholar]
- 22. Fukuda K, Yao H, Ibayashi S, et al. Ovariectomy exacerbates and estrogen replacement attenuates photothrombotic focal ischemic brain injury in rats. Stroke. 2000; 31: 155– 160. [DOI] [PubMed] [Google Scholar]
- 23. Mendelowitsch A, Ritz MF, Ros J, Langemann H, Gratzl O. 17beta-Estradiol reduces cortical lesion size in the glutamate excitotoxicity model by enhancing extracellular lactate: a new neuroprotective pathway. Brain Res. 2001;. 901: 230– 236. [DOI] [PubMed] [Google Scholar]
- 24. Behl C, Widmann M, Trapp T, Holsboer F. 17-beta Estradiol protects neurons from oxidative stress-induced cell death in vitro. Biochem Biophys Res Commun. 1995;. 216: 473– 482. [DOI] [PubMed] [Google Scholar]
- 25. Behl C, Skutella T, Lezoualc'h F, et al. Neuroprotection against oxidative stress by estrogens: structure–activity relationship. Mol Pharmacol. 1997;. 51: 535– 541. [PubMed] [Google Scholar]
- 26. Sawada H, Ibi M, Kihara T, Urushitani M, Akaike A, Shimohama S. Estradiol protects mesencephalic dopaminergic neurons from oxidative stress-induced neuronal death. J Neurosci Res. 1998;. 54: 707– 719. [DOI] [PubMed] [Google Scholar]
- 27. Singer CA, Rogers KL, Dorsa DM. Modulation of bcl-2 expression: a potential component of estrogen protection in NT2 neurons. Neuroreport. 1998;. 9: 2565– 2568. [DOI] [PubMed] [Google Scholar]
- 28. Moosmann B, Behl C. The antioxidant neuroprotective effects of estrogens and phenolic compounds are independent from their estrogenic properties. Proc Natl Acad Sci USA. 1999;. 96: 8867– 8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Green PS, Perez EJ, Calloway T, Simpkins JW. Estradiol attenuation of beta-amyloid-induced toxicity: a comparison. J Neurocytol. 2000;. 29: 419– 423. [DOI] [PubMed] [Google Scholar]
- 30. Bishop J, Simpkins JW. Estradiol treatment increases viability of glioma and neuroblastoma cells in vitro. Mol Cell Neurosci. 1994;. 5: 303– 308. [DOI] [PubMed] [Google Scholar]
- 31. Green PS, Bishop J, Simpkins JW. 17α-Estradiol exerts neuroprotective effects on SK-N-SH cells. J Neurosci. 1997; 17: 511– 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Green PS, Gordon K, Simpkins JW. Phenolic A ring requirement for the neuroprotective effects of steroids. J Steroid Biochem Mol Biol. 1997; 63: 229– 235. [DOI] [PubMed] [Google Scholar]
- 33. Bae YH, Hwang JY, Kim YH, Koh JY. Anti-oxidative neuroprotection by estrogens in mouse cortical cultures. J Korean Med Sci. 2000;. 15: 327– 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Regan RF, Guo Y. Estrogens attenuate neuronal injury due to hemoglobin, chemical hypoxia, and excitatory amino acids in murine cortical cultures. Brain Res. 1997; 764: 133– 140. [DOI] [PubMed] [Google Scholar]
- 35. Wilson ME, Dubal DB, Wise PM. Estradiol protects against injury-induced cell death in cortical explant cultures: a role for estrogen receptors. Brain Res. 2000;. 873: 235– 242. [DOI] [PubMed] [Google Scholar]
- 36. Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid beta-peptide toxicity in hippocampal neurons. J Neurochem. 1996;. 66: 1836– 1844. [DOI] [PubMed] [Google Scholar]
- 37. Singer CA, Rogers KL, Strickland TM, Dorsa DM. Estrogen protects primary cortical neurons from glutamate toxicity. Neurosci Lett. 1996;. 212: 13– 16. [DOI] [PubMed] [Google Scholar]
- 38. Zaulyanov LL, Green PS, Simpkins JW. Glutamate receptor requirement for neuronal death from anoxia-reoxygenation: an in vitro model for assessment of the neuroprotective effects of estrogens. Cell Mol Neurobiol. 1999;. 19: 705– 718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Green PS, Simpkins JW. Neuroprotective effects of estrogens: potential mechanisms of action. Int J Dev Neurosci. 2000;. 18: 347– 358. [DOI] [PubMed] [Google Scholar]
- 40. Chen JQ, Cammarata PR, Baines CP, Yager JD. Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim Biophys Acta. 2009;. 1793: 1540– 1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Simpkins JW, Singh M. More than a decade of estrogen neuroprotection. Alzheimers Dement. 2008;. 4: S131– S136. [DOI] [PubMed] [Google Scholar]
- 42. Simpkins JW, Yi KD, Yang SH. Role of protein phosphatases and mitochondria in the neuroprotective effects of estrogens. Front Neuroendocrinol. 2009;. 30: 93– 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Simpkins JW, Yang SH, Sarkar SN, Pearce V. Estrogen actions on mitochondria—physiological and pathological implications. Mol Cell Endocrinol. 2008;. 290: 51– 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prokai L, Prokai-Tatrai K, Perjesi P, et al. Quinol-based cyclic antioxidant mechanism in estrogen neuroprotection. Proc Natl Acad Sci USA. 2003;. 100: 11741– 11746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Prokai L, Prokai-Tatrai K, Perjesi P, Zharikova AD, Simpkins JW. Quinol-based metabolic cycle for estrogens in rat liver microsomes. Drug Metab Dispos. 2003;. 31: 701– 704. [DOI] [PubMed] [Google Scholar]
- 46. Wang X, Simpkins JW, Dykens JA, Cammarata PR. Oxidative damage to human lens epithelial cells in culture: estrogen protection of mitochondrial potential, ATP, and cell viability. Invest Ophthalmol Vis Sci. 2003;. 44: 2067– 2075. [DOI] [PubMed] [Google Scholar]
- 47. Wang J, Green PS, Simpkins JW. Estradiol protects against ATP depletion, mitochondrial membrane potential decline and the generation of reactive oxygen species induced by 3-nitroproprionic acid in SK-N-SH human neuroblastoma cells. J Neurochem. 2001;. 77: 804– 811. [DOI] [PubMed] [Google Scholar]
- 48. Simpkins JW, Yang SH, Liu R, et al. Estrogen-like compounds for ischemic neuroprotection. Stroke. 2004; 35: 2648– 2651. [DOI] [PubMed] [Google Scholar]
- 49. Vina J, Sastre J, Pallardo FV, Gambini J, Borras C. Role of mitochondrial oxidative stress to explain the different longevity between genders: protective effect of estrogens. Free Radic Res. 2006;. 40: 1359– 1365. [DOI] [PubMed] [Google Scholar]
- 50. Borras C, Gambini J, Gomez-Cabrera MC, et al. 17beta-Oestradiol up-regulates longevity-related, antioxidant enzyme expression via the ERK1 and ERK2[MAPK]/NFkappaB cascade. Aging Cell. 2005;. 4: 113– 118. [DOI] [PubMed] [Google Scholar]
- 51. Perez E, Liu R, Yang SH, Cai ZY, Covey DF, Simpkins JW. Neuroprotective effects of an estratriene analog are estrogen receptor independent in vitro and in vivo. Brain Res. 2005; 1038: 216– 222. [DOI] [PubMed] [Google Scholar]
- 52. Kobayashi K, Kobayashi H, Ueda M, Honda Y. Estrogen receptor expression in bovine and rat retinas. Invest Ophthalmol Vis Sci. 1998;. 39: 2105– 2110. [PubMed] [Google Scholar]
- 53. Ivanova MM, Mazhawidza W, Dougherty SM, Klinge CM. Sex differences in estrogen receptor subcellular location and activity in lung adenocarcinoma cells. Am J Respir Cell Mol Biol. 2010;. 42: 320– 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. de Cupis A, Noonan D, Pirani P, Ferrera A, Clerico L, Favoni RE. Comparison between novel steroid-like and conventional nonsteroidal antioestrogens in inhibiting oestradiol- and IGF-I-induced proliferation of human breast cancer-derived cells. Br J Pharmacol. 1995;. 116: 2391– 2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Klein ML, Mauldin WM, Stoumbos VD. Heredity and age-related macular degeneration. Observations in monozygotic twins. Arch Ophthalmol. 1994;. 112: 932– 937. [DOI] [PubMed] [Google Scholar]
- 56. Seddon JM, Ajani UA, Mitchell BD. Familial aggregation of age-related maculopathy. Am J Ophthalmol. 1997;. 123: 199– 206. [DOI] [PubMed] [Google Scholar]
- 57. Ishida BY, Bailey KR, Duncan KG, et al. Regulated expression of apolipoprotein E by human retinal pigment epithelial cells. J Lipid Res. 2004;. 45: 263– 271. [DOI] [PubMed] [Google Scholar]
- 58. Beatty S, Koh H, Phil M, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;. 45: 115– 134. [DOI] [PubMed] [Google Scholar]
- 59. Cai J, Nelson KC, Wu M, Sternberg P Jr, Jones DP. Oxidative damage and protection of the RPE. Prog Retin Eye Res. 2000;. 19: 205– 221. [DOI] [PubMed] [Google Scholar]
- 60. Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;. 119: 1417– 1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Seddon JM, Willett WC, Speizer FE, Hankinson SE. A prospective study of cigarette smoking and age-related macular degeneration in women. JAMA. 1996; 276: 1141– 1146. [PubMed] [Google Scholar]
- 62. Smith W, Assink J, Klein R, et al. Risk factors for age-related macular degeneration: pooled findings from three continents. Ophthalmology. 2001;. 108: 697– 704. [DOI] [PubMed] [Google Scholar]
- 63. Dykens JA, Carroll AK, Wiley S, et al. Photoreceptor preservation in the S334ter model of retinitis pigmentosa by a novel estradiol analog. Biochem Pharmacol. 2004; 68: 1971– 1984. [DOI] [PubMed] [Google Scholar]
- 64. Kaja S, Yang SH, Wei J, et al. Estrogen protects the inner retina from apoptosis and ischemia-induced loss of Vesl-1L/Homer 1c immunoreactive synaptic connections. Invest Ophthalmol Vis Sci. 2003; 44: 3155– 3162. [DOI] [PubMed] [Google Scholar]
- 65. Nakazawa T, Takahashi H, Shimura M. Estrogen has a neuroprotective effect on axotomized RGCs though ERK signal transduction pathway. Brain Res. 2006; 1093: 141– 149. [DOI] [PubMed] [Google Scholar]
- 66. Lebesgue D, Traub M, De Butte-Smith M, et al. Acute administration of non-classical estrogen receptor agonists attenuates ischemia-induced hippocampal neuron loss in middle-aged female rats. PLoS One. 2010; 5: e8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Richardson TE, Yang SH, Wen Y, Simpkins JW. Estrogen protection in Friedreich's ataxia skin fibroblasts. Endocrinology. 2011; 152: 2742– 2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gingerich S, Kim GL, Chalmers JA, et al. Estrogen receptor alpha and G-protein coupled receptor 30 mediate the neuroprotective effects of 17beta-estradiol in novel murine hippocampal cell models. Neuroscience. 2010; 170: 54– 66. [DOI] [PubMed] [Google Scholar]