

Abstract
Scaling of a microphysiological system (MPS) or physiome-on-a-chip is arguably two interrelated, modeling-based activities: on-platform scaling and in vitro-in vivo translation. This dual approach reduces the need to perfectly rescale and mimic in vivo physiology, an aspiration that is both extremely challenging and not substantively meaningful because of uncertain relevance of any specific physiological condition. Accordingly, this perspective offers a tractable approach for designing interacting MPSs and relating in vitro results to analogous context in vivo.
Significant efforts to develop single and interacting microphysiological systems (MPSs)1—engineered, in vitro human tissues that mimic human physiology—have been underway for several decades. Sometimes called “body-on-a-chip,” we prefer “physiome-on-a-chip” to emphasize function rather than literal reproduction of the body. Interest has accelerated in response to advances in culture of human primary cells and induced pluripotent stem cells, tissue engineering entering the clinic, and the ongoing need for improved preclinical-to-clinical translation in drug development. Serious attempts at integrating these physiologically representative MPSs on a single platform are just recently underway.1
A fundamental challenge is “scaling”—the specification of tissue dimensions and operating conditions to capture desired physiology and enable studies of interest. Several approaches include allometric scaling, histological sections, and functional scaling.2 Currently, the most favored is functional scaling—designing the MPS to reproduce tissue or organ functions, such as metabolism rate, blood residence time, or oxygen transport.1–3 Exactly which functions should be recapitulated is a matter of planned platform use and a researcher's vision. A frequent conception is that proper scaling will sufficiently reproduce in vivo physiology such that “the same” measurements of response will be obtained in vitro as in vivo without further translational analysis.
Our “physiome-on-a-chip” experience has driven us to pragmatically modify our objective to developing MPSs and platforms from which results are understandable within the platform's context and are translatable to in vivo physiology using mathematical models. Practically, this splits scaling into two interrelated activities: “on-platform scaling,” the specification of MPS and platform characteristics and operating parameters, and in vitro-in vivo translation (IVIVT), the process of relating platform results to in vivo physiology (Figure 1). On-platform scaling must reproduce biological functions of a tissue well enough to allow observation and measurement, but not identically to an organ. IVIVT then uses mathematical models to relate in vitro results to the analogous in vivo context, preferably on a mechanistic, not just correlative or extrapolative, basis. Relevant in vitro to in vivo differences are accounted for in this process. Approaches to IVIVT depend on on-platform scaling choices, so while their aims differ (design vs. translation) both must be considered throughout platform development and may well use the same mathematical models.
Figure 1.
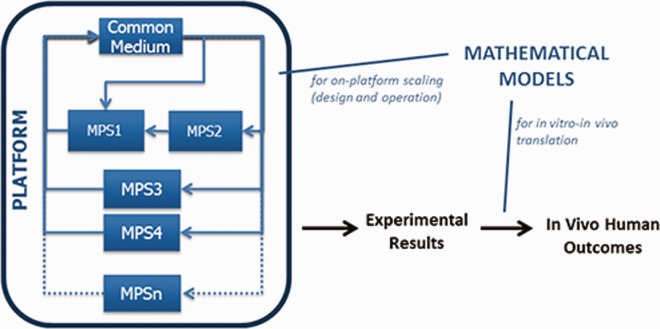
Multi-MPS platforms will require mechanistic mathematical models for both on-platform scaling and in vitro-in vivo translation. The models for each are likely to have common elements, while also being specialized for each intended use.
ON-PLATFORM SCALING
Because the purpose of MPS development is to create better in vitro models of human function, scaling based on functions rather than dimensions is sensible. It begs the question, however, of which functions take precedence, especially in multi-MPS platforms. Although a common default has been to select an organ's most defining functions, an alternate is to consider a function's purpose on the platform: does it provide a crucial operational or regulatory role (“maintenance functions”), or simply an indication of MPS activity (“output functions”). For example, if a lung MPS is responsible for oxygenating a platform, then its oxygen transport properties and operating conditions (e.g., surface area, diffusivity, and oxygen concentration) must be designed accordingly, but if oxygen transport is simply an indicator of MPS health, then design need only enable proper measurement. The practical implications are that design requirements for maintenance and output purposes will frequently be different, and, interestingly, there is no single correct scaling for a given function.
Another consideration is that platforms must recapitulate multiple functions simultaneously, so a systemic scaling approach using multivariate optimization is highly desirable. Such on-platform scaling must enable desired functions while accounting for relative tissue and medium dimensions, dynamics, MPS-MPS interactions (e.g., via released mediators or drug metabolites), and desired research applications. We recently attempted this to revise a liver MPS (comprising human hepatocytes and Kupffer cells in a 3D structure) to improve pharmacokinetic (PK) measurability and pharmacodynamic (PD) response. We wished to simultaneously attain certain biological function (e.g., viability, oxygen utilization, cytochrome P450 activity) and PK and PD responses (e.g., drug and hormone metabolism, autocrine activity, inflammation), while constrained by an established tissue mass and requirements for nutrients, oxygenation, and medium change interval. Design parameters included medium volume relative to tissue mass, medium circulation rate, and surface area for oxygen exchange. With this problem definition, we utilized a systems pharmacology mathematical model including flow; kinetics of drugs, hormones, and tissue-produced factors; oxygen utilization; and nutrient requirements to identify ranges of design parameter values that best met functional and operational requirements. Although not all features were simultaneously feasible with current hardware, the analysis provides a basis for future design iterations to better accommodate all constraints.
Extension of this approach to multi-MPS platforms should be reasonably straightforward using appropriate systems pharmacology models4 to identify key modulators of basic biology, PK and PD within desired operational schemas of an MPS or multi-MPS platform. Such a functional approach to on-platform scaling requires careful consideration of both organ functions of interest and desired platform uses; different uses may result in differently specified platforms.
IVIVT
Although recapitulating human organ physiology is a key MPS goal, even ideally designed multi-MPS platforms cannot represent the complete human context. Additional interpretive steps—IVIVT—will be needed to predict human responses from platform results to account for missing organs and functions, organ and media size mismatches, and drug exposure differences. Especially challenging will be accounting for variations in physiological and pathological states because human responses frequently vary due to exercise, feeding, and rest; acute and chronic illnesses; and aging.
On-platform scaling of MPSs is still an immature endeavor and IVIVT even more so. Nonetheless, it is useful to consider what will be required for MPSs to be more predictive than existing preclinical models. Where substantial mechanistic understanding exists, such as for PK and absorption, distribution, metabolism, and excretion, translational mathematical models will likely suffice. A critical consideration is what metrics to translate; quantifying fundamental aspects of biological functions rather than representing them by phenomenological parameters seems necessary. For example, our measurements of typical PK parameters (drug half-life, clearance) in our liver MPS and multi-MPS platforms are different than in humans because they reflect major system characteristics: our MPS medium-to-tissue ratio is larger than blood-to-liver ratio in vivo so drug disappearance is slower on-platform for the same initial drug concentration. To alleviate this, we are developing more mechanistic models for both in vitro and in vivo systems that isolate elimination processes from system characteristics, thereby defining tissue-specific elimination rate parameters. Such parameters should be better comparators from in vitro and in vivo, with a major outstanding challenge being measurement or derivation of them in in vivo systems. Similar approaches should also hold for IVIVT of PD, toxicodynamics, and therapeutic window in which enough mechanistic information exists to separate mechanism from system context.
Practically, validation of an IVIVT approach will require in vivo data. For pharmacological uses, this implies focusing on drugs already studied in humans, as done for development of liver microsomes and hepatocyte cultures as predictive systems for PK.5 Nonetheless, research on fundamental biology in MPSs will be fruitful regardless of extensive IVIVT validation.
In contrast to PK and absorption, distribution, metabolism, and excretion, areas of lesser understood biology, disease processes, and drug targets may require significant rethinking of how biology itself is observed and interpreted long before translational work is attempted. Even for in vitro studies, the systems biology field has learned that “information-centric” rather than “mechanism-centric” models are generally required to predict complex cell behavioral responses. For such behaviors as proliferation, migration, and cytokine production, the dynamics of multiple signaling pathways must be integrated, but insufficient understanding of regulatory biochemistry frequently impedes construction of experimentally verifiable mathematical models.6 In contrast, computational frameworks that are relational (e.g., multilinear regression or partial least-squares regression) or logical (e.g., Boolean logic or fuzzy logic) in nature have shown the most powerful predictive capabilities across physiologically diverse contexts despite experimental measurement limitations.7 These frameworks provide input-output “transfer function” models similar to PD modeling but comprise many more variables, as required for a systems perspective.8 Similar approaches are likely required to understand the complex biology in MPS platforms, much less predict in vivo responses from them.
Unfortunately, measurement types obtainable in vivo to populate relational or logical PD models are much more limited than in vitro. There might, however, be sufficient measurements offering surrogate information suitable to construct comparable input-output transfer function models for translation between in vitro and in vivo, as evidenced by equally valid alternative models for functional responses based on different kinds of molecular data, including mRNA, miRNA, protein, phosphoprotein, and protein activities.9 To discern key mechanisms of the invasive inflammatory disease endometriosis, for example, experimental measurements of cytokines, growth factors, and protease activities from human patient peritoneal fluid samples have been used in multivariate classification and network models.9,10 To study this disease in vitro, these cellular and molecular properties could be similarly quantified in an endometrium MPS and analogous models constructed; translation between in vitro and in vivo models using “transfer functions” is envisioned. Figure 2 illustrates the idea of utilizing system models to capture and translate between in vivo and in vitro systems based on measurements feasible in both, buttressed by additional knowledge enabled by superior measurement and manipulation capabilities in vitro.
Figure 2.
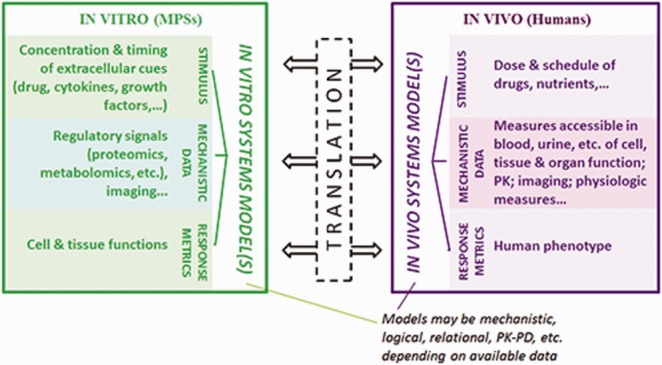
Translation from in vitro studies to in vivo studies will require computational systems models for each that relate stimuli, mechanism, and outcomes. Translation will then occur between the two respective models. These models are most likely to involve quantitative multivariate relational algorithms, relating molecular-/cellular-level measurements to tissue-level phenotypic measurements, although more mechanistic models or simpler PK-PD models may be appropriate at times. For in vitro studies, the molecular-/cellular-level measurements can frequently derive from highly invasive techniques, whereas minimally invasive measurements are generally required for in vivo studies. Hence, the types of mechanistic data used in the models will need to be accessible in both environments, most likely by providing surrogate information about more proximal regulatory molecular/cellular processes.
SUMMARY
Our perspective is that “scaling” of MPSs comprises more than system specifications. Multivariate on-platform scaling is critical to provide appropriate mechanistic interactions within and between MPS units; however, relating observations from in vitro platforms to observations in the related in vivo systems also requires translation using mathematical models. Observations in vitro need not, and likely do not, mimic those in vivo because of overwhelming uncertainty about contextual variables and variation in living organisms. However, insights gained from in vitro systems should be translatable to insights into in vivo systems by means of appropriate modeling relationships.
Acknowledgments
We gratefully acknowledge support from the DARPA Microphysiological Systems Program (W911NF-12-2-0039), the NIH Microphysiological Systems Program (4-UH3-TR000496-03), and the Institute for Collaborative Biotechnologies through grant W911NF-09-0001 from the U.S. Army Research Office.
Author Contributions: C.L.S., M.C., and D.A.L. wrote the manuscript and developed the perspectives within.
Conflict of Interest: The authors declared no conflict of interest.
References
- Sung JH, et al. Microfabricated mammalian organ systems and their integration into models of whole animals and humans. Lab Chip. 2013;13:1201–1212. doi: 10.1039/c3lc41017j. ) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikswo JP, et al. Scaling and systems biology for integrating multiple organs-on-a-chip. Lab Chip. 2013;13:3496–3511. doi: 10.1039/c3lc50243k. ) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes C, Labuz JM, Leung BM, Inoue M, Chun TH. Takayama S. On being the right size: scaling effects in designing a human-on-a-chip. Integr. Biol. (Camb.) 2013;5:1149–1161. doi: 10.1039/c3ib40040a. & ) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, et al. Quantitative systems pharmacology approaches applied to microphysiological systems (MPS): data interpretation and multi-MPS integration. CPT Pharmacometrics Syst. Pharmacol. doi: 10.1002/psp4.12010. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown HS, Griffin M. Houston JB. Evaluation of cryopreserved human hepatocytes as an alternative in vitro system to microsomes for the prediction of metabolic clearance. Drug Metab. Dispos. 2007;35:293–301. doi: 10.1124/dmd.106.011569. & ) [DOI] [PubMed] [Google Scholar]
- Janes KA. Lauffenburger DA. A biological approach to computational models of proteomic networks. Curr. Opin. Chem. Biol. 2006;10:73–80. doi: 10.1016/j.cbpa.2005.12.016. & ) [DOI] [PubMed] [Google Scholar]
- Terfve C, et al. CellNOptR: a flexible toolkit to train protein signaling networks to data using multiple logic formalisms. BMC Syst. Biol. 2012;6:133. doi: 10.1186/1752-0509-6-133. ) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirouac DC. Onsum MD. Using network biology to bridge pharmacokinetics and pharmacodynamics in oncology. CPT Pharmacometrics Syst. Pharmacol. 2013;2:e71. doi: 10.1038/psp.2013.38. & ) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beste MT, et al. Molecular network analysis of endometriosis reveals a role for c-Jun-regulated macrophage activation. Sci. Transl. Med. 2014;6:222ra216. doi: 10.1126/scitranslmed.3007988. ) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MA, et al. ADAM-10 and −17 regulate endometriotic cell migration via concerted ligand and receptor shedding feedback on kinase signaling. Proc. Natl. Acad. Sci. USA. 2013;110:E2074–E2083. doi: 10.1073/pnas.1222387110. ) [DOI] [PMC free article] [PubMed] [Google Scholar]