

The preemptive quality control (pQC) pathway participates in the unfolded protein response regulating ER homeostasis, yet many components are not known. The role of p97 and its adaptor, AIRAPL, in proteasomal processing of pQC substrates is shown, and an insulin-processing mutant (R6C) is identified as a pQC substrate.
Abstract
The initial folding of secreted proteins occurs in the ER lumen, which contains specific chaperones and where posttranslational modifications may occur. Therefore lack of translocation, regardless of entry route or protein identity, is a highly toxic event, as the newly synthesized polypeptide is misfolded and can promiscuously interact with cytosolic factors. Mislocalized proteins bearing a signal sequence that did not successfully translocate through the translocon complex are subjected to a preemptive quality control (pQC) pathway and are degraded by the ubiquitin-proteasome system (UPS). In contrast to UPS-mediated, ER-associated degradation, few components involved in pQC have been identified. Here we demonstrate that on specific translocation inhibition, a p97–AIRAPL complex directly binds and regulates the efficient processing of polyubiquitinated pQC substrates by the UPS. We also demonstrate p97’s role in pQC processing of preproinsulin in cases of naturally occurring mutations within the signal sequence of insulin.
INTRODUCTION
It is estimated that up to one-third of all newly synthesized polypeptides enter the endoplasmic reticulum (ER), the entry gate into the secretory pathway (Wallin and von Heijne, 1998). Entry into the ER is an elaborate and well-regulated process in order to ensure the efficient entry of newly synthesized proteins into the correct folding milieu (Shao and Hegde, 2011; Ast and Schuldiner, 2013). Failure to properly translocate will result in a misfolded polypeptide, further emphasizing the importance of proper and efficient translocation into the ER. Recognition of mislocalized proteins, regardless of their entry route into the ER, is achieved by cytosolic factors that deliver the mislocalized proteins for degradation (Rane et al., 2008; Rodrigo-Brenni et al., 2014).
The main entry port into the ER lumen is the Sec61 translocon, which provides an aqueous pore through the ER membrane for polypeptides to enter the ER lumen (Deshaies and Schekman, 1987; Gorlich et al., 1992; Park and Rapoport, 2012). The conducting channel is the entry site for N-terminal signal sequence–bearing polypeptides for both cotranslational and posttranslational insertions. Whereas both modes of insertion require the Sec61 translocon, cotranslational insertion is mediated through the initial recognition of the nascent chains emerging out of the ribosomal exit tunnel by signal peptide recognition particle (SRP; Shan and Walter, 2005). Translational pausing and ribosomal localization to the SRP receptor–translocon complex ensures that the emerging nascent chain is immediately inserted into the translocon. The translocon channel enables either transition through the hydrophobic surrounding of the membrane or immediate lateral movement of a hydrophobic segment into the ER membrane. Nascent chains bearing a signal peptide can fail to be recognized by the SRP due to features within the signal sequence itself (lower hydrophobicity; Ng et al., 1996) or early termination of translation (70–80 amino acids), which will not enable SRP binding at the ribosomal exit site before translational termination (Schlenstedt et al., 1992). In these cases of SRP-independent, Sec61-dependent ER insertions, additional accessory factors are required to maintain solubility and localization to the translocon (Plath and Rapoport, 2000).
Regardless of the insertion mode through the translocon, the signal sequence is endoproteolyticaly processed by the signal peptidase almost immediately upon entry to the ER lumen (Fujimoto et al., 1984; Evans et al., 1986). ER entrance through the translocon is a dynamic process, as it can be fine tuned to reduce translocation during ER stress in a signal sequence–selective manner (Kang et al., 2006). This quality control process has been termed preemptive quality control (pQC) and can be therefore viewed as part of the unfolded protein response (UPR) required for maintaining ER homeostasis (Kang et al., 2006; Rane et al., 2008). Investigation of several branches of the UPR, such as ER-associated degradation (ERAD; Christianson and Ye, 2014), ER stress–triggered transcriptional and translational reprograming (Ron and Walter, 2007), and ER stress–triggered mRNA membrane release and cleavage (Hollien et al., 2009), have identified many of the molecular components involved in these quality control processes. In contrast to this situation, the molecular entities and events governing ER homeostasis sensing by the pQC process are poorly understood. In fact, only a subset of the molecular components involved in pQC have been identified (Hessa et al., 2011; Rodrigo-Brenni et al., 2014). We envision that many mechanistic steps required for ERAD substrate processing by the ubiquitin proteasome, such as recognition, ubiquitination, extraction, and proteasomal delivery, are also required in the pQC, yet the identities of many of these components remain to be found.
Previously we showed that arsenite-inducible, RNA-associated protein–like (AIRAPL) is an ER membrane protein that binds ubiquitin, p97, and the 26S proteasome (Yun et al., 2008; Glinka et al., 2014). RNA interference of the Caenorhabditis elegans homologue of AIRAPL (aip-1) gave rise to various impairments in protein-folding homeostasis (Yun et al., 2008), whereas no effect on ERAD substrate proteolysis was observed. Nevertheless, the translocation rate into the ER lumen of several ERAD substrates was responsive to AIRAPL knockdown in a signal sequence–dependent manner (Glinka et al., 2014). On the basis of these observations, we decided to investigate the role of p97 and AIRAPL in pQC processing of polyubiquitinated, mislocalized proteins.
RESULTS
Selective recruitment of p97 to mislocalized vascular cell adhesion molecule 1
To evaluate the possible interaction between the p97–AIRAPL complex and a mislocalized protein that is subjected to the pQC pathway, we used CAM741, a previously characterized selective ER translocational inhibitor of vascular cell adhesion molecule 1 (VCAM-1; Besemer et al., 2005). VCAM-1 is usually detected as a high–molecular weight (HMW) glycosylated protein (compare to the migration of the untreated and PNGase F–treated sample; Figure 1A). Although no impairment in HMW migration was noted upon in vitro deubiquitination of the sample (Figure 1A, Usp2) or upon proteasomal inhibition (Figure 1A, Velcade), proteasomal inhibition enabled the detection of two additional low–molecular weight (LMW) bands. Upon CAM741 treatment, HMW glycosylated VCAM-1 was completely undetectable, attesting to the lack of ER translocation, whereas a faint LMW band was detected (Figure 1A, CAM741). This species could be readily detected upon addition of a proteasome inhibitor (Figure 1A, CAM741 + Velcade). Thus the VCAM-1 CAM741 system enables rerouting of VCAM-1 into the pQC pathway (Besemer et al., 2005).
FIGURE 1:

(A) VCAM1-HA was transfected into cells, and 48 h posttransfection, VCAM1 content was evaluated by HA immunoblots. Cell lysates were analyzed directly or after in vitro PNGase or Usp2 treatment. Where indicated, cells were treated overnight with Velcade (100 nM) and/or CAM741 (250 nM) as indicated. Asterisk marks an additional LMW band of VCAM-1. Glycosylated VCAM-1 is labeled Glc-Nac-VCAM-1. (B) Cells expressing VCAM1 were treated with Velcade in the presence or absence of CAM741 as indicated. Cell lysates were evaluated directly (input) or subjected to a HA IP to evaluate Bag6 and p97 interaction. (C) VCAM-1 IP in the presence of CAM741 and Velcade as in B was subjected to an in vitro deubiquitination by Usp2 as indicated. (D) Recombinant P97 was layered under a discontinuous sucrose gradient in the absence of membranes (–M) or presence of ribosome-stripped PK-RMs or purified reconstituted trimeric Sec61-PLs. After centrifugation, fractions were collected from top to bottom and immunoblotted against p97. The membrane binding of p97 is evident by its flotation together with the membranes to the top of the gradient.
To evaluate whether p97 interaction could be detected and whether this interaction is selective to nontranslocated VCAM-1, we immunoprecipitated VCAM-1 (VCAM-1 IP) from control and CAM741–treated cells. As a positive control for selective interaction of mislocalized VCAM-1 with a pQC component, we used Bag6, which was shown previously to interact directly with a pQC substrate (Hessa et al., 2011). As seen in Figure 1B, CAM741 treatment readily inhibited VCAM-1 glycosylation, and copurification of Bag6 and p97 could be detected only with the mislocalized species of VCAM-1. This result implies a selective recruitment of both Bag6 and p97 to a mislocalized protein. Bag6 is considered a holdase, as it has been reported to engage the exposed hydrophobic segments of a mislocalized substrate (both in ERAD and in pQC; Hessa et al., 2011; Wang et al., 2011) in order to increase substrate solubility in the cytosol. Because mislocalized VCAM-1 is readily degraded by the 26S proteasome (Besemer et al., 2005; Figure 1A), we hypothesized that its targeting to the proteasome is ubiquitin dependent and requires an ER membrane–localized ubiquitin-binding protein and a recruitment cofactor for p97. This hypothesis is based on the fact that mislocalized VCAM-1 is initially still ER membrane localized to the Sec61 translocon channel, the binding site for CAM741 and the signal peptide (Besemer et al., 2005). This hypothesis would also predict that p97 interaction with VCAM-1 is after polyubiquitination, whereas Bag6 interaction is independent of the ubiquitination status of VCAM-1. Repeating the purification of mislocalized VCAM-1 in the presence or absence of a deubiquitinating enzyme indicated the complete dependence of p97 recruitment on VCAM-1 polyubiquitination, as opposed to the marginal effect this treatment had on Bag6 interaction (Figure 1C).
To evaluate the ability of p97 to interact with microsomal membrane fractions, we used an in vitro binding flotation assay and evaluated the ability of p97 to float with the membrane fractions above a dense sucrose cushion. To this end, recombinant p97 was mixed with ribosome-stripped dog pancreatic rough microsomes (PK-RMs) or purified reconstituted trimeric Sec61 complex proteoliposomes (Sec61-PLs). After a short incubation, samples were equilibrated to 2.1 M sucrose and layered beneath a discontinuous sucrose gradient. Samples were centrifuged, and p97 localization throughout the gradient was evaluated. As seen in Figure 1D, p97 was able to bind ribosome-stripped PK-RMs or purified reconstituted trimeric Sec61-PLs, as evident from its redistribution into the top fractions. This redistribution was not apparent if no microsomes were present (–M). Thus p97 can directly interact with components of the protein translocation channel in pQC sites.
p97 role in processing mislocalized VCAM-1
Although our results confirmed the proteasomal degradation of mislocalized VCAM-1 upon CAM741 treatment, we could also detect the appearance of another LMW species upon proteasomal inhibition treatment (Figure 1A, asterisk; previously described in Garrison et al., 2005). We reasoned that an additional discriminating feature between ER-localized and -mislocalized VCAM-1 pQC substrate could be the presence of the signal sequence. This feature would also discriminate between VCAM-1 as an ERAD or pQC substrate. To this end, we inserted a Flag tag into the 5′ end of the VCAM-1 signal sequence (see illustration in Figure 2A) and noted that this insertion did not perturb the ability of VCAM-1 to translocate into the ER or the ability of CAM741 to inhibit this translocation (Figure 2B). First we evaluated VCAM-1 status by immunoblotting (IB) against the C-terminal epitope hemagglutinin (HA; as in Figure 1A) and toward the signal sequence Flag epitope (Figure 2B). In the presence of wild-type p97-untreated cells, we could detect the glycosylated isoform of VCAM-1 only using the C-terminal tag, whereas the signal sequence tag could barely be detected, as expected from an efficient translocation and signal-sequence removal event (Figure 2B, lane 1, HA and Flag IB). CAM741 treatment eliminated the presence of VCAM-1 in both IBs (Figure 2B, lane 2), and Velcade treatment in the presence of CAM741 dramatically increased the reactivity of both HA and Flag IBs (Figure 2B, lane 3). Whereas proteasomal inhibition induced the accumulation of two LMW species (Figure 2B, lane 3, HA IB), only the slower-migrating form seemed to contain the signal sequence, as evident from the Flag IB (Figure 2B, lane 3, Flag IB). We assume that the LMW band represents a small fraction of VCAM-1 that was able to translocate into the ER and was subjected to proteasomal degradation through the ERAD pathway, thus explaining the Velcade sensitivity and the lack of signal sequence reactivity in the Flag IBs. We performed the same experiments in the presence of a p97 mutant that retains substrate binding but cannot hydrolyze ATP, thus functioning as a dominant negative (p97QQ; Ye et al., 2003). We noted that p97QQ expression by itself stabilized the nontranslocated form of VCAM-1 and had hardly any effect on the mislocalized VCAM-1 that could be observed upon Velcade addition (Figure 2B, lanes 5 and 6 vs. lanes 2 and 3). This result confirms a role for p97 in processing of VCAM-1 as a pQC substrate and implies a role for p97 in pQC proteasomal degradation, as observed in ERAD substrates.
FIGURE 2:
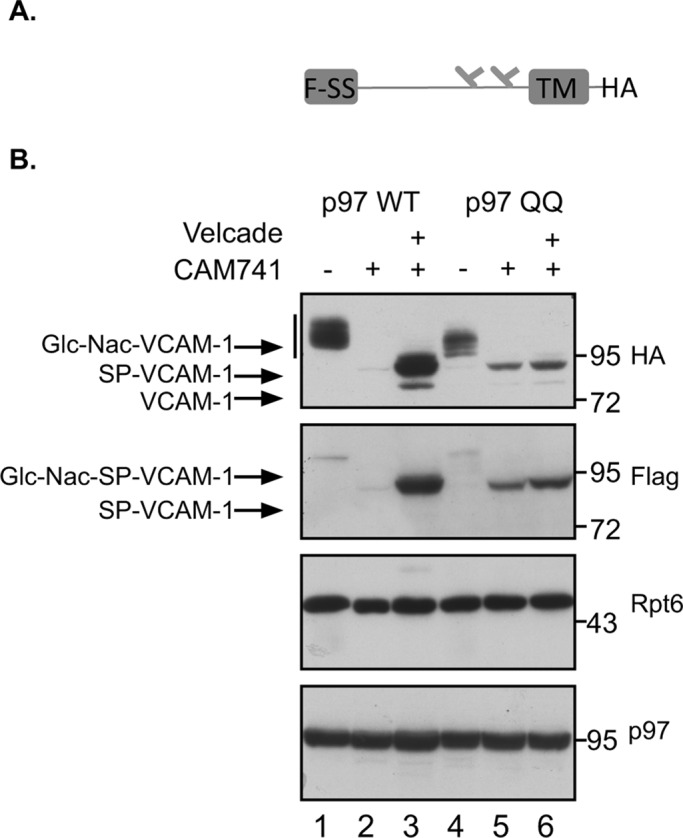
(A) Illustration of VCAM-1 containing the Flag signal sequence (F-SS), the glycosylation sites, transmembrane domain (TM), and C-terminus HA tag. (B) Cells expressing the Flag-SS VCAM-1 and the indicated p97 were treated with Velcade and CAM741 as indicated, and VCAM-1 content was evaluated by Flag and HA IB. Cellular expression levels of p97 are indicated, as well as Rpt6 levels serving as a loading control. Glycosylated VCAM-1 is labeled Glc-Nac-VCAM-1, signal peptide–bearing VCAM-1 is labeled SP-VCAM-1, and signal peptide–bearing glycosylated VCAM-1 is labeled Glc-Nac-SP-VCAM-1. VCAM-1 that does not contain a signal peptide is labeled as VCAM-1.
Ubiquitin-dependent binding of AIRAPL to mislocalized VCAM-1
Mislocalized proteins often expose hydrophobic segments due to lack of membrane insertion. Therefore one of the earliest events required for processing a pQC substrate is the ability to maintain it in a soluble form in order to be processed by the ubiquitin proteasome system (Hessa et al., 2011; Wang et al., 2011). Furthermore, previous reports demonstrated the role of hydrophobic segments as elements required in-cis for targeting mislocalized proteins for proteasomal degradation (Hessa et al., 2011). Bag6 is one of the few identified components required for efficient pQC processing, and our data indicated the lack of ubiquitin dependence in this interaction (Figure 1C). We reasoned that AIRAPL binding to a pQC substrate should enable copurification with Bag6. If this association is mediated by the polyubiquitinated pQC substrate, then this interaction should be dependent on the ability of AIRAPL to bind ubiquitin. We therefore expressed wild-type (WT) and ubiquitin-binding mutant forms of AIRAPL (UIM mut), treated cells with CAM741 and Velcade (to stabilize nontranslocated VCAM-1), and evaluated the ability of AIRAPL to bind the pQC substrate. As seen in Figure 3A (left), AIRAPL copurified with VCAM-1 in a functional ubiquitin-interacting motif (UIM)–dependent manner and highly enriched a HMW polyubiquitinated form of the pQC substrate. This HMW band completely disappeared upon incubation with a deubiquitinating enzyme (Usp2), and a discrete LMW band corresponding to VCAM-1 was enhanced upon this treatment (Supplemental Figure S1). This result confirms the identity of the HMW VCAM-1 band as a polyubiquitinated form of the protein. Furthermore, the binding of AIRAPL to p97 did not show any dependence on UIM functionality or deubiquitination treatment (Glinka et al., 2014).
FIGURE 3:

(A) Left, cellular lysates of CAM741- and Velcade-treated cells expressing VCAM-1 and AIRAPL WT or UIM mutant were subjected to an AIRAPL IP. Copurification of VCAM-1 is observed to be UIM dependent, whereas p97 copurification is not UIM dependent. Right, AIRAPL IP was performed from cellular lysates of cells treated with Velcade or usp2 as indicated. Bag6 and ubiquitin copurification was evaluated by the indicated immunoblots. (B) Cells expressing AIRAPL and the indicated Bag6 were treated with CAM741 with or without Velcade (as indicated), and cellular lysates were subjected to an AIRAPL IP. Copurification of Bag6 and VCAM-1 was evaluated by immunoblots, and increased levels of VCAM-1 upon Bag6 88–387 expression are noted (input). Note the reactive band at the size of the Bag6 mutant that appears even in the absence of Bag6 mutant expression, indicating a nonspecific or partial Bag6 product expressed at this molecular weight. Nevertheless, the reduced amount of Bag6 mutant copurified upon AIRAPL IP (compare to WT) implies a role for additional Bag6-mediated processes in AIRAPL binding. Long exposures of cellular input content reveal the reduced levels of HMW VCAM-1 upon Bag6 mutant expression. (C) Cells treated with CAM741 and Velcade expressing the indicated AIRAPL isoform were subjected to an AIRAPL IP, and VCAM-1, Bag6, ubiquitin, and p97 interaction was evaluated. Longer exposure of VCAM-1 content in the AIRAPL IP reveals the presence of a HMW VCAM-1 isoform in the WT and VIM mutants of AIRAPL (bottom, long exp.).
To evaluate AIRAPL association with Bag6 and its dependence on pQC substrate ubiquitination status, we affinity purified AIRAPL in the presence of Velcade or in the presence of a deubiquitinating enzyme (Usp2). As seen in Figure 3A (right), we were able to copurify Bag6 with AIRAPL, and this copurification was responsive to Velcade and usp2 treatments, thus demonstrating that it is a polyubiquitin-mediated interaction. Previous analysis demonstrated that the ubl domain of Bag6 is not required for substrate interaction but is for E3 recruitment and substrate ubiquitination (Hessa et al., 2011; Rodrigo-Brenni et al., 2014) and that further deletion of the Bag6 ZF and C-term PR domains (Bag6 88–387) reduces the ability of Bag6 to form a hexamer (Xu et al., 2013). Because Bag6 mutant 88–387 can still bind the pQC substrate, it acts as a dominant negative, as noted from the elevated VCAM-1 levels (Figure 3B, input). Unlike Velcade treatment, VCAM-1 accumulation upon Bag6 mutant expression did not include a polyubiquitin HMW form, as it cannot support pQC substrate ubiquitination (Figure 3B, long exposure). The reduced HMW form of VCAM-1 upon Bag6 mutant expression is not explained by increased proteasomal turnover, as a short-lived proteasomal substrate (ATF4) was found to be elevated (and not reduced) upon Bag6 mutant expression (Supplemental Figure S2). Furthermore, the Bag6 mutant reduced the association of AIRAPL with Bag6 (Figure 3B, AIRAPL IP). Thus we conclude that Bag6 functionality is required for AIRAPL association and efficient turnover of VCAM-1 upon mislocalization.
Our previous characterization of AIRAPL identified a UIM required for polyubiquitin binding, a VCP-interacting motif (VIM) required for p97 interaction, and a CAAX domain required for ER membrane localization (Yun et al., 2008; Glinka et al., 2014). To address the requirement of all of these AIRAPL domains for pQC substrate interaction, we affinity purified AIRAPL (WT or the indicated mutant) from CAM741 and Velcade-treated cells and monitored the binding of VCAM-1, Bag6, p97, and ubiquitin. As seen from Figure 3C, AIRAPL association with VCAM-1 and Bag6 was mainly dependent on a functional UIM and decreased upon SAAX mutation, whereas no impairment was observed in a VIM mutation. This result indicates the ability of AIRAPL to bind the pQC substrate directly without p97 assistance and the partial ability of a mislocalized pQC substrate to interact also with a cytosolic form of AIRAPL, implying a possible rerouting of the pQC substrate to the cytosol (Figure 4). Furthermore, the cytosolic mutant form of AIRAPL (SAAX mutant) was preferentially impaired for Bag6 interaction and less impaired for substrate and ubiquitin interaction (Figure 3C; compare Bag6 to ubiquitin and VCAM-1 IB of SAAX), implying a role for Bag6–AIRAPL interaction occurring mainly on the ER membrane, presumably at initial substrate mislocalization. Longer exposure also indicated the enrichment of HMW isoforms of VCAM-1 upon AIRAPL WT and VIM mutant purifications, most likely polyubiquitinated VCAM-1 (Figure 3C, long exposure).
FIGURE 4:
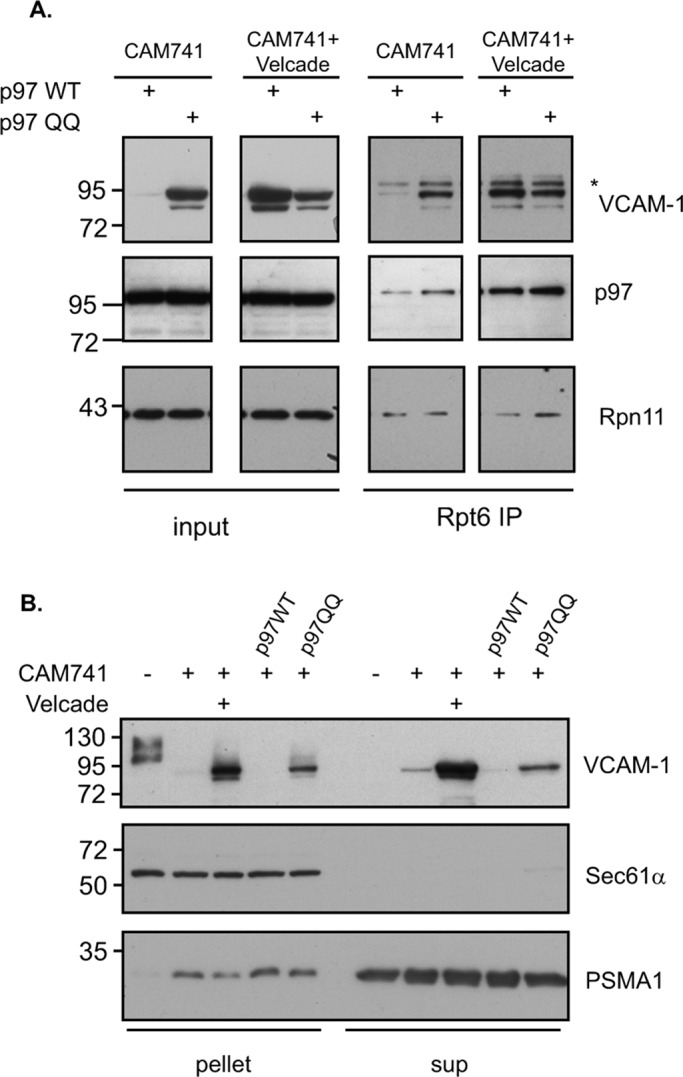
(A) Cells expressing VCAM-1 and the indicated p97 isoform were treated with CAM741 alone or with Velcade as indicated. Cellular lysate content was evaluated directly (input) or subjected to a Rpt6 IP to evaluate proteasomal association (Rpn11) of VCAM-1 and p97. The asterisk represents a nonspecific band in the Rpt6 IP. (B) VCAM-1 and p97 expressing cells were treated with CAM741 or Velcade as indicated. Cellular lysates were fractionated into supernatant and pellet and VCAM-1, and proteasomal (PSMA1) content distribution was evaluated by IB. Sec61 immunoblot serves as a positive marker for ER membrane distribution.
Proteasomal recruitment of mislocalized VCAM-1
Previously we demonstrated the direct recruitment of p97 to the 26S proteasome upon protein degradation impairment (Isakov and Stanhill, 2011). Our results (Figure 2) imply a role for p97 in the processing of a pQC substrate, but it remains to be established whether the observed impairment in pQC processing in the p97QQ cells is due to lack of efficient relocalization of the pQC substrate from the ER membrane or impairment in substrate release from p97 to the proteasome that may occur in the cytosol or on the ER membrane itself (Kalies et al., 2005). To address this point, we evaluated the proteasomal recruitment status of VCAM-1 upon mislocalization by affinity purifying proteasomes (Rpt6 IP) and evaluating VCAM-1 content (Figure 4A). As noted, p97QQ increased steady-state levels of VCAM-1 upon CAM741 treatment (Figure 4A, input). We could not observe any impairment in proteasomal recruitment of VCAM-1 (Rpt6 IP) and in fact observed elevated purification of the pQC substrate, probably reflecting the increase in the steady-state levels of VCAM-1 upon p97QQ expression. Thus p97QQ does not perturb proteasomal recruitment and functions upstream to proteasomal degradation. The timely release of VCAM-1 from p97 occurring on the proteasome seems to be impaired in the p97QQ cells.
Although membrane extraction of ERAD substrates may largely depend on p97 activity (Ye et al., 2003), it is possible that mislocalized proteins that are more loosely bound to the membrane do not require extraction or may occur in a p97-independent manner. We further evaluated the localization of VCAM-1 and its accumulation upon p97QQ expression or Velcade treatment, using a biochemical fractionation assay. As seen in Figure 4B, glycosylated VCAM-1 entirely partitioned into the microsomal pellet fraction. However, upon CAM741 treatment, it was found mainly in the cytosolic supernatant fraction, indicating relocalization of VCAM-1 from the microsomal pellet. The relocalization of VCAM-1 to the cytosolic fraction in the p97QQ cells indicates that relocalization only partially requires p97 activity (Figure 4B). Furthermore, our analysis of proteasomal localization (Figure 4B, PSMA1 IB) revealed an increase in proteasomal association with the microsomal pellet upon CAM741 treatment, in accordance with the reported direct binding of the proteasome and translocon complexes (Kalies et al., 2005). We do not attribute the increased proteasomal VCAM-1 association in the p97QQ cells (Figure 4A, Rpt6 IP) to the increase in proteasomal microsomal recruitment upon CAM741 treatment (Figure 4B, PSMA1 IB), as p97QQ expression was still able to relocalize VCAM-1 to the supernatant fractions (Figure 4B, VCAM-1 IB). Our data support a scenario in which the increased association of VCAM-1 with proteasomes is due to impaired release of the pQC substrate from p97QQ, an event that occurs on the proteasome itself (Figure 4A) mainly in the cytosol (Figure 4B). This scenario is further supported by the findings that the main interaction of p97 with mislocalized VCAM-1 is cytosolic (supernatant fraction), and only a small amount is membrane localized (microsomal pellet fraction, Supplemental Figure S3).
pQC proteasomal degradation of insulin bearing a signal peptide mutant
Our data until now relied on pharmacological impairment in ER translocation imposed artificially by CAM741. pQC processes have been implicated in various pathological conditions (Rane et al., 2008). Therefore we wanted to expand our observations to include a natural variant that is largely subjected to pQC processing. It is worth noting that whereas VCAM-1 translocation is an efficient process, a small fraction is naturally mislocalized, as evident from accumulated VCAM-1 in Velcade-treated cells (Besemer et al., 2005; Garrison et al., 2005; Figure 1A).
Late onset of diabetes type I is found in individuals bearing mutations within the INS gene (Boesgaard et al., 2010; Meur et al., 2010) leading to beta-cell destruction and is therefore viewed as a protein-misfolding disease. A specific R6C mutation within the insulin signal sequence impairs insulin’s initial ER translocation (Guo et al., 2014). To address a possible role for the UPS and p97 in correct processing of insulin R6C as a pQC substrate, we evaluated the steady-state levels of WT and R6C insulin in control and Velcade-treated cells (Figure 5A). Using a C-peptide antibody that detects only preproinsulin (untranslocated, bearing the signal sequence) or proinsulin (ER/Golgi localized insulin lacking the signal sequence) indicated that whereas insulin WT is not subjected to UPS processing, as evident from the lack of any effect of Velcade on the proinsulin product, the R6C insulin mutant can only be detected as a preproinsulin upon Velcade treatment (Figure 5, compare lanes 1 and 2 to lanes 5 and 6). Furthermore, the expression of p97QQ together with the insulin R6C mutant led to accumulation of preproinsulin even without Velcade treatment (Figure 5A, lane 7). This situation resembled our results obtained with VCAM-1 upon CAM741 treatment (Figure 2). The apparent increase of proinsulin R6C only upon p97QQ expression and not Velcade treatment (Figure 5A, lanes 6 and 7) is compatible with the partial and not complete impairment of insulin R6C translocation (Guo et al., 2014) and with the role of autophagy in insulin quality control processing of translocated insulin (Gil Leibowitz, personal communication). This is further evident from the accumulation of WT proinsulin in the p97QQ cells (Figure 5A, lanes 3 and 4).
FIGURE 5:
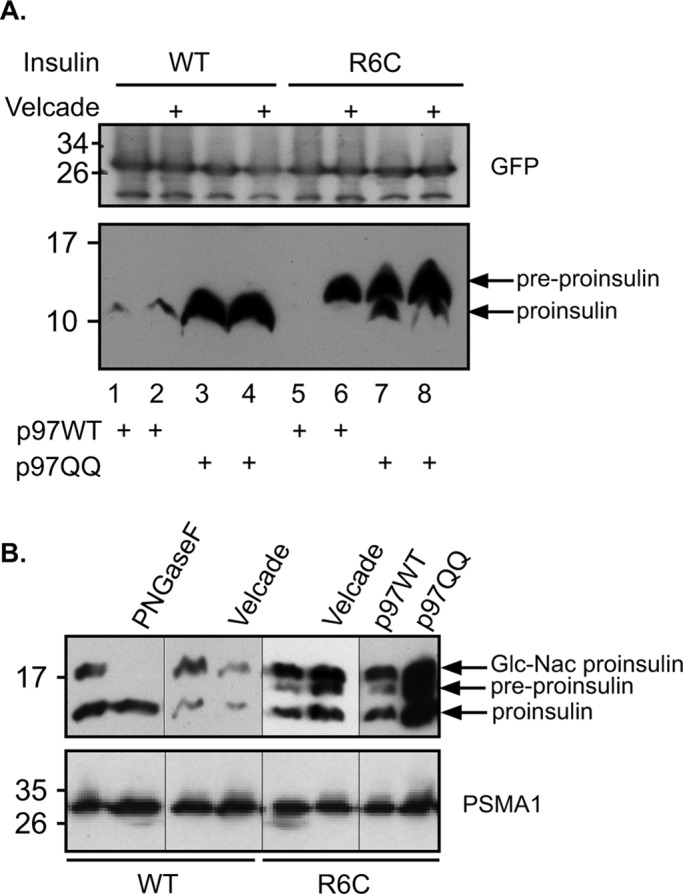
(A) Cells expressing p97 and insulin isoforms (as indicated) were treated with Velcade or left untreated as indicated. Preproinsulin and proinsulin migration is indicated, and GFP expression was evaluated to demonstrate equal transfection and loading of the gel. (B) N-glycosylation–bearing insulin was expressed in cells, and cellular content was resolved to discriminate between the indicated insulin isoforms. Velcade treatment and p97 expression were performed as in A. PNGaseF treatment was performed on cellular lysate, and PSMA1 content served as a loading control for cell lysates.
To demonstrate further the selective role of proteasomal degradation with regard to nontranslocated preproinsulin, we inserted into the C-peptide region of insulin an N-glycosylation site to enable discrimination between ER and nontranslocated proinsulin. As seen in Figure 5B, both insulin WT and R6C mutant appear in a HMW glycosylated form that is PNGaseF sensitive. However, the R6C mutant appeared as an additional band that is not sensitive to PNGaseF (unpublished data) and accumulates in response to both Velcade and p97QQ (Figure 5B), whereas the WT isoform did not accumulate upon Velcade treatment, thus demonstrating that it is the nontranslocated preproinsulin that is subjected to proteasomal degradation in a p97-dependent manner. Furthermore, insulin R6C degradation was substantially impaired upon Bag6 knockdown (Supplemental Figure S4), providing further evidence for the involvement of pQC components in insulin turnover upon ER mislocalization.
DISCUSSION
Surveillance mechanisms toward polypeptides that were designated to enter the secretory pathway ensure that mislocalized proteins will not perturb cytosolic homeostasis. This is achieved by rerouting mislocalized proteins for proteasomal degradation (Kang et al., 2006). Components that ensure solubility and ubiquitinate mislocalized proteins are essential components in such surveillance systems. The roles of two such components, Bag6 and RNF126, have been demonstrated (Hessa et al., 2011; Rodrigo-Brenni et al., 2014), whereas the identity and role of additional components in this quality control process remain to be discovered. In this article, we demonstrate the direct binding of p97 and its ubiquitin-binding adaptor, AIRAPL, to the pQC substrate VCAM-1. Our data indicate that Bag6 binding to the pQC substrate is independent of the substrate’s ubiquitination status (Figure 1), consistent with the ability of Bag6 to directly bind hydrophobic segments of a pQC substrate (Hessa et al., 2011). In contrast to this situation, AIRAPL and p97 binding is dependent on substrate ubiquitination (Figures 1 and 3). This result implies the required activity of an E3 ligase before p97 binding. This requirement in turn suggests the recruitment of p97 by a ubiquitin-binding protein (AIRAPL) at stages in which the substrate’s fate (translocation vs. degradation) has already been determined, consistent with the binding of AIRAPL to proteasomes and polyubiquitin chains (Yun et al., 2008). In contrast to this situation, Bag6 binding is required due to biophysical hydrophobic features of the substrate and does not necessarily indicate the substrate’s fate. This scenario resembles the role of Bag6 in the ER translocation of tail-anchored proteins in which Bag6 binding is required for solubility regardless of its final fate (Leznicki et al., 2010; Mariappan et al., 2010; Lee and Ye, 2013). One possible scenario is the initial recruitment of Bag6 to the mislocalized protein and subsequent recruitment of AIRAPL and p97 postubiquitination. Additional time-resolved analysis would be required to further test this scenario.
Numerous p97 cofactors have been identified (Alexandru et al., 2008; Yeung et al., 2008), and a selective combination of p97 with AIRAPL has been identified (Glinka et al., 2014), including Npl4-Ufd1 and Ubxd8 (an ER membrane–bound Bag6 interactor; Xu et al., 2013). The wide range of p97 adaptors and the fact that p97 is a hexamer enable a wide range of adaptor combinations, but a complex hierarchy is apparent (Alexandru et al., 2008). The fact that Bag6 is also a hexamer (Xu et al., 2013) with several identified cofactors (Lee and Ye, 2013) increases the range of possible p97–Bag6 cofactors combinations. Furthermore, it is possible that p97 interaction with Bag6 in several cellular scenarios (ribosome, ERAD, and pQC) will give rise to several different combinations of adaptor proteins. Our findings regarding AIRAPL interaction with Bag6 and p97, as well as the previous identification of Ubxd8 as an additional ER membrane–bound protein that binds both hexameric proteins (Bag6 and p97; Alexandru et al., 2008; Xu et al., 2013), enable the following complex scenario (Figure 6): the mislocalization of a pQC substrate on the ER membrane will first recruit the Bag6 complex to exposed hydrophobic segments, which will enable the binding of Ubxd8 via the Ubl domain of Bag6 and the UBA domain within Ubxd8 (Xu et al., 2013). After ubiquitination by an E3 ligase, an event that is Bag6 ubl domain dependent (Figure 3B; Rodrigo-Brenni et al., 2014), AIRAPL will bind the ubiquitinated pQC substrate and recruit p97, which can also bind its additional adaptor Ubxd8. This sequence of events is consistent with the ability of a Bag6 mutant that does not enable Ubl-mediated interaction (Bag6 88–387; Figure 3B) to stabilize the pQC substrate and impair its interaction with AIRAPL and also with the ability of an AIRAPL mutant that fails to bind p97 to bind the pQC substrate (Figure 3C). Furthermore, the selective reduction in AIRAPL SAAX mutant ability to bind Bag6 but not VCAM-1 (Figure 3C) may also indicate that, after p97 recruitment, Bag6 may not be required for complete escort to the proteasome as long as p97–substrate engagement is in place. Further kinetic experiments are required in order to establish the relationship between Bag6 and p97 in pQC substrate proteasomal processing.
FIGURE 6:
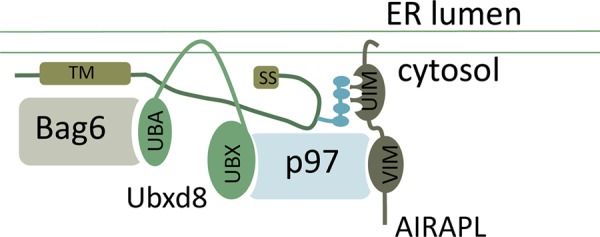
Based on the presented data, a plausible scenario can be drawn in which a mislocalized protein (MLP) that has not translocated into the ER is bound to the ER membrane by virtue of hydrophobic segments of its transmembrane (TM) domain and/or hydrophobic segments within its signal sequence (SS). Bag6 interaction with hydrophobic segments within the MLP would then enable recruitment of the p97 adaptor Ubxd8 (via its UBA domain). Polyubiquitination of the MLP (by virtue of the ribosome quality control ligase LTN1, RNF126, or another E3 ligase) would enable recruitment of AIRAPL, an additional p97 adaptor to the polyubiquitinated MLP.
Both p97 and Bag6 hexameric solubilizing complexes will retain the pQC UPS substrate in a soluble proteasome-accessible form in order to facilitate its degradation. Our findings with the p97QQ mutant clearly indicate the functional role of p97 in pQC substrate degradation by the proteasome (Figures 2, 4, and 5) but indicate no impairment in pQC substrate recruitment to the proteasome (Figure 4A). This result fits well with previous results indicating the enhanced recruitment of p97 to the 26S proteasome upon UPS impairment (Isakov and Stanhill, 2011) and implies that the substrate relay between p97 and the 26S proteasome requires both ATPase hydrolysis by p97 (Figure 4A) and catalytic activity of the proteasome (Isakov and Stanhill, 2011). In spite of the increase in proteasomal recruitment to microsomal fractions observed upon CAM741 treatment (Figure 4B), we still conclude that enhanced proteasomal recruitment of VCAM-1 upon p97QQ expression is not due the relocalization of proteasomes (Figure 4B), as CAM741 treatment by itself enhances VCAM-1 levels only in the supernatant fraction and not in the microsomal pellet fractions (Figure 4B, VCAM-1 IB), thus excluding proteasomal recruitment as the sole explanation for the enhanced recruitment of VCAM-1 to the proteasome. Furthermore, close examination of VCAM-1 in p97QQ and Velcade-expressing cells indicates a HMW smear in the pellet membrane fractions that is absent from the cytosolic supernatant fractions (Figure 4B). This observation is consistent with the initial recruitment of p97 to the polyubiquitinated pQC substrate that occurs on the membrane (pellet fractions). The stabilization of the pQC substrate in the p97QQ or Velcade-treated cells occurs on the proteasome itself (Figure 4A), in close proximity to proteasomal deubiquitinating enzymes. Further analysis is required to evaluate the exact relay steps of mislocalized membrane substrates to the 26S proteasome.
We also addressed the role of p97 in a physiological pQC substrate and identified preproinsulin R6C mutant as a pQC substrate (Figure 5), implying a role for p97 in late onset of diabetes type I. This role of p97 in insulin processing is probably not restricted to preproinsulin as a pQC substrate because even translocated proinsulin is dependent on proper activity of p97 (Figure 5A); however, only the preproinsulin was sensitive to proteasomal inhibition (Figure 5A, lanes 1 and 2 vs. lanes 5 and 6). These observations are consistent with a role for p97 in autophagy (Ju et al., 2009; Dargemont and Ossareh-Nazari, 2012) and a role for autophagy in proinsulin homeostasis (Gil Leibowitz, personal communication). Whereas mutations within the signal sequence of α1 anti-trypsin and pre–proparathyroid hormone have also been observed in emphysema and hypoparathyroidism (Arnold et al., 1990; Gooptu et al., 2009), systematic identification of pQC substrates and their role in p97 related pathologies remains to be addressed.
MATERIALS AND METHODS
Cell culture, transfection, lysis, and protein purification
HEK293 cells were cultured in complete medium composed of DMEM supplemented with 1% penicillin–streptomycin solution, 55 μM β-mercaptoethanol, 1% nonessential amino acids solution, and 10% heat-inactivated fetal bovine serum. Plasmids expressing human VCAM-1 with a C-terminal HA tag were constructed by subcloning the human VCAM-1 cDNA (using as a template an expression vector kindly provided by Novartis; Besemer et al., 2005) into pCDNA3.1 with a C-terminus HA tag. The N-terminus Flag tag was inserted into the foregoing plasmid by PCR. All constructs were verified by sequencing. Expression constructs of p97 and AIRAPL, WT, and mutant forms have been previously described (Glinka et al., 2014). Expression constructs of human Inulin WT and R6C were kindly provided by Ming Liu (University of Michigan Medical School, Ann Arbor, MI; Guo et al., 2014), and N-glycosylation–site insertion into the foregoing plasmids was performed by overlapping PCR. Where indicated, cells were treated with Velcade (100 nM) and/or CAM741 (250 nM) overnight, and cell lysis was performed in TNH buffer (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.9, 100 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1.5 mM MgCl2, 1 mM dithiothreitol [DTT], and protease inhibitors) and clarified at 20,000 × g for 10 min. Where indicated, cell lysates were directly incubated with PNGase F (NEB [Ipswich, MA] P0704) or Usp-2 (Baker et al., 2005). Transient transfections were performed using the calcium phosphate precipitation method, and protein immunopurifications were performed using the indicated antibodies. For direct proteasome purifications, cell lysates were immunopurified against Rpt6 in the presence of 1 mM ATP. Immunopurified material was extensively washed with lysis buffer and subsequently eluted with Laemmli buffer. Cellular fractionation was performed as previously described. Briefly, cells (2 × 106) were resuspended in 100 μl of 0.01% digitonin in HCN buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 2 mM CaCl2, and protease inhibitors), incubated on ice for 10 min, and centrifuged at 16,000 × g for 10 min. Microsomal pellet fraction content was extracted by resuspension in TNH buffer.
Antibodies and Western blots
Rpn11 polyclonal antiserum was produced by immunizing rats against the full-length human Rpn11. GFP, p97, and AIRAPL polyclonal rabbit antisera were previously described (Yun et al., 2008; Isakov and Stanhill, 2011; Glinka et al., 2014), Bag6 polyclonal serum was a kind gift from Yihong Ye (National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD; Wang et al., 2011), and Rpt6 hybridoma cell line was a kind gift from Shigeo Murata (University of Tokyo, Tokyo, Japan; Kaneko et al., 2009). Sec61 was detected with peptide-specific antibody against the C-terminus (Gorlich et al., 1992). The commercial sources for antibodies were as follows: ubiquitin (Zymed-Invitrogen, Waltham, MA), Flag M2 (Sigma-Aldrich, St. Louis, MO), HA (Covance, Princeton, NJ), p97 in flotation assays (PROGEN Biotechnik, Germany), and insulin C-peptide (GNID4 hybridoma supernatant from the Developmental Studies Hybridoma Bank, Department of Biology, University of Iowa, Iowa City, IA). IBs toward insulin were performed using tricine–SDS–PAGE urea gels (Schagger, 2006), and all other separations were performed on standard glycine SDS–PAGE.
Membranes and flotation assay
Dog pancreas microsomes stripped of ribosomes by a treatment with puromycin and high salt (PK-RMs) were prepared as in Gorlich et al. (1992). Proteoliposomes (PLs) containing the purified Sec61 complex were prepared as previously described (Kalies et al., 1994). The Sec61p complex was purified on the basis of an immunoisolation procedure, starting with ribosome-stripped microsomes and using an antibody directed against the β-subunit of the Sec61 complex (Gorlich and Rapoport, 1993). The His-tagged recombinant p97 was a kind gift of Tom A. Rapoport (Harvard Medical School, Boston, MA). P97 binding assays were performed essentially as previously described (Kalies et al., 2005). Briefly, ribosome-stripped microsomes (PK-RMs) or proteoliposomes containing the purified Sec61 complex corresponding to 17 equivalents (for definition, see Walter and Blobel, 1983) of the original rough microsomes were mixed with 5 pmol of p97 in a final volume of 30 μl of binding buffer (120 mM potassium acetate, 5 mM magnesium acetate, 20 mM HEPES/KOH, pH 7.5, 1 mM DTT, 5 mM ATP, and 250 mM sucrose). Samples were incubated for 20 min at 0°C and 10 min at room temperature and mixed with 270 μl of the same buffer containing 2.3 M sucrose and ammonium acetate instead of potassium acetate. Samples were layered under a 800 μl of 1.8 M sucrose cushion in the same buffer and overlaid with 200 μl of binding buffer. After centrifugation in a TLS-55 rotor for 1 h at 4°C at 55,000 rpm, 130-μl fractions were collected from the top and analyzed by SDS–PAGE and immunoblotting.
Supplementary Material
Acknowledgments
We thank Yihong Ye for Bag6 antisera and expression and knockdown plasmids, Tom Rapoport for Sec61 antisera and recombinant p97, Oberhauser Berndt and Novartis for CAM741, Ming Liu for insulin-expressing plasmids, Gil Leibowitz for sharing unpublished data, and Tom Schultheiss for assistance during the revision process and critical reading of the manuscript. The research was supported by Israeli Science Foundation Grant 438/14 to A.S. and the Schwarzbach Medical Research Fund to K.U.K.
Abbreviations used:
- AIRAPL
arsenite inducible regulatory particle associated protein-like
- ERAD
endoplasmic reticulum–associated degradation
- MLP
mislocalized protein
- pQC
pre-emptive quality control
- SRP
signal peptide recognition particle
- UIM
ubiquitin interacting motif
- UPR
unfolded protein response
- UPS
ubiquitin proteasome system
- VCAM-1
vascular cell adhesion molecule 1
- VIM
valosin-containing protein–interacting motif.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-02-0085) on September 2, 2015.
REFERENCES
- Alexandru G, Graumann J, Smith GT, Kolawa NJ, Fang R, Deshaies RJ. UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell. 2008;134:804–816. doi: 10.1016/j.cell.2008.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold A, Horst SA, Gardella TJ, Baba H, Levine MA, Kronenberg HM. Mutation of the signal peptide-encoding region of the preproparathyroid hormone gene in familial isolated hypoparathyroidism. J Clin Invest. 1990;86:1084–1087. doi: 10.1172/JCI114811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ast T, Schuldiner M. All roads lead to Rome (but some may be harder to travel): SRP-independent translocation into the endoplasmic reticulum. Crit Rev Biochem Mol Biol. 2013;48:273–288. doi: 10.3109/10409238.2013.782999. [DOI] [PubMed] [Google Scholar]
- Baker RT, Catanzariti AM, Karunasekara Y, Soboleva TA, Sharwood R, Whitney S, Board PG. Using deubiquitylating enzymes as research tools. Methods Enzymol. 2005;398:540–554. doi: 10.1016/S0076-6879(05)98044-0. [DOI] [PubMed] [Google Scholar]
- Besemer J, Harant H, Wang S, Oberhauser B, Marquardt K, Foster CA, Schreiner EP, de Vries JE, Dascher-Nadel C, Lindley IJ. Selective inhibition of cotranslational translocation of vascular cell adhesion molecule 1. Nature. 2005;436:290–293. doi: 10.1038/nature03670. [DOI] [PubMed] [Google Scholar]
- Boesgaard TW, Pruhova S, Andersson EA, Cinek O, Obermannova B, Lauenborg J, Damm P, Bergholdt R, Pociot F, Pisinger C, et al. Further evidence that mutations in INS can be a rare cause of Maturity-Onset Diabetes of the Young (MODY) BMC Med Genet. 2010;11:42. doi: 10.1186/1471-2350-11-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Ye Y. Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat Struct Mol Biol. 2014;21:325–335. doi: 10.1038/nsmb.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dargemont C, Ossareh-Nazari B. Cdc48/p97, a key actor in the interplay between autophagy and ubiquitin/proteasome catabolic pathways. Biochim Biophys Acta. 2012;1823:138–144. doi: 10.1016/j.bbamcr.2011.07.011. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ, Schekman R. A yeast mutant defective at an early stage in import of secretory protein precursors into the endoplasmic reticulum. J Cell Biol. 1987;105:633–645. doi: 10.1083/jcb.105.2.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans EA, Gilmore R, Blobel G. Purification of microsomal signal peptidase as a complex. Proc Natl Acad Sci USA. 1986;83:581–585. doi: 10.1073/pnas.83.3.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto Y, Watanabe Y, Uchida M, Ozaki M. Mammalian signal peptidase: partial purification and general characterization of the signal peptidase from microsomal membranes of porcine pancreas. J Biochem. 1984;96:1125–1131. doi: 10.1093/oxfordjournals.jbchem.a134930. [DOI] [PubMed] [Google Scholar]
- Garrison JL, Kunkel EJ, Hegde RS, Taunton J. A substrate-specific inhibitor of protein translocation into the endoplasmic reticulum. Nature. 2005;436:285–289. doi: 10.1038/nature03821. [DOI] [PubMed] [Google Scholar]
- Glinka T, Alter J, Braunstein I, Tzach L, Wei Sheng C, Geifman S, Edelmann MJ, Kessler BM, Stanhill A. Signal-peptide-mediated translocation is regulated by a p97-AIRAPL complex. Biochem J. 2014;457:253–261. doi: 10.1042/BJ20130710. [DOI] [PubMed] [Google Scholar]
- Gooptu B, Ekeowa UI, Lomas DA. Mechanisms of emphysema in alpha1-antitrypsin deficiency: molecular and cellular insights. Eur Respir J. 2009;34:475–488. doi: 10.1183/09031936.00096508. [DOI] [PubMed] [Google Scholar]
- Gorlich D, Prehn S, Hartmann E, Kalies KU, Rapoport TA. A mammalian homolog of SEC61p and SECYp is associated with ribosomes and nascent polypeptides during translocation. Cell. 1992;71:489–503. doi: 10.1016/0092-8674(92)90517-g. [DOI] [PubMed] [Google Scholar]
- Gorlich D, Rapoport TA. Protein translocation into proteoliposomes reconstituted from purified components of the endoplasmic reticulum membrane. Cell. 1993;75:615–630. doi: 10.1016/0092-8674(93)90483-7. [DOI] [PubMed] [Google Scholar]
- Guo H, Xiong Y, Witkowski P, Cui J, Wang LJ, Sun J, Lara-Lemus R, Haataja L, Hutchison K, Shan SO, et al. Inefficient translocation of preproinsulin contributes to pancreatic beta cell failure and late-onset diabetes. J Biol Chem. 2014;289:16290–16302. doi: 10.1074/jbc.M114.562355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessa T, Sharma A, Mariappan M, Eshleman HD, Gutierrez E, Hegde RS. Protein targeting and degradation are coupled for elimination of mislocalized proteins. Nature. 2011;475:394–397. doi: 10.1038/nature10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakov E, Stanhill A. Stalled proteasomes are directly relieved by P97 recruitment. J Biol Chem. 2011;286:30274–30283. doi: 10.1074/jbc.M111.240309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju JS, Fuentealba RA, Miller SE, Jackson E, Piwnica-Worms D, Baloh RH, Weihl CC. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol. 2009;187:875–888. doi: 10.1083/jcb.200908115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalies KU, Allan S, Sergeyenko T, Kroger H, Romisch K. The protein translocation channel binds proteasomes to the endoplasmic reticulum membrane. EMBO J. 2005;24:2284–2293. doi: 10.1038/sj.emboj.7600731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalies KU, Gorlich D, Rapoport TA. Binding of ribosomes to the rough endoplasmic reticulum mediated by the Sec61p-complex. J Cell Biol. 1994;126:925–934. doi: 10.1083/jcb.126.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko T, Hamazaki J, Iemura S, Sasaki K, Furuyama K, Natsume T, Tanaka K, Murata S. Assembly pathway of the mammalian proteasome base subcomplex is mediated by multiple specific chaperones. Cell. 2009;137:914–925. doi: 10.1016/j.cell.2009.05.008. [DOI] [PubMed] [Google Scholar]
- Kang SW, Rane NS, Kim SJ, Garrison JL, Taunton J, Hegde RS. Substrate-specific translocational attenuation during ER stress defines a pre-emptive quality control pathway. Cell. 2006;127:999–1013. doi: 10.1016/j.cell.2006.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JG, Ye Y. Bag6/Bat3/Scythe: a novel chaperone activity with diverse regulatory functions in protein biogenesis and degradation. BioEssays. 2013;35:377–385. doi: 10.1002/bies.201200159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leznicki P, Clancy A, Schwappach B, High S. Bat3 promotes the membrane integration of tail-anchored proteins. J Cell Sci. 2010;123:2170–2178. doi: 10.1242/jcs.066738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariappan M, Li X, Stefanovic S, Sharma A, Mateja A, Keenan RJ, Hegde RS. A ribosome-associating factor chaperones tail-anchored membrane proteins. Nature. 2010;466:1120–1124. doi: 10.1038/nature09296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meur G, Simon A, Harun N, Virally M, Dechaume A, Bonnefond A, Fetita S, Tarasov AI, Guillausseau PJ, Boesgaard TW, et al. Insulin gene mutations resulting in early-onset diabetes: marked differences in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention. Diabetes. 2010;59:653–661. doi: 10.2337/db09-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng DT, Brown JD, Walter P. Signal sequences specify the targeting route to the endoplasmic reticulum membrane. J Cell Biol. 1996;134:269–278. doi: 10.1083/jcb.134.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Rapoport TA. Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annu Rev Biophys. 2012;41:21–40. doi: 10.1146/annurev-biophys-050511-102312. [DOI] [PubMed] [Google Scholar]
- Plath K, Rapoport TA. Spontaneous release of cytosolic proteins from posttranslational substrates before their transport into the endoplasmic reticulum. J Cell Biol. 2000;151:167–178. doi: 10.1083/jcb.151.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane NS, Kang SW, Chakrabarti O, Feigenbaum L, Hegde RS. Reduced translocation of nascent prion protein during ER stress contributes to neurodegeneration. Dev Cell. 2008;15:359–370. doi: 10.1016/j.devcel.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigo-Brenni MC, Gutierrez E, Hegde RS. Cytosolic quality control of mislocalized proteins requires RNF126 recruitment to Bag6. Mol Cell. 2014;55:227–237. doi: 10.1016/j.molcel.2014.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Schagger H. Tricine-SDS–PAGE. Nat Protocols. 2006;1:16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- Schlenstedt G, Gudmundsson GH, Boman HG, Zimmermann R. Structural requirements for transport of preprocecropinA and related presecretory proteins into mammalian microsomes. J Biol Chem. 1992;267:24328–24332. [PubMed] [Google Scholar]
- Shan SO, Walter P. Co-translational protein targeting by the signal recognition particle. FEBS Lett. 2005;579:921–926. doi: 10.1016/j.febslet.2004.11.049. [DOI] [PubMed] [Google Scholar]
- Shao S, Hegde RS. Membrane protein insertion at the endoplasmic reticulum. Annu Rev Cell Dev Biol. 2011;27:25–56. doi: 10.1146/annurev-cellbio-092910-154125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Blobel G. Preparation of microsomal membranes for cotranslational protein translocation. Methods Enzymol. 1983;96:84–93. doi: 10.1016/s0076-6879(83)96010-x. [DOI] [PubMed] [Google Scholar]
- Wang Q, Liu Y, Soetandyo N, Baek K, Hegde R, Ye Y. A ubiquitin ligase-associated chaperone holdase maintains polypeptides in soluble states for proteasome degradation. Mol Cell. 2011;42:758–770. doi: 10.1016/j.molcel.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Liu Y, Lee JG, Ye Y. A ubiquitin-like domain recruits an oligomeric chaperone to a retrotranslocation complex in endoplasmic reticulum-associated degradation. J Biol Chem. 2013;288:18068–18076. doi: 10.1074/jbc.M112.449199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol. 2003;162:71–84. doi: 10.1083/jcb.200302169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung HO, Kloppsteck P, Niwa H, Isaacson RL, Matthews S, Zhang X, Freemont PS. Insights into adaptor binding to the AAA protein p97. Biochem Soc Trans. 2008;36:62–67. doi: 10.1042/BST0360062. [DOI] [PubMed] [Google Scholar]
- Yun C, Stanhill A, Yang Y, Zhang Y, Haynes CM, Xu CF, Neubert TA, Mor A, Philips MR, Ron D. Proteasomal adaptation to environmental stress links resistance to proteotoxicity with longevity in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2008;105:7094–7099. doi: 10.1073/pnas.0707025105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.