

Through characterization of ccq1 mutants that disrupt Ccq1-Tpz1TPP1 interaction, the authors establish that Ccq1-Tpz1TPP1 interaction contributes to optimal binding of the Ccq1-SHREC complex and is required for Ccq1 Thr93 phosphorylation and telomerase recruitment.
Abstract
Evolutionarily conserved shelterin complex is essential for telomere maintenance in the fission yeast Schizosaccharomyces pombe. Elimination of the fission yeast shelterin subunit Ccq1 causes progressive loss of telomeres due to the inability to recruit telomerase, activates the DNA damage checkpoint, and loses heterochromatin at telomere/subtelomere regions due to reduced recruitment of the heterochromatin regulator complex Snf2/histone deacetylase–containing repressor complex (SHREC). The shelterin subunit Tpz1TPP1 directly interacts with Ccq1 through conserved C-terminal residues in Tpz1TPP1, and tpz1 mutants that fail to interact with Ccq1 show telomere shortening, checkpoint activation, and loss of heterochromatin. While we have previously concluded that Ccq1-Tpz1TPP1 interaction contributes to Ccq1 accumulation and telomerase recruitment based on analysis of tpz1 mutants that fail to interact with Ccq1, another study reported that loss of Ccq1-Tpz1TPP1 interaction does not affect accumulation of Ccq1 or telomerase. Furthermore, it remained unclear whether loss of Ccq1-Tpz1TPP1 interaction affects SHREC accumulation at telomeres. To resolve these issues, we identified and characterized a series of ccq1 mutations that disrupt Ccq1-Tpz1TPP1 interaction. Characterization of these ccq1 mutants established that Ccq1-Tpz1TPP1 interaction contributes to optimal binding of the Ccq1-SHREC complex, and is critical for Rad3ATR/Tel1ATM-dependent Ccq1 Thr93 phosphorylation and telomerase recruitment.
INTRODUCTION
To ensure stable maintenance of linear eukaryotic chromosomes, the telomere protection complex shelterin must carefully coordinate telomere replication by replicative DNA polymerases, telomere extension by telomerase, and DNA damage responses against DNA ends at telomeres (Palm and de Lange, 2008; Moser and Nakamura, 2009). Using its RNA subunit as a template, telomerase adds repetitive GT-rich sequences de novo to chromosome ends to counter telomere erosion caused by the inability of replicative DNA polymerases to fully replicate ends of linear DNA molecules, known as “end replication problem” (Blackburn et al., 2006).
Telomeric repeats consist mostly of double-stranded DNA (dsDNA) but terminate with a 3′ single-stranded DNA overhang, often referred to as a G-tail (Figure 1A). In fission yeast, the shelterin subunit Taz1 (orthologue of mammalian TRF1 and TRF2) binds directly to the dsDNA portion of telomeric repeats (Cooper et al., 1997), while another shelterin subunit Pot1 binds directly to the G-tail (Baumann and Cech, 2001). Through Taz1-Rap1, Rap1-Poz1, Poz1-Tpz1, and Tpz1-Pot1 interactions, the remaining shelterin subunits Rap1, Poz1 (analogue of mammalian TIN2), and Tpz1 (orthologue of mammalian TPP1) connect Taz1 to Pot1, to allow assembly of the fission yeast shelterin complex (Miyoshi et al., 2008; Moser and Nakamura, 2009; Figure 1A). In addition, Ccq1, which directly interacts with Tpz1TPP1, plays an essential role in telomerase recruitment in fission yeast (Miyoshi et al., 2008; Tomita and Cooper, 2008). The ability of Ccq1 to recruit telomerase to telomeres depends on phosphorylation of Ccq1 Thr93 by two redundant checkpoint kinases Rad3ATR and Tel1ATM, since the telomerase regulatory subunit Est1 specifically recognizes phosphorylated Ccq1 Thr93 to promote telomerase association with telomeres (Moser et al., 2011; Figure 1A).
FIGURE 1:

C-terminal truncation analysis of Ccq1. (A) A working model of telomere maintenance regulation in fission yeast (Moser et al., 2011). Negative regulators of telomere extension (Taz1, Rap1, and Poz1) fulfill their function by limiting the accumulation of Rad3ATR kinase at telomeres in late S/G2 through regulation of replicative DNA polymerases (Chang et al., 2013). Rad3ATR/Tel1ATM-dependent Ccq1 Thr93 phosphorylation facilitates Ccq1-Est1 interaction and promotes telomerase complex recruitment (Moser et al., 2011). (B) Summary of protein–protein interaction domains for Est1, Tpz1TPP1, Ccq1, and Poz1 (Moser et al., 2011; Harland et al., 2014). Previous studies identified 1) Tpz1TPP1 Tyr439, Leu445, and Leu449 as critical for Ccq1-Tpz1TPP1 interaction; 2) Tpz1TPP1 Trp498 and Ile501 as critical for Poz1-Tpz1TPP1 interaction (Harland et al., 2014); and 3) Ccq1 Thr93 and Est1 Arg79 and Arg180 as critical for Ccq1-Est1 interaction (Moser et al., 2011). A series of Ccq1 truncation constructs characterized in this study are indicated below. (C) Telomere length determination by Southern blot analysis for indicated Ccq1 truncation mutants. Genomic DNA was prepared after indicated strains were successively restreaked on yeast extract with supplement (YES) plates as indicated. (D) Western blot analysis of Ccq1 truncation mutants with anti-FLAG antibody. (E) Examination of Ccq1-Tpz1TPP1 interaction by co-IP. The amount of whole cell extract (WCE) utilized was adjusted as indicated to compensate for significantly reduced expression of Ccq11-500 compared with other Ccq1 truncation mutants. Cdc2 Western blot was included to serve as loading control. (F) Effects of Ccq1 truncation mutants on transcriptional silencing of his3+ marker inserted adjacent to telomeric repeats (Nimmo et al., 1998). A fivefold serial dilution series of indicated strains was spotted on YES or Pombe Medium Glutamate supplemented with uracil, leucine, and adenine (PMG ULA) (−histidine) plates.
Ccq1 is important for preventing checkpoint activation and recombination at telomeres (Miyoshi et al., 2008; Tomita and Cooper, 2008). In addition, Ccq1 interacts with the Snf2/HDAC-containing repressor complex (SHREC), which consists of the histone deacetylase (HDAC) Clr3, the SNF2 family chromatin-remodeling factor Mit1, and two additional silencing factors, Clr1 and Clr2 (Sugiyama et al., 2007). A previous study has found that Ccq1 and Taz1 are important for promoting localization of SHREC at telomeres and subtelomeric regions and for transcriptional silencing of marker genes inserted adjacent to telomeric repeats (Sugiyama et al., 2007).
Sequence analysis of Ccq1 indicated that the N-terminal domain harbors homology to HDAC complex subunit 2/3 (HDAC2/3), while the C-terminal domain shows homology to a coiled-coil domain found in SMC (Structural Maintenance of Chromosomes; Sonnhammer et al., 1997; Marchler-Bauer et al., 2002; Flory et al., 2004; Moser et al., 2011; Figure 1B). However, the functional significance of these predicted domains within Ccq1 has not yet been established, since previous studies have mostly utilized ccq1Δ strains to investigate Ccq1 functions. Therefore we decided to investigate the contribution of domains and conserved sequence motifs within Ccq1 in regulating telomere stability and telomeric heterochromatin formation by carrying out domain truncation analysis and mutational analysis of conserved amino acid residues. These studies revealed that conserved amino acid residues within the HDAC2/3-like domain are important for mediating Ccq1-Tpz1TPP1 interaction, Ccq1 Thr93 phosphorylation, telomerase recruitment, efficient accumulation of Ccq1 and SHREC at telomeres, and transcriptional silencing at telomeres.
RESULTS
Characterization of C-terminal Ccq1 truncation mutants
To define functional domains critical for Ccq1 function, we generated a series of fission yeast strains that express C-terminally truncated Ccq1, integrated at its endogenous locus (Figure 1B). We found the C-terminal amino acid residues 501–735, which include a conserved SMC-like domain, to be dispensable for maintenance of telomeres, based on telomere Southern blot analysis of strains expressing Ccq11-632, Ccq11-583, and Ccq11-500 mutants (Figure 1C), even though protein expression levels for these truncation mutants were substantially reduced compared with full-length Ccq1 (Figure 1D). All three mutant strains also grew robustly, and did not show cell elongation indicative of checkpoint activation.
We find it remarkable that even Ccq11-500, which showed the lowest expression level with > 10-fold reduction compared with full-length Ccq1, can maintain nearly wild-type steady-state telomere length. Thus it appears that fission yeast cells express Ccq1 at much higher levels than necessary to maintain normal telomere length. Indeed, comparative Western blot analysis of 13×myc-tagged Ccq1 and Tpz1TPP1 revealed that Ccq1 expression is ∼10-fold higher than Tpz1TPP1 (Supplemental Figure S1), consistent with the notion that only a small fraction of Ccq1 can participate in telomere regulation as part of the shelterin complex in fission yeast.
In contrast, a mutant strain expressing Ccq11-436 exhibited progressive telomere shortening, eventual telomere loss and chromosome circularization (Figure 1C and Supplemental Figure S2), much like ccq1Δ cells (Miyoshi et al., 2008; Tomita and Cooper, 2008; Harland et al., 2014). Expression of Ccq11-436 was also reduced to an extent similar to Ccq11-632 and Ccq11-583, but significantly higher than Ccq11-500 (Figure 1D), suggesting that reduced expression cannot be the sole cause of the telomere maintenance defect observed in ccq11-436 cells. Coimmunoprecipitation (co-IP) analysis revealed that Ccq1-Tpz1TPP1 interaction was disrupted for Ccq11-436 (Figure 1E, lane 6), while Ccq11-632, Ccq11-583, and Ccq11-500 maintained Ccq1-Tpz1TPP1 interaction (Figure 1E, lanes 2, 4, and 8). Thus we speculated that loss of Ccq1-Tpz1TPP1 interaction could be a major contributing factor in the Ccq11-436 truncation mutant protein’s inability to maintain telomeres.
We also monitored the ability of the Ccq1 truncation mutants to form heterochromatin at telomeres by monitoring transcriptional silencing of a his3+ marker inserted adjacent to telomeric repeats (Nimmo et al., 1998) and found that ccq11-500 and ccq11-436, much like ccq1Δ cells, failed to repress his3+ expression, while ccq11-632 and ccq11-583 cells maintained transcriptional repression at telomeres (Figure 1F). Thus the ccq11-500 mutant, despite maintaining normal telomere length, showed a defect in telomeric silencing comparable with ccq1Δ cells. It is currently unclear whether this defect in telomeric silencing may be caused by very low expression or misfolding of Ccq11-500 or, alternatively, whether amino acid residues 501–583 play an important role in heterochromatin formation, since Ccq11-583 can still maintain intact transcriptional silencing at telomeres.
Identification of Ccq1 amino acid residues critical for mediating stable Ccq1-Tpz1TPP1 interaction
To define amino acid residues that are critical for Ccq1 function, we generated a series of fission yeast strains carrying various Ccq1 point mutations for amino acid residues conserved among four Schizosaccharomyces species (Figure 2A and Supplemental Figure S3). Among all mutants tested, we identified five point mutations within the HDAC2/3-like domain of Ccq1 (C163R, L253R, L257R, G360R, and A363R) that caused telomere shortening when C-terminally FLAG-tagged mutant ccq1 alleles were integrated at the ccq1 locus (Figure 2B). Conversely, ccq1-R522A,R523A mutations within the SMC-like domain caused slight elongation of telomeres, while ccq1-R150, ccq1-Y561A,R562A, and ccq1-E645A,E646A showed no obvious change in steady-state telomere lengths (Figure 2B).
FIGURE 2:

Determination of Ccq1 amino acid residues critical for mediating Ccq1-Tpz1TPP1 interaction. (A) A schematic representation of Ccq1, marked with locations of mutated amino acid residues. Residues colored in red caused telomere shortening, residues colored in blue caused telomere elongation, and residues colored in black did not affect telomere length. (B) Telomere length determination by Southern blot analysis for indicated ccq1 mutants. Genomic DNA was prepared after indicated strains were successively restreaked on YES plates as indicated. (C) Expression levels of ccq1 mutants examined by anti-FLAG Western blot analysis. For ccq1-T93A, -C163R, -G360R, and -A363R alleles, Ccq1 protein was hyperphosphorylated (marked with asterisk *). (D) Examination of Ccq1-Tpz1TPP1 interaction by co-IP analysis for indicated ccq1 mutants. (E) Y2H assays for Tpz1-FL (full length), Ccq1-FL, Tpz1421-485, or Ccq1123-436 for indicated ccq1 mutant alleles. Positive interaction between Ccq1 and Tpz1 allowed cells to grow on plates that lack histidine (−His) and contain 5 mM 3AT.
Characterization of these point mutant alleles by anti-FLAG Western blot analysis revealed that L253R and L257R mutant proteins are expressed at lower levels than wild-type Ccq1 or other Ccq1 point mutants (Figure 2, C and D). In addition, Western blot analysis detected a slow-mobility Ccq1 band on SDS–PAGE corresponding to hyperphosphorylated Ccq1 (Moser et al., 2011) for three ccq1 mutants with shorter telomeres (ccq1-C163R, ccq1-G360R, and ccq1-A363R; Figure 2C). The hyperphosphorylation of Ccq1 in these mutant cells is likely induced by telomere shortening, since we have previously found that telomere shortening (in trt1Δ and ccq1-T93A mutant cells) leads to increased recruitment of the Rad3ATR-Rad26ATRIP DNA damage checkpoint kinase complex and hyperphosphorylation of Ccq1 (Moser et al., 2011; Chang et al., 2013).
Co-IP analysis revealed that all ccq1 mutants that cause telomere shortening (C163R, L253R, L257R, G360R, and A363R) also disrupt Ccq1-Tpz1TPP1 interaction, while ccq1-T93A and all other mutants retain robust interaction with Tpz1TPP1 (Figure 2D). On the other hand, yeast two-hybrid (Y2H) assays found that the interaction between full-length Ccq1 and Tpz1TPP1 is disrupted only for L253R and L257R mutants, while C163R, G360R, and A363R mutants retained Ccq1-Tpz1TPP1 interaction (Figure 2E). Because Y2H assays can often detect weak protein–protein interactions that cannot be easily detected by co-IP assays, it is possible that C163R, G360R, and A363R mutants retain residual weak Ccq1-Tpz1TPP1 interaction not detected by co-IP. On the other hand, loss of Ccq1-Tpz1TPP1 Y2H interaction for L253R and L257R mutants could simply reflect the fact that these mutant proteins may not fold properly or express at lower levels in budding yeast, since both mutants showed reduced expression in fission yeast (Figure 2, C and D).
When we tested Ccq1-Tpz1TPP1 interaction by Y2H using the minimum Ccq1-Tpz1TPP1 interaction domains Ccq1123-436 and Tpz1421-485, we found that Ccq1-C163R and Ccq1-A363R disrupted Ccq1-Tpz1TPP1 interaction, suggesting that Cys163 and Ala363 play critical roles in promoting interaction between the minimum Ccq1-Tpz1TPP1 binding domains of Ccq1 and Tpz1TPP1 (Figure 2E). On the other hand, since ccq1-C163R and ccq1-A363R did not disrupt Ccq1-Tpz1TPP1 Y2H interaction in the context of full-length proteins, it appears that Ccq1 and Tpz1TPP1 can also interact outside of the minimum Ccq1-Tpz1TPP1 binding domains previously identified by Y2H and in vitro binding assays (Jun et al., 2013; Harland et al., 2014).
In any case, we were able to successfully identify multiple Ccq1 mutations that disrupt (or substantially weaken) Ccq1-Tpz1TPP1 interaction and cause telomere shortening in fission yeast cells, much like the ccq11-436 truncation mutant that affected Ccq1-Tpz1TPP1 interaction (Figure 1E). Taken together, these analyses highlight the importance of Ccq1-Tpz1TPP1 interaction for maintaining telomeres at the wild-type length in fission yeast.
Loss of Ccq1-Tpz1TPP1 interaction causes defects in telomere function similar to ccq1Δ
We have recently reported the characterization of fission yeast cells expressing tpz1 mutant alleles (tpz1-L449R, tpz1-L449A, tpz1-Y439R,L445R, and tpz1-Y439R,L445R,L449R) that also disrupt Ccq1-Tpz1TPP1 interaction (Harland et al., 2014; Figure 1B). These mutant cells showed nearly identical phenotypes as ccq1Δ cells, that is, 1) formation of heterogeneous size colonies and preferential circularization of chromosomes among smaller colonies upon successive restreaks on plates, 2) synergistic severe cell death and chromosome circularization in combination with poz1Δ, and 3) inability to repress a checkpoint response at telomeres (Harland et al., 2014). Therefore we decided to also monitor these phenotypes for our new Ccq1-Tpz1TPP1–interaction disruption mutants of ccq1.
For a detailed characterization of telomere maintenance defects and epistasis analysis with poz1Δ, we decided to focus on ccq1-C163R, since this mutant allele did not affect Ccq1 expression, but caused loss of Ccq1-Tpz1TPP1 interaction in vivo and between the minimum Ccq1-Tpz1TPP1 interaction domains in Y2H assays (Figure 2, C–E). Just as we had previously reported for three Ccq1-Tpz1TPP1–interaction disruption mutant alleles of tpz1 (Harland et al., 2014), ccq1-C163R cells formed heterogeneously sized colonies, and selecting for a single small colony during successive restreaks resulted in telomere loss and isolation of circular chromosome survivor cells for ccq1-C163R (fifth restreak results in Figure 3A, lanes 10 and 13, and Figure 3C, lanes 9 and 12). On the other hand, when single large colonies were picked during successive restreaks (fifth restreak results in Figure 3A, lanes 11 and 14, and Figure 3C, lanes 10 and 13) or when a mixture of large and small colonies were used in successive restreaks (Figure 3A, lanes 5–8, and Figure 3C, lanes 4–7), ccq1-C163R cells were able to maintain short but stable telomeres, even after extensive restreaks on plates (16th restreak corresponds to ∼400 cell divisions). Taken together, these results and our previous analysis of tpz1 mutants (Harland et al., 2014) support the notion that loss of Ccq1-Tpz1TPP1 interaction causes heterogeneous phenotypes with either 1) immediate loss of cell viability due to elevated telomere fusions or 2) frequent emergence of more rapidly growing cells that manage to maintain short telomeres for many generations.
FIGURE 3:

Characterization of ccq1-C163R for its effect on telomere maintenance. (A) Southern blot analysis of telomere length for indicated mutant strains. For lanes 5–8, a mixture of faster and slower growing colonies were used in successive restreaks. For lanes 10 and 13, small (s) colonies were selectively used in successive restreaks. For lanes 11 and 14, big (b) colonies were selectively used in successive restreaks. (B) A NotI restriction map of S. pombe chromosomes. Telomeric NotI fragments (C, I, L, and M) are indicated with black boxes. (C) Pulsed-field gel analysis of telomere fusions for ccq1-C163R mutant restreaks. For lanes 4–7, a mixture of faster and slower growing colonies were used in successive restreaks. For lanes 9 and 12, small (s) colonies were selectively used in successive restreaks. For lanes 10 and 13, big (b) colonies were selectively used in successive restreaks. (D) Pulsed-field gel analysis for ccq1-C163R poz1Δ cells. Samples were prepared from early (2×) generation cells after strains were generated by genetic cross of parental haploid strains and dissection of resulting double-mutant spores.
When the ccq1-C163R mutation was combined with poz1Δ by genetic crosses of single mutant haploid cells, we found that most ccq1-C163R poz1Δ double-mutant cells were inviable and unable to form colonies on plates. However, we were able to observe occasional formation of microcolonies and found that all ccq1-C163R poz1Δ survivor cells had completely lost their telomeric repeats (Figure 3A, lanes 15 and 16) and carried circular chromosomes (Figure 3D, lanes 5 and 6). These results indicate that, much like ccq1Δ, tpz1-L449R, and tpz1-L449A mutants (Harland et al., 2014), ccq1-C163R cells are unable to maintain telomeres in the absence of Poz1. Thus our current results provide further support for our previous conclusion that Ccq1-Tpz1TPP1 and Poz1-Tpz1TPP1 interactions redundantly fulfill an essential telomere protection function of the shelterin complex.
In our previous study, we also found that tpz1 mutants that disrupt Ccq1-Tpz1TPP1 interaction cause cell elongation and activate the G2 DNA damage checkpoint (Harland et al., 2014). Likewise, our ccq1 mutants that disrupt Ccq1-Tpz1TPP1 interaction showed extensive cell elongation, suggesting activation of the G2 DNA damage checkpoint (Figure 4A). Consistently, we found that these ccq1 mutant cells show hyperphosphorylation of the DNA damage checkpoint kinase Chk1 (Figure 4B), and increased binding of the Rad3ATR regulatory subunit Rad26ATRIP to telomeres, based on chromatin immunoprecipitation (ChIP) assays (Figure 4C). Thus our results further confirm our earlier conclusion that Ccq1-Tpz1TPP1 interaction is critical for preventing DNA damage checkpoint activation at telomeres.
FIGURE 4:

Ccq1-Tpz1TPP1 interaction is required to prevent checkpoint activation. (A) Microscopic analysis of early generation (20–40 cell divisions) wild-type (wt), ccq1Δ, and various indicated ccq1 point mutant cells grown in liquid YES culture. White scale bar: 10 μm. (B) Western blot analysis of Chk-myc for indicated strains. Wild-type cells were also exposed to 100 Gy of gamma irradiation to induce DNA damage and Chk1 phosphorylation (*). (C) Effect of disrupting Ccq1-Tpz1TPP1 interaction on telomere association for Rad26ATRIP, monitored by ChIP assays. Error bars represent SEM from at least three experimental replicates. Statistical analysis of ChIP data by two-tailed Student’s t test is shown in Supplemental Table S4. Expression level of myc-tagged Rad26 was monitored by Western blot analysis. Anti-Cdc2 blot served as loading control.
Ccq1-Tpz1TPP1 interaction contributes to efficient accumulation of Ccq1 at telomeres and plays an essential role in Ccq1 Thr93 phosphorylation and telomerase recruitment
Our previous analysis of tpz1 mutants that disrupt Ccq1-Tpz1TPP1 interaction (tpz1-L449R, tpz1-L449A, tpz1-Y439R,L445R, and tpz1-Y439R,L445R,L449R) revealed that, although these mutants had substantial reduction in Ccq1 accumulation at telomeres, they still retained significant binding. This suggested that Ccq1-Tpz1TPP1 interaction promotes optimal Ccq1 binding to telomeres, but an alternative Ccq1-Tpz1TPP1 interaction-independent Ccq1 recruitment mechanism is likely to exist in fission yeast cells (Harland et al., 2014). Strikingly, loss of Ccq1-Tpz1TPP1 interaction eliminated Ccq1 Thr93 phosphorylation and, as a consequence, led to loss of telomerase recruitment to telomeres (Harland et al., 2014). These findings disagree with findings from another recent study, which concluded that 1) Ccq1-Tpz1TPP1 interaction does not contribute to Ccq1 association with telomeres, and 2) Ccq1-Tpz1TPP1 interaction plays no significant role in telomerase recruitment (Jun et al., 2013).
To resolve these conflicting conclusions, we therefore monitored by ChIP assays how our new Ccq1-Tpz1TPP1–interaction disruption mutations in ccq1 affect association of Ccq1 and Trt1TERT with telomeres. We found that C163R, G360R, and A363R caused similar reduction in Ccq1 association with telomeres, while L253R and L257R reduced Ccq1 binding even further (Figure 5A), which could also be caused by reduced expression of these two mutant proteins (Figures 2, C and D, and 5A). We further found that C163R and A363R mutations greatly reduced but did not eliminate binding of Ccq1 to subtelomeric regions 2–18 kb away from telomeric repeats (Supplemental Figure S4).
FIGURE 5:
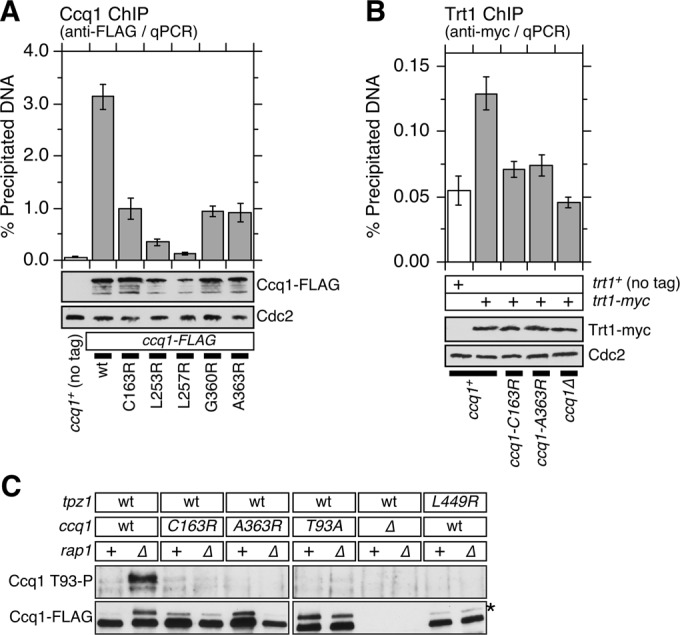
Ccq1-Tpz1TPP1 interaction contributes to optimal Ccq1 accumulation and is required for Ccq1 Thr93 phosphorylation and telomerase recruitment. (A and B) Effects of disrupting Ccq1-Tpz1TPP1 interaction on telomere association for (A) Ccq1 or (B) Trt1TERT, monitored by ChIP assays. Error bars represent SEM from at least three experimental replicates for Ccq1 ChIP and at least seven experimental replicates for Trt1TERT ChIP. Statistical analysis of ChIP data by two-tailed Student’s t test is shown in Supplemental Table S4. Expression levels of FLAG-tagged Ccq1 or myc-tagged Trt1TERT were monitored by Western blot analysis. Anti-Cdc2 blots served as loading control. (C) Ccq1 Thr93 phosphorylation, detected by phospho-(Ser/Thr) ATM/ATR substrate antibody (Moser et al., 2011), is abolished in Ccq1-Tpz1TPP1–interaction disruption mutants. Asterisk (*) indicates hyperphosphorylated form of Ccq1.
ChIP assays for Trt1TERT revealed that both ccq1-C163R and ccq1-A363R mutations cause loss of Trt1TERT binding to telomeres, much like the ccq1Δ mutation (Figure 5B). Consistently, we also found that both ccq1-C163R and ccq1-A363R, much like tpz1-L449R (Harland et al., 2014), cause loss of Ccq1 Thr93 phosphorylation, even in rap1Δ background, which we have previously established to strongly induce Ccq1 Thr93 phosphorylation (Moser et al., 2011; Chang et al., 2013; Figure 5C). Taken together, our current and previous studies utilizing ccq1 or tpz1 mutants that disrupt Ccq1-Tpz1TPP1 interaction firmly establish that Ccq1-Tpz1TPP1 interaction is required for optimal Ccq1 accumulation, Ccq1 Thr93 phosphorylation, and telomerase recruitment.
Ccq1-Tpz1TPP1 interaction contributes to heterochromatin formation and association of the SHREC subunit Clr3 with telomeres
Although we observed significant reduction in Ccq1 binding to telomeres in Ccq1-Tpz1TPP1–interaction disruption mutants of ccq1 (-C163R, -G360R, and -A363R; Figure 5A) and tpz1 (-L449R, -Y439R,L445R, and -Y439R,L445R,L449R; Harland et al., 2014), these mutant cells retained substantial residual Ccq1 binding, suggesting that fission yeast cells can recruit Ccq1 to telomeres even in the absence of Ccq1-Tpz1TPP1 interaction. An earlier study has shown that the heterochromatin silencing effector complex SHREC is recruited to telomeric and subtelomeric regions in a Ccq1- and Taz1-dependent manner and that Ccq1 associates with the SHREC subunit Clr3, an HDAC (Sugiyama et al., 2007). Therefore we previously speculated that residual binding of Ccq1 to telomeres in the absence of Ccq1-Tpz1TPP1 interaction might be facilitated by the SHREC complex itself or by SHREC-dependent heterochromatin formation (Harland et al., 2014).
We found that all ccq1 mutants that disrupt Ccq1-Tpz1TPP1 interaction (C163R, L253R, L257R, G360R, and A363R) were defective in transcriptional repression of the his3+ gene inserted adjacent to telomeric repeats (Figure 6A), much like ccq1Δ and tpz1 mutants that disrupt Ccq1-Tpz1TPP1 interaction (Harland et al., 2014). Thus intact Ccq1-Tpz1TPP1 interaction is critical for the formation of heterochromatin at telomeres, and heterochromatin formation per se is unlikely to play a critical part in the residual binding of Ccq1 in the absence of Ccq1-Tpz1TPP1 interaction.
FIGURE 6:

Ccq1-Tpz1TPP1 interaction contributes to heterochromatin formation and localization of the SHREC subunit Clr3 to telomeres. (A) Effects of Ccq1-Tpz1TPP1 interaction disruption mutants on transcriptional silencing of his3+ marker inserted adjacent to telomeric repeats (Nimmo et al., 1998). A fivefold serial dilution series of indicated strains was spotted on YES or PMG ULA (−histidine) plates. (B and C) Effects of disrupting Ccq1-Tpz1TPP1 interaction and elimination of factors essential for heterochromatin formation on telomere association for (B) Ccq1 or (C) Clr3, monitored by ChIP assays. Error bars represent SEM from at least four experimental replicates for Ccq1 and at least three experimental replicates for Clr3. Statistical analysis of ChIP data by two-tailed Student’s t test is shown in Supplemental Table S4. Expression levels of FLAG-tagged Ccq1 or Clr3 were monitored by Western blot analysis. Anti-Cdc2 blots served as loading control. (D) Examination of Ccq1-Clr3 interaction by co-IP indicated that Ccq1-Tpz1TPP1 interaction is dispensable for Ccq1-Clr3 interaction. (E) Y3H assay for Clr3, Ccq1, and Tpz1. Expression of Ccq1 was able to bridge interaction between GBD-Clr3 and GAD-Tpz1 to allow growth on plates that lacked histidine (−His) and contained 1 mM 3AT. As expected, disruption of Ccq1-Tpz1TPP1 interaction by tpz1-L449R mutation eliminated positive Y3H signal.
Next we monitored the association of Ccq1 with telomeres in the absence of Clr3 and the histone H3 methyltransferase Clr4 by ChIP assay (Figure 6B). In clr3Δ and clr4Δ cells, Ccq1 binding to telomeres was only slightly reduced, while Ccq1 binding was significantly more reduced in ccq1-C163R cells, indicating that, although heterochromatin formation at telomeres is required for optimal Ccq1 binding, Ccq1-Tpz1TPP1 interaction is more important for the efficient accumulation of Ccq1 at telomeres. Importantly, we found that loss of Clr3 or Clr4 does not cause any further reduction in Ccq1 binding when clr3Δ or clr4Δ are combined with ccq1-C163R (Figure 6B). Thus residual telomere association of Ccq1 observed in ccq1-C163R cells does not depend on Clr3 or Clr4, supporting the notion that the residual Ccq1 binding found in Ccq1-Tpz1TPP1–interaction disruption mutant cells are indeed independent of heterochromatin formation.
To gain a more mechanistic insight into why Ccq1-Tpz1TPP1 interaction is required to establish heterochromatin at telomeres, we also monitored telomere association of the HDAC Clr3. Previous studies have found that Clr3 is required for heterochromatin formation at both telomeric and nontelomeric sites, where it directly interacts with Ccq1, and that ccq1Δ cells fail to efficiently recruit Clr3 to telomeres (Sugiyama et al., 2007; Miyoshi et al., 2008).
ChIP assays for Clr3 indicated that both ccq1-C163R and ccq1-A363R cells show substantial reduction in Clr3 binding to telomeres (Figure 6C) but retain more binding compared with ccq1Δ cells, consistent with the finding that Ccq1 is still recruited to telomeres in C163R and A363R cells (Figure 5A). On the other hand, elimination of heterochromatin at telomeres by deletion of Clr4 only moderately reduced Clr3 association with telomeres. Furthermore, we found that Ccq1 still interacts with Clr3 in C163R and A363R mutant cells in co-IP assays (Figure 6D). Interestingly, while we and others have not been able to detect the complex that contains Tpz1, Ccq1, and Clr3 by co-IP analysis (Miyoshi et al., 2008; Moser et al., 2011), we were able to obtain positive yeast three-hybrid (Y3H) interaction among GAD-Tpz1, DBD-Clr3, and untagged Ccq1 (Figure 6E), suggesting that the shelterin and SHREC complexes may be able to interact through their common subunit Ccq1, even though shelterin–SHREC interaction may be too weak or transient to be detected by co-IP assays. Taken together, results from our current study establish that Ccq1-Tpz1TPP1 interaction plays a major role in efficient accumulation of the Ccq1-SHREC complex at telomeres, while Clr3- and Clr4-dependent formation of heterochromatin also contributes to further enhance the optimal association of Ccq1-SHREC with telomeres.
DISCUSSION
In this paper, we investigated the functional significance of the Ccq1-Tpz1TPP1 interaction in fission yeast by isolating a series of ccq1 HDAC2/3-like domain mutants that disrupt Ccq1-Tpz1TPP1 interaction based on co-IP assays. Consistent with our previous study, in which we analyzed tpz1 mutants that disrupt Ccq1-Tpz1TPP1 interaction (Harland et al., 2014), ccq1 mutational analyses described in this study have further established that Ccq1-Tpz1TPP1 interaction is required for optimal localization of Ccq1 at telomeres, Rad3ATR/Tel1ATM-dependent phosphorylation of Ccq1 Thr93, and hence telomerase recruitment to telomeres (Figure 5). Failure to efficiently recruit telomerase then leads to defects in telomere length maintenance and chromosome fusions (Figures 2 and 3). Our analysis further confirmed that disruption of Ccq1-Tpz1TPP1 interaction causes more accumulation of the Rad3ATR-Rad26ATRIP kinase complex at telomeres and activation of the G2 checkpoint kinase Chk1, much like ccq1Δ (Figure 4). We have previously shown that Rad3ATR and Tel1ATM redundantly contribute to Ccq1 accumulation at telomeres and suggested the existence of a regulatory loop wherein dysfunctional telomeres accumulate more Rad3ATR, which in turn would ensure restoration of telomere protection by promoting Ccq1 accumulation at telomeres (Moser et al., 2009b). Because rad3Δ tel1Δ cells show greater reduction in Ccq1 accumulation than Tpz1TPP1 at telomeres based on ChIP assays (Moser et al., 2009b), it is possible that Rad3ATR/Tel1ATM kinases promote Ccq1 accumulation at telomeres by stabilizing the Ccq1-Tpz1TPP1 interaction.
We find it remarkable that, despite increased accumulation of the Rad3ATR-Rad26ATRIP kinase complex, Rad3ATR/Tel1ATM-dependent phosphorylation of Ccq1 Thr93 (Moser et al., 2011; Yamazaki et al., 2012) is eliminated when Ccq1-Tpz1TPP1 interaction is disrupted, while Ccq1 remains hyperphosphorylated on other (not yet identified) sites (Harland et al., 2014; Figure 2C). Thus we speculate that Tpz1TPP1 binding to Ccq1 may cause a conformational change within Ccq1 to allow for optimal phosphorylation of Ccq1 Thr93, while such Ccq1-Tpz1TPP1 interaction–dependent conformational change is not required for Rad3ATR/Tel1ATM-dependent phosphorylation of other Ccq1 phosphorylation sites. Because all mutants that disrupt Ccq1-Tpz1TPP1 interaction from our current ccq1 mutagenesis study and previous tpz1 mutagenesis study lost Ccq1 Thr93 phosphorylation that is required for efficient telomerase recruitment, it is reassuring to see that Trt1TERT recruitment is essentially eliminated in all those mutants (Harland et al., 2014; Figure 5, B and C). Conversely, the claim that Ccq1-Tpz1TPP1 interaction does not contribute to telomerase recruitment (Jun et al., 2013) seems unlikely, based on the observed loss of Ccq1 Thr93 phosphorylation upon disruption of Ccq1-Tpz1TPP1 interaction.
The current study has also established for the first time that Ccq1-Tpz1TPP1 interaction contributes to the optimal localization of the HDAC Clr3 at telomeres but is dispensable for Ccq1-Clr3 interaction (Figure 6). Therefore, much like Ccq1, substantial residual binding of Clr3 to telomeres can be observed in cells that lack Ccq1-Tpz1TPP1 interaction. We have also established that the residual binding of Ccq1 to telomeres observed in the absence of Ccq1-Tpz1TPP1 interaction is independent of the histone H3 methyltransferase Clr4, but Clr4/Clr3-dependent formation of heterochromatin at telomeres is needed to achieve optimal localization of Ccq1 and Clr3 in wild-type cells with intact Ccq1-Tpz1TPP1 interaction (Figure 6B). Our results also strongly implicate the existence of unidentified mechanism(s) that allow Ccq1-Clr3 localization independent of both the Ccq1-Tpz1TPP1 interaction and histone H3 K9 methylation-dependent heterochromatin formation. Because previous studies have found physical and genetic interactions between Ccq1 and the condensin complex (Motwani et al., 2010; Reyes et al., 2015), it is tempting to speculate that Ccq1–condensin interaction may allow localization of Ccq1 at telomeres in the absence of Ccq1-Tpz1TPP1 interaction and heterochromatin. However, it remains to be tested whether condensin or other unknown factor(s) contribute to optimal Ccq1 and SHREC association at telomeres. It would be a major challenge for future investigations to decipher how multiple mechanisms that promote Ccq1-SHREC accumulation work collaboratively to ensure telomerase recruitment, checkpoint inhibition, and heterochromatin formation at fission yeast telomeres.
MATERIALS AND METHODS
Yeast strains and plasmids
Standard techniques and protocols were used to generate and culture fission yeast and budding yeast strains (Alfa et al., 1993; Amberg et al., 2005). The status of heterochromatin-dependent transcriptional silencing at telomeres in various ccq1 mutants was monitored by utilizing his3+::tel(1L) strains that carry an integrated his3+ marker adjacent to telomeric repeats of the chromosome I left arm, as previously described (Nimmo et al., 1998).
Fission yeast strains used in this study are listed in Supplemental Table S1. For ccq1∆::hphMX, poz1∆::natMX6, rap1∆::ura4+, and clr4Δ::kanMX6, original deletion strains were described previously (Kanoh and Ishikawa, 2001; Moser et al., 2009b; Khair et al., 2010). For tpz1-myc, trt1-myc, ccq1-myc, ccq1-FLAG, chk1-myc, and clr3-FLAG, original tagged strains were described previously (Lopez-Girona et al., 2001; Miyoshi et al., 2008; Webb and Zakian, 2008; Moser et al., 2009a, b). Strain construction for ccq1-T93A-FLAG and tpz1-L449R-myc were also previously described (Moser et al., 2011; Harland et al., 2014).
Various FLAG-tagged ccq1 mutant alleles were integrated at the endogenous ccq1+ locus. Point mutations and C-terminal truncations for ccq1 were introduced by Phusion site-directed mutagenesis (F-541; New England Biolabs, Ipswich, MA) or QuikChange Lightning site-directed mutagenesis (210513; Agilent Technologies, Santa Clara, CA) into ccq1 Y2H plasmids, and these mutant ccq1 plasmids were used as templates for two-step stitching PCR to generate ccq1-FLAG-kanMX integration constructs to be used in transformation of fission yeast cells. After confirmation of correct integration of mutant ccq1 alleles at the ccq1 locus, the ccq1 open reading frame, epitope tag, and 5′- and 3′-flanking regions of the transformants were sequenced to confirm that desired mutations were correctly integrated. Strains carrying various ccq1 mutations without epitope tags were subsequently generated by transforming ccq1-FLAG mutant strains with a C-terminal ccq1 fragment marked with natMX within the 3′ untranslated region of the ccq1+ gene. Correct removal of tag and lack of undesired mutations were then confirmed by sequencing.
Plasmids used in Y2H or Y3H assays are listed in Supplemental Table S2. Y2H assays were performed by mating Saccharomyces cerevisiae MATa (Y2HGold: MATa trp1-901 leu2-3,-112 ura3-52 his3-200 LYS2::GAL1(UAS)-GAL1(TATA)-HIS3 GAL2(UAS)-GAL2(TATA)-ADE2 gal4Δ gal80Δ URA3::MEL1(UAS)-MEL1(TATA)-AUR1-C MEL1) strains harboring GAL4-DBD (DNA-binding domain) plasmids with MATα (Y187: MATα trp1-901 leu2-3,-112 ura3-52 his3-200 ade2-101 gal4Δ gal80Δ met− URA3::GAL1(UAS)-GAL1(TATA)-LacZ MEL1) strains harboring GAL4-AD (activation domain) plasmids, as previously described (Moser et al., 2011). Positive interactions were identified by spotting mated cells onto synthetic defined -His -Trp -Leu (SD-HTL) plates supplemented with either 1 or 5 mM 3-amino-1,2,4-triazole (3AT), as indicated in the figure legends.
Co-IP and Western blot analysis
Co-IP experiments were performed as previously described (Moser et al., 2009b, 2011). Monoclonal anti-FLAG (M2-F1804; Sigma, St. Louis, MO), anti-myc (9B11; Cell Signaling, Danvers, MA) or anti-Cdc2 (y100.4; Abcam, Cambridge, MA) were used as primary antibodies for Western blot analysis. Cdc2 was used as the loading control. For detection of Ccq1 Thr93 phosphorylation by phospho-(Ser/Thr) ATM/ATR substrate antibody (2851; Cell Signaling), Ccq1 was first immunopurified from whole-cell extract using anti-FLAG antibody (Moser et al., 2011). Either horseradish peroxidase (HRP)-conjugated (goat) anti-mouse (31430; Thermo Fisher Scientific, Waltham, MA) or HRP-conjugated (goat) anti-rabbit (31460; Thermo Fisher Scientific) was used as the secondary antibody.
Pulsed-field gel electrophoresis and Southern blot analysis
Pulsed-field gel electrophoresis of NotI-digested chromosomal DNA was performed with the CHEF-DR III system (6 V/cm [200 V] and a pulse time of 60–120 s for 24 h in 0.5×TAE buffer at 14°C; Bio-Rad [Hercules, CA]; Nakamura et al., 2002). For telomere length analysis by Southern blotting, EcoRI-digested genomic DNA was separated on a 1% agarose gel and probed with a telomeric DNA probe as previously described (Nakamura et al., 2002; Moser et al., 2014).
ChIP assay, quantitative PCR, and statistical analysis
Cells were processed for ChIP using monoclonal anti-Myc (9B11) or anti-FLAG (M2-F1804; Sigma) antibodies and analyzed by quantitative real-time PCR with primers annealing to a region adjacent to the telomeric repeat or subtelomeric regions, as previously described (Moser et al., 2011, 2014). Primers used in ChIP assays are listed in Supplemental Table S3. Statistical analysis of ChIP data by two-tailed Student’s t test is summarized in Supplemental Table S4.
Supplementary Material
Acknowledgments
We thank Fuyuki Ishikawa, Junko Kanoh, Virginia A. Zakian, Robin Allshire, Mark Flory, and Paul Russell for sharing published yeast strains and plasmids used in this study. We also thank Ya-Ting Chang, Lynn Khair, and Tiffani Berkel for their efforts in generating control strains that were invaluable for carrying out the current study. This work was supported by a National Institutes of Health R01 grant (GM078253).
Abbreviations used:
- 3AT
3-amino-1,2,4-triazole
- AD
activation domain
- ChIP
chromatin immunoprecipitation
- co-IP
coimmunoprecipitation
- dsDNA
double-stranded DNA
- HDAC
histone deacetylase
- HRP
horseradish peroxidase
- SHREC
Snf2/HDAC-containing repressor complex
- SMC
Structural Maintenance of Chromosomes
- Y2H
yeast two-hybrid
- Y3H
yeast three-hybrid.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-07-0481) on September 9, 2015.
REFERENCES
- Alfa C, Fantes P, Hyams J, McLoed M, Warbrick E. Experiments with Fission Yeast. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. [Google Scholar]
- Amberg DC, Burke DJ, Strathern JN. Methods in Yeast Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2005. [Google Scholar]
- Baumann P, Cech TR. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science. 2001;292:1171–1175. doi: 10.1126/science.1060036. [DOI] [PubMed] [Google Scholar]
- Blackburn EH, Greider CW, Szostak JW. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med. 2006;12:1133–1138. doi: 10.1038/nm1006-1133. [DOI] [PubMed] [Google Scholar]
- Chang YT, Moser BA, Nakamura TM. Fission yeast shelterin regulates DNA polymerases and Rad3ATR kinase to limit telomere extension. PLoS Genet. 2013;9:e1003936. doi: 10.1371/journal.pgen.1003936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JP, Nimmo ER, Allshire RC, Cech TR. Regulation of telomere length and function by a Myb-domain protein in fission yeast. Nature. 1997;385:744–747. doi: 10.1038/385744a0. [DOI] [PubMed] [Google Scholar]
- Flory MR, Carson AR, Muller EG, Aebersold R. An SMC-domain protein in fission yeast links telomeres to the meiotic centrosome. Mol Cell. 2004;16:619–630. doi: 10.1016/j.molcel.2004.10.027. [DOI] [PubMed] [Google Scholar]
- Harland JL, Chang YT, Moser BA, Nakamura TM. Tpz1-Ccq1 and Tpz1-Poz1 interactions within fission yeast shelterin modulate Ccq1 Thr-93 phosphorylation and telomerase recruitment. PLoS Genet. 2014;10:e1004708. doi: 10.1371/journal.pgen.1004708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun HI, Liu J, Jeong H, Kim JK, Qiao F. Tpz1 controls a telomerase-nonextendible telomeric state and coordinates switching to an extendible state via Ccq1. Genes Dev. 2013;27:1917–1931. doi: 10.1101/gad.219485.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanoh J, Ishikawa F. spRap1 and spRif1, recruited to telomeres by Taz1, are essential for telomere function in fission yeast. Curr Biol. 2001;11:1624–1630. doi: 10.1016/s0960-9822(01)00503-6. [DOI] [PubMed] [Google Scholar]
- Khair L, Subramanian L, Moser BA, Nakamura TM. Roles of heterochromatin and telomere proteins in regulation of fission yeast telomere recombination and telomerase recruitment. J Biol Chem. 2010;285:5327–5337. doi: 10.1074/jbc.M109.078840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Girona A, Tanaka K, Chen XB, Baber BA, McGowan CH, Russell P. Serine-345 is required for Rad3-dependent phosphorylation and function of checkpoint kinase Chk1 in fission yeast. Proc Natl Acad Sci USA. 2001;98:11289–11294. doi: 10.1073/pnas.191557598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Panchenko AR, Shoemaker BA, Thiessen PA, Geer LY, Bryant SH. CDD: a database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Res. 2002;30:281–283. doi: 10.1093/nar/30.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi T, Kanoh J, Saito M, Ishikawa F. Fission yeast Pot1-Tpp1 protects telomeres and regulates telomere length. Science. 2008;320:1341–1344. doi: 10.1126/science.1154819. [DOI] [PubMed] [Google Scholar]
- Moser BA, Chang YT, Kosti J, Nakamura TM. Tel1ATM and Rad3ATR kinases promote Ccq1-Est1 interaction to maintain telomeres in fission yeast. Nat Struct Mol Biol. 2011;18:1408–1413. doi: 10.1038/nsmb.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser BA, Chang YT, Nakamura TM. Telomere regulation during the cell cycle in fission yeast. Methods Mol Biol. 2014;1170:411–424. doi: 10.1007/978-1-4939-0888-2_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser BA, Nakamura TM. Protection and replication of telomeres in fission yeast. Biochem Cell Biol. 2009;87:747–758. doi: 10.1139/o09-037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser BA, Subramanian L, Chang YT, Noguchi C, Noguchi E, Nakamura TM. Differential arrival of leading and lagging strand DNA polymerases at fission yeast telomeres. EMBO J. 2009a;28:810–820. doi: 10.1038/emboj.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser BA, Subramanian L, Khair L, Chang YT, Nakamura TM. Fission yeast Tel1ATM and Rad3ATR promote telomere protection and telomerase recruitment. PLoS Genet. 2009b;5:e1000622. doi: 10.1371/journal.pgen.1000622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motwani T, Doris R, Holmes SG, Flory MR. Ccq1p and the condensin proteins Cut3p and Cut14p prevent telomere entanglements in the fission yeast Schizosaccharomyces pombe. Eukaryot Cell. 2010;9:1612–1621. doi: 10.1128/EC.00339-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura TM, Moser BA, Russell P. Telomere binding of checkpoint sensor and DNA repair proteins contributes to maintenance of functional fission yeast telomeres. Genetics. 2002;161:1437–1452. doi: 10.1093/genetics/161.4.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmo ER, Pidoux AL, Perry PE, Allshire RC. Defective meiosis in telomere-silencing mutants of Schizosaccharomyces pombe. Nature. 1998;392:825–828. doi: 10.1038/33941. [DOI] [PubMed] [Google Scholar]
- Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- Reyes C, Serrurier C, Gauthier T, Gachet Y, Tournier S. Aurora B prevents chromosome arm separation defects by promoting telomere dispersion and disjunction. J Cell Biol. 2015;208:713–727. doi: 10.1083/jcb.201407016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer EL, Eddy SR, Durbin R. Pfam: a comprehensive database of protein domain families based on seed alignments. Proteins. 1997;28:405–420. doi: 10.1002/(sici)1097-0134(199707)28:3<405::aid-prot10>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Cam HP, Sugiyama R, Noma K, Zofall M, Kobayashi R, Grewal SI. SHREC, an effector complex for heterochromatic transcriptional silencing. Cell. 2007;128:491–504. doi: 10.1016/j.cell.2006.12.035. [DOI] [PubMed] [Google Scholar]
- Tomita K, Cooper JP. Fission yeast Ccq1 is telomerase recruiter and local checkpoint controller. Genes Dev. 2008;22:3461–3474. doi: 10.1101/gad.498608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb CJ, Zakian VA. Identification and characterization of the Schizosaccharomyces pombe TER1 telomerase RNA. Nat Struct Mol Biol. 2008;15:34–42. doi: 10.1038/nsmb1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki H, Tarumoto Y, Ishikawa F. Tel1ATM and Rad3ATR phosphorylate the telomere protein Ccq1 to recruit telomerase and elongate telomeres in fission yeast. Genes Dev. 2012;26:241–246. doi: 10.1101/gad.177873.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.