

Abstract
Multidrug resistance (MDR) to doxorubicin (DOX) limits its effectiveness against tumor cells. Arsenic trioxide (As2O3) has been reported to reduce MDR in various types of cancer, but the mechanisms involving Ras and p-glycoprotein (P-gp) remain to be fully elucidated. The objectives of the present study were to evaluate As2O3 in reversing MDR to DOX, and to identify the association in antitumor activities between the effectiveness of DOX and Ras/phosphorylated (p-) extracellular signal-regulated kinase (ERK)1/2 signaling in SGC7901/ADM and SGC7901/S human gastric cancer cell lines. Cytotoxicity and sensitivity towards As2O3 were assessed using non-toxic and mildly-toxic concentrations (0.1 and 0.5 µM, respectively). The reversing effect of As2O3 on MDR was investigated prior to and following treatment with a cytokine activation of the recombinant human granulocyte colony stimulating factor ERK pathway. The SGC7901/ADM and SGC7901/S cells had the same sensitivity to As2O3. The SGC7901/ADM cells were resistant to DOX and As2O3 treatment reduced the level of resistance to DOX (P<0.01). The expression of P-glycoprotein (P-gp) in the SGC7901/ADM cells was higher than in the SGC7901/S cells (P<0.001). As2O3 treatment decreased the levels of P-gp in a time- and dose-dependent manner (P<0.01). The expression of Ras was higher in the SGC7901/ADM cells than in the SGC7901/S cells, while the expression of p-ERK1/2 remained the same. As2O3 decreased the levels of Ras and p-ERK1/2 (P<0.01). Following pretreatment with rhG-CSF, the levels of Ras and p-ERK1/2 were further decreased (P<0.01). Drug-resistant gastric cancer cells had higher expression levels of P-gp and Ras, but not of p-ERK1/2. Non- and mildly-toxic doses of As2O3 reduced MDR to DOX through Ras/p-ERK1/2 signaling.
Keywords: arsenic trioxide, extracellular signal-regulated mitogen-activated protein kinases, Ras, P-Glycoprotein, gastric cancer, multidrug resistance, doxorubicin
Introduction
In 2008, there were a 989,600 new cases of gastric cancer (GC) and a GC-associated mortality rate of 738,000 worldwide (1). Although the rate of GC-associated mortality has declined in the last 50 years (2), it remains the second leading cause of cancer-associated mortality worldwide, with an incidence rate ranking fourth following lung, breast and colorectal cancer (3). Systemic chemotherapy remains critical in the treatment of GC.
DOX is a well-known chemotherapeutic treatment used in Hodgkin's lymphoma, non-Hodgkin's lymphoma, multiple myeloma, acute leukemia, Kaposi's sarcoma, Ewing's sarcoma, Wilms' tumor and in solid tumors of the breast, adrenal cortex, prostate, thyroid, head and neck, endometrium, lung and ovary (4). DOX is widely used to treat GC in combination regimens with etoposide, cisplatin and 5-fluorouracil. However, drug resistance to DOX significantly limits its efficacy. Resistance to DOX is often associated with the expression of p-glycoprotein (P-gp) (4).
Multidrug resistance (MDR) is a complex process due to several mechanisms. Numerous resistance-associated proteins, including P-gp, have been found in different cancer cells, and overexpression of this transmembrane glycoprotein causes accelerated efflux of several lipid-soluble chemotherapeutic agents from the cells, decreasing their efficacy and leading to treatment failure (5). The difficulty of reversing this mechanism underlies its complexity.
Arsenic trioxide (As2O3) appears to achieve complete remission in 70% of patients with acute promyelocytic leukemia (APL) by inducing cancer cell apoptosis (6). Additional studies have confirmed that low doses of As2O3 induces complete remission in 90% of relapsed patients with APL (7–9). To date, a number of studies have provided novel insights into the pathogenesis of this agent and indicate that As2O3 may be useful in treating other types of cancer (10,11). It has become evident that the effects of As2O3 are not restricted to APL cells, but that As2O3 may also be effective in other malignant cells in several of types of cancer, including chronic myeloid leukemia, multiple myeloma, and solid prostate, esophageal and ovarian tumors (12–15). A number of mechanisms are associated with the effects of As2O3, including the induction of apoptosis, inhibition of growth, inhibition of differentiation and angiogenesis, and inhibition of mitochondrial respiration (16–18). In addition, several signaling pathways are involved in the antitumoral activity of As2O3, including inactivation of the Notch, c-Jun NH2-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK) signaling pathways (19–21). Human GC cells have been found to be sensitive to As2O3 (22).
Few studies have investigated the involvement of Ras and P-gp in MDR in GC cancer. The present study was designed to assess As2O3 in reversing DIX-induced MDR in GC cancer cell lines. The association between the antitumor effects of As2O3 and the Ras/phosphorylated (p-)ERK1/2 signaling pathway was also examined. The results of the present study aimed to provide novel insights into the treatment of gastric cancer.
Materials and methods
Chemicals and reagents
In the present study, As2O3 and 3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Cyclosporin A (CsA) was purchased from Novartis (Bale, Switzerland). RPMI 1640, fetal bovine serum (FBS), and dimethyl sulfoxide (DMSO) were purchased from Thermo Fisher Scientific (Hyclone, Waltham, MA, USA). DOX was purchased from Zhejiang Haizheng Pharmaceutical Co. (Taizhou, China). The recombinant human granulocyte colony stimulating factor (rhG-CSF) was purchased from Qilu Pharmaceutical (Jinan, China). The mouse anti-P-gp (P-170), Ras (cat. no. ZA-0343), p-ERK1/2 (cat. no. sc-7383), horseradish peroxidase (HRP)-conjugated goat-anti-mouse secondary antibody and the corresponding immunohisto-chemistry kits were purchased from Beijing Golden Bridge Biotechnology Co. (Beijing, China).
Cell culture
The parental SGC7901/S human GC cell line and the DOX-resistant SGC7901/ADM cell line were obtained from the Key Laboratory of Cancer Biology and Xijing Hospital of Digestive Diseases, Fourth Military Medical University (Xi'an, China). The SGC7901/ADM cells were obtained from the SGC7901/S cells, based on a previously described method (23). In brief, the SGC7901/ADM cells were induced in vitro by continuous exposure of SGC7901 parent cells to adriamycin (ADM) at concentrations of 0.25 µg/l to 25 µg/l. Cell lines capable of sustained growth in medium containing 25 µg/l ADM were considered to be resistant after 3 months. The SGC7901/S cells were cultured in RPMI 1640 medium supplemented with 100 U/ml penicillin, 100 µg/ml streptomycin and 10% (v/v) heat-inactivated FBS. The SGC7901/ADM cells were cultured in the same medium with 1 µg/ml DOX, and were grown without DOX for 2 weeks prior to the experiments. All the cells were cultured at 37°C in a 5% CO2 humidified atmosphere and analyses were performed in the logarithmic growth phase.
Cytotoxicity and sensitivity of SGC7901/S and SGC7901/ADM cells to As2O3
The SGC7901/S and SGC7901/ADM cells were trypsinized, centrifuged at 1,000 × g for 5 min, harvested and seeded into 96-well plates, in triplicate, at a density of 1×104 cells/well (100 µl) at 37°C. Following overnight incubation, the cells became adherent. The culture medium was removed and As2O3 was added at 12 concentrations (0.01, 0.05, 0.10, 0.25, 0.50, 0.75, 1.0, 2.5, 5.0, 10.0, 15.0 or 20.0 mM). The cells (1×104 cells/well) were incubated for 24 h, 48 h and 72 h, and were further incubated for an additional 4 h in the presence of 10 µl/well of MTT reagent, containing 5.00 mg/ml in phosphate-buffered saline (PBS). Following aspiration of the medium, the resulting formazan was dissolved with DMSO (100 µl/cell). The plates were agitated mechanically for 60 sec, and the optical density (OD) or absorbance was measured immediately at 490 nm on a microplate reader (AR2010; Bio-Rad Laboratories, Inc., Hercules, CA, USA). The cell survival rate was calculated as: (OD of the assessment group / OD of the control group) × 100%. The inhibition rate and half maximal inhibitory concentration of the reagent (IC50) were calculated based on the survival rate (SR) (24,25). The dose of As2O3, which resulted in a SR of >95% was considered non-toxic, and the dose resulting in a SR >85% was considered mildly-toxic.
Resistance of SGC7901/ADM cells and the resistance-reversing effect of As2O3
The present study included five groups, which were as follows: SGC7901/ADM, SGC7901/S, SGC7901/ADM+As2O3 (0.10 µM), SGC7901/ADM+As2O3 (0.50 µM) and SGC7901/ADM+CsA (4.00 µg/ml). These two concentrations of As2O3 were selected as the non-toxic and the mildly-toxic doses in the experiment described above. To investigate the drug resistance-reversing effect of As2O3, DOX (0.1, 0.5, 1.0, 2.5, 5.0, 10.0, 15.0 and 30.0 µg/ml) was added to the cell culture (1×104 cells/well), rather than As2O3. The cells were then incubated for 24 h and 48 h at 37°C. The drug resistance ratio was calculated as the IC50 of the drug-resistant cells divided by the IC50 of the parental cells. The reversal ratio was calculated as the IC50 of the drug-resistant cells divided by the IC50 of the drug-resistant cells treated with reversal agents.
Reversing effect of As2O3 following treatment with rhG-CSF
The SGC7901/ADM, SGC7901/S, SGC7901/ADM+0.10 µM As2O3, SGC7901/ADM+0.50 µM As2O3, and SGC7901/ADM+4.00 µg/ml CsA cells (1×104 cells/well) were incubated with rhG-CSF (1.50 µg/ml) for 3 days at 37°C, and then cultured with DOX at different concentrations (0.1, 0.5, 1.0, 2.5, 5.0, 10.0, 30.0, 40.0 and 50.0 µM) for a further 2 days at 37°C, following which the reversing effect of As2O3 was assessed. The blank control contained no cells in the culture medium, while the negative control contained medium with no reagent.
Analyzing the expression of P-gp using western blotting
The SGC7901/ADM and SGC7901/S cells (1×104 cells/well) were grouped, as described above, and were pretreated with non- and mildly-toxic concentrations of As2O3 for 24, 48 and 72 h (18). The cells were collected by centrifugation at 1,000 × g for 20 min at 4°C and washed with ice-cold PBS. The cell pellets were then resuspended in lysis buffer (Invitrogen Life Technologies, Carlsbad, CA, USA), containing 50 mM Tris-chloride (pH 7.4), 150 mM sodium chloride, 0.1% sodium dodecyl sulfate, 1% Triton-100 and 1 mM ethylenediaminetetraacetic acid (pH 8.0). Following incubation at 4°C for 40 min, the samples were centrifuged at 15,400 × g for 20 min. The supernatants containing cytosolic proteins were separated by polyacrylamide gel (Bio-Rad Laboratories, Inc.) electrophoresis at 200 V for 1–2 h, and were transferred onto a nitrocellulose membrane (EMD Millipore, Billerica, MA, USA) at 120 V for 3 h. The membrane was blocked with 5% skim milk powder at room temperature for 2 h, followed by incubation with primary monoclonal mouse anti-human antibody (P-gp, 1:50) for 2 h at room temperature. Following washing with blocking buffer, the membranes were incubated with HRP-conjugated goat-anti-mouse secondary antibody (1:1,000) for 2 h at room temperature, followed by chemiluminescence detection (Pierce Biotechnology, Inc., Rockford, IL, USA). Western blot analysis was performed using an image analysis system (VIDAS System; Biomérieux, Marcy l'Étoile, France).
Immunocytochemical assays
Expression levels of Ras and p-ERK1/2 in cells treated with As2O3
A total of six treatment groups were used to assess the levels of Ras and p-ERK1/2: SGC7901/ADM+As2O3 (0.10, 0.50, and 0.75 µM), SGC7901/ADM+CsA (4.00 µg/ml), SGC7901/ADM and SGC7901/S. The cells (1×104 cells/well) were treated, as described above, and incubated for 24, 48, and 72 h. As previously described (13), the cells were fixed in ice-cold acetone for 15 min. Following incubating with 3% hydrogen peroxide-carbinol for 10 min, the cells were incubated at 37°C for 2 h with the following primary antibodies: Mouse-anti-human monoclonal Ras or p-ERK1/2. Following incubation with Poly Helper (50 µl; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) at 37°C for 30 min, the HRP-conjugated goat-anti-mouse IgG antibody solution was added and incubated again at 37°C for 30 min. Finally, staining with 3,3′-diaminobenzidine (Sigma-Aldrich) and hematoxylin (Sigma-Aldrich) were performed. The labeled cells were analyzed using an image analysis system (VIDAS, Option Company).
Expression levels of Ras and p-ERK1/2 following treatment with an ERK activator
rhG-CSF is a cytokine, which activates a number of cellular pathways, including the ERK pathway (26,27). Therefore, the SGC7901/ADM and SGC7901/S cells (1×104 cells/well) were treated with rhG-CSF (1.50 µg/ml) for 3 days at 37°C, and then divided into the five treatment groups, described above. The cells were then incubated with the primary antibody. The experiments were repeated three times.
Statistical analysis
Data are presented as the mean ± standard deviation of three independent experiments. Student's t-test and analysis of variance were used to analyze the differences between groups, with a Student-Newman-Keuls (q-test) post hoc test. P<0.05 was considered to indicate a statistically significant difference. All data were analyzed using SPSS 17.0 (SPSS Inc., Chicago, IL, USA).
Results
Sensitivity of SGC7901/ADM and SGC7901/S cells to As2O3
In the present study, SGC7901/ADM and SGC7901/S cells were treated with different concentrations of As2O3 for 24, 48 and 72 h, following which the OD at 490 nm was measured and the IC50 and survival rates were calculated (Table I). The IC50 values of the two cell types to As2O3 decreased gradually with increasing treatment duration. However, no difference was observed between the two cell lines at any time-point (all P>0.05). Doses of 0.1 and 0.5 mM were considered non-toxic and mildly-toxic doses, respectively (Table I), and subsequent experiments were performed using these doses. These results revealed that the SGC7901/ADM and SGC7901/S cells had the same sensitivity to As2O3, which occurred in a time- and dose-dependent manner.
Table I.
IC50 volume of non- and mildly-toxic doses of arsenic trioxide at different time-points.
| Cell line | IC50µMa
|
95% >SR>85%b
|
SR>95%c
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 24 h | 48 h | 72 h | 24 h | 48 h | 72 h | 24 h | 48 h | 72 h | ||
| SGC7901/ADM | 5.57±0.36 | 2.63±0.18 | 1.42±0.09 | 0.10–0.08 | 0.10–0.75 | 0.05–0.25 | 0.01–0.01 | 0.01–0.01 | 0.01–0.05 | |
| SGC7901/S | 5.94±0.54 | 3.06±0.28 | 1.59±0.12 | 0.25–1.30 | 0.07–0.08 | 0.04–0.25 | 0.01–0.25 | 0.01–0.07 | 0.01–0.04 | |
Data are presented as the mean ± standard deviation of three experiments (n=3).
SR>95% was considered the non-toxic dose.
95% >SR>85% was considered a mildly-toxic dose. SR, survival rate; IC50, half maximal inhibitory concentration.
As2O3 reduces drug resistance of SGC7901/ADM cells to DOX, possibly through the p-ERK1/2 pathway
The IC50 to DOX was assessed in the five groups of cells incubated for 24 and 48 h. The experimental groups were: SGC7901/ADM+0.10 µM As2O3 and SGC7901/ADM+0.50 µM As2O3. The positive control group was SGC7901/ADM+4.00 µg/ml CsA. The negative control groups were SGC7901/ADM and SGC7901/S. The results revealed that the level of drug resistance between the resistant and parental cells were 6.63- and 6.85-fold at 24 h and 48 h, respectively (Table II). The IC50 values to DOX in the experimental groups were significantly lower than that in the SGC7901/ADM group when treated with 0.10 µM at 24 and 48 h (P<0.01) and with 0.50 µM at 24 and 48 h (P<0.01). The reversal in the CsA group was ~4.5-fold at different time points, while in the experimental groups, the effect was time-dependent (Table II). The IC50 of As2O3 at 0.50 µM was markedly lower than that at As2O3 at 0.10 µM, particularly at 48 h. The value was close to that in the SGC7901/S group and lower than that in the CsA group, but without statistical significance (P>0.05).
Table II.
Comparison of the IC50 values of DOX and drug-reversing ratio of As2O3 at different time-points.
| Factor | Treatment | −rhG-CSF
|
+rhG-CSF
|
|
|---|---|---|---|---|
| 24 h | 48 h | 48 h | ||
| IC50 to DOXa | As2O3 (0.1 µM) | 23.46±3.30 | 14.35±1.84 | 19.28±2.16 |
| As2O3 (0.5 µM) | 9.52±1.02 | 4.28±0.55 | 6.85±1.31 | |
| CsA (4 µg/ml) | 7.62±0.83 | 5.98±0.71 | 7.73±2.24 | |
| SGC7901/ADM | 34.31±4.38 | 26.94±3.67 | 32.39±4.52 | |
| SGC7901/S | 5.17±0.46 | 3.93±0.42 | 5.49±0.68 | |
| Drug-resistance ratiob | 6.63 | 6.85 | 5.90 | |
| Drug-reversal ratioc | As2O3 (0.1 µM) | 1.46 | 1.87 | 1.68 |
| As2O3 (0.5 µM) | 3.60 | 6.29 | 4.72 | |
| CsA (4 µg/ml) | 4.50 | 4.51 | 4.19 | |
Data are presented as the mean ± standard deviation.
Drug resistance ratio was calculated as the IC50 of resistant cells to that of SGC7901/S cells.
Reversal ratio was calculated as the IC50 of resistant cells to the reversal-agent treated cells. As2O3, arsenic trioxide; CsA, cyclosporin A; IC50, half maximal inhibitory concentration; DOX, doxorubicin; rhG-CSF, recombinant human granulocyte colony stimulating factor.
Following treatment with rhG-CSF, a cytokine that activates the ERK pathway, the IC50 at 48 h was assessed again (Table II). Compared with the cells without rhG-CSF treatment, the IC50 to DOX of As2O3 (0.50 µM) was significantly higher (P<0.05), suggesting that the ERK activator had the ability to partially reduce the drug resistance-reversing effect of As2O3.
These findings suggested that the SGC7901/ADM cells were resistant to DOX, and that As2O3 had a drug resistance-reversing effect, in a time- and dose-dependent manner. In addition, p-ERK1/2 may be involved in this mechanism, which requires further investigation.
Reduction in the expression of P-gp
The expression of P-gp was detected using western blotting (Fig. 1). The expression of P-gp was more marked in the SGC7901/ADM cells than in the SGC7901/S cells (0.38±0.025 vs. 0.19±0.012, respectively; P<0.001), without significant variation in with time (Table III). In the SGC7901/ADM cells, As2O3 at the non-toxic concentration (0.10 µM) at 24 h did not decrease the protein expression of P-gp, however, the increase in duration significantly decreased the levels of P-gp (P<0.01). Unlike the non-toxic concentration, the mildly-toxic concentration (0.50 µM) reduced the levels of P-gp even at 24 h (P<0.01). In the positive control cells, the levels of P-gp were close to that in the SGC7901/S cells, without significant changes with time (Table III). These results revealed that the expression of P-gp in the drug-resistant cells was higher than in the parental cells. CsA appeared to reduce the protein expression of P-gp, and As2O3 decreased the levels of P-gp, even at non- or mildly-toxic concentrations, in a time- and dose-dependent manner.
Figure 1.
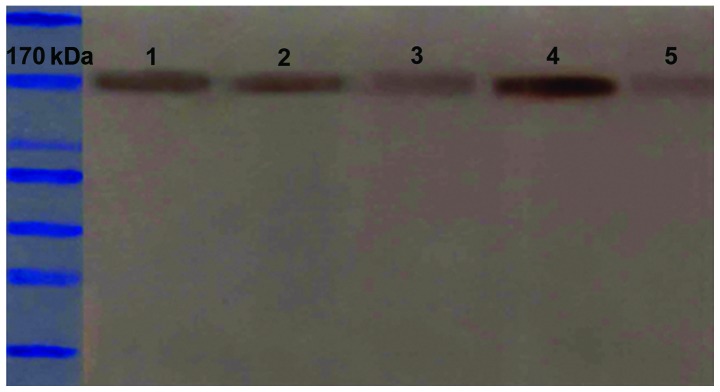
Protein expression levels of P-gp with or without arsenic trioxide treatment for 48 h, determined using western blotting. 1, protein expression of P-gp in SGC7901/ADM cells treated with 0.1 µM As2O3 for 48 h; 2, protein expression of P-gp in SGC7901/ADM cells treated with 0.5 µM As2O3 for 48 h; 3, protein expression of P-gp in SGC7901/ADM cells treated with 4 g/ml CsA for 48 h; 4, protein expression of P-gp in SGC7901/ADM cells without As2O3; 5, protein expression of P-gp in SGC7901/S without As2O3. P-gp, p-glycoprotein; CsA, cyclosporin A; As2O3, arsenic trioxide.
Table III.
Comparison of the OD values of P-gp, measured using western blot analysis, and the expression levels of Ras and p-ERK1/2, measured using immunocytochemistry, at different time-points.
| Protein | Treatment | OD value
|
||
|---|---|---|---|---|
| 24 h | 48 h | 72 h | ||
| P-gp | As2O3 (0.1 µM) | 0.349±0.001 | 0.32±0.003 | 0.29±0.002 |
| As2O3 (0.5 µM) | 0.30±0.002 | 0.28±0.002 | 0.16±0.001 | |
| CsA (4 µg/ml) | 0.16±0.004 | 0.16±0.001 | 0.15±0.002 | |
| SGC7901/ADM | 0.32±0.061 | 0.38±0.010 | 0.38±0.005 | |
| SGC7901/S | 0.14±0.003 | 0.14±0.003 | 0.14±0.002 | |
| Ras | As2O3 (0.1 µM) | 0.091±0.004 | 0.084±0.001 | 0.080±0.002 |
| As2O3 (0.5 µM) | 0.089±0.003 | 0.078±0.003 | 0.076±0.003 | |
| As2O3 (0.75 µM) | 0.081±0.004 | 0.073±0.002 | 0.073±0.002 | |
| CsA (4 µg/ml) | 0.091±0.003 | 0.093±0.003 | 0.084±0.005 | |
| SGC7901/ADM | 0.093±0.003 | 0.090±0.003 | 0.091±0.004 | |
| SGC7901/S | 0.068±0.003 | 0.071±0.005 | 0.066±0.008 | |
| p-ERK1/2 | As2O3 (0.1µM) | 0.099±0.009 | 0.086±0.002 | 0.083±0.004 |
| As2O3 (0.5 µM) | 0.086±0.007 | 0.080±0.002 | 0.070±0.001 | |
| As2O3 (0.75 µM) | 0.08±0.004 | 0.078±0.005 | 0.067±0.002 | |
| CsA (4 µg/ml) | 0.098±0.009 | 0.11±0.018 | 0.11±0.025 | |
| SGC7901/ADM | 0.11±0.008 | 0.11±0.012 | 0.11±0.014 | |
| SGC7901/S | 0.10±0.016 | 0.090±0.010 | 0.10±0.027 | |
Data are presented as the mean ± standard deviation of three experiments. As2O3, arsenic trioxide; CsA, cyclosporin A; P-gp, P-glycoprotein; p-ERK, phosphorylated extracellular signal-regulated kinase; OD, optical density.
Expression of Ras and p-ERK1/2 expression, determined using immunocytochemistry
Ras protein was predominantly localized in the membrane and partially translocated in the cytoplasm close to the membrane (Fig. 2A). The expression of Ras was higher in the SGC7901/ADM cells than in the SGC7901/S cells (P<0.01). Following treatment with As2O3, the levels of Ras were reduced in a time- and dose-dependent manner (Table III; Fig. 2B). At 24 h, only the 0.75 µM group exhibited lower expression than the SGC7901/ADM cells (P<0.01). At 48 and 72 h, the cell groups treated with As2O3 had significantly lower levels of Ras (P<0.05). The expression of Ras in the three control groups remained stable over time. These results suggested that drug-resistant cells may have a higher expression levels of Ras expression, and that As2O3 decreases these levels.
Figure 2.

Immunohistochemistry of Ras protein in (A) SGC7901/ADM cells (Ras predominantly localized in the membrane and partially translocated in the cytoplasm) and (B) SGC7901/ADM cells treated with 0.5 µM As2O3 for 48 h (Ras protein expression observed to be reduced) (magnification, ×400). As2O3, arsenic trioxide.
Expression of p-ERK1/2 expression was detected in the cytoplasm surrounding the nucleus of cells (Fig. 3A and B). No significant differences were identified in the levels of p-ERK1/2 between the SGC7901/ADM, SGC7901/S and CsA-treated cells. Following treatment with As2O3, the protein expression of p-ERK1/2 decreased (Table III). At 24 h, the levels of p-ERK1/2 were decreased at all As2O3 concentrations, without statistical significance (P>0.05). However, in the 0.50 and 0.75 µM As2O3 groups, the expression of p-ERK1/2 was decreased, particularly in the As2O3 0.75 µM group at 72 h (P<0.01). These results suggested that the Ras/p-ERK1/2 signaling pathway may be involved in the mechanism of reducing drug resistance to DOX by As2O3 in GC cells.
Figure 3.

Immunohistochemistry of (A) p-ERK1/2 protein in the SGC7901/ADM cells and (B) SGC7901/ADM cells treated with 0.5 µM As2O3 for 48 h (magnification, ×400). Expression of p-ERK1/2 was detected in the cytoplasm surrounding the nucleus of the cells. p-ERK, phosphorylated-extracellular signal-regulated kinase; As2O3, arsenic trioxide.
Ras/p-ERK1/2 signaling pathway is involved in reducing drug resistance
The cells were incubated with non- and mildly-toxic concentrations of As2O3 for 48 h and, following pretreatment with rhG-CSF, cytokines activating the ERK pathway, Ras and p-ERK1/2 were assessed using immunocytochemistry.
Following rhG-CSF treatment, the expression of Ras was similar in all groups (P>0.05). No difference was identified in the levels of Ras between the SGC7901/ADM and CsA-treated cells (P>0.05; Table IV; Fig. 4A and B). Compared with the cells without rhG-CSF treatment, the levels of Ras were not different (P=0.12) in the 0.1 µM As2O3-treated groups, but were higher in the As2O3 0.5 µM As2O3-treated groups (P<0.01).
Table IV.
Protein expression levels of Ras and p-ERK1/2 with or without treatment of rhG-CSF, determined using immunocytochemistry (optical density).
| Protein | Treatment | rhG-CSF/− | rhG-CSF/+ |
|---|---|---|---|
| Ras b | As2O3 (0.1 µM)b | 0.084±0.001 | 0.093±0.008 |
| As2O3 (0.5 µM) | 0.078±0.003 | 0.089±0.001 | |
| CsA (4 µg/ml) | 0.093±0.003 | 0.099±0.005 | |
| SGC7901/ADM | 0.094±0.006 | 0.10±0.008 | |
| p-ERK1/2a | As2O3 (0.1 µM) | 0.086±0.002 | 0.093±0.002 |
| As2O3 (0.5 µM) | 0.080±0.002 | 0.091±0.001 | |
| CsA (4 µg/ml) | 0.11±0.018 | 0.12±0.019 | |
| SGC7901/ADMc | 0.10±0.010 | 0.14±0.003 |
Data are presented as the mean ± standard deviation of three experiments.
Cells were incubated with non- and midly-toxic concentrations of As2O3.
Compared with cells without rhG-CSF, the levels of Ras levels not significantly different (P=0.12) in the As2O3 (0.1 µM) groups, but were higher in the As2O3 (0.5 µM) groups (P<0.01).
Expression of p-ERK1/2 in the SGC7901/ADM cells increased following rhG-CSF treatment (P<0.01). As2O3, arsenic trioxide; rhG-CSF, recombinant human granulocyte colony stimulating factor; p-ERK, phosphorylated extracellular signal-regulated kinase; CsA, cyclosporin A.
Figure 4.
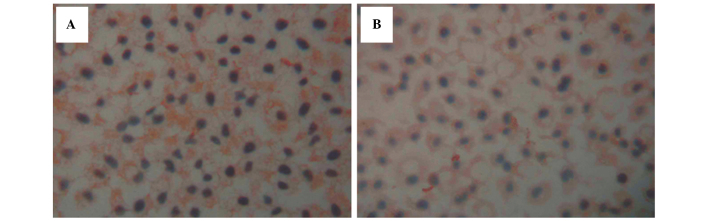
Immunohistochemistry of (A) Ras protein in the SGC7901/ADM cells following treatment with 0.1 µM As2O3 and rhG-CSF for 48 h. (B) Ras protein in the SGC7901/ADM cells following treatment with 0.5 µM As2O3 and rhG-CSF for 48 h (magnification, ×400). No differences were identified in the levels of Ras between the SGC7901/ADM- and CsA-treated cells. Compared with the cells without rhG CSF treatment, the levels of Ras were not significantly different (P=0.12) in the 0.1 µM As2O3-treated groups, however were greater in the 0.5 µM As2O3-treated groups (P<0.01). rhG-CSF, recombinant human granulocyte colony stimulating factor; As2O3, arsenic trioxide.
The levels of p-ERK1/2 in all the cells increased following rhG-CSF treatment (Table IV; P<0.01). Treatment with 0.10 and 0.50 µM As2O3 reduced the levels of p-ERK1/2, compared with the negative control group (P<0.05; Fig. 5A and B), whereas the levels of p-ERK1/2 increased significantly at the same doses of As2O3 following rhF-CSF treatment (P<0.01). Similar to Ras, these results suggested that the expression of p-ERK1/2 decreased following treatment with the ERK activator.
Figure 5.
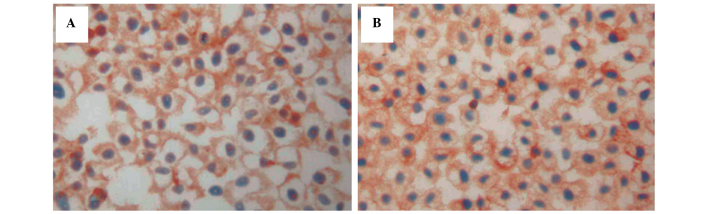
Immunohistochemistry of (A) p-ERK1/2 protein in the SGC7901/ADM cells following treatment with As2O3 (0.1 µM) and rhG-CSF for 48 h; (B) p-ERK1/2 protein in the SGC7901/ADM cells following treatment with 0.5 µM As2O3 and rhG-CSF for 48 h (magnification, ×400). Treatment with 0.10 and 0.50 µM As2O3 reduced the levels of p-ERK1/2, compared with the negative control group. p-ERK, phosphorylated extracellular signal-regulated kinase; rhG-CSF, recombinant human granulocyte colony stimulating factor; As2O3, arsenic trioxide.
Discussion
GC is one of the most common types of cancer, and generally has a poor prognosis. DOX is used as one of the key components in a number of chemotherapeutic regimens against GC, however, MDR eventually limits its use. Several types of cancer cell, including non-acute myeloid leukemia, multiple myeloma, breast cancer and APL, have been found to be sensitive to As2O3 through cell growth inhibition and apoptosis (12,20,28,29). However, few studies have examined the effects of As2O3 in GC cells, and these studies reported a number of different mechanisms (30–32). In the present study, the human GC cell lines, SGC7901/S and SGC7901/ADM, were investigated. The latter was induced by long-term continuous exposure to DOX in a step-wise increment of various concentrations. The high IC50 of SGC7901/ADM, but not of SGC7901/S, confirmed the resistance to DOX. The doses of 0.10 and 0.50 µM of As2O3 were defined as non- and mildly-toxic concentrations, respectively. This was different from the study by Zhao et al (22), which reported that 2 µM was the non-toxic concentration. In the MTT assay for the detection of cytotoxicity and sensitivity to As2O3, cell growth was inhibited without any treatment at 72 h. This was possibly the result of the toxic metabolic accumulation and nutrient deficiency. Previously, it was found that As2O3 induced APL cells to enter apoptosis (6). The present study demonstrated that As2O3 partially reduced in vitro drug-resistance, even at a non-toxic dose (0.10 µM), in a time- and dose-dependent manner.
A number of mechanisms are involved in MDR, and the ovexpression of P-gp is one of the key mechanisms. Agents to reduce MDR have been investigated for a number of years (33,34). A novel inhibitor of P-gp-mediated MDR, OC144-093, was reported in 2000 (28). In the present study, the expression of P-gp in SGC7901/ADM was higher than in the parental cells and, following As2O3 treatment, the levels of P-gp decreased significantly. The non- and mildly-toxic doses of As2O3 were selected with the purpose of using them in clinical practice without serious side effects.
Several studies have reported the in vitro effects of As2O3 in the inhibition of different types of solid tumor, through a number of different mechanisms. Eguchi et al (19) demonstrated that apoptosis induced in human mesothelioma cells is accompanied by the activation of JNK1/2 and ERK1/2 (19). In breast cancer, As2O3 inhibits cell growth through the inactivation of the Notch signaling pathway (20). To investigate the role of the Ras/p-ERK1/2 signaling pathway in the mechanism of As2O3, the rhG-CSF cytokine, which activates the ERK pathway was used to treat the cells.
Ras is a monomeric GTP-coupled protein encoded by the Ras gene, which is important in cell growth regulation (35). It was reported in 1993 that Ras has direct interactions with the RAF1 serine/threonine kinase, which was the first mammalian Ras effector to be identified (36). ERK1 and ERK2, also termed mitogen-activated protein kinase (MAPK)1 and MAPK2 are MAPK isomers, which are widely expressed in eukaryotic cells. In addition, the identification of B-Raf mutations in cancer emphasize the importance for aberrant Raf-MEK-ERK signaling in oncogenesis (36). In the present study, the levels of Ras and p-ERK were determined using immunocytochemistry. The levels of Ras were higher in the SGC7901/ADM cells than in the SGC7901/S cells, however, no significant difference was observed in the levels of p-ERK1/2. The results suggested that p-ERK1/2 was not involved in the MDR mechanisms in these cells, but indicated that Ras was involved. Following treatment with As2O3, the levels of Ras levels reduced, and the protein levels of p-ERK1/2 decreased until intervention with mildly-toxic concentrations of As2O3 (0.50 and 0.75 µM).
Following pretreatment with rhG-CSF, no change was observed in the levels of Ras in cells from the control groups, however, levels increased in the 0.50 µM As2O3 treatment group. The expression of p-ERK1/2 increased in all cells, suggesting that rhG-CSF activated p-ERK1/2. As2O3 reduced the levels of p-ERK1/2, compared with the negative control group. Thus, a p-ERK activator may partially inhibit the drug resistance-reversing effects of As2O3. A phase II trial revealed that As2O3 is inactive in patients with pancreatic cancer, who develop a progressive disease following gemcitabine treatment (37). This failure may be due to the lack of co-treatment with other chemotherapeutics. As2O3 has been demonstrated to be effective in treating APL without significant side effects (8,9).
The present study involved the examination of MDR induced by DOX. There are several other chemotherapeutic agents, which are actively used in the treatment of GC in a number of combination regimens (38). Further investigations are required to examine the effect of As2O3 in reversing the MDR induced by these other agents. In addition, it is important to further investigate the effects of As2O3 in GC, compared with other types of cancer. Although the present study clearly suggested the involvement of Ras in the MDR of GC, further investigations are required to clarify whether mutations in Raf are involved in drug-resistant cells. Finally, the rhG-CSF cytokine, used in the present study to activate the ERK pathway, also activates a number of other cell mechanisms, which require further investigations, including the PI3K/AKT pathway (39,40).
In conclusion, the results of the present study revealed that As2O3 had the ability to reverse MDR in human GC cells. This mechanism may be relevant to reduce the expression of P-gp. Drug-resistant cells may have higher expression levels of Ras, and As2O3 may decrease these levels. The Ras/p-ERK1/2 signal transduction pathway may be involved in this mechanism. Further investigations, involving a combination of chemotherapeutics and arsenic trioxide, are essential.
Acknowledgments
This study was supported by ths Qingdao Municipal Science and Technology Commission (grant no. 09-1-3-75-jch). The authors would like to thank Dr Jing Dong for his critical reading of the manuscript, and the Central Laboratory of the Affiliated Hospital of the Medical College Qingdao University for providing cell lines.
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Matsuda A, Machii R. Trends in stomach cancer mortality rates in Japan, USA, UK, France and Korea based on the WHO mortality database. Jpn J Clin Oncol. 2012;42:154. doi: 10.1093/jjco/hys004. [DOI] [PubMed] [Google Scholar]
- 3.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 4.Zheng LH, Bao YL, Wu Y, Yu CL, Meng X, Li YX. Cantharidin reverses multidrug resistance of human hepatoma HepG2/ADM cells via down-regulation of P-glycoprotein expression. Cancer Lett. 2008;272:102–109. doi: 10.1016/j.canlet.2008.06.029. [DOI] [PubMed] [Google Scholar]
- 5.Volm M, Mattern J. Resistance mechanisms and their regulation in lung cancer. Crit Rev Oncog. 1996;7:227–244. doi: 10.1615/CritRevOncog.v7.i3-4.50. [DOI] [PubMed] [Google Scholar]
- 6.Mervis J. Ancient remedy performs new tricks. Science. 1996;273:578. doi: 10.1126/science.273.5275.578. [DOI] [PubMed] [Google Scholar]
- 7.Zhu J, Chen Z, Lallemand-Breitenbach V, de Thé H. How acute promyelocytic leukaemia revived arsenic. Nat Rev Cancer. 2002;2:705–713. doi: 10.1038/nrc887. [DOI] [PubMed] [Google Scholar]
- 8.Zhang TD, Chen GQ, Wang ZG, Wang ZY, Chen SJ, Chen Z. Arsenic trioxide, a therapeutic agent for APL. Oncogene. 2001;20:7146–7153. doi: 10.1038/sj.onc.1204762. [DOI] [PubMed] [Google Scholar]
- 9.Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, Ferrara F, Fazi P, Cicconi L, Di Bona E, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369:111–121. doi: 10.1056/NEJMoa1300874. [DOI] [PubMed] [Google Scholar]
- 10.Murgo AJ. Clinical trials of arsenic trioxide in hematologic and solid tumors: Overview of the National Cancer Institute Cooperative Research and Development Studies. Oncologist. 2001;6(Suppl 2):22–28. doi: 10.1634/theoncologist.6-suppl_2-22. [DOI] [PubMed] [Google Scholar]
- 11.Lu J, Chew EH, Holmgren A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc Natl Acad Sci USA. 2007;104:12288–12293. doi: 10.1073/pnas.0701549104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Munshi NC. Arsenic trioxide: An emerging therapy for multiple myeloma. Oncologist. 2001;(6 Suppl 2):17–21. doi: 10.1634/theoncologist.6-suppl_2-17. [DOI] [PubMed] [Google Scholar]
- 13.Maeda H, Hori S, Nishitoh H, Ichijo H, Ogawa O, Kakehi Y, Kakizuka A. Tumor growth inhibition by arsenic trioxide (As2O3) in the orthotopic metastasis model of androgen-independent prostate cancer. Cancer Res. 2001;61:5432–5440. [PubMed] [Google Scholar]
- 14.Shen ZY, Zhang Y, Chen JY, Chen MH, Shen J, Luo WH, Zeng Y. Intratumoral injection of arsenic to enhance antitumor efficacy in human esophageal carcinoma cell xenografts. Oncol Rep. 2004;11:155–159. [PubMed] [Google Scholar]
- 15.Bornstein J, Sagi S, Haj A, Harroch J, Fares F. Arsenic Trioxide inhibits the growth of human ovarian carcinoma cell line. Gynecol Oncol. 2005;99:726–729. doi: 10.1016/j.ygyno.2005.07.125. [DOI] [PubMed] [Google Scholar]
- 16.Woo SH, Park IC, Park MJ, Lee HC, Lee SJ, Chun YJ, Lee SH, Hong SI, Rhee CH. Arsenic trioxide induces apoptosis through a reactive oxygen species-dependent pathway and loss of mitochondrial membrane potential in HeLa cells. Int J Oncol. 2002;21:57–63. [PubMed] [Google Scholar]
- 17.Miller WH, Jr, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002;62:3893–3903. [PubMed] [Google Scholar]
- 18.Pelicano H, Feng L, Zhou Y, Carew JS, Hileman EO, Plunkett W, Keating MJ, Huang P. Inhibition of mitochondrial respiration: a novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J Biol Chem. 2003;278:37832–37839. doi: 10.1074/jbc.M301546200. [DOI] [PubMed] [Google Scholar]
- 19.Eguchi R, Fujimori Y, Takeda H, Tabata C, Ohta T, Kuribayashi K, Fukuoka K, Nakano T. Arsenic trioxide induces apoptosis through JNK and ERK in human mesothelioma cells. J Cell Physiol. 2011;226:762–768. doi: 10.1002/jcp.22397. [DOI] [PubMed] [Google Scholar]
- 20.Xia J, Li Y, Yang Q, Mei C, Chen Z, Bao B, Ahmad A, Miele L, Sarkar FH, Wang Z. Arsenic trioxide inhibits cell growth and induces apoptosis through inactivation of notch signaling pathway in breast cancer. Int J Mol Sci. 2012;13:9627–9641. doi: 10.3390/ijms13089627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao Z, Ding N, Xiao G, Wang S, Wu Y, Tang L. Reversal of multidrug resistance by gefitinib via RAF1/ERK pathway in pancreatic cancer cell line. Anat Rec (Hoboken) 2012;295:2122–2128. doi: 10.1002/ar.22552. [DOI] [PubMed] [Google Scholar]
- 22.Zhao D, Jiang Y, Dong X, Liu Z, Qu B, Zhang Y, Ma N, Han Q. Arsenic trioxide reduces drug resistance to adriamycin in leukemic K562/A02 cells via multiple mechanisms. Biomed Pharmacother. 2011;65:354–358. doi: 10.1016/j.biopha.2011.04.016. [DOI] [PubMed] [Google Scholar]
- 23.Zhao Y, You H, Liu F, An H, Shi Y, Yu Q, Fan D. Differentially expressed gene profiles between multidrug resistant gastric adenocarcinoma cells and their parental cells. Cancer Lett. 2002;185:211–218. doi: 10.1016/S0304-3835(02)00264-1. [DOI] [PubMed] [Google Scholar]
- 24.Che XF, Nakajima Y, Sumizawa T, Ikeda R, Ren XQ, Zheng CL, Mukai M, Furukawa T, Haraguchi M, Gao H, et al. Reversal of P-glycoprotein mediated multidrug resistance by a newly synthesized 1,4-benzothiazipine derivative, JTV-519. Cancer Lett. 2002;187:111–119. doi: 10.1016/S0304-3835(02)00359-2. [DOI] [PubMed] [Google Scholar]
- 25.Wang R, Xia L, Gabrilove J, Waxman S, Jing Y. Downregulation of Mcl-1 through GSK-3β activation contributes to arsenic trioxide-induced apoptosis in acute myeloid leukemia cells. Leukemia. 2013;27:315–324. doi: 10.1038/leu.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brandstetter T, Ninci E, Falken U, Wagner E, Hess R, Bauknecht T. rhG-CSF affects genes involved in mitogen signalling and early gene expression in the ovarian cancer cell line HEY. Int J Cancer. 1998;75:847–854. doi: 10.1002/(SICI)1097-0215(19980316)75:6<847::AID-IJC6>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 27.Ricciardi MR, McQueen T, Chism D, Milella M, Estey E, Kaldjian E, Sebolt-Leopold J, Konopleva M, Andreeff M. Quantitative single cell determination of ERK phosphorylation and regulation in relapsed and refractory primary acute myeloid leukemia. Leukemia. 2005;19:1543–1549. doi: 10.1038/sj.leu.2403859. [DOI] [PubMed] [Google Scholar]
- 28.Newman MJ, Rodarte JC, Benbatoul KD, Romano SJ, Zhang C, Krane S, Moran EJ, Uyeda RT, Dixon R, Guns ES, et al. Discovery and characterization of OC144-093, a novel inhibitor of P-glycoprotein-mediated multidrug resistance. Cancer Res. 2000;60:2964–2972. [PubMed] [Google Scholar]
- 29.Li Y, Qu X, Qu J, Zhang Y, Liu J, Teng Y, Hu X, Hou K, Liu Y. Arsenic trioxide induces apoptosis and G2/M phase arrest by inducing Cbl to inhibit PI3K/Akt signaling and thereby regulate p53 activation. Cancer Lett. 2009;284:208–215. doi: 10.1016/j.canlet.2009.04.035. [DOI] [PubMed] [Google Scholar]
- 30.Chen X, Zhang M, Liu LX. The overexpression of multidrug resistance-associated proteins and gankyrin contribute to arsenic trioxide resistance in liver and gastric cancer cells. Oncol Rep. 2009;22:73–80. [PubMed] [Google Scholar]
- 31.Liu Y, Zhang W, Zhang X, Qi Y, Huang D, Zhang Y. Arsenic trioxide inhibits invasion/migration in SGC-7901 cells by activating the reactive oxygen species-dependent cyclo-oxygenase-2/matrix metalloproteinase-2 pathway. Exp Biol Med (Maywood) 2011;236:592–597. doi: 10.1258/ebm.2011.010276. [DOI] [PubMed] [Google Scholar]
- 32.Zhang G, Liu J, Zhang Y, Qu J, Xu L, Zheng H, Liu Y, Qu X. Cbl-b-dependent degradation of FLIP(L) is involved in ATO-induced autophagy in leukemic K562 and gastric cancer cells. FEBS Lett. 2012;586:3104–3110. doi: 10.1016/j.febslet.2012.07.067. [DOI] [PubMed] [Google Scholar]
- 33.Baguley BC. Multiple drug resistance mechanisms in cancer. Mol Biotechnol. 2010;46:308–316. doi: 10.1007/s12033-010-9321-2. [DOI] [PubMed] [Google Scholar]
- 34.Gillet JP, Gottesman MM. Mechanisms of multidrug resistance in cancer. Methods Mol Biol. 2010;596:47–76. doi: 10.1007/978-1-60761-416-6_4. [DOI] [PubMed] [Google Scholar]
- 35.Wong KA, Russo A, Wang X, Chen YJ, Lavie A, O'Bryan JP. A new dimension to Ras function: A novel role for nucleotide-free Ras in Class II phosphatidylinositol 3-kinase beta (PI3KC2β) regulation. PLoS One. 2012;7:e45360. doi: 10.1371/journal.pone.0045360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karnoub AE, Weinberg RA. Ras oncogenes: Split personalities. Nat Rev Mol Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kindler HL, Aklilu M, Nattam S, Vokes EE. Arsenic trioxide in patients with adenocarcinoma of the pancreas refractory to gemcitabine: A phase II trial of the University of Chicago Phase II Consortium. Am J Clin Oncol. 2008;31:553–556. doi: 10.1097/COC.0b013e318178e4cd. [DOI] [PubMed] [Google Scholar]
- 38.NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines)Gastric Cancer. 3rd edition. National Comprehensive Cancer Network; Fort Washington: 2015. [Google Scholar]
- 39.Guo H, Sun F, Huang W, Liu Z, Zhang S, Zhou Q, Liang C. The effect of rhG-CSF on spleen transcriptome in mouse leukopenia model induced by cyclophosphamide. Immunopharmacol Immunotoxicol. 2014;36:114–123. doi: 10.3109/08923973.2013.869696. [DOI] [PubMed] [Google Scholar]
- 40.Kumar J, Fraser FW, Riley C, Ahmed N, McCulloch DR, Ward AC. Granulocyte colony-stimulating factor receptor signalling via Janus kinase 2/signal transducer and activator of transcription 3 in ovarian cancer. Br J Cancer. 2014;110:133–145. doi: 10.1038/bjc.2013.673. [DOI] [PMC free article] [PubMed] [Google Scholar]