

Abstract
Aquaporin 4 (AQP4) is a widely distributed membrane protein, which is found in glial cells, ependymocytes and capillary endothelial cells in the brain, and particularly in the choroid plexus. AQP4 is a key regulator of water metabolism, and changes in its expression following brain injury are associated with pathological changes in the damaged side of the brain; however, the effects of brain injury on AQP4 and injury-induced pathological changes in the contralateral non-damaged side of the brain remain to be fully elucidated. In the present study, male Sprague-Dawley rats were subjected to traumatic brain injury (TBI) and changes in brain water content, the expression of AQP4 expression and pathological characteristics in the damaged and contralateral non-damaged sides of the brain were examined. In the damaged side of the brain, vasogenic edema appeared first, followed by cellular edema. The aggravated cellular edema in the damaged side of the brain resulted in two periods of peak edema severity. Pathological changes in the contralateral non-damaged side of the brain occurred later than those in the damaged side; cellular edema appeared first, followed by vasogenic edema, which was alleviated earlier than the cellular edema. AQP4 was downregulated during vasogenic edema, and upregulated during cellular edema. Taken together, these results suggested that the downregulation of AQP4 was a result of vasogenic edema and that the upregulation of AQP4 may have induced cellular edema.
Keywords: traumatic brain injury, cerebral edema, histopathology, aquaporin 4, contralateral non-damaged side, vasogenic edema
Introduction
With rapid development of the transportation and construction industries, the occurrence of brain trauma caused by traffic accidents and falling from heights is increasing, and these types of brain injuries account for >50% of all accidental injuries (1). Cerebral contusion is usually accompanied by cerebral edema of varying extent, in which there is substantial water accumulation in the intracellular and extracellular spaces, which leads to increases in intracranial volume, intracranial hypertension and cerebral herniation, and subsequently to cellular dysfunction and the death of neurons and glia (2–4). There are no effective clinical treatment measures for traumatic cerebral edema as its precise pathological mechanisms remain to be fully elucidated (5).
The pathological changes associated with traumatic cerebral edema are complex (6,7). Reports indicate that edema during the early stage of brain trauma (<24 h) is predominantly comprised of the secondary vasogenic and intracellular types (8,9). During the early stage of brain trauma, vasogenic edema occurs first, following which the expression of intracranial aquaporin 4 (AQP4) is upregulated and cellular edema in brain cells occurs. The co-existence of these two types of edema aggravates the traumatic cerebral edema associated with acute brain trauma (4).
In previous investigations, attention has focused predominantly on the damaged side of the brain during the early stage of injury (<24 h), whereas the intermediate (24–72 h) and late stages (72–168 h), and the contralateral non-damaged side of the brain have received less attention (10). As a result, the pathological mechanisms that operate following the first day of recovery from brain injury, which lead to edema and other complications, remain to be fully elucidated, and clinical treatment strategies during this period are not guided by appropriate empirical data.
The brain acts as an integrated control center for the body via its abundant neural connections and humoral regulatory mechanisms. When an area of the brain is damaged, pathological changes appear in the contralateral non-damaged side, in addition to the damaged side, which affect clinical treatment. However, the mechanisms leading to pathological changes, including edema, in the contralateral non-damaged side of the brain following the early stage of traumatic brain injury (TBI) remain to be elucidated. In the present study, pathomorphological analysis of the damaged areas and contralateral non-damaged areas of rat brains, as well as analysis of the expression of AQP4 at different time-points, were performed following TBI, to clarify the precise molecular mechanisms associated with pathological changes following brain trauma.
Materials and methods
Experimental animals and grouping
A total of 190 male Sprague-Dawley (SD) rats weighing 300–350 g were purchased from the Experimental Animal Center of Sichuan University (license no. SCXK (Chuan) 2008-24). The rats were housed separately at 22–25°C under a 12 h:12 h light-cycle, with free access to food and water. The rats were randomly divided into a sham operation (SO) group (n=10) and a trauma group in which TBI was performed. The TBI group (n=60) was subdivided into two groups, which were used for the comparative analysis of the damaged side (TBI-D) and the contralateral non-damaged side (TBI-C) of the brain, which were further divided into six subgroups, according to the time-point post-contusion at which the rats were evaluated (1, 6, 24, 48, 72, and 168 h), with 10 rats in each group. In each group, one sample was used for pathological observation following hematoxylin and eosin (HE) staining and immunohistochemical double-staining, one sample was used for electron microscopic observation and eight samples were used for the detection of the protein expression of AQP4 using semi-quantitative western blotting. The present study was approved by the ethics committee of the Affiliated Haikou Hospital of Xiangya School of Medicine Central South University (Haiku, China).
Establishment of the animal model
The brain trauma model was established using a modified version of Feeney's method (11) and a PinPoint™ Precision Cortical Impactor (Hatteras Instruments, Inc., Cary, NC, USA). The rats were anesthetized by intraperitoneal injection of 10% chloral hydrate (0.4 ml/100 g). The head of the rat was fixed in a prone position on a stereotaxic apparatus (ST-7; Setagaya-Ku, Tokyo, Japan). The fur was shaved and routine disinfection was performed. A 2 cm incision was made on the scalp along the median line of the head. A table electric dental engine (307-2B; Shenzhen Ward Life Science and Technology Co., Ltd., Shanghai, China) with a 1.5 mm drill bit (rotational speed, 4,000 r/min) was used to bore a hole in the skull between the anterior and posterior fontanel, 2.5 mm away from the median line at the right side. With this hole as the center, a round bony window with a diameter of 5 mm was made using a mosquito clamp (Shenzhen Ward Life Science and Technology Co., Ltd.). Whilst preserving the integrity of the dura mater, the bony window was impinged in a precise manner to establish moderate right brain trauma. The following parameters were selected according to a preliminary experiment: Impinging velocity, 2.5 m/s; impinging depth, 4 mm; duration, 0.85 sec; diameter of impinging bit, 4 mm). The trauma was accompanied by cortical and medullary injury, but without intracerebral hematoma. The bony window was sealed using bone wax and the scalp was sutured following local hemostasis. With the exception of the impingement, the surgical procedures performed on the trauma group were also performed on the sham operation group.
Measurement of brain water content (BWC)
The rats (n=120) were sacrificed with 10% chloral hydrate, and the brains were removed and cleaned of surface blood using filter paper. The wet weight of each whole brain was recorded, following which it was dried at 105°C for 48 h inside an electrothermal constant temperature dry box (Shenzhen Ward Life Science and Technology Co., Ltd.). The BWC was calculated using the Elliot equation (12), as follows:
Wwet denotes the wet weight of the brain and Wdry denotes the weight following drying.
Pathomorphological observation
The rats were anesthetized using 10% chloral hydrate (0.4 ml/100 g). The left ventricle was infused with 4% paraformaldehyde (Guangzhou Chemical Reagent Factory, Guangzhou, China) until transparent liquid flowed from the right ventricle. The brain was harvested following cervical dislocation and fixed in 4% paraformaldehyde for 24 h. Coronal sections of the damaged brain regions were obtained, including the side contralateral to the injury. Routine paraffin embedding (Guangzhou Chemical Reagent Factory, Guangzhou, China) and slicing were performed following routine HE staining, the slices were observed under an optical microscope (Olympus BX50; Olympus Corporation, Toyko, Japan) and images were captured.
From each group, one animal was selected and anesthetized with 10% chloral hydrate (0.4 ml/100 g). The brain was harvested following cervical dislocation and the damaged area was cut into small cuboids (1×1×3 mm), which were immediately placed in 3% glutaraldehyde (Guangzhou Chemical Reagent Factory, Guangzhou, China) for fixation. Subsequently, the tissue samples were fixed in 1% osmium tetroxide and dehydrated using different concentrations of acetone (70, 80, 90 and 100%). The samples were embedded in Epon812 medium (Guangzhou Chemical Reagent Factory, Guangzhou, China) and serial sections were prepared for optical localization. The sections were then treated with lead-uranium double staining. Finally, the neurons, organelle morphology and the structure of the blood-brain barrier were observed and images were captured under an EX-2000 transmission electron microscope (JEOL, Ltd., Tokyo, Japan). The HE staining and the subsequent preparation, observation and imaging were also performed on tissue samples from the control group after 24 h.
AQP4 detection using immunohistochemistry (double labeling)
Prior to dewaxing, the paraffin section was placed at room temperature for 60 min or heated in an incubator at 60°C for 20 min. The section was immersed in dimethylbenzene (45–70% meta-xylene, 15–25% paraxylene and 10–15% o-xylene) for 10 min, which was then repeated using fresh dimethylbenzene, followed by immersion in absolute ethyl alcohol for 5 min, 95% alcohol for 5 min and 70% alcohol for 5 min. Thermal remediation was performed, as follows: Following the heating of 0.01 M sodium citrate buffer solution (pH 6.0) to ~95°C on an electric stove or in a water bath, the tissue section was placed into the solution and heated for 10–15 min. Subsequently, goat serum was added to the section at room temperature for 20 min. The surplus liquid was removed, and 50 µl monoclonal rabbit anti-mouse AQP4 (1:200; cat. no. 16473-AP; Wuhan Sanying Biological Technology Co., Ltd., Wuhan, China) primary antibody was added, followed by incubation of at 4°C overnight. Following incubation, the section was rewarmed at 37°C for 45 min, rinsed with phoshpate-buffered saline (PBS) three times (5 min each time), and 40–50 µl secondary monoclonal goat anti-rabbit AQP4 antibody (1:200; cat. no. ZB-2301; Wuhan Sanying Biological Technology Co., Ltd.) was added prior to incubation at 37°C for 1 h. The section was then rinsed with PBS three times (5 min each time), and DAPI fluorescence dye was added. Finally, the section was sealed between a slide and cover slip, and observed under a fluorescence microscope (YYF-300; Shanghai Yiyuan Optical Instrument Co., Ltd., Shanghai, China; GFAP, green; AQP4, red; nuclei of glia, blue).
Detection of the protein expression of AQP4 using western blotting
The damaged side of the brain was collected and ground in liquid nitrogen (Shanghai Yiyuan Optical Instrument Co., Ltd). A radioimmunoprecipitation assay (Shanghai Tong Good Biological Technology Co., Ltd, Shanghai, China) was performed to extract the total protein from the samples of each group, and the total protein concentration was detected using a bicinchoninic acid method (Shanghai Tong Good Biological Technology Co., Ltd.). Gel separation was performed by 10% SDS-PAGE (Kangwei Century Technology, Co., Ltd., Beijing, China) using 80 µg sample and the protein on the gel was transferred onto a polyvinylidene fluoride membrane (Hangzhou LeQian Scientific Instrument Co, Ltd., Hangzhou, China). The primary antibody (monoclonal rabbit anti-mouse AQP4; cat. no. ZB-2301; 1:200), was added, the membrane was washed with PBS, and secondary antibody was added (monoclonal goat anti-rabbit AQP4; cat. no. ZB-2301; 1:200). The density (D) of the target band was analyzed using a Bio-Rad Gel Imaging system (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
All data are expressed as the mean ± standard deviation. SPSS 13.0 (SPSS, Inc., Chicago, IL, USA) was used to performed t-tests and one-way analysis of variance. Comparisons between groups were performed using the Student-Newman-Keuls method. P<0.05 was considered to indicate a statistically significant difference.
Results
Changes in BWC at different time-points in the TBI and SO groups
The average BWC in the SO group was 79.17±4.28%. The BWC in the TBI rats increased to 79.25±4.01% after 1 h, 81.62±5.12% after 6 h, and reached a first peak level at 24 h (85.65±6.12%). The BWC then decreased marginally at 48 h to 84.09±7.06%, and reached a second peak level at 72 h (86.25±6.46%), prior to decreasing to 82.17±4.36% at 168 h. The BWC of the TBI-A rats at the 6 h, 24 h, 48 h, and 72 h time-points was significantly higher, compared with the BWC of the SO group at each corresponding time-point (P<0.05), however, no significant differences were observed at 1, 6 or 168 h (P>0.05). The BWC results are shown in Fig. 1.
Figure 1.
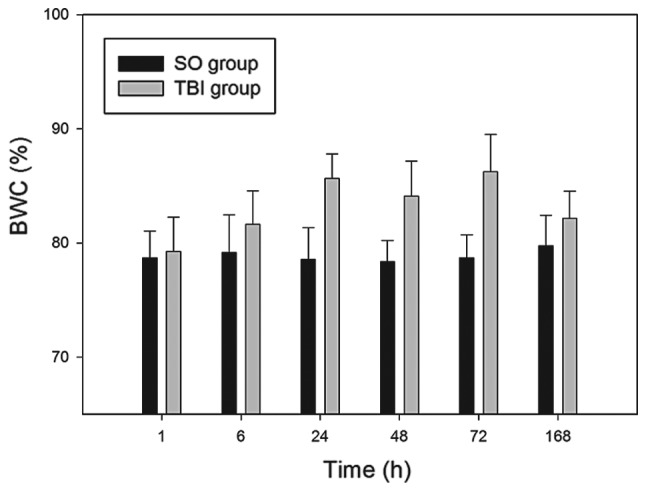
Changes in BWC at different time-points in TBI rats and SO rats. The percentage BWC was determined at the post-TBI time-points of 1, 6, 24, 48, 72 and 168 h. Y-axes:Data are expressed as the mean ± standard deviation (n=10). BWC, brain water content; TBI, traumatic brain injury; SO, sham operation.
Pathological observation of the damaged area and the contralateral non-damaged area
The two sides of the brain in the SO group exhibited a normal structure. At 1 h post-trauma in the TBI-D group, punctate hemorrhage occurred around the blood vessels in the damaged area of the brain, accompanied by broadening of the interstitial space, paling of stained eosinophilic cells and vasogenic edema with a broken blood-brain barrier, which were observed under the electron microscope. At 6 h post-trauma in the TBI-D group, glial cells began to swell, and the color of the cytoplasm was light following staining, exhibiting a balloon-like appearance. Signs of intracellular edema, including swelling of glial nuclei and organelles were observed under the electron microscope, and vasogenic edema was present. At 24 h, intracellular edema was aggravated and accompanied by the infiltration of inflammatory cells and the proliferation of microglia. The first edema peak appeared at the 24 h time-point. After 48 h, composite edema was alleviated, compared with the appearance at 24 h. At 72 h, intracellular edema was aggravated further, and a second edema peak appeared, while vasogenic edema was alleviated. At 168 h, intracellular edema was alleviated, while vasogenic edema was marked and was accompanied by tissue necrosis, infiltration of inflammatory cells and glial scar-like changes.
After 1 h, no abnormality in the contralateral non-damaged area of the brains of TBI-C rats was observed in pathological observation. At 6 h, intracellular edema occurred, and after 24 h, composite brain edema occurred, with intracellular edema in dominance. After 48 h, intracellular edema was present in the contralateral non-damaged area of the brains of the TBI-C rats, and vasogenic edema was alleviated. After 72 h, intracellular edema remained present and after 168 h, intracellular edema was alleviated (Figs. 2–4).
Figure 2.

Ultrastructure of tissue sections exhibiting edema under an electron microscope (magnification, ×6,000). (A) Blood-brain barrier was complete (red arrow) in the sham operation group. (B) Vasogenic edema in the damaged area 1 h post-trauma. Vascular endothelial cells were swollen and the basilar membranes were thickened (red arrow). (C) Intracellular edema in the contralateral non-damaged area 6 h post-trauma. Mitochondria were swollen and the endoplasmic reticulum was expanded (red arrow).
Figure 3.
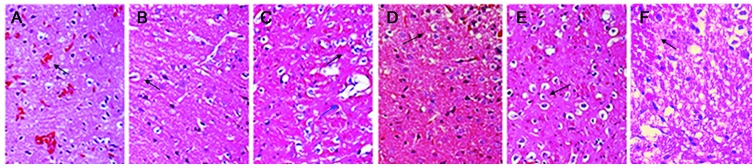
Microscopic observation of the damaged side of the brain at each time-point using hematoxylin and eosin staining. (A) Vasogenic edema after 1 h; Punctate hemorrhage occurred around the microvessels (black arrow), accompanied by broadening of the interstitial space and paling of stained eosinophilic cells. (B) Intracellular edema after 6 h; glial cells were swollen and the color of the cytoplasm was light following staining, with balloon-like changes (black arrow) accompanied by vasogenic edema (blue arrow). (C) After 24 h, composite edema was aggravated, with intracellular edema in dominance (first edema peak, blue arrow). (D) After 48 h, composite edema was alleviated, compared with the 24 h time-point (black arrow). (E) After 72 h, intracellular edema was aggravated further (second edema peak, black arrow), while vasogenic edema was alleviated. (F) After 168 h, intracellular edema was alleviated, while vasogenic edema was apparent and was accompanied by tissue necrosis, infiltration of inflammatory cells and glial scar-like changes (black arrow). Images were captured under a light microscope (magnification, ×200).
Figure 4.
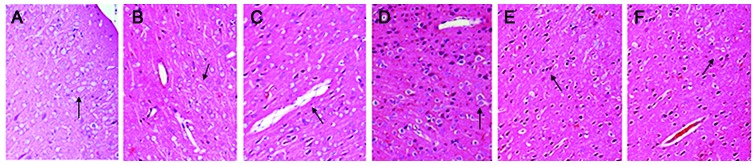
Microscopic observation of the contralateral non-damaged side of the brain at each time-point via hematoxylin and eosin staining. (A) Cellular structure was normal after 1 h. (B) After 6 h, glial cells were swollen and the color of the cytoplasm was light following staining, exhibiting intracellular edema and a loose cytoplasm (black arrow). (C) After 24 h, composite brain edema occurred, with intracellular edema in dominance (black arrow). (D) After 48 h, intracellular edema persisted and vasogenic edema was alleviated (black arrow). (E) Intracellular edema remained after 72 h. (F) After 168 h, intracellular edema was alleviated considerably (black arrow). Images were captured under a light microscope (magnification, ×200).
Detection of the distribution of AQP4 using immunohistochemistry (double labeling)
In the SO group, AQP4 was primarily expressed in the glial cells, vascular endothelial cells and podocytes of the glial cells. In the damaged areas of the TBI-D rats, the expression of AQP4 around the blood vessels increased after 1 h; the expression of AQP4 in the glial cells was abundant after 6 h; and the blood vessels and glial cells exhibited marked expression levels of AQP4 after 24 h and 72 h, while the two areas exhibited reduced expression after 48 h and 168 h (Fig. 4). In the contralateral non-damaged areas of the TBI-C rats, AQP4 was expressed in the glial cells, vascular endothelial cells and the podocytes of glial cells after 1 h, and he expression of AQP4 in this area of the glial cells increased after 6 h. AQP4 was observed in the blood vessels and glial cells, with higher levels in the glial cells) in the contralateral non-damaged areas of the TBI-C rats after 24 h and 72 h, however, these levels declined after 48 h and 168 h (Fig. 5).
Figure 5.

AQP4 distribution in double-labeled immunofluorescence images of the damaged area and contralateral non-damaged area of the brain (magnification, ×1.7). (A) Images from the sham operation rats. (B–D) Images from the damaged side of the brain at 6, 24, and 72 h, respectively. (E–G) Images from the contralateral non-damaged side of the brain at 6, 24, and 72 h, respectively. The expression of AQP4 appeared red, the stained glial fibrillary acidic protein (a glial cell indicator) appeared green, and stained nuclei of glial cells appeared blue. In the sham operation group, AQP4 was predominantly expressed in the glial cells, vascular endothelial cells and the podocytes of the glial cells (white arrow in A). In the damaged area, glial cells exhibited abundant expression of AQP4 after 6 h (white arrow in B), and after 24 h and 72 h, the blood vessels and glial cells exhibited significant AQP4 expression (white arrow in C and D). In the contralateral non-damaged area, the expression of AQP4 in the glial cells increased after 6 h (white arrow in E); AQP4 was abundant in the blood vessels and glial cells after 24 h and 72 h (white arrows in F and G), which was higher in the glial cells. AQP4, aquaporin 4.
Detection of the expression of AQP4 using western blotting at different time-points following TBI in rat brains
The expression level of AQP4 was 1.379±0.03 in the SO group. The expression of AQP4 decreased 1 h following TBI (1.154±0.068) in the damaged areas of the TBI-D group, reached a minimum at 6 h (0.922±0.078), and then increased to reach a first peak at 24 h (2.848±0.102). The expression of AQP4 decreased at 48 h (2.566±0.052) and reached a second peak level at 72 h (2.936±0.090), followed by a decrease at 168 h (2.258±0.104). The expression levels of AQP4 in the damaged areas of the TBI-D groups at each time-point were significantly higher than the expression levels in the SO groups at each corresponding time-point (P<0.01), with the exception of the 1 and 6 h time-points (P>0.05). In the TBI-C rats, AQP4 was upregulated in the glial cells and vascular endothelial cells in the contralateral non-damaged side of the brain, The expression of AQP4 increased slowly from the 6 h time-point (1.846±0.088) and peaked at 72 h (2.662±0.116) prior to decreasing to normal levels at 168 h (1.652±0.052). The expression levels of AQP4 in the TBI-C groups at each time-point were significantly higher than the expression levels in the SO groups at each corresponding time-point (P<0.01), with the exception of the 1 and 168 h time-points (P>0.05; Fig. 6A and B).
Figure 6.
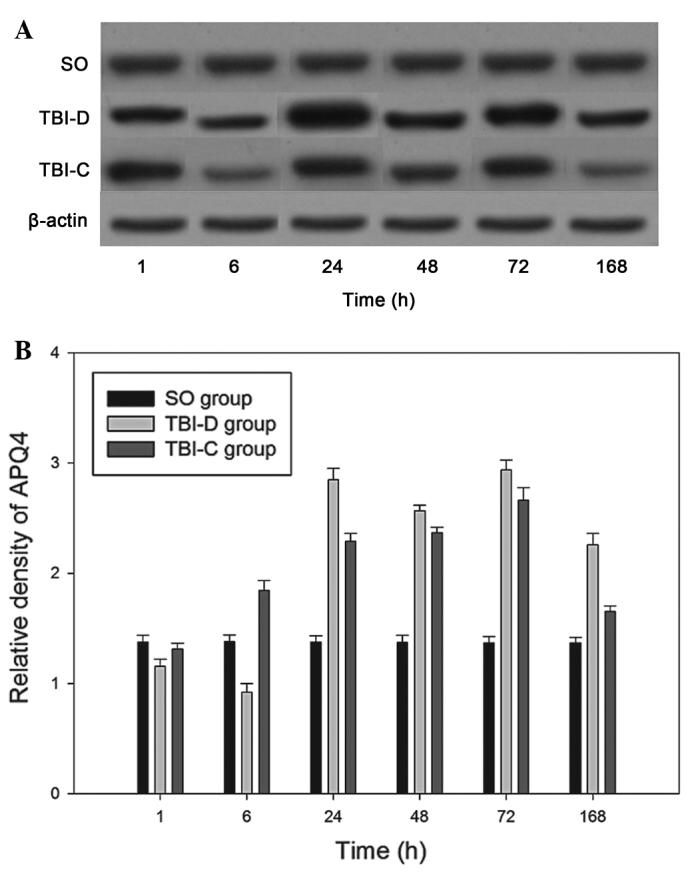
Detection of the expression of AQP4 using western blotting in TBI rat brains at different time-points. (A) Rat brain homogenates of different groups were recognized via probing with rabbit anti-rat AQP4 antibody. (B) Relative densities of AQP4 at different time-points following injury (1, 6, 24, 48, 72 and 168 h) were determined using a Bio-Rad Gel Imaging system. Data are expressed as the mean ± standard deviation (n=10). AQP4, aquaporin 4; TBI, traumatic brain injury; SO, brain homogenates of sham operation rats; TBI-D, brain homogenates of the damage side of TBI rats; TBI-C, brain homogenates of the contralateral non-damaged side of TBI rats.
Discussion
Pathological changes in the damaged area and contralateral non-damaged area of the brain following brain trauma
Cerebral edema is the accumulation of water molecules in brain cells and the extracellular space, which causes a pathological increase in brain volume and is one of the most serious complications of traumatic brain injury (13,14). Cerebral edema can be divided into cytotoxic brain edema (intracellular), vasogenic brain edema, interstitial brain edema and osmotic brain edema subtypes. Severe cerebral edema results in increased intracranial pressure and brain herniation, which are risk factors for mortality and disability following trauma (15). The precise molecular mechanisms acting following TBI remain to be elucidated. As time following trauma progresses, composite cerebral edema appears, aggravating the disease status and causing irreversible brain trauma.
It has been reported that vasogenic edema first occurs during the early stage of brain trauma (<24 h) (16). Immunohistochemical staining and semi-quantitative analysis of brain sections revealed that injury to the blood-brain barrier (BBB) was a key factor in traumatic brain edema. It was previously suggested that, at the early stage of trauma, the primary site of brain trauma predominantly exhibits cytotoxic edema and the surrounding region is dominated by vasogenic edema (17). In the present study, evaluations of rat brain injury were divided by clinical stages: Early stage (<24 h), intermediate stage (24–72 h) and late stage (72–168 h). Pathological changes in the damaged side and contralateral non-damaged side of the brain were evaluated at the various stages. At the early stage (<24 h), vasogenic edema with a broken BBB was observed in the damaged area, followed by intracellular edema. At the intermediate stage (24–72 h), composite edema with intracellular edema was dominant. At the late stage (72–168 h), intracellular edema was markedly alleviated, however, vasogenic edema persisted. There were two edema peaks at 24 h and 72 h, respectively, which were predominantly caused by the aggravation of intracellular edema. These results were consistent with those of previous reports (17,18).
In the contralateral non-damaged area, no abnormalities were detected in the brain after 1 h; intracellular edema occurred after 6 h; composite edema occurred after 24 h; and intracellular edema persisted at 48 h, but vasogenic edema was alleviated. Intracellular edema remained in the contralateral non-damaged area after 72 h, but was alleviated after 168 h. Compared with the damaged area, the edema status in the contralateral non-damaged area was less severe; no typical edema peak was observed, and the alleviation of vasogenic edema occurred earlier, compared with that of intracellular edema. Therefore, it was hypothesized that, following brain injury to one side of the brain, the contralateral non-damaged side exhibits corresponding pathological changes, which are delayed, compared with those of the damaged side and are characterized by different types of edema. The present study is the first, to the best of our knowledge, to report observations of this type.
Expression of AQP4 in the damaged side of the brain and the contralateral non-damaged area, and its correlation with cerebral edema
As a membrane protein, AQP4 is widely distributed in the brain in glial cells, ependymal cells and capillary endothelial cells, and particularly in the choroid plexus, where it regulates water metabolism (3). Previous experiments on cerebral ischemia, cerebral hemorrhage, brain trauma and in vitro glial cell hypoxia have demonstrated that AQP4 upregulation is closely associated with intracellular edema (19–22). In addition, it has been indicated that destruction of the BBB results in the upregulation of AQP4 (23). It has been reported that vasogenic edema is aggravated in rats with reduced AQP4 abundance; therefore, it is suggested that AQP4 is responsible for the formation of vasogenic edema (23). Certain reports have also indicated that damage to the BBB and AQP4 downregulation are associated with the alleviation of vasogenic edema and the generation of intracellular edema (24). Therefore, the precise nature of the association between AQP4 and edema in TBI remain to be fully elucidated.
In the present study, a varied time-course of changes in BWC were detected in the TBI rats. The BWC began to increase in the TBI rats at 1 h, was significantly increased at 6 h, reached two peak levels at 24 and 72 h, and was significantly decreased at 168 h. Notably, in the damaged and contralateral non-damaged areas of the TBI rats, a similar time-course was observed in the expression of AQP4 to that of BWC, in which the expression of AQP4 reached two peak levels at 24 and 72 h, and was significantly decreased at 168 h. In the brains of the TBI rats, the expression of AQP4 in the damaged side decreased at 6 h (0.922±0.078), but increased (1.846±0.088) in the contralateral non-damaged side. The expression of AQP4 in the whole brain of the TBI rats was increased, a change that was also similar to that observed in BWC at 6 h. These results indicated that changes in BWC levels were consistent with changes in the expression of AQP4 in the brains of TBI rats, suggesting that AQP4 may have acted as a molecular regulator of edema in TBI.
Pathological observation revealed that vasogenic edema was the predominant type of edema between 1 and 6 h following brain trauma. Intracellular edema was the predominant pathological change at the two AQP4 expression peaks at 24 and 72 h. BWC was significantly increased 6 h following brain trauma, while general damage to the BBB was observed 1 h following brain trauma, suggesting that BBB damage occurred earlier than brain edema. Therefore, it was hypothesized that vasogenic edema due to BBB damage resulted in AQP4 downregulation, while the AQP4 upregulation was caused by intracellular edema. When the BBB is damaged, the down-regulation of AQP4 can alleviate vasogenic edema by reducing water permeation from the blood vessels to the interstitial space. As harmful substances, including injury-associated factors, enter cells, the upregulation of AQP4 can promote water to enter the cells, diluting the harmful substances and protecting the cells (18). This suggested that the expression and regulation of AQP4 following brain trauma are organismal defense mechanisms.
In the contralateral non-damaged area, no AQP4 downregulation was observed following brain trauma, and the expression level after 1 h (1.313±0.052) was not significantly different from that of the SO group. After 6 h, the expression of AQP4 gradually increased and peaked at 72 h (2.662±0.116). Subsequently, the expression of AQP4 declined until the 168 h time-point. The expression levels at each time-point in the contralateral non-damaged area were all lower than those in the damaged area. Intracellular edema was the predominant pathological change in the contralateral non-damaged area, following which vasogenic edema soon appeared. These results demonstrated that the upregulation of AQP4 was associated with the severity of intracellular edema. It was hypothesized that damage to one side of the brain induces the transmission of a signal to upregulate the expression of AQP4 in the contralateral side via the neural-humoral regulation system, resulting in intracellular edema.
The pathological process of traumatic brain edema is complex, with alternation of vasogenic edema and intracellular edema, the interaction of which aggravates secondary brain injury. AQP4 may be involved in the occurrence of traumatic brain edema via changes in expression, which act as a defense response. A more detailed evaluation of AQP4 gene function may be achieved in future investigations using cellular or animal models, in which AQP4 is reduced or absent.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (grant. no.81160181) and the Hainan Provincial International Corporation Project of China (grant. no. 2012-GH016).
References
- 1.Sabre L, Tomberg T, Kõrv J, Kepler J, Kepler K, Linnamägi U, Asser T. Brain activation in the acute phase of traumatic spinal cord injury. Spinal Cord. 2013;51:623–629. doi: 10.1038/sc.2013.41. [DOI] [PubMed] [Google Scholar]
- 2.Suzuki R, Okuda M, Asai J, Nagashima G, Itokawa H, Matsunaga A, Fujimoto T, Suzuki T. Astrocytes co-express aquaporin-1-4 and vascular endothelial growth factor in brain edema tissue associated with brain contusion. Acta Neurochir. 2006;(Suppl 96):S398–S401. doi: 10.1007/3-211-30714-1_82. [DOI] [PubMed] [Google Scholar]
- 3.Tait MJ, Saadoun S, Bell BA, Papadopoulos MC. Water movements in the brain: Role of aquaporins. Trends Neurosci. 2008;31:37–43. doi: 10.1016/j.tins.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Yao X, Hrabetová S, Nicholson C, Manley GT. Aquaporin-4-deficient mice have increased extracellular space without tortuosity change. J Neurosci. 2008;28:5460–5464. doi: 10.1523/JNEUROSCI.0257-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Papadopoulos MC, Verkman AS. Aquaporin-4 and brain edema. Pediatr Nephrol. 2007;22:778–784. doi: 10.1007/s00467-006-0411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Badaut J, Fukuda AM, Jullienne A, Petry KG. Aquaporin and brain diseases. Biochim Biophys Acta. 2014;1840:1554–1565. doi: 10.1016/j.bbagen.2013.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lescot T, Fulla-Oller L, Po C, Chen XR, Puybasset L, Gillet B, Plotkine M, Meric P, Marchand-Leroux C. Temporal and regional changes after focal traumatic brain injury. J Neurotrauma. 2010;27:85–94. doi: 10.1089/neu.2009.0982. [DOI] [PubMed] [Google Scholar]
- 8.Easton AS. Regulation of permeability across the blood-brain barrier. Adv Exp Med Biol. 2012;763:1–19. doi: 10.1007/978-1-4614-4711-5_1. [DOI] [PubMed] [Google Scholar]
- 9.Zhang L, Yuan F. Enhanced expression of aquaporin-4 aggravates brain edema after traumatic brain injury in rat. Journal of Capital Medical University. 2008;29:732–736. In Chinese. [Google Scholar]
- 10.Imam AM, Jin G, Sillesen M, Duggan M, Jepsen CH, Hwabejire JO, Lu J, Liu B, DeMoya MA, Velmahos GC, Alam HB. Early treatment with lyophilized plasma protects the brain in a large animal model of combined traumatic braininjury and hemorrhagic shock. J Trauma Acute Care Surg. 2013;75:976–983. doi: 10.1097/TA.0b013e31829e2186. [DOI] [PubMed] [Google Scholar]
- 11.Feeney DM, Boyeson MG, Linn RT, Murray HM, Dail WG. Responses to cortical injury I. Methodology and local effects of contusions in the rat. Brain Res. 1981;211:67–77. doi: 10.1016/0006-8993(81)90067-6. [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Wang G, Gao C, Shao G, Kang N. Effects of hyperbaric oxygen on MMP-2 and MMP-9 expression and spinal cord edema after spinal cord injury. Life Sci. 2013;93:1033–1038. doi: 10.1016/j.lfs.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 13.Hirt L, Ternon B, Price M, Mastour N, Brunet JF, Badaut J. Protective role of early aquaporin 4 induction against post-ischemic edema formation. J Cereb Blood Flow Metab. 2009;29:423–433. doi: 10.1038/jcbfm.2008.133. [DOI] [PubMed] [Google Scholar]
- 14.Kim JY, Lee YW, Kim JH, Lee WT, Park KA, Lee JE. Agmatine Attenuates Brain Edema and Apoptotic Cell Death after Traumatic Brain Injury. J Korean Med Sci. 2015;30:943–952. doi: 10.3346/jkms.2015.30.7.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lo Pizzo M, Schiera G, Di Liegro I, Di Liegro CM, Pál J, Czeiter E, Sulyok E, Dóczi T. Aquaporin-4 distribution in control and stressed astrocytes in culture and in the cerebrospinal fluid of patients with traumatic brain injuries. Neurol Sci. 2013;34:1309–1314. doi: 10.1007/s10072-012-1233-4. [DOI] [PubMed] [Google Scholar]
- 16.Liu H, Sun S, Yang M. Role of aquaporin-4 in the formation of brain edema after mild traumatic brain injury in rats. Chinese Journal of Anatomy. 2009;32:68–72. In Chinese. [Google Scholar]
- 17.Wei XE, Zhang YZ, Li YH, Li MH, Li WB. Dynamics of rabbit brain edema in focal lesion and perilesion area after traumatic brain injury: A MRI study. J Neurotrauma. 2012;29:2413–2420. doi: 10.1089/neu.2010.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu H, Lei XY, Hu H, He ZP. Relationship between AQP4 expression and structural damage to the blood-brain barrier at early stages of traumatic brain injury in rats. Chin Med J (Engl) 2013;126:4316–4321. [PubMed] [Google Scholar]
- 19.Ding Z, Zhang J, Huang G. Studies on siRNA interferes with aquaporin-4 expression and influences on traumatic brain injury. Chinese Journal of Difficult and Complicated Cases. 2013;12:950–952. In Chinese. [Google Scholar]
- 20.Hu H, Lu H, He Z, Han X, Chen J, Tu R. Gene interference regulates aquaporin-4 expression in swollen tissue of rats with cerebral ischemic edema: correlation with variation in apparent diffusion coefficient. Neural Regen Res. 2012;7:1659–1666. doi: 10.3969/j.issn.1673-5374.2012.21.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hui H, Hong L, Zhanping H. AQP4 expression in hypoxic cultured astrocytes can be inhibited by AQP4 mRNA silence in vitro. Chinese Journal of Anatomy. 2011;34:47–50. In Chinese. [Google Scholar]
- 22.Ren Z, Iliff JJ, Yang L, Yang J, Chen X, Chen MJ, Giese RN, Wang B, Shi X, Nedergaard M. 'Hit & Run' model of closed-skull traumatic brain injury (TBI) reveals complex patterns of post-traumatic AQP4 dysregulation. J Cereb Blood Flow Metab. 2013;33:834–845. doi: 10.1038/jcbfm.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukuda AM, Adami A, Pop V, Bellone JA, Coats JS, Hartman RE, Ashwal S, Obenaus A, Badaut J. Posttraumatic reduction of edema with aquaporin-4 RNA interference improves acute and chronic functional recovery. J Cereb Blood Flow Metab. 2013;33:1621–1632. doi: 10.1038/jcbfm.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bao HJ, Wang T, Zhang MY, Liu R, Dai DK, Wang YQ, Wang L, Zhang L, Gao YZ, Qin ZH, Chen XP, Tao LY. Poloxamer-188 attenuates TBI-induced blood-brain barrier damage leading to decreased brain edema and reduced cellular death. Neurochem Res. 2015;37:2856–2867. doi: 10.1007/s11064-012-0880-4. [DOI] [PubMed] [Google Scholar]