

Abstract
The inflammatory reaction is important in secondary injury following traumatic brain injury (TBI). Rapamycin has been demonstrated as a neuroprotective agent in a mouse model of TBI, however, there is a lack of data regarding the effects of rapamycin on the inflammatory reaction following TBI. Therefore, the present study was designed to assess the effects of treatment with rapamycin on inflammatory reactions and examine the possible involvement of microglial activation following TBI. Male imprinting control region mice were randomly divided into four groups: Sham group (n=23), TBI group (n=23), TBI + dimethyl sulfoxide (DMSO) group (n=31) and TBI + rapamycin group (n=31). Rapamycin was dissolved in DMSO (50 mg/ml) and injected 30 min after TBI (2 mg/Kg; intraperitoneally). A weight-drop model of TBI was induced, and the brain tissues were harvested 24 h after TBI. The findings indicated that the administration of rapamycin following TBI was associated with decreased levels of activated microglia and neuron degeneration at the peri-injury site, reduced levels of proinflammatory cytokines and increased neurobehavioral function, possibly mediated by inactivation of the mammalian target of rapamycin pathway. The results of the present study offer novel insight into the mechanisms responsible for the anti-neuroinflammatory effects of rapamycin, possibly involving the modulation of microglial activation.
Keywords: rapamycin, brain contusion, inflammatory reaction, microglial activation, proinflammatory cytokines
Introduction
Traumatic brain injury (TBI) causes primary neuronal degeneration in the contusion site and also initiates secondary injury in the surrounding tissue (pericontusion). The secondary nonmechanical injury is progressive and can last for days, weeks or months (1). Several pathological processes are reported to be responsible for the neuronal cell death, which occurs as a result of secondary damage following TBI, including inflammation, apoptosis and oxidative stress (2–4). Among these, inflammation is key in secondary injury (3,5). Microglial activation has been widely-accepted as histological evidence of brain injury (6). It is also considered to be the major source of proinflammatory cytokines following TBI (7–9). A number of investigations have specifically targeted microglial activation in order to retard the pathological processes associated with inflammation following TBI (10,11).
Mammalian target of rapamycin (mTOR) is a rapamycin sensitive serine/threonine protein kinase, which is involved in modulating the initiation of protein synthesis (12). Growth factors, mitogens, hormones and nutrients are able to activate the phosphoinositide 3-kinase pathway, leading to the phosphorylation and activation of mTOR (13,14). Activated mTOR exerts its functions in stimulating translation through the phosphorylation and activation of the p70S6K S6 kinase, which phosphorylates the S6RP ribosomal protein (15). The phosphorylation of S6RP is a critical effector of mTOR in the regulation of protein synthesis. The mTOR pathway is important in the synthesis of proinflammatory cytokines. Inactivation of the mTOR pathway attenuates the expression of proinflammatory cytokines, including interleukin (IL)-1β and tumor necrosis factor-α (TNF-α), which promote adipose-derived stromal cell viability and antiapoptotic/proangiogenic efficacy in vivo (16). Thus, the mTOR pathway may be a significant proinflammatory factor due to its ability to induce proinflammatory cytokines, and targeting this pathway may offer a reasonable therapeutic strategy against the inflammatory response following TBI.
Rapamycin is a macrolide antibiotic product from Streptomyces hygroscopicus, which specifically inhibits the activity of mTOR. To exert its inhibitory effect on mTOR, rapamycin binds to immunophilin FK-506-binding protein 12, which prevents the phosphorylation of mTOR (17,18). The function of rapamycin has been an intensive area of investigation, and a focus of attention in cancer, development, metabolism and central nervous system diseases (14). Rapamycin has also been investigated for its neuroprotective effects in closed head injury TBI models. The injection of rapamycin increases the survival of neurons and significantly improves the functional recovery of the brain (19). However, the effect of rapamycin on the inflammatory reaction, which follows TBI remains to be fully elucidated. Thus, in the present study, the possible involvement of rapamycin on the decreased expression of proinflammatory cytokines was examined in a mouse TBI model. In addition, the functional significance of rapamycin on the neurological outcome, associated with attenuating the inflammatory reaction following TBI, was assessed.
Materials and methods
Animals
Male imprinting control region (ICR) mice (Experiment Animal Centre of Nanjing Medical University, Jiangsu, China) aged between 6–8 weeks, weighing 28–32 g were used in the present study. The experimental procedures were approved by the Animal Care and Use Committee of Nanjing Jinling Hospital and conformed to the Guide for the Care and Use of Laboratory Animals by the National Institutes of Health (Bethesda, MD, USA). The mice were housed on a 12 h light/dark cycle with ad libitum access to food and water.
Model of TBI
The model of TBI used in the present study was a weight-drop model (Beyotime Institute of Biotechnology, Shanghai, China), as described by Flierl et al (20). The mice were anesthetized with an intraperitoneal (i.p.) injection of chloral hydrate (1%; 5 ml/kg; Beyotime Institute of Biotechnology) and then placed onto a platform directly below the weight of the weight-drop device. A 1.5 cm midline longitudinal scalp incision was made and the skull was exposed. Subsequent to locating the left anterior frontal area (1.5 mm lateral to the midline on the mid-coronal plane) as the impact area, a 200 g weight was released and dropped onto the skull from a height of 2.5 cm. The mortality rate resulting from apnea was reduced by early respiratory support. The scalp wound was closed using standard suture material, and the mice were returned to cages, where they had ad libitum access to water and food. Sham-injured animals underwent the same procedures, but did not undergo the weight-drop.
Experimental procedure
The male ICR mice were divided into four groups (n=45 per group): Sham group, TBI group, TBI + dimethyl sulfoxide (DMSO) group and TBI + rapamycin group. In the TBI + rapamycin group, rapamycin (cat. no. S1039; Selleckchem, Munich, Germany) was dissolved in DMSO (50 mg/ml; Sigma-Aldrich, St. Louis, MO, USA) and injected into the mice 30 min after TBI (2 mg/kg; i.p.). The mice in the TBI + DMSO group received equal volumes of vehicle (5% DMSO) at 30 min subsequent to TBI.
Brain tissue processing
For isolation of the proteins, the animals were anesthetized with a solution of chloral hydrate (1%, 5 ml/kg) 24 h after TBI and were perfused intracardially with 30–40 ml cold (4°C) heparinized 0.9% saline (Beyotime Institute of Biotechnology). The left, ipsilateral, cerebral cortex (pericontusion) was collected, immediately frozen in liquid nitrogen and then transferred to a −80°C freezer until use. For immunohistochemical analysis, the animals were sacrificed 24 h after TBI in the following way. Following anesthesia, induced with chloral hydrate (1%, 5 ml/kg), the animals were intracardially perfused with 30–40 ml cold heparinized 0.9% saline followed by 20–30 ml cold 4% paraformaldehyde (Beyotime Institute of Biotechnology). The whole brain was removed and immersed in 4% paraformaldehyde overnight at 4°C. For immunofluorescence, the brain was subsequently immersed in 20% sucrose (Beyotime Institute of Biotechnology) followed by 30% sucrose.
Western blot analysis
The protein concentrations were determined using the Bradford assay (Beyotime Institute of Biotechnology) (21). Equal quantities of protein (50 µg/per lane) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel (Beyotime Institute of Biotechnology) electrophoresis and transferred onto polyvinylidene-difluoride membranes (EMD Millipore, Billerica, MA, USA). The membranes were blocked for 2 h in blocking buffer (Tris-buffered saline/0.05% Tween 20; TBST; Beyotime Institute of Biotechnology) containing 5% skim milk) and were incubated overnight at 4°C with the following primary antibodies in blocking buffer: Rabbit monoclonal phospho-mTOR (Ser2448) (cat..no. #5536; 1:1,000; Cell Signaling Technology, Danvers, MA, USA), rabbit monoclonal phospho-S6 ribosomal protein (Ser235/236) (cat..no. #4858; 1:2,000; Cell Signaling Technology) and rabbit polyclonal GAPDH (cat. no. AP0063; 1:5,000; Bioworld Technology, Minneapolis, MN, USA). Following washing of the membranes three times with TBST (10 min each), the membranes were incubated with polyclonal goat anti-rabbit horseradish peroxidase conjugated immunoglobulin G (cat. no. BS13278; 1:5,000; Bioworld Technology) for 2 h at room temperature. The protein bands were visualized using enhanced chemiluminescence western blotting detection reagents (EMD Millipore) and exposure to X-ray film (Carestream, Xiamen, China). The developed films were digitized using an Epson Perfection 2480 scanner (Seiko Corp., Nagano, Japan). The band density was quantified using Un-Scan-It 6.1 software (Silk Scientific Inc., Orem, UT, USA) and the data were normalized to GAPDH.
Immunofluorescence
For immunofluorescence, serial 8-µm coronal sections were obtained using a cryostat (RM2235; Leica Microsystems GmbH, Wetzlar, Germany). A total of four sets of five evenly spaced (300 µm apart) sections, spanning the injured cortex, were collected from each brain. Based on established immunostaining procedures (22), slides (Beyotime Institute of Biotechnology) were incubated in blocking buffer, containing 10% normal goat serum in phosphate-buffered saline (PBS) and 0.1% Triton X-100 (Beyotime Institute of Biotechnology) for 2 h, followed by incubation at 4°C overnight with the primary antibody, rabbit anti-ionized calcium-binding adapter molecule 1 (IBA-1; cat. no. 019-19741; 1:5,000; Wako, Osaka, Japan). On the following day, the slides were washed with PBS three times for 5 min and incubated with the appropriate secondary antibodies for 1 h at room temperature. The slides were then washed three times in PBS. Cover slips were applied using mounting medium. Images of the immunofluorescence were captured using an Axio Observer A1 microscope system (Carl Zeiss, Oberkochen, Germany) and analyzed using Image-Pro Plus 6.0 software (Media Cybernetics, Rockville, MD, USA). The specificity of the immunofluorescence reaction was evaluated by replacement of the primary antibody with PBS. A total of six visual fields (magnification, ×200) surrounding the contusion in each coronary section were randomly selected, and the mean number of microglia in the six fields was calculated. A total of four sections from each animal were used for quantification. The data for each sample was the mean number of microglia in the four sections. Data are presented as the mean number of microglia per magnification field (magnification, ×200). All analyses were performed by two investigators in a blinded-manner.
Enzyme-linked immunosorbent assay (ELISA)
The expression levels of interluekin (IL)-1β and TNF-α were analyzed using an ELISA (Biocalvin Company, Suzhou, China), according to the manufacturer's instructions. The protein concentrations were measured using a Bradford assay. Equal quantities of lysate were used for the analyses of TNF-α and IL-1β, with values expressed as pg/mg protein.
Cresyl violet (Nissl) staining
Tissue sections were stained with Cresyl Violet (Nissl; Sigma-Aldrich), as described previously (23). Normal neurons have relatively large cell bodies and are rich in cytoplasm, with one or two large round nuclei. By contrast, damaged cells exhibit shrunken cell bodies, condensed nuclei, a dark cytoplasm and numerous empty vesicles (23). The counting of cells was restricted to the lesion boundary zone (where the most damage was observed). A total of six high-power fields (magnification, ×400) in each coronary section were randomly selected, and the mean number of surviving neurons in the six views were calculated for each section using the Axio Observer A1 microscope system. A total of four sections from each animal were used for quantification and the average quantity of the four sections was calculated for each sample. Data are presented as the quantity of neurons per high-power field. All analyses were performed by two investigators in a blinded-manner.
Neurobehavioral evaluation
The neurological statuses of the mice were evaluated 24 h and 72 h after TBI using a grip test and neurological severity score (NSS). The grip test was developed to assess the gross vestibulomotor function (24). The mouse was placed on a thin, horizontal metal wire, measuring 45 cm in length, which was suspended 45 cm above a foam pad between two vertical poles. Each mouse was graded on its ability to grip, attach and move, as described in Table I. The grip test was performed in triplicate, with the total score calculated for each mouse. In the NSS, the ability of each mouse to perform 10 different tasks, which demonstrate motor function, balance and alertness, was evaluated. A single point was scored for failing to perform each of the tasks; thus, 0=minimum deficit and 10=maximum deficit (Table II) (3,20). The severity of the injury was defined by the initial NSS, evaluated 1 h after TBI, providing a reliable predictor of the later outcome. All neurobehavioral assessments were performed by two investigators in a blinded-manner.
Table I.
Grip test scoring system.
| Task | Score |
|---|---|
| Unable to grasp wire for 30 sec | 0 |
| Grip wire for 30 sec with one or two paws | 1 |
| Jump up and grasp wire with four paws | 2 |
| Grasp wire with four paws and wrap tail around | 3 |
| Crawl along the wire for ≥5 cm | 4 |
| Crawl along the wire to the end and dismount | 5 |
| Maximum score | 5 |
Table II.
Neurological severity score scoring system.
| Task | Number of points (success/failure) |
|---|---|
| Exit a circle of 30 cm diameter within 3 min | 0/1 |
| Paresis of upper and/or lower limb of the contralateral side | 0/1 |
| Able to walk straight | 0/1 |
| Presence of startle reflex | 0/1 |
| Presence of seeking behavior | 0/1 |
| Able to balance on a beam of 7 mm width for 10 sec | 0/1 |
| Able to balance on a round stick of 5 mm diameter for 10 sec | 0/1 |
| Able to cross a 30 cm long beam of 3 cm width | 0/1 |
| Able to cross a 30 cm long beam of 2 cm width | 0/1 |
| Able to cross a 30 cm long beam of 1 cm width | 0/1 |
| Maximum total | 10 |
Statistical analysis
Each experiment was repeated at least three times and the data are expressed as the mean ± standard error of the mean. For the behavioral assessment, two-way analysis of variance was used followed by a Bonferroni post hoc test. For the other assays, one-way analysis of variance was used followed by Tukey's test. SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA) was used for the statistical analyses. P<0.05 was considered to indicate a statistically significant difference.
Results
Rapamycin improves the neurobehavioral scores of mice following TBI
In the grip test, the scores of the rapamycin-treated mice were significantly improved, compared with those of the vehicle-treated mice 24 h and 72 h after TBI (P<0.01 and P<0.05, respectively; Fig. 1A). As shown in Fig. 1B, the NSS scores of the TBI and the TBI + DMSO groups were lower at 72 h than at 24 h. At 24 h following TBI, the NSS scores of the rapamycin-treated mice were significantly lower compared with those of the vehicle treated mice (P<0.01), and at 72 h, the scores of the two groups had improved, maintaining a significant difference (P<0.05).
Figure 1.
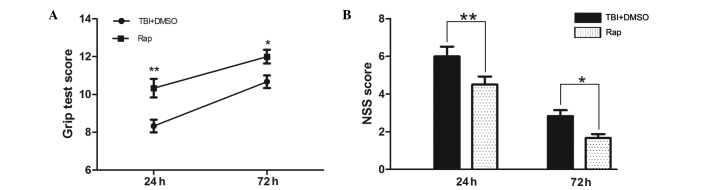
Rapamycin improves the neurobehavioral performance of mice following TBI. At 24 h and 72 h following TBI, the mice pretreated with rapamycin had significantly improved neurological functions compared with the mice pretreated with DMSO, as assessed by a (A) grip test and (B) NSS. Data are presented as the mean ± standard error of the mean (n=8). **P<0.01 and *P<0.05, compared with the sham group, nsP>0.05 compared with the TBI group, #P<0.05, compared with the TBI + DMSO group. TBI, traumatic brain injury; DMSO, dimethyl sulfoxide; NSS, neurological severity score; Rap, rapamycin.
Rapamycin protects neurons in the pericontusional region from degeneration following TBI
Nissl staining was used to evaluate neuronal survival in the pericontusional cortex 24 h after TBI (Fig. 2). Compared with the sham group, TBI decreased the number of neurons in the pericontusional cortex (P<0.01). A large proportion of neurons in the TBI group were damaged, exhibiting extensive degenerative changes, which included sparse cellular arrangements, loss of integrity, shrunken cytoplasma and misshapen nuclei. The sham group was observed to contain clear and intact neurons. Rapamycin significantly increased the proportion of surviving neurons at 24 h after TBI (P<0.05) and may, therefore, have alleviated the severity of neuronal degeneration.
Figure 2.

Rapamycin protects pericontusional neurons from degeneration 24 h after TBI. (A) Representative photomicrographs of Nissl staining of the pericontusional cortex in the sham group, TBI, TBI + DMSO and TBI + rapamycin groups. Scale bar=50 µm at magnification, ×200; scale bar=25 µm at magnification, ×400. (B) Total neurons in each visual field (magnification, ×400). Compared with the sham group, TBI decreased the number of neurons in the pericontusional cortex. Rapamycin treatment significantly increased the proportion of surviving neurons 24 h after TBI. (C) Diagram of a coronal mouse brain section, revealing the association between the lesion cavity (solid black) and the regions where the images were captured (squares). Data are expressed as the mean ± standard error of the mean (n=6). **P<0.01, compared with sham group; nsP>0.05, compared with TBI group; #P<0.05, compared with TBI + DMSO group. TBI, traumatic brain injury; Nissl, cresyl violet; DMSO, dimethyl sulfoxide; Rap, rapamycin; ns, not significant.
Rapamycin treatment decreases the production of proinflammatory cytokines in the injured cortex following TBI
To evaluate the association between rapamycin and proinflammatory cytokines, an ELISA was used to detect the protein levels of proinflammatory factors 24 h after TBI (Fig. 3). The expression levels of IL-1β and TNF-α increased significantly following TBI compared with the sham group (P<0.01 and P<0.001, respectively). No difference was observed between the TBI + DMSO group and the TBI group (P>0.05). The expression levels of IL-1β and TNF-α were reduced in the rapamycin-treated groups compared with those in the DMSO-treated group (P<0.05).
Figure 3.
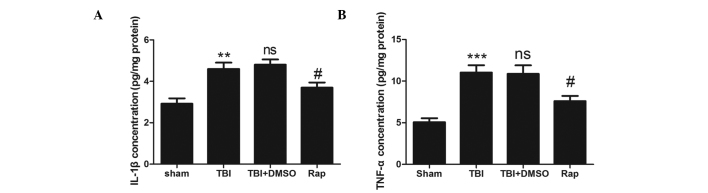
Rapamycin attenuates the upregulation of proinflammatory cytokines in injured brain tissue following TBI. (A) Concentration of IL-1β was increased significantly in the TBI and TBI + DMSO groups compared with the sham group. Treatment with rapamycin significantly attenuated the levels of IL-1β in the ipsilateral cortex compared with DMSO treatment. (B) Concentration if TNF-α was increased significantly in the TBI and TBI + DMSO groups compared with the sham group. Treatment with rapamycin significantly attenuated the levels of TNF-α in the ipsilateral cortex compared with DMSO treatment. Data are expressed as the mean ± standard error of the mean (n=6). ***P<0.001 and **P<0.01, compared with the sham group; nsP>0.05, compared with the TBI group, #P<0.05, compared with the TBI + DMSO group. TBI, traumatic brain injury; DMSO, dimethyl sulfoxide; Rap, rapamycin; IL, interleukin; TNF, tumor necrosis factor; ns, not significant.
Rapamycin suppresses microglial activation in the pericontusional cortex following TBI
The levels of microglial activation were investigated by detecting the immunofluorescence of IBA-1. As shown in Fig. 4, the microglia in the sham group exhibited long branching processes and small cellular bodies. In response to brain injury, the number of microglia increased significantly (P<0.01) compared with the sham group, and the branches of the IBA-1-stained microglia became short, retracted and thick, indicating activated microglia. Compared with the TBI and TBI + DMSO groups, rapamycin injection decreased the number of IBA-1-stained cells (P<0.05) and microglia exhibited smaller cellular bodies.
Figure 4.

Rapamycin decreases microglial activation 24 h following TBI. (A–D) Representative photomicrographs of immunofluorescence staining for IBA-1 in the pericontusive cortex. (A) Immunofluorescence indicated that microglia of the sham group exhibited long branching processes and a small cellular body. (B and C) In response to TBI, the number of microglia increased and the branches of the microglia became short, retracted and thick. (D) Rapamycin injection decreased the number of IBA-1-stained cells, and the microglia exhibited a smaller cellular body. Scale bar=50 µm. (E) Quantification of immunofluorescence. Data are expressed as the mean ± standard error of the mean (n=6). **P<0.01, compared with the sham group; nsP>0.05, compared with the TBI group; #P<0.05, compared with the TBI + DMSO group. TBI, traumatic brain injury; IBA-1, ionized calcium-binding adapter molecule 1DMSO, dimethyl sulfoxide; Rap, rapamycin; IBA-1, ionized calcium-binding adapter molecule 1; ns, not significant.
Rapamycin dephosphorylates the mTOR pathway following TBI
To confirm the inactivation of the mTOR pathway induced by rapamycin, the protein expression levels of phosphorylated mTOR and S6RP were investigated using western blotting. As shown in Fig. 5, the expression levels of p-mTOR and p-S6RP were significantly increased in the pericontusional cortex 24 h after TBI (P<0.01). No statistically significant differences were identified between the TBI group and the TBI + DMSO group (P>0.05). Compared with the TBI + DMSO group, rapamycin administration markedly decreased the expression levels of p-mTOR and p-S6RP (P<0.05).
Figure 5.

Dephosphorylation of mTOR pathway by rapamycin at 24 h after TBI. (A) Upper: representative western blotting demonstrating P-mTOR protein expression in cerebral cortex in all groups. GAPDH was used as the loading control. Bottom: graphic representations of the ratios of p-mTOR/GAPDH. (B) Upper: representative western blotting demonstrating p-S6RP protein expression in cerebral cortex in all groups. GAPDH was used as the loading control. Bottom: graphic representations of the ratios of p-S6RP/GAPDH. Data are expressed as the mean ± standard error of the mean (n=5). **P<0.01 compared with the sham group, nsP>0.05 compared with the TBI group, #P<0.05 compared with the TBI + DMSO group. TBI, traumatic brain injury; DMSO, dimethyl sulfoxide; Rap, rapamycin; mTOR, mamaalian target of rapamycin; p, phosphorylated; S6RP, S6 ribosomal protein; ns, not significant.
Discussion
In the present study, the effects of rapamycin on the upregulation of proinflammatory cytokines and the activation of microglia were evaluated following TBI in a mouse model. The data revealed that rapamycin administration significantly inhibited microglial activation and decreased the expression levels of IL-1β and TNF-α. Furthermore, rapamycin treatment increased the number of neurons in the pericontusional cortex and improved neurological function following TBI.
Neuroinflammation is important in the pathogenesis of secondary brain injury following TBI (25,26). Elevated levels of proinflammatory cytokines in the injured cortex, including TNF-α and IL-1β, are considered to contribute to the level of cerebral damage (27–30). TNF-α has been observed as a major initiator of neuroinflammation and is released early following an inflammatory stimulus (31). In inflammatory states, TNF-α, in addition to a number of other proinflammatory mediators, is produced predominantly by activated microglia (5). The contribution of TNF-α to tissue damage has been assessed in animal models, whereby recombinant TNF injected into the brain induced cerebral inflammation, breakdown of the blood-brain barrier and the recruitment of intracranial leukocytes (29). IL-1β is an important initiator of the immune response, which is involved in the onset and development of a complex inflammatory cascade. Elevated levels of IL-1β have been detected in the cerebrospinal fluid and brain tissue within the early hours following brain injury (32), and neuronal damage is attenuated when recombinant IL-1 receptor antagonist is injected intracerebroventricularly following brain injury in rats (33). In the present study, the increased number of neurons in the pericontusional cortex and the improved neurological status following rapamycin administration may result from the ability of rapamycin to decrease the expression levels of IL-1β and TNF-α, thereby inhibiting the inflammatory reaction following TBI.
As proinflammatory cytokines are predominantly secreted by microglia, and activated microglia are important in the progression of the inflammatory reaction following TBI, the effect of rapamycin on microglial activation was further investigated. The data revealed that rapamycin injection markedly suppressed the levels of microglial activation following TBI. The activation of microglia is widely accepted as histological evidence of brain injury (34). Activated microglia produce cytotoxic molecules, including nitric oxide, oxygen radicals, arachidonic acid derivatives and cytokines (35), which induce microglial-mediated neurotoxicity and aggravate the secondary injury following TBI. Microglial activation leads to progressive and cumulative neuronal cell loss (14), thereby inducing irreversible neurological dysfunction. Therefore, in the present study, the downregulated levels of proinflammatory cytokines may have resulted from the inactivation of microglia, induced by rapamycin.
The inner mechanisms of microglial activation following treatment with various compounds have been reported in a number of studies associated with microglial activation in vitro (36–40). mTOR is key in microglial viability (40) and the process of microglial activation (39,40). The mTOR pathway is an essential cellular signaling pathway involved in a number of important physiological functions, including cell growth, proliferation, metabolism, protein synthesis and autophagy (41). Under basal conditions, the inhibition of mTOR significantly reduces microglial viability, and following activation of microglial cells using a mixture of proinflammatory cytokines, mTOR inhibition reduces microglial proliferation in a significant and dose-dependent manner (40). In addition, the mTOR pathway is important in the progression of protein synthesis. The overexpression of p70S6K increases cell size (42), whereas p70S6K-knockout mice or the inhibition of mTOR using rapamycin results in smaller cell sizes (42,43). Thus, in the present study, the activation of microglia following TBI may have resulted from cell proliferation or enlargement, induced by phosphorylation and activation of the mTOR pathway. Similar to previous studies, the present data demonstrated that the mTOR pathway was important for microglial activation, and inactivation of the mTOR pathway by rapamycin resulted in a significant decrease in the activation of microglia.
In conclusion, the present study demonstrated that administration of rapamycin following TBI resulted in a significantly decreased quantity of activated microglia in the pericontusional cortex, suggesting that the mTOR pathway is important in the process of microglial activation. However, there were certain limitations of the present study, including the limited investigation of the mechanism underlying the activation if microglia by mTOR, short-term and single time-point observationa following TBI, and the lack of investigation of the long-term effects of rapamycin injection. Therefore, the long-term consequences of rapamycin treatment following TBI requires further investigation.
Acknowledgments
The present study was supported by the Project Planning of Nanjing General Hospital (grant no. 2013067).
References
- 1.Cernak I. Animal models of head trauma. NeuroRx. 2005;2:410–422. doi: 10.1602/neurorx.2.3.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yatsiv I, Grigoriadis N, Simeonidou C, et al. Erythropoietin is neuroprotective, improves functional recovery and reduces neuronal apoptosis and inflammation in a rodent model of experimental closed head injury. FASEB J. 2005;19:1701–1703. doi: 10.1096/fj.05-3907fje. [DOI] [PubMed] [Google Scholar]
- 3.Xu J, Wang H, Ding K, et al. Inhibition of cathepsin S produces neuroprotective effects after traumatic brain injury in mice. Mediators Inflamm. 2013;2013:187873. doi: 10.1155/2013/187873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cornelius C, Crupi R, Calabrese V, et al. Traumatic brain injury: oxidative stress and neuroprotection. Antioxid Redox Signal. 2013;19:836–853. doi: 10.1089/ars.2012.4981. [DOI] [PubMed] [Google Scholar]
- 5.Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 2010;7:22–30. doi: 10.1016/j.nurt.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Venkatesan C, Chrzaszcz M, Choi N, Wainwright MS. Chronic upregulation of activated microglia immunoreactive for galectin-3/Mac-2 and nerve growth factor following diffuse axonal injury. J Neuroinflammation. 2010;7:32. doi: 10.1186/1742-2094-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao T, Thomas TC, Ziebell JM, Pauly JR, Lifshitz J. Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience. 2012;225:65–75. doi: 10.1016/j.neuroscience.2012.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelley BJ, Lifshitz J, Povlishock JT. Neuroinflammatory responses after experimental diffuse traumatic brain injury. J Neuropathol Exp Neurol. 2007;66:989–1001. doi: 10.1097/NEN.0b013e3181588245. [DOI] [PubMed] [Google Scholar]
- 9.Zhang R, Liu Y, Yan K, et al. Anti-inflammatory and immunomodulatory mechanisms of mesenchymal stem cell transplantation in experimental traumatic brain injury. J Neuroinflammation. 2013;10:106. doi: 10.1186/1742-2094-10-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ng SY, Semple BD, Morganti-Kossmann MC, Bye N. Attenuation of microglial activation with minocycline is not associated with changes in neurogenesis after focal traumatic brain injury in adult mice. J Neurotrauma. 2012;29:1410–1425. doi: 10.1089/neu.2011.2188. [DOI] [PubMed] [Google Scholar]
- 11.D'Avila JC, Lam TI, Bingham D, et al. Microglial activation induced by brain trauma is suppressed by post-injury treatment with a PARP inhibitor. J Neuroinflammation. 2012;9:31. doi: 10.1186/1742-2094-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dazert E, Hall MN. mTOR signaling in disease. Curr Opin Cell Biol. 2011;23:744–755. doi: 10.1016/j.ceb.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 13.Reynolds TH, IV, Bodine SC, Lawrence JC., Jr Control of Ser2448 phosphorylation in the mammalian target of rapamycin by insulin and skeletal muscle load. J Biol Chem. 2002;277:17657–17662. doi: 10.1074/jbc.M201142200. [DOI] [PubMed] [Google Scholar]
- 14.Chong ZZ, Shang YC, Zhang L, Wang S, Maiese K. Mammalian target of rapamycin: hitting the bull's-eye for neurological disorders. Oxid Med Cell Longev. 2010;3:374–391. doi: 10.4161/oxim.3.6.14787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park J, Zhang J, Qiu J, et al. Combination therapy targeting Akt and mammalian target of rapamycin improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2012;32:330–340. doi: 10.1038/jcbfm.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan W, Cheng K, Qin X, et al. mTORC1 and mTORC2 play different roles in the functional survival of transplanted adipose-derived stromal cells in hind limb ischemic mice via regulating inflammation in vivo. Stem Cells. 2013;31:203–214. doi: 10.1002/stem.1265. [DOI] [PubMed] [Google Scholar]
- 17.Vignot S, Faivre S, Aguirre D, Raymond E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann Oncol. 2005;16:525–537. doi: 10.1093/annonc/mdi113. [DOI] [PubMed] [Google Scholar]
- 18.Hosoi H, Dilling MB, Shikata T, et al. Rapamycin causes poorly reversible inhibition of mTOR and induces p53-independent apoptosis in human rhabdomyosarcoma cells. Cancer Res. 1999;59:886–894. [PubMed] [Google Scholar]
- 19.Erlich S, Alexandrovich A, Shohami E, Pinkas-Kramarski R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol Dis. 2007;26:86–93. doi: 10.1016/j.nbd.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 20.Flierl MA, Stahel PF, Beauchamp KM, Morgan SJ, Smith WR, Shohami E. Mouse closed head injury model induced by a weight-drop device. Nat Protoc. 2009;4:1328–1337. doi: 10.1038/nprot.2009.148. [DOI] [PubMed] [Google Scholar]
- 21.Ding K, Wang H, Wu Y, Zhang L, Xu J, Li T, Ding Y, Zhu L, He J. Rapamycin protects against apoptotic neuronal death and improves neurologic function after traumatic brain injury in mice via modulation of the mTOR-p53-Bax axis. J Surg Res. 2015;194:239–247. doi: 10.1016/j.jss.2014.09.026. [DOI] [PubMed] [Google Scholar]
- 22.Wang J, Doré S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain. 2007;130:1643–1652. doi: 10.1093/brain/awm095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhuang Z, Zhou Ml, You WC, et al. Hydrogen-rich saline alleviates early brain injury via reducing oxidative stress and brain edema following experimental subarachnoid hemorrhage in rabbits. BMC Neurosci. 2012;13:47. doi: 10.1186/1471-2202-13-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bermpohl D, You Z, Korsmeyer SJ, Moskowitz MA, Whalen MJ. Traumatic brain injury in mice deficient in Bid: effects on histopathology and functional outcome. J Cereb Blood Flow Metab. 2006;26:625–633. doi: 10.1038/sj.jcbfm.9600258. [DOI] [PubMed] [Google Scholar]
- 25.Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun. 2012;26:1191–1201. doi: 10.1016/j.bbi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 26.Lu J, Goh SJ, Tng PY, Deng YY, Ling EA, Moochhala S. Systemic inflammatory response following acute traumatic brain injury. Front Biosci (Landmark Ed) 2009;14:3795–3813. doi: 10.2741/3489. [DOI] [PubMed] [Google Scholar]
- 27.Rothwell NJ. Annual review prize lecture cytokines-killers in the brain? J Physiol. 1999;514:3–17. doi: 10.1111/j.1469-7793.1999.003af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramilo O, Saez-Llorens X, Mertsola J, et al. Tumor necrosis factor alpha/cachectin and interleukin 1 beta initiate meningeal inflammation. J Exp Med. 1990;172:497–507. doi: 10.1084/jem.172.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim KS, Wass CA, Cross AS, Opal SM. Modulation of blood-brain barrier permeability by tumor necrosis factor and antibody to tumor necrosis factor in the rat. Lymphokine Cytokine Res. 1992;11:293–298. [PubMed] [Google Scholar]
- 30.Smith RM, Giannoudis PV. Trauma and the immune response. J R Soc Med. 1998;91:417–420. doi: 10.1177/014107689809100805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hesse DG, Tracey KJ, Fong Y, et al. Cytokine appearance in human endotoxemia and primate bacteremia. Surg Gynecol Obstet. 1988;166:147–153. [PubMed] [Google Scholar]
- 32.Winter CD, Iannotti F, Pringle AK, Trikkas C, Clough GF, Church MK. A microdialysis method for the recovery of IL-1beta, IL-6 and nerve growth factor from human brain in vivo. J Neurosci Methods. 2002;119:45–50. doi: 10.1016/S0165-0270(02)00153-X. [DOI] [PubMed] [Google Scholar]
- 33.Loddick SA, Rothwell NJ. Neuroprotective effects of human recombinant interleukin-1 receptor antagonist in focal cerebral ischaemia in the rat. J Cereb Blood Flow Metab. 1996;16:932–940. doi: 10.1097/00004647-199609000-00017. [DOI] [PubMed] [Google Scholar]
- 34.Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T. Modulation of immune response by head injury. Injury. 2007;38:1392–1400. doi: 10.1016/j.injury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 35.Min KJ, Jou I, Joe E. Plasminogen-induced IL-1beta and TNF-alpha production in microglia is regulated by reactive oxygen species. Biochem Biophys Res Commun. 2003;312:969–974. doi: 10.1016/j.bbrc.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 36.Dean JM, Wang X, Kaindl AM, et al. Microglial MyD88 signaling regulates acute neuronal toxicity of LPS-stimulated microglia in vitro. Brain Behav Immun. 2010;24:776–783. doi: 10.1016/j.bbi.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 37.Pinteaux-Jones F, Sevastou IG, Fry VA, Heales S, Baker D, Pocock JM. Myelin-induced microglial neurotoxicity can be controlled by microglial metabotropic glutamate receptors. J Neurochem. 2008;106:442–454. doi: 10.1111/j.1471-4159.2008.05426.x. [DOI] [PubMed] [Google Scholar]
- 38.Pais TF, Figueiredo C, Peixoto R, Braz MH, Chatterjee S. Necrotic neurons enhance microglial neurotoxicity through induction of glutaminase by a MyD88-dependent pathway. J Neuroinflammation. 2008;5:43. doi: 10.1186/1742-2094-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dello RC, Lisi L, Feinstein DL, Navarra P. mTOR kinase, a key player in the regulation of glial functions: relevance for the therapy of multiple sclerosis. Glia. 2013;61:301–311. doi: 10.1002/glia.22433. [DOI] [PubMed] [Google Scholar]
- 40.Dello Russo C, Lisi L, Tringali G, Navarra P. Involvement of mTOR kinase in cytokine-dependent microglial activation and cell proliferation. Biochem Pharmacol. 2009;78:1242–1251. doi: 10.1016/j.bcp.2009.06.097. [DOI] [PubMed] [Google Scholar]
- 41.Wong M. Mammalian target of rapamycin (mTOR) pathways in neurological diseases. Biomed J. 2013;36:40–50. doi: 10.4103/2319-4170.110365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002;16:1472–1487. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J. 1998;17:6649–6659. doi: 10.1093/emboj/17.22.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]