

Abstract
Cyclin-dependent kinase 2 (CDK2) has been reported to be overexpressed in human colorectal cancer; it is responsible for the G1-to-S-phase transition in the cell cycle and its deregulation is a hallmark of cancer. The present study was the first to use idock, a free and open-source protein-ligand docking software developed by our group, to identify potential CDK2 inhibitors from 4,311 US Food and Drug Administration-approved small molecular drugs with a re-purposing strategy. Among the top compounds identified by idock score, nine were selected for further study. Among them, adapalene (ADA; CD271,6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphtoic acid) exhibited the highest anti-proliferative effects in LoVo and DLD1 human colon cancer cell lines. Consistent with the expected properties of CDK2 inhibitors, the present study demonstrated that ADA significantly increased the G1-phase population and decreased the expression of CDK2, cyclin E and retinoblastoma protein (Rb), as well as the phosphorylation of CDK2 (on Thr-160) and Rb (on Ser-795). Furthermore, the anti-cancer effects of ADA were examined in vivo on xenograft tumors derived from DLD1 human colorectal cancer cells subcutaneously inoculated in BALB/C nude mice. ADA (20 mg/kg orally) exhibited marked anti-tumor activity, comparable to that of oxaliplatin (40 mg/kg), and dose-dependently inhibited tumor growth (P<0.05), while combined administration of ADA and oxaliplatin produced the highest therapeutic effect. To the best of our knowledge, the present study was the first to indicate that ADA inhibits CDK2 and is a potential candidate drug for the treatment of human colorectal cancer.
Keywords: adapalene, cycling-dependent kinase 2 inhibition, colorectal carcinoma
Introduction
Cyclin-dependent kinase (CDK2) is a serine/threonine protein kinase and regulates the cell cycle transition from G1- to S-phase. It is therefore a key factor in the control of cell proliferation (1–3). Overexpression of CDK2 has been reported in numerous types of human neoplasia, including colorectal, ovarian, breast and prostate cancers (4,5). Therefore, CDK2 inhibitors have the potential to be effective anti-cancer agents. Numerous CDK2 inhibitors have been reported in the literature, including flavopiridol, roscovitine and olomoucine (6–8). However, to date, CDK2 inhibitors are not available for clinical use due to their high toxicity and low selectivity.
The present study used the free and open-source protein-ligand docking software idock (9,10) to identify US Food and Drug Administration (FDA)-approved small molecular drugs with the ability to inhibit CDK2. Among the top compounds identified by their idock score, nine were selected for further study. Among them, adapalene (ADA, CD271,6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphtoic acid) exhibited the highest anti-cancer effects in the LoVo and DLD1 human colorectal cell lines.
ADA is a third-generation synthetic retinoid. At present, it is mainly used for topical therapy of acne vulgaris (11). Its anti-proliferative and pro-apoptotic effects were first reported by Ocker et al (12) in vitro in colon carcinoma (CC-531, HT-29 and LoVo) and hepatoma (HepG2, Hep1B) cell lines; these effects were based on increasing the activity of caspase-3 via upregulating B-cell lymphoma-2 (Bcl-2)-associated X (Bax) and down-regulating Bcl-2 (12,13).
The present study assessed the effects of ADA on the viability and cell cycle of colorectal cancer cells, as well as the expression of CDK2, cyclin E and retinoblastoma protein (Rb), and the phosphorylation of CDK2 (on Thr-160) and Rb (on Ser-795). Furthermore, ADA was evaluated in vivo in a BALB/C nude mouse xenograft model using a DLD1 human colorectal cancer cells alone or in combination with oxaliplatin. As ADA is an FDA-approved drug, its clinical use is facilitated compared with that of novel drugs; therefore, its potential use as a drug for the treatment of human colorectal cancer, particularly in combination with oxaliplatin, should be further investigated.
Materials and methods
Docking
A total of 44 X-ray crystallographic structures of CDK2 in complex with a ligand were collected from the Protein Data Bank (PDB) (14). The co-crystallized ligands and water molecules were manually removed. The structures of FDA-approved drugs were collected from the Drug Bank-approved (DBAP) and FDA catalogs of the ZINC database (15,16). The DBAP catalog (version 2014-03-19) comprising 1,738 compounds and the FDA catalog (version 2012-07-25) comprising 3,176 compounds were downloaded. The 44 CDK2 structures in PDB format and the 4,914 compounds in Mol2 format were then converted into PDBQT format using AutoDockTools (17). The free and open-source docking software idock v2.1.2 (9,10) developed by our group was then applied to dock all of the 4,914 compounds onto all of the 44 CDK2 structures, and to predict their binding conformations as well as their binding affinities. Finally, the compounds were sorted in an ascending order according to their predicted binding free energy averaged across the 44 CDK2 structures, and the top nine commercially available compounds were purchased (Sigma-Aldrich, St. Louis, MO, USA) and biologically evaluated.
Chemicals and antibodies
ADA, oxaliplatin, nilotinib, LS-194959, estradiol benzoate, nandrolone phenylpropionate, vilazodone, azelastine hydrochloride, latuda and paliperidone were purchased from Sigma-Aldrich. Anti-cyclin D, -B1 and -E as well as anti-CDK2, -Rb, phosphorylated (pho)-CDK2 (Thr-160), pho-Rb (Ser-795) and GAPDH were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Cell lines and cell culture
The colorectal cancer cell lines LoVo and DLD1 were obtained from the American Type Culture Collection (Manassas, VA, USA). These cell lines were cultured in RPMI 1640 medium (GE Healthcare Life Sciences, Shanghai, China) containing 10% fetal bovine serum (FBS) (Invitrogen Life Technologies, Carlsbad, CA, USA) at 37°C in 5% CO2 and 95% humidified air. The present study was approved by the ethics committee of the Kunming Medical University (Kunming, China).
Cell culture experimental conditions
Cells were plated in 96-, 24-, or six-well plates (Corning Incorporated, Corning, NY, USA) with medium containing 0.125% FBS for 24 h and then treated with medium containing 10% FBS and the test compounds at various concentrations as indicated (1, 3, 10 and 30 µM), and incubated for 6, 12, 24, 48 or 72 h.
MTT assay
For the MTT assay (Sigma-Aldrich), cells were plated at an initial density of 9×103 cells/well in 96-well plates and incubated with 0.5 mg/ml MTT (Sigma-Aldrich) for 4 h. The medium was then discarded and 200 µl dimethyl-sulfoxide (Sigma-Aldrich) was added to dissolve the formed formazan crystals. The absorbance was measured at 570 nm with a Synergy 2 microplate reader (Bio-Tek Instruments, Inc., Winooski, VT, USA) according the standard protocol.
Cell cycle analysis
LoVo or DLD1 cells (4×104) were seeded in 24-well plates in RPMI 1640 medium containing 0.125% FBS, and cultured for 24 h. The cells were incubated in medium containing 10% FBS and various doses of ADA (1, 3, 10 or 30 µM) for 6, 12 or 24 h at 37°C, then fixed in ice-cold 70% ethanol and stained using a Coulter DNA-Prep Reagents kit (Beckman Coulter, Brea, CA, USA). Cellular DNA content of 1×104 cells from each sample was determined using an EPICS xL4 flow cytometer (Beckman Coulter). The cell cycle phase distribution was analyzed using ModFit LT 2.0 software (Verity Software House, Topsham, ME, USA). All data were obtained from two separate experiments of which each was performed in triplicate.
Western blot analysis
Cells were lysed in radioimmunoprecipitation assay buffer (Beijing Solarbio Science & Technology Co., Ltd., Beijing, China) containing 1 mM phenylmethanesulfonylfluoride (Beijing Solarbio Science & Technology Co., Ltd.) and protease inhibitor cocktail (Beijing Solarbio Science & Technology Co., Ltd.) for 30 min at 4°C. After centrifugation for 15 min at 5,668 × g, the supernatants were recovered and the protein concentration was measured using a bicinchoninic acid Protein Assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of cell lysates were resolved using 10% SDS-PAGE and transferred onto nitrocellulose membranes (Sigma-Aldrich). After blocking, the membranes were incubated sequentially with the following primary antibodies (Cell Signaling Technology, Inc.) in 5% w/v bovine serum albumin (Sigma-Aldrich), 1X Tris-buffered saline, and 0.1% Tween® 20 (MP Biomedicals, Illkirch, France) at 4°C overnight, with gentle agitation: Rabbit monoclonal anti-cyclin D1 (cat. no. 2978), rabbit monoclonal anti-cyclin B1 (cat. no. 12231), mouse monoclonal anti-cyclin E (cat. no. 4129), rabbit monoclonal anti-CDK2 (cat. no. 2546), mouse monoclonal anti-Rb (cat. no. 9313), polyclonal anti-phospho-CDK2 (cat. no. 2561), polyclonal anti-Rb (cat. no. 9301), rabbit monoclonal anti-GAPDH (cat. no. 5174; 1:1,000 dilution for all antibodies). The membranes were then incubated with the appropriate secondary antibodies, including goat anti-rabbit IgG horseradish peroxidase (HRP)-linked antibody (Cell Signalling Technology, Inc.; cat. no. 7074; 1:1,000–3,000 dilution) and horse anti-mouse IgG HRP-linked antibody (Cell Signalling Technology, Inc.; cat. no. 7076; 1:1,000–3,000 dilution). Proteins were detected using an enhanced chemiluminescence detection system (Amersham, Piscataway, NJ, USA). To ensure equal loading of the samples, the membranes were re-probed with an anti-GAPDH antibody (Cell Signalling Technology, Inc.).
Evaluation of ADA in vivo in nude mice xenografted with colorectal cancer DLD1 cells
Female BALB/C nude mice (n=50; weighing 15 g; 4–5 weeks old; Vital River Laboratory Technology Co. Ltd., Beijing, China), were housed under specific pathogen-free conditions with a 12 h light/dark cycle, in an environment containing 50–80% humidity at 15–27°C, and cared for in accordance with the guidelines of the laboratory animal ethics committee of Kunming University (Kunming, China). The cages, food, and water of the mice were sterilized. In order to establish the xenograft model, 1×106 DLD1 cells in 0.2 ml phosphate-buffered saline were injected subcutaneously into the right flank of the mice (n=3) and the tumor size was measured every day using a caliper. One week after inoculation, when the tumors grew to a volume of 80–100 m3, the mice were randomly divided into groups (5 mice/group) and gavaged daily for 21 days with 0.5% carboxymethylcellulose (CMC)-NaCl (Sigma-Aldrich) containing various doses of ADA (15, 20, 65 and 100 mg/kg) and oxaliplatin (Sigma-Aldrich; 40 mg/kg). Mice were sacrificed by cervical dislocation, tumors were excised and weighed, and their images were captured. The tumor volume was calculated using the formula V=ab2/2 (a=longest axis; b=shortest axis).
Statistical analysis
Data were obtained from at least three experiments. Values are expressed as the mean ± standard deviation. Statistical analysis was performed by Student's t-test, and the results were analyzed using SPSS 16.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference between values.
Results
Selection of candidate inhibitors of CDK2
A total of 4,914 FDA-approved drugs were extracted from the DBAP and FDA catalogs of the ZINC database. The drugs were docked onto CDK2 and ranked according to their average predicted binding affinity. Nine top-scoring compounds were selected based on their commercial availability and further examined (Table I) (18–26). The selected drugs were nilotinib, LS-194959, ADA, estradiol benzoate, nandrolone phenylpropionate, vilazodone, azelastine hydrochloride, latuda and paliperidone.
Table I.
Nine candidate cyclin-dependent kinase 2 inhibitors selected from US Food and Drug Administration-approved drugs using structure-based virtual screening by idock.
| Name | ZINC ID | idock score (kcal/mol)a | Clinical application | Ref. |
|---|---|---|---|---|
| Nilotinib | 6716957 | −10.46 | Chronic myeloid leukemia | 18 |
| LS-194959 | 3830332 | −10.43 | Food, drug additive | 19 |
| Adapalene | 3784182 | −10.38 | Acne | 20 |
| Estradiol benzoate | 3830768 | −10.23 | Estrogen | 21 |
| Nandrolone phenylpropionate | 3881613 | −10.08 | Osteoporosis | 22 |
| Vilazodone | 1542113 | −10.06 | Major depressive disorder | 23 |
| Azelastine hydrochloride | 897240 | −10.01 | Seasonal allergic rhinitis and perennial | |
| Allergic rhinitis | 24 | |||
| Latuda | 33974796 | −9.98 | Schizophrenia | 25 |
| Paliperidone | 1481956 | −9.95 | Schizophrenia | 26 |
The idock score is an estimation of binding free energy in units kcal/mol. A more negative value implies a higher predicted binding affinity.
ADA decreases the viability of LoVo and DLD1 colorectal cancer cell lines
The present study first evaluated the anti-cancer effects of the nine compounds using MTT assays. The nine compounds all decreased the viability of LoVo and DLD1 cells. The IC50-values were calculated using GraphPad Prime5 (GraphPad, Inc., La Jolla, CA, USA). Among them, ADA had the lowest IC50 (4.43 µM for DLD1 and 7.135 µM for LoVo) (Fig. 1A). The growth inhibitory effect of ADA was dose- and time-dependent (P<0.05; Fig. 1B), with marked inhibition observed at concentrations >3 µM.
Figure 1.

Comparison of the effects of nine candidate cyclin-dependent kinase 2 inhibitors on the viability of LoVo and DLD1 colorectal cancer cells. (A) As determined using an MTT assay, nine compounds inhibited the viability of LoVo and DLD1 cell lines at various concentrations, among which adapalene decreased the cell viability most markedly compared with that of the control group. (B) Adapalene dose- and time-dependently decreased the viability of LoVo and DLD1 cell lines compared with that of the control group at concentrations of >3 µM. Values are expressed as the mean ± standard deviation of at least three independent experiments. ※P<0.05 vs control group.
ADA causes cell cycle arrest in G1 phase
In order to assess whether ADA inhibited the activity of CDK2 in colorectal cancer cells, LoVo or DLD1 cells were treated with ADA (3, 10 or 30 µM) for 6, 12 or 24 h, and its effects on the cell cycle profile were assessed using flow cytometry. ADA significantly increased the G1-phase population as compared to that in the control group in a dose- and time-dependent manner (P<0.05) (Fig. 2A). After 24 h of incubation with ADA, increases in the G1-phase populations accompanied by simultaneous and significant decreases in the S- and G2/M-phase populations were apparent (Fig. 2B).
Figure 2.

Effects of adapalene on the cell cycle distribution of LoVo and DLD1 colorectal cancer cells. LoVo and DLD1 cells were treated with adapalene at various concentrations (3, 10 and 30 µM) for 6, 12 or 24 h, and cell cycle distributions were determined using flow cytometry. (A) Adapalene treatment dose- and time-dependently increased the percentage of cells in G1 phase. With 30 µM adapalene, the G1-phase population was maximal at 6–10 h, and with 10 µM adapalene, the G1-phase population increased continuously over 24 h, as compared to that of the control group. (B) Cell cycle distributions. The bar graphs indicate the G1-, G2- and S-phase populations at 24 h after adapalene treatment. Values are expressed as the mean ± standard deviation of at least three independent experiments. ※P<0.05 vs. control.
ADA treatment decreases the expression of CDK2, Rb, cyclin E, pho-CDK2 and pho-Rb, but not cyclin D and cyclin B1 in LoVo and DLD1 cells
The present study investigated the effects of ADA on the expression of significant proteins involved in G1-to-S-phase transition, including CDK2, cyclin E, Rb, pho-CDK2 and pho-Rb by western blot analysis in LoVo and DLD1 cells. As shown in Fig. 3A and B, ADA reduced the expression of CDK2, Rb, pho-CDK2, pho-Rb and cyclin E (Fig. 3). By contrast, the expression of cyclin D1 and cyclin B1 was not affected. This observed activity pattern was typical for CDK2 inhibitors (27).
Figure 3.

Effects of adapalene (3, 10 and 30 μM) on the expression of cyclins, CDK2 and Rb in LOVO and DLD1 cells. Western blot analysis showed that adapalene treatment significantly reduced the expression of CDK2, Rb, pho-CDK2, pho-Rb and cyclin E in LOVO and DLD1 cells. By contrast, the expression levels of cyclin D1 and cyclin B1 were not affected. The numbers in between the blots indicate the ratio between the indicated protein and GAPDH gray values. The blots were representative of experiments repeated three times. CDK, cyclin-dependent kinase; pho-Rb, phosphorylated retinoblastoma protein.
Daily oral ADA administration reduces the growth of DLD1 cell-derived xenograft tumors in BALB/C nude mice in vivo
In order to assess the inhibitory potential of adapalene on the growth of colorectal carcinoma in vivo, DLD1 cell-derived xenograft tumors were established in BALB/C nude mice. Carcinoma volumes were measured every 3–4 days after the appearance of the tumors. At 7 days after tumor inoculation, the volume of the tumors reached 80–100 mm3, and animals were administered various doses of ADA (15, 65 or 100 mg/kg in 0.5% CMC-NaCl) daily for 21 days by oral gavage. In a separate experiment, the efficacy of ADA (20 mg/kg), oxaliplatin (40 mg/kg) and the combination of ADA (20 mg/kg) plus oxaliplatin (40 mg/kg) was compared.
The results showed that oral administration of ADA significantly inhibited tumor growth (P<0.05). Following 21 days of treatment a dose of ADA of as low as 15 mg/kg achieved a significant reduction in tumor weight and volume as compared with that in the control (P<0.05) (Fig. 4A–C). There was no significant difference between 15 and 65 mg/kg ADA treatment.
Figure 4.
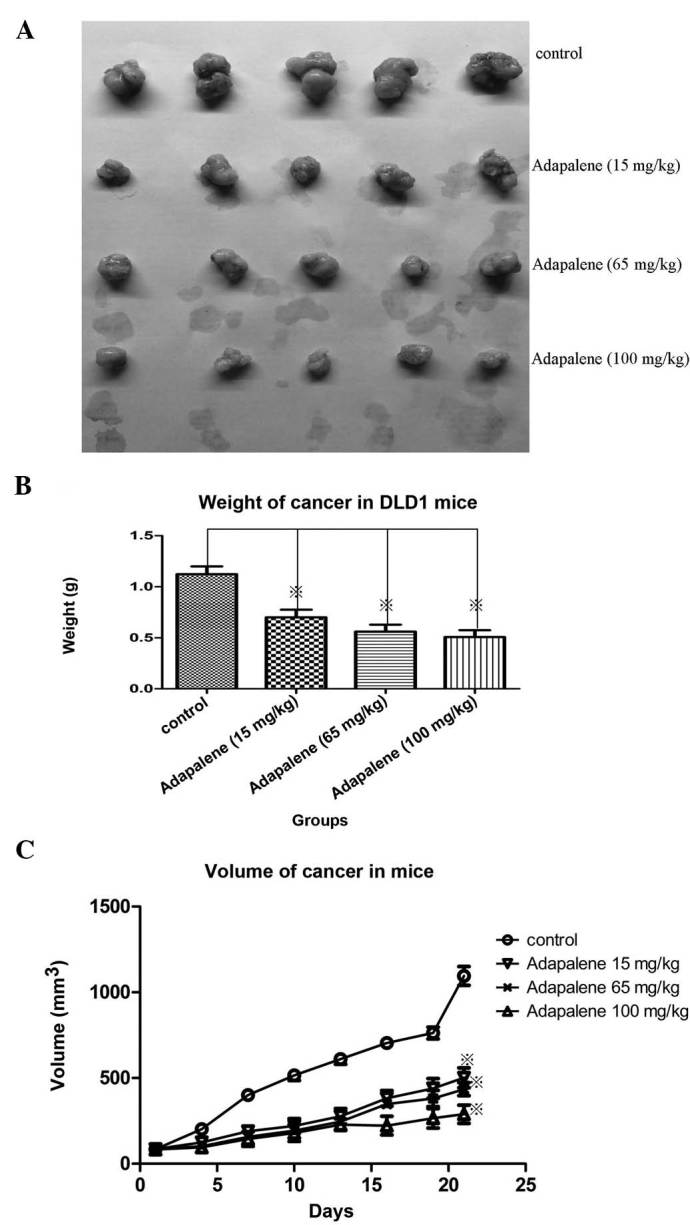
Oral administration of adapalene significantly reduced tumor growth in vivo in nude mice xenografted with DLD1 cells. (A) Daily adapalene treatment (from days 1–21) dose-dependently (15, 65 and 100 mg/kg) reduced the tumor volume. (B) Significant reductions in tumor weight were observed following treatment with adapalene for 21 days at concentrations as low as 15 mg/kg compared with the control group. (C) Increases in tumor volume were significantly decreased following adapalene treatment at 15–100 mg/kg for 21 days compared with the control group. Values are expressed as the mean ± standard deviation of at least three independent experiments. ※P<0.05 vs. control.
In addition, the potency of of ADA (20 mg/kg) was similar to that of oxaliplatin (40 mg/kg). Of note, combined administration produced the highest therapeutic effect (Fig. 5A–C). To the best of our knowledge, the present study was the first to demonstrate the anti-cancer activity of ADA in vivo, which may be a suitable CDK2-targeting drug for the treatment of human colorectal cancer.
Figure 5.
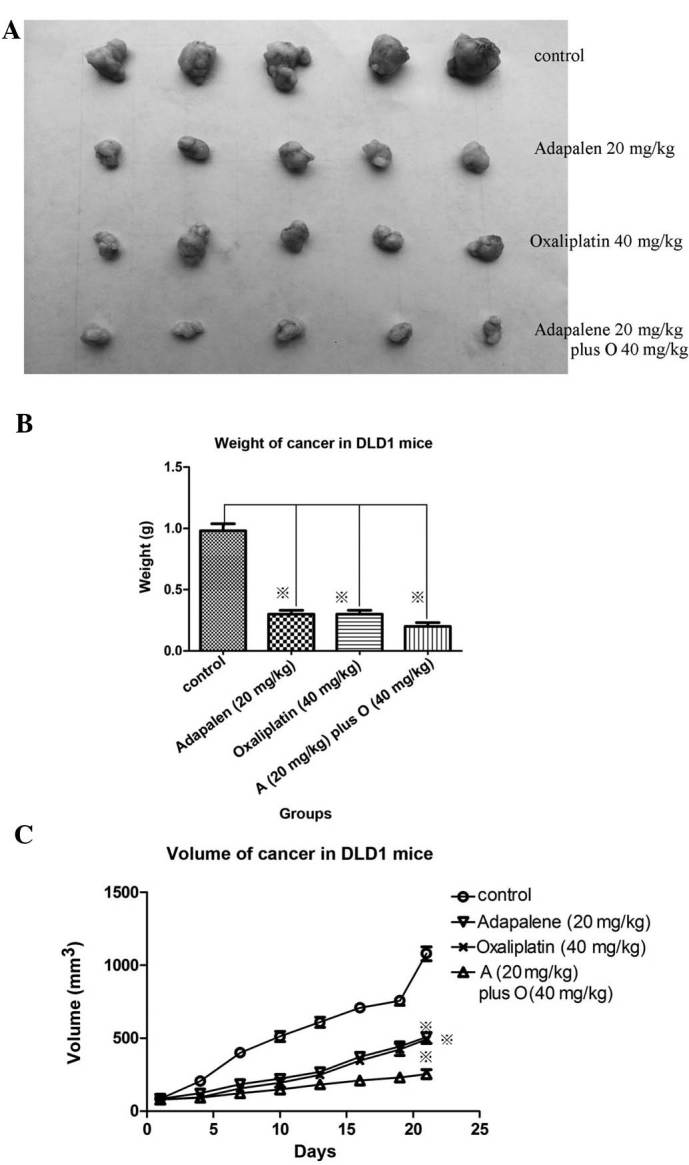
Oral administration of adapalene combined with oxaliplatin significantly reduced tumor growth in vivo in nude mice xenografted with DLD1 cells. (A) Daily administration of adapalene (20 mg/kg), (oxaliplatin (40 mg/kg) or their combined treatment (from days 1–21) reduced the tumor volume. (B) A significant reduction in tumor weight was observed following combined adapalene and oxaliplatin treatment for 21 days compared with that in the control group. (C) The tumor volume significantly decreased following combined adapalene and oxaliplatin treatment for 21 days compared with that in the groups treated with either adapalene or oxaliplatin alone and that in the control group. Values are expressed as the mean ± standard deviation of at least three independent experiments ※P<0.05 vs. control. O, oxaliplatin.
Structural analysis of the predicted conformation of ADA docked onto CDK2
Fig. 6A illustrates the conformation of CDK2 in complex with ADA in a three-dimensional manner predicted by iview (28). Fig. 6B shows the intermolecular interaction diagram in a two-dimensional manner using PoseView (29). ADA was predicted to reside in the adenosine triphosphate-binding site of CDK2 and interact with CDK2 mainly through hydrophobic contacts with Phe82, Ile10, Leu134, Lys33 and His84.
Figure 6.
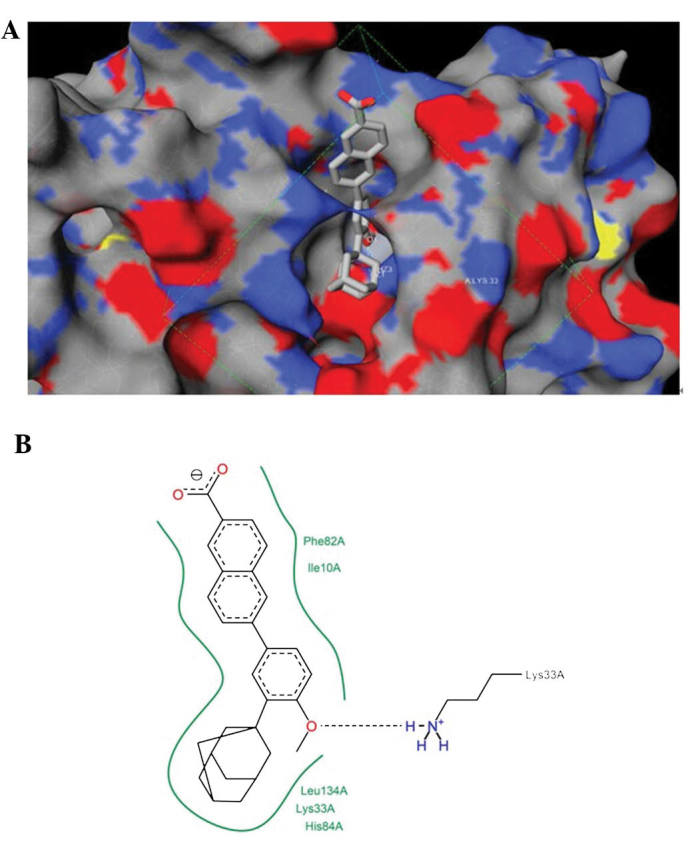
Docking of adapalene on CDK2. (A) The predicted conformation of CDK2 in complex with adapalene was visualized in a three-dimensional manner using iview 27. The blue, red and yellow colors represent the nitrogen atom, oxygen atom and sulphur atom, respectively. (B) The intermolecular interaction diagram of adapalene with amino acids of CDK2 was illustrated in a two-dimensional manner using PoseView28. Adapalene was predicted to reside in the adenosine triphosphate-binding site of CDK2 and interact with CDK2 mainly through hydrophobic contacts with Phe82, Ile10, Leu134, Lys33 and His84. CDK2, cyclin-dependent kinase 2.
Discussion
The present study adopted the computational methodology of structure-based virtual screening (SBVS) by protein-ligand docking to shortlist candidates from FDA-approved small molecular drugs. SBVS has become a routine task in pharmaceutical institutions.
CDK2 is an important target for cancer therapy. Through the interaction of CDKs and cyclins, cell cycle progression is regulated in a sequential and highly organized manner (30). Various cyclin/CDK complexes are activated at different stages of the cell cycle (31–33). During G1-to-S-phase transition, the cyclinD1-CDK4/6 and cyclinE-CDK2 complexes are sequentially activated and Rb is hyperphosphorylated on serine and threonine residues (34,35). Upon hyperphosphorylation, Rb stimulates the release of E2F transcription factors, which in turn facilitates the transcription of numerous genes required for G1-to-S transition and S-phase progression (36). A large number of CDK2 inhibitors have been reported by previous studies (Table II) (6–8,37–45). However, to date, due to drug toxicity and low selectivity, these compounds are currently not available for clinical use.
Table II.
Cyclin-dependent kinase 2 inhibitors in the literature.
| Name | Research institution | Ref. |
|---|---|---|
| AG-24322 | Agouron | 36 |
| AT-7519 | Astex | 37 |
| AT-9311 | Astex | 38 |
| AZD-5438 | AstraZeneca | 39 |
| AZD-5597 | AstraZeneca | 40,41 |
| Compound 6b | Palacký University | 42 |
| SCH-727965 | Schering-Plough | 43,44 |
| Flavopiridol | Sanofi-Aventis | 6 |
| Roscovitine | Emory University and Imperial College | 7 |
| Olomoucine | Laboratoire de PhysiologieVégétaleMoléculaire CNRS | 8 |
The present study was the first to perform a successful prospective application of idock (9,10) in identifying CDK2 inhibitors using a re-purposing strategy. idock is a novel and promising software developed by our group, which is free and open source under a permissive license, and which has been demonstrated to outperform the state-of-the-art docking software AutoDock Vina (46) in terms of docking speed by 8.69–37.51 times, while maintaining comparable re-docking success rates (10). Due to its free availability, users from industry as well as academia can utilize idock for protein-ligand docking projects.
idock was designed to be user-friendly by featuring input terms and output results similar to those of AutoDock Vina, allowing existing users to easily adapt to using idock and benefit from the considerably increased speed of the SBVS performance. In addition, to facilitate prospective SBVS by idock, a web server named istar (10) was developed by our group. istar is freely available at http://istar.cse.cuhk.edu.hk and contains 23,129,083 purchasable small molecular compounds ready for docking against any protein provided by the user. Therefore, idock (9) and istar (10) are able to supplement the efforts of medicinal chemists in drug discovery research.
In the present study, ADA was selected out of nine compounds for in-depth study, as its IC50-value was <10 µmol/l according to an MTT assay. At present, ADA is used for topical application for the treatment of acne vulgaris (11). ADA has previously been shown to have anti-proliferative and pro-apoptotic effects in colon carcinoma (CC-531, HT-29 and LoVo) and hepatoma (HepG2, Hep1B) cell lines by increasing the activity of caspase-3 via upregulating Bax and decreasing levels of Bcl2 (12,13). To the best of our knowledge, the present study was the first to report that ADA inhibits CDK2, and that oral administration of ADA (20 mg/kg) exhibited significant anti-cancer efficacy, which was comparable to that of the clinically used anti-cancer drug oxaliplatin (40 mg/kg) DLD1 cell-derived xenograft tumors in nude mice in vivo. Of note, the combination of an effective dose of ADA and oxaliplatin enhanced their therapeutic effects, suggesting that ADA, which has a different mechanism of action from that of oxaliplatin, may be combined with other chemotherapeutic drugs to maximize therapeutic effects.
A previous study indicated that ADA had no obvious toxic effects following either intraperitoneal (i.p.) injection at 100 mg/kg in rats with carrageenan-induced paw oedema or topical use at 10% (10 mg/ml) on guinea pigs with ultraviolet light-induced erythema (47). The present study did not observe any significant changes in the body weight of the nude mice orally administered with ADA (15–100 mg/kg) over 21 days. All of these results suggested that oral administration or i.p. injection of ADA is relatively safe. Tashiro et al (48) reported that i.p. injection of 5 and 10 mg/kg oxaliplatin on day 2 in B6D2F mice subcutaneously xenografted with colon 38 carcinoma cells significantly reduced the tumor weight to 16 and 38%, respectively, of that in the control group following 21 days of treatment. In the present study oxaliplatin was administered by oral gavage doses of 10, 20 and 40 mg/kg, which significantly reduced the tumor weight to 28% of that in the control group on following 21 days of treatment without showing any changes in body weight, suggesting that oral administration of oxaliplatin is relatively safe and effective.
In recent years, a large number of CDK inhibitors have been reported by previous studies. However, due to high toxicity and low selectivity, they have not been made available for clinical use. As an FDA-approved drug, ADA alone or in combination with other chemotherapeutic drugs may be suitable for the treatment of colorectal neoplasms and other cancer types, which should be further evaluated in future studies.
Acknowledgments
The present study was supported by grants from the Hsiang-fu Kung academician workstation of Kunming Medical University, (no. NSFC 81272549), the Key Lab project of Shenzhen of the P.R. China (no. ZDSY 20130329101130496), the Direct Grant from the Chinese University of Hong Kong and the GRF Grant from the Research Grants Council of Hong Kong SAR (project nos. 414413, 772910 and 470911).
References
- 1.Morgan DO. Cyclin-depend kinases: engines, clocks, and microprocessors. Ann Rev Cell Dev Biol. 1997;13:261–291. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- 2.Sherr CJ, Roberts JM. CDK inhibitor: Positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 3.Murray AW. Recycling the cell cycle: Cyclins revisited. Cell. 2004;116:221–234. doi: 10.1016/S0092-8674(03)01080-8. [DOI] [PubMed] [Google Scholar]
- 4.Carnero A. Targeting the cell cycle for cancer therapy. Br J Cancer. 2002;87:129–133. doi: 10.1038/sj.bjc.6600458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Webster KR, Kimball D. Novel drugs targeting the cell cycle. Emerging Drugs. 2000;5:45–59. doi: 10.1517/14728214.5.1.45. [DOI] [Google Scholar]
- 6.Senderowicz AM. Flavopiridol: The first cyclin-dependent kinase inhibitor in human clinical trials. Invest New Drugs. 1999;17:313–320. doi: 10.1023/A:1006353008903. [DOI] [PubMed] [Google Scholar]
- 7.De Azevedo WF, Leclerc S, Meijer L, Havlicek L, Strnad M, Kim SH. Inhibition of cyclin-dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine. Eur J Biochem. 1997;243:518–526. doi: 10.1111/j.1432-1033.1997.0518a.x. [DOI] [PubMed] [Google Scholar]
- 8.Glab N, Labidi B, Qin LX, Trehin C, Bergounioux C, Meijer L. Olomoucine, an inhibitor of the cdc2/cdk2 kinases activity, blocks plant cells at the G1 to S and G2 to M cell cycle transitions. FEBS Lett. 1994;353:207–211. doi: 10.1016/0014-5793(94)01035-8. [DOI] [PubMed] [Google Scholar]
- 9.Li H, Leung KS, Wong MH. idock: A multithreaded virtual screening tool for flexible ligand docking; Proceedings of the 2012 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB); San Diego. 2012. pp. 77–84. [Google Scholar]
- 10.Li H, Leung KS, Ballester PJ, Wong MH. Istar: A web platform for large-scale protein-ligand docking. PLoS One. 2014;9:e85678. doi: 10.1371/journal.pone.0085678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Millikan LE. Adapalene: An update on newer comparative studies between the various retinoids. Int J Dermatol. 2000;39:784–788. doi: 10.1046/j.1365-4362.2000.00050.x. [DOI] [PubMed] [Google Scholar]
- 12.Ocker M, Herold C, Ganslmayer M, Hahn EG, Schuppan D. The synthetic retinoid adapalene inhibites proliferation and induces apoptosis in colorectal cancer cells in vitro. Int J Cancer. 2003;107:453–459. doi: 10.1002/ijc.11410. [DOI] [PubMed] [Google Scholar]
- 13.Ocker M, Herold C, Ganslmayer M, Zopf S, Hahn EG, Schuppan D. Potentiated anticancer effects on hepatoma cells by the retinoid adapalene. Cancer Lett. 2004;208:51–58. doi: 10.1016/j.canlet.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 14.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irwin JJ, Shoichet BK. ZINC-a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: A free tool to discover chemistry for biology. J Chem Inf Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weisberg E, Manley PW, Breitenstein W, Brüggen J, Cowan-Jacob SW, Ray A, Huntly B, Fabbro D, Fendrich G, Hall-Meyers E, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129–141. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 19.Abrahart EN. Dyes and their intermediates. Pergamon Press; Oxford: 1968. [Google Scholar]
- 20.Bernard BA. Adapalene, a newchemical entity with retinoid activity. Skin Pharmacol. 1993;6(Suppl 1):S61–S69. doi: 10.1159/000211165. [DOI] [PubMed] [Google Scholar]
- 21.Fremont-Smith M, Meigs JV, Graham RM, Gilbert HH. Cancer of endometrium and prolonged estrogen therapy. J Am Med Assoc. 1946;131:805–808. doi: 10.1001/jama.1946.02870270005002. [DOI] [PubMed] [Google Scholar]
- 22.Overbeek GA, de Visser. A comparison of the myotrophic and androgenic activities of the phenylpropionates and decanoates of testosterone and nandrolone. Acta Endocrinol (Copenh) 1961;38:285–292. doi: 10.1530/acta.0.0380285. [DOI] [PubMed] [Google Scholar]
- 23.Bartoszyk GD, Hegenbart R, Ziegler H. EMD 68843, a serotonin reuptake inhibitor with selective presynaptic 5-HT1A receptor agonistic properties. Eur J Pharmacol. 1997;322:147–153. doi: 10.1016/S0014-2999(96)00999-5. [DOI] [PubMed] [Google Scholar]
- 24.Tasaka K, Akagi M. Anti-allergic properties of a new histamine antagonist, 4-(p-chlorobenzyl)-2-[N-methyl-perhy-droazepinyl-(4)]-1-(2H)-phthalazinone hydrochloride (azelastine) Arzneimittelforschung. 1079;29:488–493. [PubMed] [Google Scholar]
- 25.Ishiyama T, Tokuda K, Ishibashi T, Ito A, Toma S, Ohno Y. Lurasidone (SM-13496), a novel atypical antipsychotic drug, reverses MK-801-induced impairment of learning and memory in the rat passive-avoidance test. Eur J Pharmacol. 2007;572:160–170. doi: 10.1016/j.ejphar.2007.06.058. [DOI] [PubMed] [Google Scholar]
- 26.Kramer M, Simpson G, Maciulis V, Kushner S, Vijapurkar U, Lim P, Eerdekens M. Paliperidone extended-release tablets for prevention of symptom recurrence in patients with schizophrenia: a randomized, double-blind, placebo-controlled study. J Clin Psychopharmacol. 2007;27:6–14. doi: 10.1097/JCP.0b013e31802dda4a. [DOI] [PubMed] [Google Scholar]
- 27.Dolman ME, Poon E, Ebus ME, den Hartog IJ, van Noesel CJ, Jamin Y, Hallsworth A, Robinson SP, Petrie K, Sparidans RW, et al. Cyclin-dependent kinase inhibitor AT7519 as a potential drug for MYCN-dependent neuroblastoma. Clin Cancer Res. 2015 doi: 10.1158/1078-0432.CCR-15-0313. pii: clincanres.0313.2015 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Leung KS, Nakane T, Wong MH. iview: An interactive WebGL visualizer for protein-ligand complex. BMC Bioinformatics. 2014;15:56. doi: 10.1186/1471-2105-15-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stierand K, Rarey M. PoseView-molecular interaction patterns at a glance. J Cheminform. 2010;2(Suppl 1):P50. doi: 10.1186/1758-2946-2-S1-P50. [DOI] [Google Scholar]
- 30.Pines J. Four-dimensional control of the cell cycle. Nat Cell Biol. 1999;1:E73–E79. doi: 10.1038/11041. [DOI] [PubMed] [Google Scholar]
- 31.Furuno N, den Elzen N, Pines J. Human cyclin A is required for mitosis until mid prophase. J Cell Biol. 1999;147:295–306. doi: 10.1083/jcb.147.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Juan G, Cordon-Cardo C. Intranuclear compartmentalization of cyclin E during the cell cycle: Disruption of the nucleoplasm-nucleolar shuttling of cyclin E in bladder cancer. Cancer Res. 2001;61:1220–1226. [PubMed] [Google Scholar]
- 33.Ohi R, Gould KL. Regulating the onset of mitosis. Curr Opin Cell Biol. 1999;11:267–273. doi: 10.1016/S0955-0674(99)80036-2. [DOI] [PubMed] [Google Scholar]
- 34.Reed SI. Control of the G1/S transition. Cancer Surv. 1997;29:7–23. [PubMed] [Google Scholar]
- 35.Cobrinik D. Pocket proteins and cell cycle control. Oncogene. 2005;24:2796–2809. doi: 10.1038/sj.onc.1208619. [DOI] [PubMed] [Google Scholar]
- 36.Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA. Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nat Cell Biol. 1999;1:88–93. doi: 10.1038/10054. [DOI] [PubMed] [Google Scholar]
- 37.Agouron 3,5-Disubstituted indazole compounds. Pharmaceutical compositions, or method for mediating or inhibiting cell proliferation. 0142345. US. 2006
- 38.Astex Preparation of 3,4-disubstituted 1 h-pyrazole compounds and their use as cyclin dependent kinases (cdk) and glycogen synthase kinase-3 (gsk-3) modulators. 012256. WO. 2005
- 39.Astex Preparation of 3,4-disubstituted pyrazoles as inhibitors of cyclin dependent kinases (CDK), Aurora A kinase or glycogen synthase kinase 3 (GSK3) 003440. WO. 2006
- 40.Boss DS, Schwartz GK, Middleton MR, Amakye DD, Swaisland H, Midgley RS, Ranson M, Danson S, Calvert H, Plummer R, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of the oral cyclin-dependent kinase inhibitor AZD5438 when administered at intermittent and continuous dosing schedules in patients with advanced solid tumours. Ann Oncol. 2010;21:884–894. doi: 10.1093/annonc/mdp377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones CD, Andrews DM, Barker AJ, Blades K, Byth KF, Finlay MR, Geh C, Green CP, Johannsen M, Walker M, Weir HM. Imidazole pyrimidine amides as potent, orally bioavailable cyclin-dependent kinase inhibitors. Bioorg Med Chem Lett. 2008;18:6486–6489. doi: 10.1016/j.bmcl.2008.10.075. [DOI] [PubMed] [Google Scholar]
- 42.Jones CD, Andrews DM, Barker AJ, Blades K, Daunt P, East S, Geh C, Graham MA, Johnson KM, Loddick SA, et al. The discovery of AZD5597, a potent imidazole pyrimidine amide CDK inhibitor suitable for intravenous dosing. Bioorg Med Chem Lett. 2008;18:6369–6373. doi: 10.1016/j.bmcl.2008.10.102. [DOI] [PubMed] [Google Scholar]
- 43.Gucký T, Jorda R, Zatloukal M, Bazgier V, Berka K, Řezníčková E, Béres T, Strnad M, Kryštof V. A novel series of highly potent 2,6,9-trisubstituted purine cyclin-dependent kinase inhibitors. J Med Chem. 2013;56:6234–6247. doi: 10.1021/jm4006884. [DOI] [PubMed] [Google Scholar]
- 44.Shapiro GI, Bannerji R, Small K, et al. A phase I dose-escalation study of the safety, pharmacokinetics (PK) and pharmacodynamics (PD) of the novel cyclin-dependent kinase inhibitor SCH 727965 administered every 3 weeks in subjects with advanced malignancies. J Clin Oncol (Meeting Abstracts) 2008;26:3532. [Google Scholar]
- 45.Nemunaitis J, Saltzman M, Rosenberg MA, et al. A phase I dose-escalation study of the safety, pharmacokinetics (PK) and pharmacodynamics (PD) of SCH 727965, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies. J Clin Oncol (Meeting Abstracts) 2009;27:3535. [Google Scholar]
- 46.Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hensby C, Cavey D, Bouclier M, Chatelus A, Algate D, Eustache J, Shroot B. The in vivo and in vitro anti-inflammatory activity of CD271: A new retinoid-like modulator of cell differentiation. Agents Actions. 1990;29:56–58. doi: 10.1007/BF01964720. [DOI] [PubMed] [Google Scholar]
- 48.Tashiro T, Kawada Y, Samurai Y, Kidan Y. Antitumor activity of a new platinum complex, Oxalato(trans-l-1,2-diaminocyclohexane) platinum(II): New experimental data. Bioed Pharmacother. 1989;43:251–260. doi: 10.1016/0753-3322(89)90004-8. [DOI] [PubMed] [Google Scholar]