

Abstract
CLN3 is a recently identified anti-apoptotic gene, which has been demonstrated to be highly expressed in a diverse range of cancer cell lines, including ovarian cancer. In the present study, RNA interference, mediated by a lentivirus expressing CLN3 short hairpin RNA (shRNA) was utilized to knockdown the expression of CLN3 in the A2780 human ovarian cancer cell line, and its cisplatin-resistant and carboplatin-resistant sublines, A2780/DDP and A2780/CBP cells. It was revealed that the mRNA and protein expression levels of CLN3 were significantly reduced in the CLN3-specific shRNA-transduced cells, compared with the untransduced and control shRNA-transduced cells. In addition, specific knockdown of CLN3 in these cells inhibited cell proliferation and led to cell cycle arrest at the G0/G1 phase, with eventual apoptosis. CLN3 knockdown caused increases in the levels of Bax, FAX, cleaved-caspase 3, cleaved-caspase 8 and cleaved-RARP, but decreased the level of Bcl-2. Finally, it was observed that CLN3 depletion markedly reduced the half maximum inhibitory concentration in the A2780/DDP and A2780/CBP cells. Taken together, these data suggested that CLN3 is involved in tumorigenesis and drug resistance in ovarian cancer, and may serve as a promising therapeutic target for its treatment.
Keywords: CLN3, ovarian cancer, RNA interference, proliferation, apoptosis, chemosensitivity
Introduction
Ovarian cancer is the most life-threatening gynecologic malignancy, accounting for a mortality rate of ~140,200 in 2008 worldwide (1). Despite significant advances in surgical techniques and chemotherapy regimens, the 5-year survival rate for patients with the advanced disease is ~13%, primarily due to recurrence and drug resistance (2). In general, the majority of patients with ovarian cancer initially respond to surgery and standard chemotherapy; however, the majority ultimately develop recurrence, and resistance to chemotherapeutic drugs is frequently observed in recurrent disease (3). Therefore, understanding the molecular mechanisms involved in chemotherapy resistance is urgently required to develop novel therapeutic strategies and improve clinical outcomes of this life-threatening malignancy.
CLN3 is a recently identified defective gene in juvenile Batten disease, an inherited neurodegenerative disease of childhood resulting from accelerated apoptotic death of photoreceptors and neurons (4). The CLN3 gene encodes a hydrophobic transmembrane protein, which is involved in intracellular trafficking and regulation in neuronal and non-neuronal cells (5–8). Previous studies have indicated that CLN3 has anti-apoptotic properties in NT2 neuronal precursor cells and cancer cells (9,10). In particular, Rylova et al reported that CLN3 mRNA and protein are abundantly expressed in various cancer cell lines, including glioblastoma, neuroblastoma, and prostate, ovarian, breast and colon cancer (11,12). In addition, a series of functional investigations have revealed that the knockdown of CLN3 by RNA interference (RNAi) inhibits the proliferation and/or induces apoptosis in several cancer cells (11,12). CLN3 has, therefore, been indicated as a potential molecular target for future cancer drug discovery (11). However, the potential role of CLN3 in ovarian cancer remains to be fully elucidated.
In the present study, an RNAi-based approach, specifically targeting CLN3 mRNA, was used to investigate the effects of CLN3 knockdown on the proliferation, apoptosis and chemosensitivity of the A2780 human ovarian cancer cell line, and cells of its cisplatin-resistant (A2780/DDP) and carboplatin-resistant (A2780/CBP) sublines.
Materials and methods
Cell culture
Human ovarian carcinoma SW626, OVCaR-3 and SK-OV-3 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; GE Healthcare, Logan, UT, USA). Human ovarian carcinoma HO-8910, COC1, HO-8910PM cells were grown in RPMI 1640 medium (Gibco Life Technologies) containing 10% fetal calf serum (GE Healthcare). Human ovarian carcinoma ES-2 cells were maintained in McCoy's 5A medium (Gibco Life Technologies) containing 10% FBS. All cells were cultured at 37°C in a 5% CO2 atmosphere. The A2780 human ovarian carcinoma cell line, and its cisplatin-resistant (A2780/DDP) and carboplatin-resistant (A2780/CBP) sub-lines were obtained from KeyGen Biotech, Co., Ltd. (Nanjing, China) and cultured in DMEM, containing 10% FBS, at 37°C in a 5% CO2 atmosphere. The resistant sub-lines were established by exposure of the A2780 cells to stepwise increasing cisplatin or carboplatin (Qilu Pharmaceutical Co., Ltd., Jinan, China) concentrations, respectively. To ensure maintenance of the resistant phenotype, the culture medium of the A2780/DDP and A2780/CBP cells were treated with 1 mg/l cisplatin and 0.1 µmol/l carboplatin, respectively. The cisplatin and carboplatin were removed from the growth medium 2 weeks prior to all subsequent experiments. Normal ovarian tissues were provided by the Department of Gynaecology and Obstetrics, The Fourth Affiliated Hospital of Harbin Medical University (Harbin, China) and the Department of Gynaecology, The Second Affiliated Hospital of Harbin Medical University (Harbin, China) following surgery or a laparoscopy. All patients provided written informed consent for the use of their tissues, and the use of tissue specimens was approved by the Ethics Committee of Harbin Medical University. Following primary culture, the cells were grown in DMEM supplemented with 10% FBS.
Construction and transfection of the CLN3 shRNA lentiviral vector
Oligonucleotides corresponding to CLN3 mRNA or control mRNA were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China) as follows: CLN3, sense 5′-CCU UGG UCG UAG UUU ACU UTT-3′ and antisense 5′-AAG UAA ACU ACG ACC AAG GTT-3′; and control, sense 5′-UUC UCC GAA CGU GUC ACG UTT-3 and antisense 5′-ACG UGA CAC GUU CGG AGA ATT-3′. The annealed double-stranded oligonucleotides were cloned into the pGCSIL-green fluorescent protein (GFP) viral vector. The recombinant plasmid was verified using PCR and DNA sequencing. Recombinant lentiviral particles were prepared via the co-transfection of subconfluent 293T cells with the modified pGCSIL-GFP viral vector and the pHelper 1.0 and pHelper 2.0 helper plasmids using the Lipofectamine™ 2000 reagent (Invitrogen Life Technologies, Carlsbad, CA, USA). For lentiviral transfection, the cells were seeded at a density of 5×104 into six-well plates and transduced with the modified pGCSIL-GFP (KeyGen Biotech Co., Ltd.) viral vector when they reached 30–40% confluence. At 3 days post-transfection, the expression of GFP was examined under a fluorescence microscope, and the cells were harvested for the subsequent assays.
Reverse transcription-quantitative polymerase chain reaction (RT-q)PCR
Total RNA was extracted from the cultured cells using TRIzol reagent (Invitrogen Life Technologies) and reverse-transcribed to synthesize cDNA using a Maxima® First Strand cDNA Synthesis kit (Thermo Fisher Scientific, Rockford, IL, USA), according to the manufacturers' instructions. Subsequently, qPCR reactions were performed using SYBR Green Real-time PCR Master Mix (Toboyo, Co., Ltd., Osaka, Japan) with 2 µg total RNA on an Applied Biosystems Step One Plus Real-Time PCR system (Applied Biosystems Life Technologies, Foster City, CA, USA). Following 15 sec at 95°C to activate DNA polymerase, the cycling parameters were as follows: 40 cycles of denaturation at 95°C for 5 sec, and annealing and extension at 60°C for 30 sec. The relative mRNA expression levels were calculated from the threshold cycle (Ct) value of each PCR product and normalized to β-actin using the comparative Ct method. The primer sets used for qPCR were as follows: CLN3, forward 5-′GGTGGACAGTATTCAAGGG-3′ and reverse: 5′-CTTGGCAGAAAGACGAAC-3′; Bcl-2, forward 5′-ACGACTTCTCCCGCCGCTAC-3′ and reverse 5′-CTGAAGAGCTCCTCCACCAC-3′; caspase-3, forward 5′-CATGGAAGCGAATCAATGGACT-3′ and reverse 5′-CTGTACCAGACCGAGATGTCA-3′; caspase-8, forward 5′-CTCCAAATGCAAACTGGATG-3′ and reverse 5′-TGTTGATTTGGGCACAGACT-3′; and β-actin, forward 5′-GGCGGCACCACCATGTACCCT-3′ and reverse 5′-AGG GGC CGG ACT CGT CAT ACT-3′. Primers were synthesized by Sangon Biotech Co., Ltd.
Western blot analysis
The cells were washed twice with ice-cold phosphate-buffered saline (PBS) and lysed in lysis buffer (KeyGen Biotech Co., Ltd.) for 30 min. Following centrifugation at 8,000 × g for 5 min at 4°C, the soluble proteins were boiled at 95–100°C for 5 min, separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (KeyGen Biotech Co., Ltd.) at a 70 V constant voltage, and transferred electrophoretically onto polyvinylidene difluoride membranes (EMD Millipore, Bedford, MA, USA). The membranes were blocked with 5% non-fat milk at room temperature for 1.5 h and immunoblotted at 37°C for 1.5 h with the following antibodies: Polyclonal goat anti-human CLN3 (cat. no. sc-49627; 1:200; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), poly-clonal mouse anti-human cleaved caspase-8 (cat. no. sc-81656; 1:200; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), polyclonal rabbit anti-human Bcl-2 (cat. no. KG22169; 1:500; KeyGen Biotech Co., Ltd.), polyclonal rabbit anti-human Bax (cat. no. KG22165; 1:500; KeyGen Biotech Co., Ltd.), polyclonal rabbit anti-human cleaved PARP (cat. no. YC0073; 1:1,000; ImmunoWay Biotechnology, Newark, DE, USA) and poly-clonal rabbit anti-human cleaved caspase-3 (cat. no. KG22205; 1:200; KeyGen Biotech Co., Ltd.), followed by incubation with horseradish peroxidase-linked secondary antibody (1:10,000; KeyGen Biotech Co., Ltd.) at 37°C for 40 min. Finally, the protein bands were visualized using an enhanced chemiluminescence detection system (KeyGen Biotech Co., Ltd.).
Cell proliferation assay
Cell proliferation was analyzed using a Cell Counting Kit-8 (CCK-8; KeyGen Biotech Co., Ltd.), according to the manufacturer014's instructions. Briefly, the cells were seeded into 96-well plates at a density of 2×102 cells/well and cultured for 0, 12, 24, 48 or 72 h. Subsequently, 10 µl CCK-8 reagent was added to each well, and the cells were incubated at 37°C for 2 h. Finally, the absorbance at 450 nm was determined using a microplate reader (Bio-Rad Laboratories, Inc., San Diego, CA, USA).
Apoptosis and cell cycle analysis using flow cytometry
For cell cycle analysis, the cells were washed twice with PBS, fixed overnight in 70% ethanol, and stained with propidium iodide (PI; KeyGen Biotech Co., Ltd.) in the presence of RNase A (Takara Biotechnology Co., Ltd., Dalian, China) at 4°C for 30 min. The DNA contents were determined using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA).
The number of apoptotic cells were quantified by flow cytometry using an Annexin V-FITC/PI Apoptosis Detection kit (KeyGen Biotech Co., Ltd.). In brief, the cells were washed with PBS and incubated with 500 µl binding buffer (KeyGen Biotech Co., Ltd.). Subsequently, 5 µl fluorescein isothiocya-nate (FITC)-labeled Annexin V and 5 µl PI were added, and the samples were incubated at room temperature for 5–15 min in the dark. The percentage of apoptotic cells was determined using flow cytometry.
Cytotoxicity assay
In vitro cytotoxicity was evaluated using a CCK-8 assay. Briefly, the cells were trypsinized and plated in 96-well plates at a density of 2×102 cells/well. Following treatment with different concentrations of cisplatin or carboplatin, the CCK-8 assay was performed, as described above. The half maximum inhibitory concentration (IC50) values were calculated by linear interpolation with SPSS 19.0 software (SPSS Inc., Chicago, IL, USA).
Statistical analysis
The results are expressed as the mean ± standard deviation, and raw data were analyzed using Student's t-test with SPSS 19.0 software. P<0.05 was considered to indicate a statistically significant difference.
Results
Overexpression of CLN3 in ovarian cancer cells
Firstly, the mRNA levels of CLN3 in normal ovarian cells and A2780, COC1, ES-2, HO-8910, HO-8910PM, OVCaR-3, SK-OV-3, SW626 ovarian cancer cell lines were detected using RT-qPCR. Compared with the normal ovarian cells, significantly higher levels of CLN3 were observed in the A2780, COC1, HO-8910, OVCaR-3, SK-OV-3 and SW626 cells, but not in the ES-2 and HO-8910PM cells. Among these, the SW626 and A2780 exhibited the highest endogenous mRNA levels of CLN3 (Fig. 1). These data suggested a possible role of CLN3 in the development and progression of ovarian cancer.
Figure 1.

Reverse transcription-quantitative polymerase chain reaction analysis of the expression of CLN3 in a panel of ovarian cancer cell lines. The results are expressed as the mean ± standard deviation. *P<0.05 vs. normal ovarian cells.
Knockdown of CLN3 by shRNA in the A2780 cell line and its drug-resistant sub-lines
To investigate whether CLN3 is required for the maintenance of drug-resistance in ovarian cancer cells, modified lentiviral pGCSIL-GFP vectors expressing CLN3 or control shRNA were transduced into the A2780 cell line and its cisplatin-resistant (A2780/DDP) and carboplatin-resistant (A2780/CBP) sub-lines. RT-qPCR revealed that there was ~70% decrease in the mRNA level of CLN3 in the cells transduced with the vector expressing CLN3 shRNA, compared with that in the cells transduced with the control vector. No significant difference in the mRNA expression of CLN3 was observed between the control vector-transduced cells and untransduced cells (Fig. 2A; P<0.05). The knockdown of CLN3 was further confirmed using western blot analysis, which demonstrated a decrease of ~66% in the protein level of CLN3, compared with the control cells (Fig. 2B; P<0.05). These results revealed that CLN3-specific shRNA effectively knocked down the expression of CLN3 in the ovarian cancer cells at the mRNA and protein levels.
Figure 2.
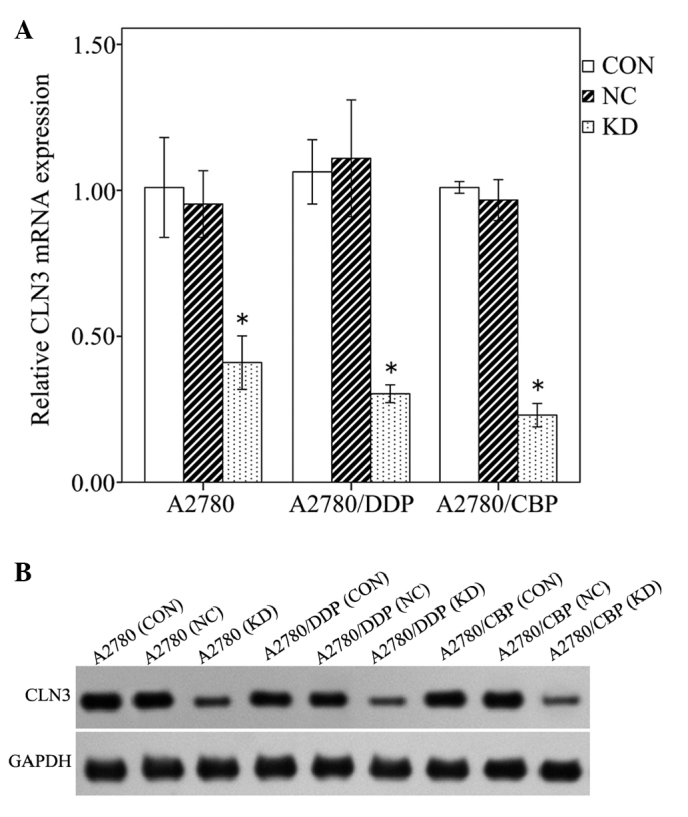
Specific knockdown of the expression of CLN3 in lentiviral vector-transduced ovarian cancer cells, analyzed using (A) reverse transcription-quantitative polymerase chain reaction and (B) western blot analysis. The results are expressed as the mean ± standard deviation. *P<0.05 vs. CON. CON, control; NC, negative control; KD, knockdown.
CLN3 knockdown inhibits cell proliferation and induces G0/G1 cell cycle arrest in the A2780 cell line and its drug-resistant sub-lines
To evaluate the effects of CLN3 knockdown on ovarian cancer cell proliferation, a CCK-8 assay was performed with the modified lentiviral pGCSIL-GFP vector transduced A2780, A2780/DDP and A2780/CBP cells. As shown in Fig. 3, CLN3 shRNA transfection in the parental A2780 cells significantly decreased cell viability on days 2 and 3, compared with the control vector-transduced cells (P<0.05). Similar results were observed in the drug-resistant A2780/DDP and A2780/CBP cells. As cell cycle progression is a critical determinant of cell growth, further cell cycle analysis was performed using flow cytometry. The results demonstrated that CLN3 knockdown in the A2780, A2780/DDP and A2780/CBP cells significantly increased the percentage of G0/G1 phase cells, with a concomitant fall in the percentage of S phase cells (P<0.05; Fig. 4). These data suggested that growth inhibition in the CLN3-knockdown drug-resistant ovarian cancer cells may have been due to the arrest at the G0/G1 transition of the cell cycle.
Figure 3.
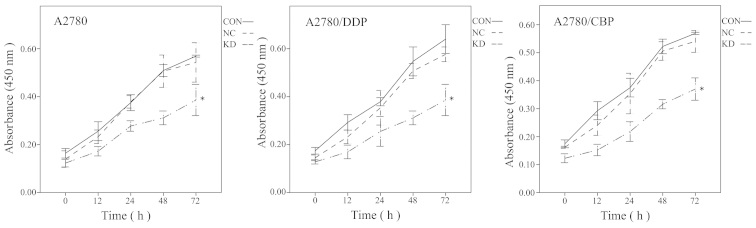
Effects of CLN3 knockdown on in vitro ovarian cancer cell proliferation, determined using a Cell Counting Kit-8 assay. The results are expressed as the mean ± standard deviation. *P<0.05 vs. CON. DDP, cisplatin-resistant; CBP, carboplatin-resistant; CON, control; NC, negative control; KD, knockdown.
Figure 4.

Effects of CLN3 knockdown on cell cycle in ovarian cancer cells. (A) Cells were stained with propidium iodide and quantified to determine cycle distribution using flow cytometry. (B) Percentages of cells in each cell phase were determined. DDP, cisplatin-resistant; CBP, carboplatin-resistant; NC, negative control; KD, knockdown.
CLN3 knockdown induces cell apoptosis in the A2780 cell line and its drug-resistant sub-lines
Flow cytometry was used to determine the effects of CLN3 knockdown on apoptosis in the A2780, A2780/DDP and A2780/CBP cells. As shown in Fig. 5, CLN3 knockdown significantly increased (35.55%) the percentage of apoptotic cells in the A2780 cells, compared with the control cells. Similarly, apoptotic cells were significantly increased by transfection with the vector expressing CLN3 shRNA in the A2780/DDP cells, between 11.26±1.05 and 59.17±2.25%, and the A2780/CBP cells, between 13.18±1.50 and 72.11±2.65%.
Figure 5.

Effects of CLN3 knockdown on cell apoptosis in ovarian cancer cells. (A) Percentages of apoptotic cells were evaluated using flow cytometric analysis with Annexin V-PI staining. (B) Graph of the percentage of apoptotic cells, measured using flow cytometry. The results are expressed as the mean ± standard deviation. *P<0.05 vs. NC. DDP, cisplatin-resistant; CBP, carboplatin-resistant; PI, propidium iodide; NC, negative control; KD, knockdown.
CLN3 knockdown enhances the chemosensitivity of ovarian cancer cells
To assess whether knockdown of CLN3 in ovarian cancer cells affects their chemosensitivity, the CLN3 shRNA- and control shRNA-transduced cells were treated with different concentrations of cisplatin or carboplatin, and a CCK-8 assay was used to determine cell viability. Compared with cisplatin treatment alone, combined CLN3 shRNA and cisplatin treatment caused a significant decrease in the IC50 value of the A2780/DDP cells, between 0.469±0.009 and 0.196±0.01 µg/ml. Similarly, in the A2780/CBP cells, the IC50 in the control shRNA cells was 0.657±0.05 µg/ml, and decreased significantly to 0.343±0.042 µg/ml in CLN3 shRNA cells (P<0.05).
CLN3 knockdown affects apoptosis-associated gene expression in the A2780 cell line and its drug-resistant sub-lines
The mRNA and protein expression levels of several apoptosis-associated factors in the A2780, A2780/DDP and A2780/CBP cells were analyzed using RT-PCR and western blot analysis, respectively. Compared with the control cells, the mRNA expression levels of Bax and FAX were significantly increased, however, the mRNA expression level of Bcl-2 was markedly reduced in the CLN3 shRNA-transduced cells (P<0.05; Fig. 6). The changes in these apoptosis-associated factors observed using western blot analysis were consistent with the results of the RT-qPCR (P<0.05; Fig. 7). Additionally, the protein levels of cleaved-caspase 3, cleaved-caspase 8 and cleaved-RARP were significantly increased in the CLN3 shRNA transduced cells, compared with the control cells (P<0.05; Fig. 6).
Figure 6.

Effects of CLN3 knockdown on the expression of apoptosis-associated mRNA in ovarian cancer cells, detected using reverse transcription-quantitative polymerase chain reaction. The results are expressed as the mean ± standard deviation. *P<0.05 vs. NC. DDP, cisplatin-resistant; CBP, carboplatin-resistant; NC, negative control; KD, knockdown.
Figure 7.

Effects of CLN3 knockdown on the expression of apoptosis-associated proteins in ovarian cancer cells, detected using western blot analysis. DDP, cisplatin-resistant; CBP, carboplatin-resistant; NC, negative control; KD, knockdown.
Discussion
Previous evidence indicates that CLN3 is highly expressed in ovarian cancer cells, however, the potential role of CLN3 in ovarian cancer cell growth and drug toxicity remains to be elucidated (11). In the present study, the expression of CLN3 was examined in the SW626, SK-OV-3, HO-8910, ES-2, COC1, A2780, OVCaR-3, HO-8910PM human ovarian cancer cell lines. Compared with normal ovarian cells, the SW626, SK-OV-3, HO-8910, COC1, A2780 and OVCaR-3 cells exhibited markedly higher expression levels of CLN3, suggesting a possible role of CLN3 in the development and progression of ovarian cancer. To further characterize the role of CLN3 in the chemoresistance of ovarian cancer, a lentivirus-based RNAi strategy was employed to transiently inhibit the endogenous expression of CLN3 in ovarian cancer A2780 cells and their drug-resistant A2780/DDP and A2780/CBP sub-lines. RT-qPCR and western blot analysis confirmed that the vector expressing CLN3 shRNA was able to effectively eliminate the expression of CLN3 in these cells. Further experiments demonstrated that CLN3 knockdown in the ovarian cancer cells significantly inhibited cell proliferation. In addition, flow cyto-metric analysis revealed that CLN3 knockdown in the A2780, A2780/DDP and A2780/CBP cells significantly increased the percentage of cells in the G0/G1 phase, with a concomitant fall in the percentage of cells in the S phase, indicating G0/G1 cell cycle arrest. These data are consistent with a previous study by Zhu et al, which reported that CLN3 silencing induces G0/G1 cell cycle arrest in HCT116 colorectal cancer cells (12). CLN3 has been previously shown to be an anti-apoptotic gene in NT2 neuronal precursor cells and several types of cancer (9,10,13). The present study demonstrated that reduced expression of CLN3 significantly induced apoptotic cell death in the A2780, A2780/DDP and A2780/CBP cells. In addition, CLN3 knockdown caused increases in the levels of Bax, FAX, cleaved-caspase 3, cleaved-caspase 8 and cleaved-RARP, and a decrease in the level of Bcl-2. Concordant with these results, Zhu et al reported that inhibiting of the expression of CLN3 promotes cell apoptosis in HCT116 cells (12). The findings of the present study, together with previous data, provide further support for CLN3 as an anti-apoptotic gene in cancer cells. However, the precise mechanisms by which knockdown of CLN3 causes cancer cell growth inhibition, cell cycle arrest and apoptosis require further elucidation.
Currently, resistance to chemotherapy and/or radiotherapy remains a major obstacle for the successful treatment of ovarian cancer. CLN3 has been suggested as a potential molecular target for the development of cancer drugs in the future (11), however, whether CLN3 contributes to chemoresis-tance in ovarian cancer cells remains to be elucidated. In the present study, A2780/DDP and A2780/CBP cells transduced with the CLN3 or scramble control were treated with various concentrations of cisplatin and carboplatin, respectively. It was observed that CLN3 knockdown significantly decreased the IC50 in the two types of cell, suggesting that CLN3 inhibition enhanced chemosensitivity in the drug-resistant ovarian cancer cells. ATP-binding cassette (ABC) transporters are a large family of transmembrane proteins, which transport various substrates across extracellular and intracellular membranes in an energy-dependent manner (14). In cancer cells, overexpression of these proteins is frequently associated with a multidrug resistance (MDR) phenotype (15–18). P-gp, a glycoprotein encoded by the MDR1 gene, is one of the most well-characterized ABC transporters, which pumps a variety of chemotherapeutics across the cell membrane and out of the cells, thereby contribute to drug resistance (19–21). A study by Tecedor et al demonstrated that CLN3 is required for normal trafficking of caveolin-1, syntaxin-6 and MDR1 microdo-main-associated proteins in brain endothelial cells (22). Thus, it seems reasonable to suggest that the increased chemosensitivity in drug-resistant ovarian cancer cells by CLN3 knockdown may be associated with the decrease in MDR1-mediated drug resistance. However, further investigation is required.
In conclusion, the present study demonstrated that knockdown of CLN3 effectively inhibited cell growth, arrested the cell cycle at the G0/G1 phase, promoted spontaneous apoptosis and enhanced chemosensitivity in drug-resistant ovarian cancer cells. These findings suggest that CLN3 may serve as a promising therapeutic target for the future treatment of ovarian cancer.
Acknowledgments
This study was supported by grants from the Science Foundation for Youths of Heilongjiang Province (grant. no. QC2011C122), the Science and Technology Foundation of Department of Education, Heilongjiang Province (grant. no. 11551161) and the Foundation of Department of Health, Heilongjiang Province (grant. no. 2007-429).
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Heintz AP, Odicino F, Maisonneuve P, Beller U, Benedet JL, Creasman WT, Ngan HY, Pecorelli S. Carcinoma of the ovary. Int J Gynaecol Obstet. 2003;83(Suppl 1):135–166. doi: 10.1016/S0020-7292(03)90118-4. [DOI] [PubMed] [Google Scholar]
- 3.Jelovac D, Armstrong DK. Recent progress in the diagnosis and treatment of ovarian cancer. CA Cancer J Clin. 2011;61:183–203. doi: 10.3322/caac.20113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Isolation of a novel gene underlying Batten disease, CLN3. The International Batten Disease Consortium. Cell. 1995;82:949–957. doi: 10.1016/0092-8674(95)90274-0. [DOI] [PubMed] [Google Scholar]
- 5.Rakheja D, Narayan SB, Pastor JV, Bennett MJ. CLN3P, the Batten disease protein, localizes to membrane lipid rafts (detergent-resistant membranes) Biochem Biophys Res Commun. 2004;317:988–991. doi: 10.1016/j.bbrc.2004.03.146. [DOI] [PubMed] [Google Scholar]
- 6.Phillips SN, Benedict JW, Weimer JM, Pearce DA. CLN3, the protein associated with batten disease: Structure, function and localization. J Neurosci Res. 2005;79:573–583. doi: 10.1002/jnr.20367. [DOI] [PubMed] [Google Scholar]
- 7.Metcalf DJ, Calvi AA, Seaman M, Mitchison HM, Cutler DF. Loss of the Batten disease gene CLN3 prevents exit from the TGN of the mannose 6-phosphate receptor. Traffic. 2008;9:1905–1914. doi: 10.1111/j.1600-0854.2008.00807.x. [DOI] [PubMed] [Google Scholar]
- 8.Cortese A, Tucci A, Piccolo G, Galimberti CA, Fratta P, Marchioni E, Grampa G, Cereda C, Grieco G, Ricca I, et al. Novel CLN3 mutation causing autophagic vacuolar myopathy. Neurology. 2014;82:2072–2076. doi: 10.1212/WNL.0000000000000490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puranam KL, Guo WX, Qian WH, Nikbakht K, Boustany RM. CLN3 defines a novel antiapoptotic pathway operative in neurodegeneration and mediated by ceramide. Mol Genet Metab. 1999;66:294–308. doi: 10.1006/mgme.1999.2834. [DOI] [PubMed] [Google Scholar]
- 10.Narayan SB, Rakheja D, Pastor JV, Rosenblatt K, Greene SR, Yang J, Wolf BA, Bennett MJ. Over-expression of CLN3P, the Batten disease protein, inhibits PANDER-induced apoptosis in neuroblastoma cells: Further evidence that CLN3P has anti-apoptotic properties. Mol Genet Metab. 2006;88:178–183. doi: 10.1016/j.ymgme.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 11.Rylova SN, Amalfitano A, Persaud-Sawin DA, Guo WX, Chang J, Jansen PJ, Proia AD, Boustany RM. The CLN3 gene is a novel molecular target for cancer drug discovery. Cancer Res. 2002;62:801–808. [PubMed] [Google Scholar]
- 12.Zhu X, Huang Z, Chen Y, Zhou J, Hu S, Zhi Q, Song S, Wang Y, Wan D, Gu W, et al. Effect of CLN3 silencing by RNA interference on the proliferation and apoptosis of human colorectal cancer cells. Biomed Pharmacother. 2014;68:253–258. doi: 10.1016/j.biopha.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 13.Wu D, Liu J, Wu B, Tu B, Zhu W, Luo J. The Batten disease gene CLN3 confers resistance to endoplasmic reticulum stress induced by tunicamycin. Biochem Biophys Res Commun. 2014;447:115–120. doi: 10.1016/j.bbrc.2014.03.120. [DOI] [PubMed] [Google Scholar]
- 14.Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156–1166. doi: 10.1101/gr.GR-1649R. [DOI] [PubMed] [Google Scholar]
- 15.Izquierdo MA, Neefjes JJ, Mathari AE, Flens MJ, Scheffer GL, Scheper RJ. Overexpression of the ABC transporter TAP in multidrug-resistant human cancer cell lines. Br J Cancer. 1996;74:1961–1967. doi: 10.1038/bjc.1996.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nooter K, Stoter G. Molecular mechanisms of multidrug resistance in cancer chemotherapy. Pathol Res Pract. 1996;192:768–780. doi: 10.1016/S0344-0338(96)80099-9. [DOI] [PubMed] [Google Scholar]
- 17.Breier A, Gibalova L, Seres M, Barancik M, Sulova Z. New insight into p-glycoprotein as a drug target. Anticancer Agents Med Chem. 2013;13:159–170. doi: 10.2174/187152013804487380. [DOI] [PubMed] [Google Scholar]
- 18.Baguley BC. Multiple drug resistance mechanisms in cancer. Mol Biotechnol. 2010;46:308–316. doi: 10.1007/s12033-010-9321-2. [DOI] [PubMed] [Google Scholar]
- 19.Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: From genomics to mechanism. Oncogene. 2003;22:7468–7485. doi: 10.1038/sj.onc.1206948. [DOI] [PubMed] [Google Scholar]
- 20.Sauna ZE, Kim IW, Ambudkar SV. Genomics and the mechanism of P-glycoprotein (ABCB1) J Bioenerg Biomembr. 2007;39:481–487. doi: 10.1007/s10863-007-9115-9. [DOI] [PubMed] [Google Scholar]
- 21.Glavinas H, Krajcsi P, Cserepes J, Sarkadi B. The role of ABC transporters in drug resistance, metabolism and toxicity. Curr Drug Deliv. 2004;1:27–42. doi: 10.2174/1567201043480036. [DOI] [PubMed] [Google Scholar]
- 22.Tecedor L, Stein CS, Schultz ML, Farwanah H, Sandhoff K, Davidson BL. CLN3 loss disturbs membrane microdomain properties and protein transport in brain endothelial cells. J Neurosci. 2013;33:18065–18079. doi: 10.1523/JNEUROSCI.0498-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]