

Abstract
The formation of a series of analogs containing a pyridine moiety in place of the natural thiazole heterocycle, based on the potent, naturally occurring HDAC inhibitor Largazole has been accomplished. The synthetic strategy was designed modularly to access multiple inhibitors with different aryl functionalities containing both the natural depsipeptide and peptide isostere variant of the macrocycle. The cytotoxicity and biochemical activity of the library of HDAC inhibitors is described herein.
Keywords: Largazole, Class I Histone Deacetylase Inhibitor, pyridine analogs, cytotoxic and biochemical assays
Graphical abstract
1. INTRODUCTION
Largazole (1) has received a great deal of attention from the synthetic and medical communities ever since its isolation from the marine cyanobacterium of the genus Symploca by the Leusch laboratory.1,2 This depsipeptide natural product has been synthesized and modified by several research groups due to its potent biochemical activity as a Class I Histone Deacetylase Inhibitor (HDACi) and consequent anticancer properties.3–5 Largazole (1) is a pro-drug with the active form being the free thiol species 2 (Figure 1) that is formed in vitro and in vivo by esterase or lipase-based cleavage of the octanoyl residue.6 It has been suggested by us that the octanoyl tail present on Largazole allows for better cell-permeability relative to the active, free thiol species.6 The nascent thiol group tightly coordinates to the Zn2+-domain within the HDAC enzyme active site resulting in tight, non-covalent binding of the drug and consequent enzymatic inhibition.
Figure 1.

Structure of Largazole and Largazole thiol and their peptide isosteres.
There are currently eighteen known HDAC enzymes present in human cells that are separated into four different classes based on their structural homology with yeast proteins.7 Of these, Class I (HDACs 1, 2, 3, and 8), Class II (HDAC 4, 5, 6, 7, 9, and 10), and Class IV (HDAC 11) are Zn2+-dependent while Class III (SirT1-7, known as the Sirtuins) are NAD+-dependent. A substantial body of data has elucidated that the inhibition of the ubiquitous Class I HDACs is a viable anticancer therapeutic target.8,9 Current research efforts in our laboratories have been to access inhibitors that are selective for Class I HDACs over the tissue-dependent Class II HDACs. The goal of our efforts has been to generate compounds based on the molecular scaffold of Largazole that exhibit superior potency, selectivity and drug-like properties relative to that of the natural product.
We have been active in pursuing new analogs of Largazole (1) since the completion of our concise total synthesis in 2008.6 The first key structural alteration we examined, was to replace the depsipeptide oxygen atom present in the macrocycle with a lactam nitrogen atom providing the corresponding more chemically robust peptide isostere 3 (Figure 1).10 We also examined other functional group changes in the macrocycle, as well as the zinc-binding domain.11,12 The present work described herein continues this exploration by investigating the exchange of the thiazole heterocycle in the macrocyclic cap group for a pyridyl residue in both the natural depsipeptide and peptide isostere variants of the macrocyclic core (Figure 2).
Figure 2.

Pyridine containing library based on Largazole.
2. RESULTS AND DISCUSSION
2.1. Design
The macrocyclic cap group of Largazole not only contributes significantly to the overall binding, but also offers unique opportunities for the design of even more potent and selective analogs because it allows the precise positioning of functional groups on the surface of the protein by appropriate functionalization of the macrocycle. The analysis of the position of Largazole in our model of the HDAC1-Largazole thiol complex13 as well as the recently published crystal structure of the HDAC8-Largazole thiol complex (pdb 3EQD)14 provide the structural basis for rational design that exploits specific surface interactions. Based on the sequence alignment of the residues surrounding the active site (Figure S1 in the Supporting Information), we envisioned the replacement of the thiazole ring by a protonated pyridine to form a charge interaction with a nearby negatively charged residue (Asp), as shown in Figure 3. In addition, the inclusion of a positive charge could modify the conformational preferences of the macrocycle, which was shown previously to be important for the relative potency of the depsipetide and its amide isostere,10 and control its overall shape, which is known to control isoform selectivity.15 With these considerations in mind, two different isomers of the pyridyl-based analogs of Largazole, compounds 6 and 8 their respective amide isosteres 10 and 12 were chosen for synthetic studies and biochemical profiling.
Figure 3.

Model of pyridyl Largazole thiol analogue bound to HDAC1
2.2. Chemistry
Our published total synthesis of Largazole (1) provided the technological framework from which we have been able to access numerous new analogs.6 The retrosynthetic analysis displayed in Scheme 1 outlines the modular nature of our approach. The key macrolactamization reaction we have found is best performed on the least-hindered amide located at the bottom of macrocycle 13. The target macrocycle has been bisected into two halves through the top amide linkage. Bottom fragment 14 is constituted from L-Valine derivative 17 and both the requisite β-hydroxy ester (16, X = OH, for the depsipeptides) and the corresponding β-amino ester (16, X = NH2, for the peptide isosteres). The thiazoline/pyridyl-containing fragment 15 will be formed through a cyclocondensation reaction between α-methyl-L-cysteine (18) and aryl nitrile 19.
Scheme 1.

Retrosynthetic analysis of library.
The synthesis of fragment 23 for the depsipeptide version of the macrocycle is displayed in Scheme 2. The formation of aldehyde 21 was achieved through a hetero-Michael addition of trityl mercaptan into acrolein followed by a Wittig olefination. The stereocenter present in alcohol 22 was set using a Crimmins’s type chiral auxiliary.16 This asymmetric aldol transformation occurred to produce alcohol 22 in very high diastereomeric purity. This material was submitted to cleavage of the chiral auxillary using 2-(trimethylsilyl)ethanol. Installation of the L-valine derived amino acid was mediated by EDCI to access depsipeptide bottom fragment 23.
Scheme 2.

Synthesis of fragment 23 for depsipeptide macrocycle. Reagents and conditions: (a) TrtSH, Et3N, CH2Cl2; (b) (formylmethylene)triphenylphosphorane, PhH, 80 ° C, 77% over 2 steps; (c) (R)-1-(4-benzyl-2-thioxothiazolidin-3-yl)ethanone, TiCl4, DIPEA, CH2Cl2, −78 ° C, 76%; (d) 2-(trimethylsilyl)ethanol, imidazole, CH2Cl2, 83%; (e) N-Fmoc-Val-OH, EDCI, DIPEA, DMAP, CH2Cl2, 77%.
Peptide precursor 28 was furnished using the previously established protocol (Scheme 3).10 Commercially available amino acid derivative 24 was converted to the primary alcohol through reduction of a mixed anhydride. Swern oxidation followed by Wittig olefination achieved formation of terminal alkene 25. Scrambling of the stereogenic center was not apparent through an aqueous KHSO4 workup of the oxidation step and submission of the aldehyde directly to the olefination reaction. The cross metathesis to construct alkene 26 proved to be troublesome in producing high yielding and reproducible results.17 The primary alcohol was converted to the protected thiol through activation with TsCl and displacement with trityl thiol anion. After methyl ester formation, the amine present on substrate 27 was unmasked using TFA and coupled with an L-valine derived amino acid to yield peptide precursor 28.
Scheme 3.
Synthesis of fragment 28 for peptide macrocycle.
Reagents and conditions: (a) 4-methylmorpholine, isobutyl chloroformate, THF, −40 ° C; NaBH4, MeOH, −20 ° C, 66%; (b) oxalyl chloride, DMSO, DIPEA, CH2Cl2, −65 ° C; (c) methyltriphenylphosphonium bromide, KHMDS, THF, −78 ° C, 80% over 2 steps; (d) 3-buten-1-ol, Grubbs catalyst, 2nd generation, CH2Cl2, 50 ° C, 25%; (e) TsCl, Et3N, DMAP, CH2Cl2, 82%; (f) TrtSH, KOtBu, THF, 87%; (g) LiOH, THF, MeOH, 50 ° C, 96%; (h) MeOH, EDCI, DIPEA, DMAP, CH2Cl2, 77%; (i) TFA, CH2Cl2; (j) N-Boc-Val-OH, PyBOP, DIPEA, CH2Cl2, 88% over 2 steps.
Aryl fragment 32 was constructed starting with dicarboxylic acid 29 (Scheme 4). Fischer esterification and reduction of one of the ethyl esters provided the requisite primary alcohol functionality. After using NH4OH to generate an amide from the corresponding carboxylic acid, POCl3 successfully dehydrated the amide to generate the requisite nitrile and chloride present in compound 30. Gabriel’s protocol was utilized to convert the chloride into an amine, which was readily protected to provide cyclization partner 31. Cyclocondensation with α-methyl-L-cysteine18 proceeded in high yields to generate the pyridyl fragment 32.
Scheme 4.
Synthesis of the pyridyl “IN” fragment 32.
Reagents and Conditions: (a) PTSA, EtOH, 80 ° C, 100%; (b) NaBH4, EtOH, 80 ° C, 53%; (c) NH4OH, EtOH; (d) POCl3, DMF, 0 ° C; (e) potassium phthalimide, DMF, 69% over three steps; (f) H2NNH2-H2O, MeOH, THF; (g) Boc2O, Et3N, CH2Cl2, 43% over 2 steps; (h) α-methyl-L-cysteine, NaHCO3, pH 6.0 buffer, MeOH, 70 ° C, 84%.
The route for the pyridyl “OUT” aryl fragment 36 is displayed in Scheme 5. The synthetic strategy follows the same method mentioned previously for the pyridine “IN” variant except dicarboxylic acid 33 was used instead.
Scheme 5.
Synthesis of pyridine “OUT” fragment.
Reagents and conditions: (a) PTSA, EtOH, 80 ° C, 100%; (b) NaBH4, CaCl2, EtOH, 80 ° C, 61%; (c) NH4OH, EtOH; (d) POCl3, DMF, 0 ° C; (e) potassium phthalimide, DMF, 50% over three steps; (f) H2NNH2-H2O, MeOH, THF; (g) Boc2O, Et3N, CH2Cl2, 84% over 2 steps; (h) α-methyl-L-cysteine, NaHCO3, pH 6.0 buffer, MeOH, 70 ° C, 85%.
The final steps to access the desired inhibitors were performed upon the successful assembly of the four key fragments. The depsipeptide class construction is shown in Scheme 6. The protected amine present in substrate 23 was unmasked using diethylamine. The successful union between the nascent amine and acid 32 or 36 was accomplished cleanly using PyBOP. The macrolactamization was performed using HATU and HOBt after exposing the requisite functionality through acid-mediated deprotection. Accessing pure material from the ring closure transformation required rigorous column chromatography. Trityl cleavage granted the formation of inhibitors 6 and 8. Unfortunately, adding the octanoyl residue was only viable using standard acid chloride procedure on pyridine “OUT” depsipeptide thiol 8. Pyridine “IN” thiol 6 gave rise to dioctanoylation when exposed to the same conditions. The N-octanoyl-imidazole reagent was prepared and then was exposed to thiol 6. The change in the reactivity of the carbonyl gave rise to only the thioester being formed.
Scheme 6.

Synthesis of depsipeptide containing analogs.
Reagents and conditions: (a) Et2NH, CH3CN; (b) 32, PyBOP, DIPEA, 79% over 2 steps; (c) 36, PyBOP, DIPEA, 81% over 2 steps; (d) TFA, CH2Cl2; (e) HATU, HOBt, DIPEA, CH3CN, 20% over 2 steps; (f) HATU, HOBt, DIPEA, CH3CN, 23% over 2 steps; (g) TFA, iPr3SiH, CH2Cl2, 82%; (h) TFA, iPr3SiH, CH2Cl2, 89%; (i) N-octanoyl–imidazole, imidazole, DMAP, THF, 50 ° C, 69%; (j) octanoyl chloride, Et3N, 49%.
The creation of the peptide class of pyridine-thiazoline inhibitors 10 and 12 followed the same method discussed for their depsipeptide counterparts. The only difference in their route arose during the deprotection steps. The same strategy was employed as previously discussed to add the octanoyl fragment to arrive at inhibitors 9 and 11.
2.3. Biochemical & Biological Evaluation
Compounds 1–12 were tested for inhibitory activity against HDACs 1–9, using an optimized homogenous assay performed in a 384-well plate, as described previously.6 The results of these studies, summarized in Table 1 and Figure S5, identify that the thiol derivatives of Largazole including compounds 2, 4, 6, 8, 10, and 12, are among the most potent with IC50 values below 25nM for HDACs 1–3. Of these thiols, the depsipeptide pyridine thiols 6, 8 show similar potency to Largazole thiol (2) and Largazole peptide isostere thiol (4) with IC50 values in the single digit nM range for HDACs 1–3. Conversely, the peptide pyridine thiols 10, 12 show a decrease in potency with IC50s in the double-digit nM range for HDACS 1–3. Largazole (1) and derivatives 3, 5, 7, 9, 11 display IC50 values in the 200nM–1μM range. Of these compounds, Largazole (1) and peptide pyridine “OUT” derivative 11 were similar in potency for HDACs 1–3 with IC50s in the high nM–μM range while the depsipeptide pyridines and peptide pyridine “IN” derivative 5, 7, 11 exhibited an increased potency with IC50s in the 200nM–350nM range for HDAC 1. These studies demonstrate that the free thiol is important for increased inhibition, as previously reported, with the depsipeptide pyridine derivatives displaying increased inhibition over the peptide isostere derivatives.
Table 1.
HDAC inhibitory activity of Largazole analogs (IC50 nM).
| Compound | HDAC1 | HDAC2 | HDAC3 | HDAC6 | HDAC8 |
|---|---|---|---|---|---|
| 1 | 10.09 | 18.65 | 9.09 | 165.6 | 1068 |
| 2 | 2.51 | 4.19 | 2.78 | 28.11 | 228.4 |
| 3 | 544.1 | 825.2 | 1151 | - | - |
| 4 | 1.95 | 3.38 | 2.59 | 102 | 255.3 |
| 5 | 203.6 | 349.5 | 332.1 | - | - |
| 6 | 2.68 | 4.39 | 3.07 | 48.55 | 341.3 |
| 7 | 340 | 655.4 | 319.5 | - | - |
| 8 | 2.20 | 4.42 | 2.31 | 35.16 | 101.8 |
| 9 | 340.3 | 471.8 | 332.4 | - | - |
| 10 | 42 | 69.8 | 42.5 | - | - |
| 11 | 816.9 | 1240 | 846.5 | - | - |
| 12 | 13.2 | 20.77 | 14.59 | 2849 | 1491 |
Next, we tested the activity of the novel analogs in the 797 and 10326 NUT midline carcinoma cell line. As can be seen in Table 2 and Figure S6, depsipeptide pyridine “OUT” 7 showed the greatest potency against both cell lines with an IC50 of 10nM. This was comparable to both Largazole and depsipeptide pyridine “IN” 1, 5 with IC50s of 20nM against both 797 and 30nM and 40nM against 10326 respectively. Compound 10 was not evaluated due to it’s low biochemical activity.
Table 2.
Activity of Largazole analogs in 797 and 10326 cell lines (IC50s μM)
| Compound | 797 | 797 LogIC50 | 10326 | 10326 LogIC50 |
|---|---|---|---|---|
|
| ||||
| 1 | 0.024 | −7.621 ± 0.06565 | 0.025 | −7.606 ± 0.02845 |
| 2 | 0.1 | −7.016 ± 0.03453 | 0.08 | −7.083 ± 0.06683 |
| 3 | 0.11 | −6.971 ± 0.04341 | 0.17 | −6.763 ± 0.06589 |
| 4 | 1.74 | −5.761 ± 0.05449 | 11.94 | −4.923 ± 0.9900 |
| 5 | 0.03 | −7.529 ± 0.05220 | 0.038 | −7.422 ± 0.04499 |
| 6 | 0.22 | −6.654 ± 0.04604 | 0.29 | −6.526 ± 0.09239 |
| 7 | 0.01 | −7.949 ± 0.07215 | 0.01 | −7.873 ± 0.04062 |
| 8 | 0.09 | −7.031 ± 0.06228 | 0.12 | −6.913 ± 0.03566 |
| 9 | 0.31 | −6.509 ± 0.02559 | 0.55 | −6.263 ± 0.07115 |
| 10 | - | - | - | - |
| 11 | 0.07 | −7.122 ± 0.05525 | 0.13 | −6.873 ± 0.06520 |
| 12 | 4.25 | −5.371 ± 0.04826 | 8.64 | −5.063 ± 0.1964 |
Conversely, the peptide isosteres 9, 11 exhibited ~10-fold decreased inhibition in comparison to the depsipeptide pyridine derivatives, demonstrating that the depsipeptide mimetic may be important for increased inhibition. Compound 11 is more active in cell culture but it appears to have less activity in the biochemical assay compared to the other prodrugs derivatives. While this is an unexpected result in cell culture as the other prodrugs and their thiols derivatives show stronger activity biochemically, it may be possible that the compound 11 is cleaved and activated within cells to a higher extent than it’s prodrug counterparts yielding higher activity in cell culture.
2.4. Computational Studies of Selectivity
The biochemical profiling provides a number of interesting results that, when combined with the results from molecular dynamics simulations, probe the structural basis for the observed selectivity. We studied Largazole thiol 2 and its analogues 6, 8, 10 and 12, which are postulated to be the active form of the corresponding Largazole derivatives,6 in our homology model of HDAC1,13 HDAC6,15 and the crystal structure of HDAC8 (pdb 3EQD).14 Here, we will discuss the cases of the largest experimentally observed selectivity in detail, while the full results for all five compounds in the three HDAC isoforms studied can be found in the Supporting Information.
We will start the discussion of the potency of Largazole thiol and its derivatives in HDAC1. Experimentally, the most notable finding is the 16-fold decrease in activity in going from 6, which is similar to Largazole thiol 2, to its amide isostere 10. Figure 4 shows snapshots from the MD simulations of 6 (blue, with protein in red) and 10 (green) in HDAC1. For comparison purposes, the previously analyzed10 position of 2 is shown in grey. As can be seen, the overall positions of 2 and 6 at the mouth of the active site pocket are similar. In addition, 6 does indeed establish an interaction between the protonated pyridyl ring and the key aspartate D99, although the NH-O distance fluctuates to up to >3.5 Å over the course of the simulation. However, the macrocyclic ring of 6 has to rotate relative to the position of 2 to enable this interaction, decreasing the covered surface area. As a result, the overall IC50 of the two compounds is similar. In contrast, 10 does not establish over the course of the simulation, presumably because it would require a ring conformation that is unfavorable for the more rigid amide isostere. Instead, the pyridyl and thiazole ring establish hydrophobic interactions at the opposite side of the binding pocket, predominately with F205 and leaving the protonated pyridyl ring solvent exposed. The resulting weaker interactions are consistent with the 16-fold decrease in binding affinity for 10.
Figure 4.

Snapshots from the MD simulations of 6 (blue, with protein in red) and 10 (green) in HDAC1. Position of 2 is shown in grey for reference purposes
Compounds 2 and 6 are experimentally found to be 11 and 18 times less potent in HDAC6 than in HDAC1 while 12 is >200 times less potent. The results for 2 and 6 can be understood in terms of the previously discussed15 differences in the shape of the protein surface between HDAC6 and the class 1 HDACs such as HDAC1 at the mouth of the active site. Specifically, HDAC6 selective inhibitors such as tubacin or WT161 have a Y-shaped cap group that is quite dissimilar to the macrocycle in the compounds studied here. As a result of this shape mismatch, significant loop reorganization in this region is necessary to establish binding contacts, as can be seen from the RMSD plots in Figure S3 in the Supporting Information.
Compounds 2 and 12 occupy very similar positions in HDAC6, as shown in the snapshots from the MD simulations of HDAC6 in Figure 5. However, the long timescale simulations of 12 bound to HDAC6 indicate a reorientation of a helix (shown on the top left in Figure 5) that is only observed in this system. It can therefore be hypothesized that it is energetically unfavorable despite the short NH-O hydrogen bond of 1.96 Å and correlates with the low activity of 12.
Figure 5.
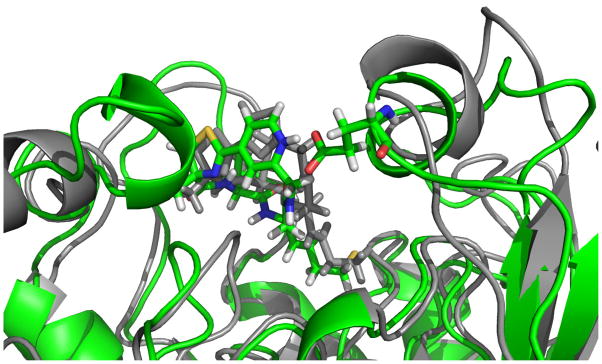
Snapshots from the MD simulations of 2 (grey) and 12 (green) in HDAC6. Position of 2 is shown in grey for reference purposes
All compounds studied are found to have a ~100:1 selectivity favoring the class I HDAC1-3 over HDAC8, another member of the same isoform class. These results can be rationalized by the computational finding that, in order to accommodate the large macrocyclic cap group, HDAC8 has to adopt the “open” conformation where the two binding sites observed in the crystal structures of HDAC8 (e.g. pdb code 1T6419) are connected through the movement of F152 and Y306, leading to a large groove resembling the one observed experimentally (pdb code 1VKG).19 This groove is stable over the course of the simulation (see Figure S4 in the Supporting Information) and specific for HDAC8.20 Compound 2 and analogs can only establish contacts to one side of the binding pocket, leading to the experimentally observed selectivity. In the representation shown in Figure 6, 2 only binds to the right side of the pocket, while the interaction with D88 leads to an interaction of 12 with the left side of the binding pocket.
Figure 6.

Snapshots from the MD simulations of 2 (grey) and 12 (green) in HDAC8.
3. CONCLUSION
Potent HDAC is have been synthesized by manipulating the macrocycle present on the natural product Largazole. Inhibitors with excellent selectivity for only HDAC1, HDAC2, and HDAC3 were formed through altering the depsipeptide framework to the peptide isostere variant and changing the thiazole to a pyridine.29 The depsipeptide pyridyl variants proved to be equipotent (pyridyl “IN”) and twice as potent (pyridyl “OUT”) when tested against two cell lines. These biological observations were further rationalized through molecular modeling.
4. EXPERIMENTAL SECTION
General experimental method
Unless otherwise noted, all reactions were run under an argon atmosphere in flame or oven dried glassware. Reactions were monitored using thin layer silica gel chromatography (TLC) using 0.25 mm silica gel 60F plates with fluorescent indicator from Merck. Plates were visualized by treatment with anisaldehyde stain with gentle heating. Products were purified via column chromatography using the solvent system(s) indicated. Silica gel 60, 230–400 mesh, was purchased from Sorbent Technologies. Tetrahydrofuran (THF), dichloromethane (CH2Cl2), acetonitrile (CH3CN), triethylamine (Et3N), toluene, diethyl ether (Et2O), and N,N-dimethylformamide (DMF) were passed through an alumina drying column (Solv-Tek Inc.) using argon pressure. All other reagents were purchased from commercial sources and used as received without additional purification. 1H NMR and 13C NMR spectra were recorded on Varian 300, 400, or 500 MHz NMR spectrometers. Chemical shifts are reported in ppm relative to CHCl3 at δ = 7.27 (1H NMR) and δ = 77.23 (13C NMR) or tetramethylsilane (TMS) δ = 0.00, where noted. Mass spectra were obtained on Fisions VG Autospec. Optical rotations were collected at 589 nm on a Rudolph Research Automatic Polarimeter Autopol III. All compounds tested in the biological assays were shown to have a purity of >95% via NMR.
(S,E)-2-(Trimethylsilyl)ethyl 3-(((S)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-methylbutanoyl)oxy)-7-(tritylthio)hept-4-enoate (23)
To a solution of 2-(trimethylsilyl)ethyl (S,E)-3-hydroxy-7-(tritylthio)hept-4-enoate6 (300 mg, 0.578 mmol) in CH2Cl2 (15.0 mL) at ambient temperature was added N-Fmoc-L-valine (981 mg, 2.89 mmol), EDCI•HCl (665 mg, 3.47 mmol), DMAP (7.1 mg, 0.058 mmol), and DIPEA (0.60 mL, 3.47 mmol). After stirring for 18 h, the reaction mixture was concentrated. The crude residue was purified using flash chromatography (1% to 20% ethyl acetate in hexanes) to provide ester (375 mg, 77%): 1H-NMR (CDCl3, 400 MHz) δ 7.71–7.75 (m, 2H), 7.56–7.58 (m, 2H), 7.18–7.38 (m, 19H), 5.58–5.69 (m, 2H), 5.34 (dd, J = 15.6, 7.6 Hz, 1H), 5.25 (d, J = 9.2 Hz, 1H), 4.30–4.40 (m, 2H), 4.25 (dd, J = 8.8, 4.4 Hz, 1H), 4.20 (t, J = 6.8 Hz, 1H), 4.12 (t, J = 8.8 Hz, 2H), 2.65 (dd, J = 16.0, 8.0 Hz, 1H), 2.52 (dd, J = 15.6, 5.2 Hz, 1H), 2.10–2.17 (m, 3H), 2.02 (q, J =6.8 Hz, 2H), 0.91–0.99 (m, 2H), 0.89 (d, J = 6.8 Hz, 3H), 0.77 (d, J = 6.8 Hz, 3H), −0.01 (s, 9H); 13C-NMR (CDCl3, 100 MHz) δ 170.9, 169.6, 156.1, 144.8, 143.9, 143.8, 141.3, 134.0, 129.6, 127.9, 127.7, 127.0, 126.6, 125.1, 120.0, 71.8, 67.0, 66.6, 63.1, 58.7, 47.2, 39.7, 31.4, 31.3, 31.1, 19.0, 17.3, −1.5; IR (neat) 3059, 2979, 1731, 1509, 1447, 1249, 1182, 1033, 858, 741; HRMS (ESI) m/z calcd for C51H57NNaO6SSi [M+Na]+ 862.3574, found 862.3583; [α]D = −15.0 (c 1.18, CHCl3).
(S,E)-Methyl 3-((S)-2-((tert-butoxycarbonyl)amino)-3-methylbutanamido)-7-(tritylthio)hept-4-enoate (28)
To a solution of Boc protected amine 2710 (0.650 g, 1.22 mmol) in CH2Cl2 (37 mL) at 0 °C was treated with TFA (0.37 mL). After 2 h, the reaction mixture was concentrated under vacuum, taken back up in toluene (15 mL), and concentrated again. In another flask, Boc-L-valine (0.531 g, 2.44 mmol), PyBOP (1.27 g, 2.44 mmol), and DIPEA (0.65 mL, 3.66 mmol) were combined in CH2Cl2 (60 mL). The freshly deprotected amine was added via CH2Cl2 (10 mL) to the flask containing the activated acid at ambient temperature. After 3 h, the resulting mixture was concentrated under reduced pressure. The crude residue was purified using flash chromatography (5% to 50% ethyl acetate in hexanes) to obtain the desired amide (0.680 g, 88%): 1H-NMR (CDCl3, 400 MHz) δ 7.15–7.37 (m, 15H), 6.49 (d, J = 8.0 Hz, 1H), 5.45 (dtd, J = 15.2, 6.4, 0.8 Hz, 1H), 5.34 (dd, J = 15.6, 6.0 Hz, 1H), 4.97–5.02 (m, 1H), 4.67–4.74 (m, 1H), 3.82–3.86 (m, 1H), 3.59 (s, 3H), 2.53 (d, J = 5.2 Hz, 2H), 2.05–2.16 (m, 3H), 2.01 (q, J = 6.8 Hz, 2H), 1.40 (s, 9H), 0.89 (d, J = 6.8 Hz, 3H), 0.83 (d, J = 6.8 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 171.6, 170.5, 155.8, 144.8, 130.5, 129.5, 127.8, 126.5, 79.7, 66.5, 59.9, 51.7, 47.2, 38.7, 31.3, 31.0, 28.3, 19.3, 17.6; IR (neat) 3304, 2965, 1738, 1685, 1650, 1522, 1492, 1365, 1172, 751; HRMS (ESI) m/z calcd for C37H46N2NaO5S [M+Na]+ 653.3025, found 653.3032; [α]D = −14.0 (c 1.21, CHCl3).
6-((((tert-Butoxy)carbonyl)amino)methyl)picolinonitrile (31)
To a solution of commercially available chloride 30 (9.66 g, 63.3 mmol) in DMF (400 mL) at ambient temperature was treated with potassium phthalimide (11.7 g, 63.3 mmol). After stirring for 5 h, the mixture was concentrated under vacuum. The remaining mixture was taken up in H2O (200 mL) and was filtered to collect the solid. The solid was washed with H2O (100 mL) and THF (100 mL) to obtain the desired phthalimide derivitive (11.5 g, 69%) and was moved forward without further purification. To a solution of the crude phthalimide derivative (5.84 g, 22.2 mmol) in THF/MeOH (200 mL, 1:1, v/v) at ambient temperature was treated with hydrazine monohydrate (1.18 mL, 24.4 mmol). After 2 h, 1.0 M HCl (24.5 mL) was added to the mixture and was stirred for another 3 h before concentrating the reaction mixture under vacuum. The remaining residue was taken up in H2O (200 mL) and the unwanted solid was removed through filtration. The filtrate was concentrated and placed under vacuum to remove the remaining H2O. The crude solid was taken up in CH2Cl2 (175 mL) and triethylamine (9.28 mL, 66.6 mmol) and Boc2O (4.86 g, 24.4 mmol) was added. After stirring for 12 h at room temperature, the reaction was quenched with a saturated solution of NaHCO3 (200 mL), extracted with CH2Cl2 (3 × 150 mL), dried over MgSO4, and concentrated under reduced pressure. The residue was purified using flash chromatography (10% to 45% ethyl acetate in hexanes) to provided the aryl pyridine “IN” fragement (2.24 g, 43%): 1H-NMR (CDCl3, 400 MHz) δ 7.78 (t, J = 7.6 Hz, 1H), 7.56 (d, J = 7.6 Hz, 1H), 7.49 (d, J = 8.0 Hz, 1H), 5.51 (s, 1H), 4.44 (d, J = 5.6 Hz, 2H), 1.43 (s, 9H); 13C-NMR (CDCl3, 100 MHz) 160.1, 155.9, 137.6, 133.0, 127.0, 125.1, 117.1, 79.9, 45.5, 28.3; IR (neat) 3347, 2979, 2934, 2239, 1699, 1518, 1453, 1250, 1170, 862; HRMS (ESI) m/z calcd for C12H15N3NaO3 [M+Na]+ 256.1062, found 256.1062.
(R)-2-(6-(((tert-Butoxycarbonyl)amino)methyl)pyridin-2-yl)-4-methyl-4,5-dihydrothiazole-4-carboxylic acid (32)
To a solution of nitrile 31 (1.50 g, 6.43 mmol) in MeOH/pH 6.0 buffer (105 mL, 3:2, v/v) was added α-methyl-L-cysteine (1.32 g, 7.72 mmol) and NaHCO3 (1.08 g, 12.9 mmol). The reaction mixture was heated to 70 °C for 48 h. The mixture was cooled to ambient temperature and the MeOH was removed under reduced pressure. The aqueous layer was extracted with diethyl ether (3 × 30 mL) before being acidified to a pH of ~2 using 1.0 M HCl. The aqueous layer was then extracted with ethyl acetate (3 × 30 mL) and the ethyl acetate layer was dried over MgSO4. The solution was concentrated to give the desired acid (1.89 g, 84%): 1H-NMR (CDCl3, 400 MHz) δ 7.97 (d, J = 7.6 Hz, 1H), 7.74 (t, J = 8.0 Hz, 1H), 7.37 (d, J = 7.6 Hz, 1H), 5.45 (bs, 1H), 4.46 (d, J = 5.2 Hz, 2H), 3.77 (d, J = 11.6 Hz, 1H), 3.29 (d, J = 11.6 Hz, 1H), 1.65 (s, 3H), 1.45 (s, 9H); 13C-NMR (CDCl3, 100 MHz) δ 176.3, 171.5, 157.5, 156.1, 149.6, 137.3, 123.9, 120.5, 84.9, 79.7, 45.3, 40.3, 28.4, 24.1; IR (neat) 3415, 2977, 1688, 1515, 1455, 1366, 1278, 1160, 754; HRMS (ESI) m/z calcd for C16H22N3O4S [M+H]+ 352.1331, found 352.1329; [α]D = −36.6 (c 0.98, CHCl3).
2-((((tert-Butoxy)carbonyl)amino)methyl)isonicotinonitrile (35)
To a solution of commercially available chloride 34 (5.84 g, 38.3 mmol) in DMF (250 mL) at ambient temperature was treated with potassium phthalimide (7.09 g, 38.3 mmol). After stirring for 5 h, the mixture was concentrated under vacuum. The remaining mixture was taken up in H2O (100 mL) and was filtered to collect the solid. The solid was washed with H2O (50 mL) and THF (50 mL) to obtain the desired phthalimide derivative (5.04 g, 50%) and was moved forward without further purification. To a solution of the crude phthalimide derivative (5.04 g, 19.1 mmol) in THF/MeOH (170 mL, 1:1, v/v) at ambient temperature was treated with hydrazine monohydrate (1.02 mL, 21.1 mmol). After 2 h, 1.0 M HCl (21.4 mL) was added to the mixture and was stirred for another 3 h before concentrating the reaction mixture under vacuum. The remaining residue was taken up in H2O (200 mL) and the unwanted solid was removed through filtration. The filtrate was concentrated and placed under vacuum to remove the remaining H2O. The crude solid was taken up in CH2Cl2 (150 mL) and triethylamine (8.00 mL, 57.4 mmol) and Boc2O (4.59 g, 21.1 mmol) was added. After stirring for 12 h at room temperature, the reaction was quenched with a saturated solution of NaHCO3 (200 mL), extracted with CH2Cl2 (3 × 150 mL), dried over MgSO4, and concentrated under reduced pressure. The residue was purified using flash chromatography (15% to 50% ethyl acetate in hexanes) to provided the aryl pyridine “IN” fragement (3.74 g, 84%): 1H-NMR (CDCl3, 400 MHz) δ 8.69 (d, J = 5.2 Hz, 1H), 7.51 (s, 1H), 7.39 (dd, J = 5.2, 0.8 Hz, 1H), 5.45 (s, 1H), 4.47 (d, J = 5.6 Hz, 2H), 1.44 (s, 9H); 13C-NMR (CDCl3, 100 MHz) 159.9, 155.9, 150.0, 123.6, 123.1, 121.1, 116.4, 80.1, 45.6, 28.3; IR (neat) 3337, 2978, 2934, 2245, 1709, 1514, 1246, 1168, 949; HRMS (ESI) m/z calcd for C12H16N3O3 [M+H]+ 234.1243, found 234.1237.
(R)-2-(2-(((tert-Butoxycarbonyl)amino)methyl)pyridin-4-yl)-4-methyl-4,5-dihydrothiazole-4-carboxylic acid (36)
To a solution of nitrile 35 (1.50 g, 6.43 mmol) in MeOH/pH 6.0 buffer (105 mL, 3:2, v/v) was added α-methyl-L-cysteine (1.32 g, 7.72 mmol) and NaHCO3 (1.08 g, 12.9 mmol). The reaction mixture was heated to 70 °C for 48 h. The mixture was cooled to ambient temperature and the MeOH was removed under reduced pressure. The aqueous layer was extracted with diethyl ether (3 × 30 mL) before being acidified to a pH of ~2 using 1.0 M HCl. The aqueous layer was then extracted with ethyl acetate (3 × 30 mL) and the ethyl acetate layer was dried over MgSO4. The solution was concentrated to give the desired acid (1.89 g, 85%): 1H-NMR (CDCl3, 400 MHz) δ 8.61 (d, J = 4.8 Hz, 1H), 7.68 (s, 1H), 7.58 (dd, J = 5.2, 1.6 Hz, 1H), 5.64 (bs, 1H), 4.48 (d, J = 4.8 Hz, 2H) 3.94 (d, J = 11.6 Hz, 1H), 3.38 (d, J = 11.6 Hz, 1H), 1.66 (s, 3H), 1.43 (s, 9H); 13C-NMR (CDCl3, 100 MHz) δ 175.5, 166.5, 158.5, 156.1, 148.7, 141.4, 121.2, 120.9, 85.0, 79.8, 45.1, 41.8, 28.3, 23.8; IR (neat) 3355, 2979, 2359, 1707, 1517, 1284, 1167, 754; HRMS (ESI) m/z calcd for C16H22N3O4S [M+H]+ 352.1331, found 352.1334; [α]D = −39.3 (c 1.08, CHCl3).
(S,E)-2-(Trimethylsilyl)ethyl 3-(((S)-2-((R)-2-(6-(((tert-butoxycarbonyl)amino)methyl)pyridin-2-yl)-4-methyl-4,5-dihydrothiazole-4-carboxamido)-3-methylbutanoyl)oxy)-7-(tritylthio)hept-4-enoate (37)
To a solution of Fmoc protected amine 23 (2.39 g, 2.85 mmol) in acetonitrile (195 mL) at ambient temperature was added diethylamine (15.0 mL). After 2 h, the reaction mixture was concentrated under reduced vacuum, taken back up in ethyl acetate (50 mL), and concentrated again. In a separate flask, acid 32 (1.00 g, 2.85 mmol), PyBOP (2.97 g, 5.70 mmol), and DIPEA (1.49 mL, 8.55 mmol) was combined in CH2Cl2 (195 mL). The freshly deprotected amine was added via CH2Cl2 (10 mL) to the flask containing the activated acid at ambient temperature. After 3 h, the resulting mixture was concentrated under reduced pressure. The crude residue was purified using flash chromatography (5% to 40% ethyl acetate in hexanes) to obtain the desired amide (2.13 g, 79%): 1H-NMR (CDCl3, 400 MHz) δ 8.04 (d, J = 8.0 Hz, 1H), 7.69 (t, J = 7.6 Hz, 1H), 7.15–7.38 (m, 16H), 5.67 (dt, J = 15.2, 6.8 Hz, 1H), 5.59–5.65 (m, 1H), 5,45–5.47 (m, 1H), 5.36 (dd, J = 15.6, 7.6 Hz, 1H), 4.45–4.48 (m, 3H), 4.14 (dd, J = 9.6, 7.6 Hz, 2H), 3.74 (d, J = 11.6 Hz, 1H), 3.28 (d, J = 12.0 Hz, 1H), 2.68 (dd, J = 15.6, 7.6 Hz, 1H), 2.54 (dd, J = 15.6, 5.6 Hz, 1H), 2.00–2.18 (m, 5H), 1.60 (s, 3H), 1.46 (s, 9H), 0.93–0.97 (m, 2H), 0.81 (d, J = 6.8 Hz, 3H), 0.74 (d, J = 6.8 Hz, 3H), 0.01 (s, 9H); 13C-NMR (CDCl3, 100 MHz) δ 174.4, 170.9, 170.4,169.6, 157.6, 156.0, 149.9, 144.8, 137.3, 133.9, 129.5, 127.8, 127.7, 126.6, 123.8, 120.0, 85.6, 79.6, 71.8, 66.6, 63.1, 56.6, 45.4, 40.5, 39.7, 31.3, 31.2, 31.0, 28.4, 24.8, 19.0, 17.4, 17.3, −1.5; IR (neat) 3393, 2960, 1736, 1684, 1508, 1446, 1390, 1249, 1171, 859; HRMS (ESI) m/z calcd for C52H66N4NaO7S2Si [M+Na]+ 973.4040, found 973.4042; [α]D = −19.0 (c 1.11, CHCl3).
(S,E)-2-(Trimethylsilyl)ethyl 3-(((S)-2-((R)-2-(2-(((tert-butoxycarbonyl)amino)methyl)pyridin-4-yl)-4-methyl-4,5-dihydrothiazole-4-carboxamido)-3-methylbutanoyl)oxy)-7-(tritylthio)hept-4-enoate (38)
To a solution of Fmoc protected amine 23 (1.55 g, 1.84 mmol) in acetonitrile (125 mL) at ambient temperature was added diethylamine (10.0 mL). After 2 h, the reaction mixture was concentrated under reduced vacuum, taken back up in ethyl acetate (50 mL), and concentrated again. In a separate flask, acid 36 (0.650 g, 1.84 mmol), PyBOP (1.92 g, 3.68 mmol), and DIPEA (0.96 mL, 5.52 mmol) was combined in CH2Cl2 (125 mL). The freshly deprotected amine was added via CH2Cl2 (10 mL) to the flask containing the activated acid at ambient temperature. After 3 h, the resulting mixture was concentrated under reduced pressure. The crude residue was purified using flash chromatography (5% to 40% ethyl acetate in hexanes) to obtain the desired amide (1.42 g, 81%): 1H-NMR (CDCl3, 400 MHz) δ 8.51–8.52 (m, 1H), 7.72–7.73 (m, 1H), 7.15–7.38 (m, 15H), 7.11 (d, J = 8.8 Hz, 1H), 5.69 (dt, J = 15.2, 6.8 Hz, 1H), 5.60–5.65 (m, 1H), 5.36 (dd, J = 15.2, 7.6 Hz, 1H), 4.54–4.56 (m, 2H), 4.48 (dd, J = 8.8, 4.4 Hz, 1H), 4.14 (dd, J = 10.0, 8.0 Hz, 2H), 3.84 (d, J = 11.6 Hz, 1H), 3.41 (d, J = 11.6 Hz, 1H), 2.67 (dd, J = 15,6, 8.0 Hz, 1H), 2.54 (dd, J = 16.0, 5.6 Hz, 1H), 2.03–2.18 (m, 5H), 1.56 (s, 3H), 1.44 (s, 9H), 0.92–0.97 (m, 2H), 0.81 (d, J = 6.8 Hz, 3H), 0.74 (d, J = 6.8 Hz, 3H), 0.01 (s, 9H); 13C-NMR (CDCl3, 100 MHz) δ 173.8, 170.3, 169.6, 167.0, 158.7, 155.9, 149.7, 144.8, 140.3, 133.9, 129.5, 127.8, 127.7, 126.6, 120.3, 119.9, 85.4, 79.6, 71.8, 66.6, 63.1, 56.8, 45.7, 41.7, 39.7, 31.3, 31.1, 31.0, 28.4, 24.5, 19.0, 17.4, 17.2, −1.5; IR (neat) 3390, 2960, 1737, 1681, 1593, 1504, 1444, 1248, 1170, 836, 744; HRMS (ESI) m/z calcd for C52H66N4NaO7S2Si [M+Na]+ 973.4040, found 973.4058; [α]D = −32.5 (c 1.03, CHCl3).
Trityl protected depsipeptide pyridyl “IN” macrocycle (39)
To a solution of depsipeptide 37 (2.10 g, 2.21 mmol) in CH2Cl2 (100 mL) at 0 °C was treated with TFA (20.0 mL) dropwise. The reaction mixture was allowed to stir for 18 h. The mixture was concentrated under reduced pressure, taken back up in toluene (50 mL), and concentrated again. The remaining residue was taken up in acetonitrile (2300 mL) and was treated with HATU (1.68 g, 4.42 mmol), HOBt (0.597 g, 4.42 mmol), and DIPEA (2.32 mL, 13.3 mmol) at room temperature. After 18 h, the reaction mixture was concentrated under reduced pressure. The crude residue was passed through a silica plug using an eluent of 10% MeOH in CH2Cl2. The filtrate was concentrated and taken up in ethyl acetate (150 mL). The solution was washed with a saturated solution of NaHCO3 (100 mL). The aqueous layer was extracted ethyl acetate (3 × 50 mL) and then the combined organic layers were washed with a saturated solution of NaCl (100 mL). After drying over Na2SO4 and concentrating under reduced vacuum, the crude mixture was purified using flash chromatography (1% to 6% MeOH in DCM) to obtain the macrocycle (0.334 g, 20%): 1H-NMR (CDCl3, 400 MHz) δ 7.78 (t, J = 7.6 Hz, 1H), 7.62 (d, J = 7.6 Hz, 1H), 7.15–7.37 (m, 16H), 7.06 (d, J = 4.8 Hz, 1H), 5.74 (dtd, J = 15.6, 6.8, 1.2 Hz, 1H), 5.62 (q, J = 6.0 Hz, 1H), 5.38 (dd, J = 15.6, 6.4 Hz, 1H), 4.97 (dd, J = 18.0, 6.8 Hz, 1H), 4.66 (dd, J = 10.0, 4.0 Hz, 1H), 4.29 (dd, J = 18.0, 2.0 Hz, 1H), 4.07 (d, J = 11.2 Hz, 1H), 3.36 (d, J = 11.6 Hz, 1H), 2.68–2.70 (m, 2H), 2.08–2.17 (m, 3H), 2.02 (q, J = 7.2 Hz, 2H), 1.82 (s, 3H), 0.73 (d, J = 7.2 Hz, 3H), 0.52 (d, J = 6.8 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 173.4, 170.8, 168.9, 168.7, 156.5, 149.4, 144.8, 137.7, 133.0, 129.4, 127.8, 127.3, 126.5, 124.1, 123.1, 84.9, 72.9, 66.5, 57.4, 43.8, 43.5, 41.4, 33.5, 31.5, 31.1, 24.7, 18.9, 16.7; IR (neat) 3368, 2960, 1735, 1675, 1575, 1512, 1444, 1242, 1037, 974, 744; HRMS (ESI) m/z calcd for C42H44N4NaO4S2 [M+Na]+ 755.2702, found 755.2696; [α]D = +27.4 (c 0.57, CHCl3).
Trityl protected depsipeptide pyridyl “OUT” macrocycle (40)
To a solution of depsipeptide 38 (2.00 g, 2.10 mmol) in CH2Cl2 (100 mL) at 0 °C was treated with TFA (20.0 mL) dropwise. The reaction mixture was allowed to stir for 18 h. The mixture was concentrated under reduced pressure, taken back up in toluene (50 mL), and concentrated again. The remaining residue was taken up in acetonitrile (2100 mL) and was treated with HATU (1.60 g, 4.20 mmol), HOBt (0.570 g, 4.20 mmol), and DIPEA (2.19 mL, 12.6 mmol) at room temperature. After 18 h, the reaction mixture was concentrated under reduced pressure. The crude residue was passed through a silica plug using an eluent of 10% MeOH in CH2Cl2. The filtrate was concentrated and taken up in ethyl acetate (150 mL). The solution was washed with a saturated solution of NaHCO3 (100 mL). The aqueous layer was extracted ethyl acetate (3 × 50 mL) and then the combined organic layers were washed with a saturated solution of NaCl (100 mL). After drying over Na2SO4 and concentrating under reduced vacuum, the crude mixture was purified using flash chromatography (1% to 6% MeOH in DCM) to obtain the macrocycle (0.350 g, 23%): 1H-NMR (CDCl3, 400 MHz) δ 8.65 (d, J = 5.2 Hz, 1H), 7.77 (s, 1H), 7.15–7.40 (m, 16H), 6.97 (d, J = 9.2 Hz, 1H), 6.33 (dd, J = 8.0, 5.2 Hz, 1H), 5.78 (dtd, J = 15.6, 6.4, 0.8 Hz, 1H), 5.71 (q, J = 6.0 Hz, 1H), 5.50 (dd, J = 15.6, 6.0 Hz, 1H), 4.97 (dd, J = 17.6, 8.0 Hz, 1H), 4.52 (dd, J = 8.8, 4.0 Hz, 1H), 4.02 (dd, J = 18.0, 5.2 Hz, 1H), 3.99 (d, J = 11.6 Hz, 1H), 3.38 (d, J = 11.6 Hz, 1H), 2.74–2.78 (m, 2H), 2.01–2.29 (m, 5H), 1.74 (s, 3H), 0.76 (d, J = 6.8 Hz, 3H), 0.66 (d, J = 6.8 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 173.1, 169.9, 168.4, 168.2, 159.3, 150.5, 144.6, 140.0, 132.6, 129.4, 128.1, 127.9, 126.7, 120.8, 116.9, 84.5, 71.5, 66.7, 58.0, 44.5, 43.5, 40.8, 33.4, 31.2, 31.0, 24.9, 18.9, 17.2; IR (neat) 3405, 2962, 1739, 1683, 1552, 1508, 1404, 1255, 1183, 1029, 748; HRMS (ESI) m/z calcd for C42H44N4NaO4S2 [M+Na]+ 755.2702, found 755.2694; [α]D = +27.6 (c 0.58, CHCl3).
Depsipeptide pyridyl “IN” thiol (6)
To a solution of trityl protected thiol 39 (300 mg, 0.409 mmol) in CH2Cl2 (55 mL) at 0 °C was treated with TFA (2.25 mL) and triisopropylsilane (0.17 mL, 0.818 mmol). The reaction mixture was warmed to ambient temperature and stirred for 2 h. The mixture was concentrated and purified using flash chromatography (1% to 6% MeOH in CH2Cl2) to obtain the desired thiol (164 mg, 82%): 1H-NMR (CDCl3, 400 MHz) δ 7.86 (t, J = 7.6 Hz, 1H), 7.68 (d, J = 7.6 Hz, 1H), 7.42 (d, J = 8.0 Hz, 1H), 7.35 (bs, 1H), 7.25 (d, J = 9.6 Hz, 1H), 5.85 (dtd, J = 15.2, 7.2, 1.2 Hz, 1H), 5.64–5.69 (m, 1H), 5.54 (ddt, J = 15.6, 6.8, 1.2 Hz, 1H), 5.11 (dd, J = 18.0, 7.6 Hz, 1H), 4.69 (dd, J = 10.0, 4.0 Hz, 1H), 4.34 (dd, J = 18.0, 1.6 Hz, 1H), 4.14 (d, J = 11.2 Hz, 1H), 3.46 (d, J = 11.6 Hz, 1H), 2.87 (dd, J = 15.2, 10.0 Hz, 1H), 2.71 (dd, J = 15.2, 2.4 Hz, 1H), 2.53 (qd, J = 7.6, 0.8 Hz, 1H), 2.31–2.37 (m, 2H), 2.09–2.17 (m, 1H), 1.91 (s, 3H), 1.40 (t, J = 8.0 Hz, 1H), 0.71 (d, J = 6.8 Hz, 3H), 0.49 (d, J = 6.8 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 174.8, 172.6, 169.7, 168.5, 156.9, 148.0, 138.3, 132.8, 128.4, 125.1, 123.7, 83.5, 72.6, 57.5, 43.7, 43.5, 41.2, 36.4, 33.3, 23.8, 23.8, 18.6, 16.5; IR (neat) 3369, 2962, 1735, 1670, 1572, 1515, 1426, 1295, 1174, 989, 799; HRMS (ESI) m/z calcd for C23H31N4O4S2 [M+H]+ 491.1787, found 491.1791; [α]D = +58.5 (c 0.53, CHCl3).
Depsipeptide pyridyl “OUT” thiol (8)
To a solution of trityl protected thiol 40 (320 mg, 0.437 mmol) in CH2Cl2 (60 mL) at 0 °C was treated with TFA (2.30 mL) and triisopropylsilane (0.18 mL, 0.873 mmol). The reaction mixture was warmed to ambient temperature and stirred for 2 h. The mixture was concentrated and purified using flash chromatography (1% to 6% MeOH in CH2Cl2) to obtain the desired thiol (188 mg, 89%): 1H-NMR (CDCl3, 400 MHz) δ 8.70 (d, J = 5.2 Hz, 1H), 7.89 (s, 1H), 7.45 (dd, J = 5.2, 1.6 Hz, 1H), 6.92 (d, J = 9.2 Hz, 1H), 6.36–6.39 (m, 1H), 5.87 (dt, J = 15.6, 6.4 Hz, 1H), 5.73–5.78 (m, 1H), 5.67 (ddt, J = 15.6, 6.4, 1.2 Hz, 1H), 5.09 (dd, J = 17.6, 8.0 Hz, 1H), 4.57 (dd, J = 9.2, 4.0 Hz, 1H), 4.37 (dd, J = 17.6, 4.8 Hz, 1H), 4.05 (d, J = 11.6 Hz, 1H), 3.40 (d, J = 11.6 Hz, 1H), 2.76–2.89 (m, 2H), 2.50–2.64 (m, 2H), 2.30–2.44 (m, 2H), 2.11–2.20 (m, 1H), 1.78 (s, 3H), 1.38 (t, J = 7.6 Hz, 1H), 0.75 (d, J = 6.8 Hz, 3H), 0.66 (d, J = 6.8 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 173.1, 169.3, 168.7, 168.4, 158.7, 149.4, 141.1, 132.4, 128.8, 121.3, 117.8, 84.5, 72.2, 57.8, 43.6, 43.6, 40.3, 36.1, 33.6, 24.9, 23.7, 18.9, 17.0; IR (neat) 3319, 2962, 1736, 1670, 1551, 1508, 1242, 1184, 1027, 971, 730; HRMS (ESI) m/z calcd for C23H31N4O4S2 [M+H]+ 491.1787, found 491.1795; [α]D = +68.3 (c 0.74, CHCl3).
Depsipeptide pyridyl “IN” (5)
To a solution of depsipeptide pyridine “IN” thiol 6 (20.0 mg, 0.041 mmol) in THF (0.8 mL) at ambient temperature was treated with N-octanoyl–imidazole (15.8 mg, 0.082 mmol), imidazole (5.6 mg, 0.082 mmol), and DMAP (0.5 mg, 0.004 mmol). The reaction mixture was warmed to 50 °C and stirred for 6 h. The mixture was cooled and concentrated and purified using flash chromatography (0.5% to 4% MeOH in CH2Cl2) to obtain the desired octanoyl masked thiol (17.4 mg, 69%): 1H-NMR (CDCl3, 400 MHz) δ 7.82 (t, J = 7.6 Hz, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.39 (d, J = 8.0 Hz, 1H), 7.30 (d, J = 9.6 Hz, 1H), 5.85 (dt, J = 15.6, 6.8 Hz, 1H), 5.63 (td, J = 16.0, 2.0 Hz, 1H), 5.52 (dd, J = 15.2, 6.8 Hz, 1H), 5.06–5.12 (m, 1H), 4.67 (dd, J = 9.6, 3.6 Hz, 1H), 4.32 (d, J = 18.0 Hz, 1H), 4.15 (d, J = 11.6 Hz, 1H), 3.42 (d, J = 11.6 Hz, 1H), 2.81–2.90 (m, 2H), 2.63 (dd, J = 14.0, 1.2 Hz, 1H), 2.47 (t, J = 7.6 Hz, 2H), 2.27 (q, J = 7.6 Hz, 2H), 2.06–2.13 (m, 1H), 1.92 (s, 3H), 1.56–1.61 (m, 2H), 1.23–1.27 (m, 10H), 0.84 (t, J = 6.8 Hz, 3H), 0.69 (d, J = 6.8 Hz, 3H), 0.49 (d, J = 6.8 Hz, 3H); 13C-NMR (CD3OD, 100 MHz) δ 199.4, 174.2, 173.7, 170.7, 168.7, 157.5, 147.7, 138.7, 132.3, 128.7, 125.0, 123.2, 83.4, 72.9, 54.1, 43.4, 42.8, 42.7, 40.2, 33.1, 31.9, 31.4, 28.6, 28.5, 27.4, 25.3, 23.0, 22.2, 18.1, 15.3, 13.0; IR (neat) 2959, 2928, 2856, 1737, 1682, 1571, 1419, 1239, 990; HRMS (ESI) m/z calcd for C31H45N4O5S2 [M+H]+ 617.2826, found 617.2832; [α]D = +125 (c 0.80, CHCl3).
Depsipeptide pyridyl “OUT” (7)
To a solution of depsipeptide pyridine “OUT” thiol 8 (85.0 mg, 0.179 mmol) in CH2Cl2 (15.0 mL) at ambient temperature was treated with octanoyl chloride (.153 mL, 0.894 mmol) and triethylamine (0.050 mL, 0.357 mmol). The reaction mixture was stirred for 2 h before cooling to 0 °C and quenching with methanol (5.0 mL). The mixture was cooled and concentrated and purified using flash chromatography (0.5% to 4% MeOH in CH2Cl2) to obtain the desired octanoyl masked thiol (54.0 mg, 49%): 1H-NMR (CDCl3, 400 MHz) δ 8.67 (d, J = 5.2 Hz, 1H), 7.91 (s, 1H), 7.45 (d, J = 4.8 Hz, 1H), 6.89 (d, J = 8.8 Hz, 1H), 6.57–6.60 (m, 1H), 5.85 (dt, J = 14.8, 7.2 Hz, 1H), 5.62–5.73 (m, 2H), 5.10 (dd, J = 18.0, 8.0 Hz, 1H), 4.56 (dd, J = 8.8, 3.6 Hz, 1H), 4.42 (dd, J = 17.6, 3.6 Hz, 1H), 4.05 (d, J = 11.6 Hz, 1H), 3.40 (d, J = 11.6 Hz, 1H) 2.74–2.90 (m, 3H), 2.49 (t, J = 7.2 Hz, 2H), 2.28–2.34 (m, 1H), 2.12–2.19 (m, 1H), 1.78 (s, 3H), 1.58–1.63 (m, 2H), 1.24–1.26 (m, 10H), 0.84 (t, J = 6.8 Hz, 3H), 0.74 (d, J = 6.8 Hz, 3H), 0.65 (d, J = 6.8 Hz, 3H); 13C-NMR (CD3OD, 100 MHz) δ 199.4, 173.9, 170.2, 169.6, 168.2, 158.6, 149.0, 141.7, 132.0, 128.5, 121.5, 117.9, 84.3, 72.4, 57.7, 43.4, 42.7, 42.7, 39.1, 33.4, 32.0, 31.4, 28.6, 28.5, 27.4, 25.3, 23.6, 22.2, 18.1, 16.1, 13.0; IR (neat) 2959, 2929, 2856, 1740, 1684, 1553, 1420, 1234, 987; HRMS (ESI) m/z calcd for C31H45N4O5S2 [M+H]+ 617.2826, found 617.2827; [α]D = +94.3 (c 0.62, CHCl3).
(S,E)-Methyl 3-((S)-2-((R)-2-(6-(((tert-butoxycarbonyl)amino)methyl)pyridin-2-yl)-4-methyl-4,5-dihydrothiazole-4-carboxamido)-3-methylbutanamido)-7-(tritylthio)hept-4-enoate (41)
To a solution of Boc protected amine 28 (500 mg, 0.793 mmol) in CH2Cl2 (30 mL) at 0 °C was treated with TFA (3.0 mL). After 2 h, the reaction mixture was concentrated under vacuum, taken back up in toluene (15 mL), and concentrated again. In another flask, acid 32 (278 mg, 0.793 mmol), PyBOP (822 mg, 1.58 mmol), and DIPEA (0.41 mL, 2.37 mmol) were combined in CH2Cl2 (35 mL). The freshly deprotected amine was added via CH2Cl2 (10 mL) to the flask containing the activated acid at ambient temperature. After 3 h, the resulting mixture was concentrated under reduced pressure. The crude residue was purified using flash chromatography (20% to 90% ethyl acetate in hexanes) to obtain the desired amide (570 mg, 84%): 1H-NMR (CDCl3, 400 MHz) δ 8.00 (d, J = 7.6 Hz, 1H), 7.73 (t, J = 7.6 Hz, 1H), 7.34–7.37 (m, 7H), 7.30 (d, J = 8.8 Hz, 1H), 7.15–7.26 (m, 9H), 6.59 (d, J = 10.0 Hz, 1H), 5.47 (dtd, J = 15.6, 6.4, 0.8 Hz, 1H), 5.36 (dd, J = 15.2, 6.0 Hz, 1H), 4.70–4.74 (m, 1H), 4.46 (d, J = 5.2 Hz, 2H), 4.17 (dd, J = 8.8, 6.0 Hz, 1H), 3.67 (d, J = 11.6 Hz, 1H), 3.61 (s, 3H), 3.28 (d, J = 11.6 Hz, 1H), 2.57 (d, J = 5.6 Hz, 2H), 2.08–2.16 (m, 3H) 2.01 (q, J =6.8 Hz, 2H), 1.56 (s, 3H), 1.45 (s, 9H), 0.82 (d, J = 6.8 Hz, 3H), 0.79 (d, J = 6.8 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 174.7, 171.5, 171.1, 169.7, 157.7, 156.0, 149.8, 144.8, 137.3, 130.6, 129.5, 127.8, 126.5, 123.8, 120.1, 85.6, 79.6, 66.5, 58.2, 51.7, 47.4, 45.4, 40.6, 38.7, 31.4, 31.3, 31.1, 28.4, 24.7, 19.3, 17.9; IR (neat) 2969, 1654, 1508, 1444, 1365, 1169, 1031, 751; HRMS (ESI) m/z calcd for C48H57N5NaO6S2 [M+Na]+ 886.3648, found 886.3639; [α]D = −30.8 (c 0.67, CHCl3).
(S,E)-Methyl 3-((S)-2-((R)-2-(2-(((tert-butoxycarbonyl)amino)methyl)pyridin-4-yl)-4-methyl-4,5-dihydrothiazole-4-carboxamido)-3-methylbutanamido)-7-(tritylthio)hept-4-enoate (42)
To a solution of Boc protected amine 28 (500 mg, 0.793 mmol) in CH2Cl2 (30 mL) at 0 °C was treated with TFA (3.0 mL). After 2 h, the reaction mixture was concentrated under vacuum, taken back up in toluene (15 mL), and concentrated again. In another flask, acid 36 (278 mg, 0.793 mmol), PyBOP (822 mg, 1.58 mmol), and DIPEA (0.41 mL, 2.37 mmol) were combined in CH2Cl2 (35 mL). The freshly deprotected amine was added via CH2Cl2 (10 mL) to the flask containing the activated acid at ambient temperature. After 3 h, the resulting mixture was concentrated under reduced pressure. The crude residue was purified using flash chromatography (20% to 100% ethyl acetate in hexanes) to obtain the desired amide (630 mg, 92%): 1H-NMR (CDCl3, 400 MHz) δ 8.61 (dd, J = 4.8, 0.8 Hz, 1H), 7.59–7.61 (m, 2H), 7.15–7.38 (m, 16H), 6.59 (d, J = 8.8 Hz, 1H), 5.47 (dtd, J = 15.6, 6.4, 0.8 Hz, 1H), 5.35 (dd, J = 15.2, 6.0 Hz, 1H), 4.69–4.75 (m, 1H), 4.48 (d, J = 5.2 Hz, 2H), 4.15 (dd, J = 8.8, 6.4 Hz, 1H), 3.79 (d, J = 11.6 Hz, 1H), 3.61 (s, 3H), 3.37 (d, J = 11.2 Hz, 1H), 2.57 (d, J = 5.2 Hz, 2H), 2.08–2.17 (m, 3H), 2.01 (q, J = 6.8 Hz, 2H), 1.55 (s, 3H), 1.45 (s, 9H), 0.82 (d, J = 6.4 Hz, 3H), 0.79 (d, J = 6.8 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 174.1, 171.6, 169.5, 167.2, 158.8, 155.9, 149.7, 144.8, 140.2, 130.6, 129.5, 129.5, 127.8, 126.5, 120.3, 120.0, 85.3, 79.6, 66.5, 58.4, 51.7, 47.3, 45.7, 41.7, 38.6, 31.4, 31.1, 31.0, 28.4, 24.4, 19.3, 17.9; IR (neat) 2969, 1654, 1511, 1443, 1390, 1366, 1168, 846, 750; HRMS (ESI) m/z calcd for C48H57N5NaO6S2 [M+Na]+ 886.3648, found 886.3661; [α]D = −29.9 (c 0.64, CHCl3).
Trityl protected peptide isostere pyridyl “IN” macrocycle (43)
To a solution of methyl ester 41 (530 mg, 0.613 mmol) in THF/H2O (15 mL, 2:1, v/v) at ambient temperature was added lithium hydroxide monohydrate (77.2 mg, 1.84 mmol). After 2 h, the reaction was cooled to 0 °C and acidified to a pH of ~3 using 1.0 M HCl. The mixture was extracted with CH2Cl2 (3 × 15 mL), dried over Na2SO4, and concentrated under vacuum. The freshly furnished acid was taken up in CH2Cl2 (20 mL) and cooled to 0 °C. The flask was treated with TFA (2.0 mL) and warmed to ambient temperature. After 2 h, the mixture was concentrated under reduced pressure, taken back up in toluene (25 mL), and concentrated again. The crude TFA salt was taken up in acetonitrile (20 mL) and was treated with DIPEA (0.68 mL, 3.90 mmol). The mixture was allowed to stir for 30 min. In another flask containing acetonitrile (610 mL) was added HATU (488 mg, 1.28 mmol), HOBt (173 mg, 1.28 mmol), and DIPEA (0.68 mL, 3.90 mmol) at room temperature. The flask containing the peptide was added to the flask containing the coupling reagents dropwise over 12 h using a syringe pump. After another 12 h, the reaction mixture was concentrated under reduced pressure. The crude residue was taken up in CH2Cl2 (15 mL) and the resulting solid was removed using filtration. The filtrate was concentrated and purified using flash chromatography (1% to 6% MeOH in DCM) to obtain the macrocycle (146 mg, 33%): 1H-NMR (CDCl3, 400 MHz) δ 7.81 (t, J = 7.6 Hz, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.16–7.37 (m, 16H), 6.92 (d, J = 6.4 Hz, 1H), 6.73 (d, J = 10.8 Hz, 1H), 6.42 (d, J = 8.4 Hz, 1H), 5.47 (dt, J = 15.6, 6.4 Hz, 1H), 5.34 (dd, J = 15.6, 5.6 Hz, 1H), 5.03 (dd, J = 18.0, 7.2 Hz, 1H), 4.83–4.90 (m, 1H), 4.60 (dd, J = 10.8, 3.2 Hz, 1H), 4.24 (dd, J = 17.6, 2.0 Hz, 1H), 3.95 (d, J = 12.0 Hz, 1H), 3.45 (d, J = 12.0 Hz, 1H), 2.67 (dd, J = 14.0, 3.6 Hz, 1H), 2.48–2.52 (m, 1H), 2.44 (dd, J = 14.4, 10.8 Hz, 1H), 2.16 (t, J = 8.0 Hz, 2H), 1.99–2.06 (m, 2H), 1.87 (s, 3H), 0.74 (d, J = 6.8 Hz, 3H), 0.32 (d, J = 6.8 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 173.8, 172.8, 169.8, 169.8, 157.5, 149.2, 144.8, 138.0, 130.3, 129.6, 129.5, 127.8, 126.6, 124.6, 122.9, 84.8, 66.5, 57.8, 48.3, 44.3, 43.7, 41.4, 31.4, 31.3, 31.1, 24.4, 19.3, 15.3; IR (neat) 2930, 1681, 1644, 1561, 1489, 1274, 1185, 1030, 842; HRMS (ESI) m/z calcd for C42H45N5NaO3S2 [M+Na]+ 754.2862, found 754.2872; [α]D = +58.0 (c 0.10, CHCl3).
Trityl protected peptide isostere pyridyl “OUT” macrocycle (44)
To a solution of methyl ester 42 (480 mg, 0.555 mmol) in THF/H2O (15 mL, 2:1, v/v) at ambient temperature was added lithium hydroxide monohydrate (69.9 mg, 1.67 mmol). After 2 h, the reaction was cooled to 0 °C and acidified to a pH of ~3 using 1.0 M HCl. The mixture was extracted with CH2Cl2 (3 × 15 mL), dried over Na2SO4, and concentrated under vacuum. The freshly furnished acid was taken up in CH2Cl2 (20 mL) and cooled to 0 °C. The flask was treated with TFA (2.0 mL) and warmed to ambient temperature. After 2 h, the mixture was concentrated under reduced pressure, taken back up in toluene (25 mL), and concentrated again. The crude TFA salt was taken up in acetonitrile (20 mL) and was treated with DIPEA (0.61 mL, 3.52 mmol). The mixture was allowed to stir for 30 min. In another flask containing acetonitrile (550 mL) was added HATU (439 mg, 1.16 mmol), HOBt (157 mg, 1.16 mmol), and DIPEA (0.61 mL, 3.52 mmol) at room temperature. The flask containing the peptide was added to the flask containing the coupling reagents dropwise over 12 h using a syringe pump. After another 12 h, the reaction mixture was concentrated under reduced pressure. The crude residue was taken up in CH2Cl2 (15 mL) and the resulting solid was removed using filtration. The filtrate was concentrated and purified using flash chromatography (1% to 6% MeOH in DCM) to obtain the macrocycle (128 mg, 32%): 1H-NMR (CDCl3, 400 MHz) δ 8.64 (dd, J = 4.8, 0.8 Hz, 1H), 7.79 (d, J = 0.8 Hz, 1H), 7.16–7.32 (m, 16H), 6.29–6.35 (m, 2H), 6.12 (d, J = 10.0 Hz, 1H), 5.54–5.57 (m, 2H) 4.81–4.86 (m, 1H), 4.67 (dd, J = 17.2, 6.4 Hz, 1H), 4.40 (dd, J = 10.0, 4.4 Hz, 1H), 4.15 (dd, J = 17.2, 5.6 Hz, 1H), 3.80 (d, J = 11.6 Hz, 1H), 3.49 (d, J = 11.6 Hz, 1H), 2.72 (dd, J = 14.8, 4.8 Hz, 1H), 2.51 (dd, J = 14.8, 5.6 Hz, 1H), 2.34–2.41 (m, 1H), 2.23 (t, J = 7.2 Hz, 2H), 1.92–2.12 (m, 2H), 1.70 (s, 3H), 0.84 (d, J = 6.8 Hz, 3H), 0.60 (d, J = 6.8 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 173.1, 170.4, 170.1, 170.1, 159.1, 150.4, 144.6, 139.7, 130.3, 130.1, 129.4, 127.9, 126.7, 120.7, 117.9, 84.5, 66.7, 59.0, 47.9, 45.1, 44.1, 41.3, 31.3, 31.2, 23.8, 23.8, 19.4, 16.5; IR (neat) 2969, 2925, 1708, 1644, 1561, 1409, 1274, 1185, 1030, 842; HRMS (ESI) m/z calcd for C42H45N5NaO3S2 [M+Na]+ 754.2862, found 754.2867; [α]D = +32.3 (c 0.13, CHCl3).
Peptide isostere pyridyl “IN” thiol (10)
To a solution of trityl protected thiol 43 (150 mg, 0.205 mmol) in CH2Cl2 (27.5 mL) at 0 °C was treated with TFA (1.12 mL) and triisopropylsilane (0.089 mL, 0.410 mmol). The reaction mixture was warmed to ambient temperature and stirred for 2 h. The mixture was concentrated and purified using flash chromatography (1% to 6% MeOH in CH2Cl2) to obtain the desired thiol (94.6 mg, 94%): 1H-NMR (CDCl3, 400 MHz) δ 7.85 (t, J = 7.6 Hz, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.40 (d, J = 8.0 Hz, 1H), 7.02 (d, J = 5.6 Hz, 1H), 6.74 (d, J = 10.8 Hz, 1H), 6.56 (d, J = 8.0 Hz, 1H), 5.60 dtd, J = 15.2, 6.8, 1.2 Hz, 1H), 5.49 (dd, J = 15.6, 6.0 Hz, 1H), 5.08 (dd, J = 17.6, 7.2 Hz, 1H), 4.89–4.96 (m, 1H), 4.63 (dd, J = 10.8, 2.8 Hz, 1H), 4.32 (dd, J = 17.6, 1.6 Hz, 1H), 3.98 (d, J = 11.6 Hz, 1H), 3.49 (d, J = 11.6 Hz, 1H), 2.69 (dd, J = 14.0, 3.2 Hz, 1H), 2.49–2.55 (m, 4H), 2.31 (q, J = 6.8 Hz, 2H), 1.91 (s, 3H), 1.38 (t, J = 8.0 Hz, 1H), 0.74 (d, J = 6.8 Hz, 3H), 0.31 (d, J = 6.8 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 174.5, 173.4, 170.1, 169.9, 157.7, 148.6, 138.2, 131.2, 129.1, 125.1, 123.2, 84.1, 57.8, 48.6, 44.3, 43.7, 41.2, 36.3, 31.2, 24.0, 24.0, 19.3, 15.2; IR (neat) 2926, 2864, 1667, 1577, 1500, 1293, 1181, 1049; HRMS (ESI) m/z calcd for C23H32N5O3S2 [M+H]+ 490.1947, found 490.1946; [α]D = +105 (c 0.08, CHCl3).
Peptide isostere pyridyl “OUT” thiol (12)
To a solution of trityl protected thiol 44 (115 mg, 0.157 mmol) in CH2Cl2 (22.0 mL) at 0 °C was treated with TFA (0.90 mL) and triisopropylsilane (0.065 mL, 0.314 mmol). The reaction mixture was warmed to ambient temperature and stirred for 2 h. The mixture was concentrated and purified using flash chromatography (1% to 6% MeOH in CH2Cl2) to obtain the desired thiol (68.4 mg, 89%): 1H-NMR (CDCl3, 400 MHz) δ 8.66 (d, J = 3.6 Hz, 1H), 7.76 (s, 1H), 7.35 (d, J = 4.4 Hz, 1H), 7.01 (d, J =8.4 Hz, 1H) 6.86–6.88 (m, 1H), 6.19 (d, J = 10.4 Hz, 1H), 5.52–5.53 (m, 2H), 4.89–4.91 (m, 1H), 4.85 (d, J = 17.6 Hz, 1H), 4.46 (dd, J = 10.4, 4.0 Hz, 1H), 4.36 (d, J = 17.2 Hz, 1H), 3.84 (d, J = 12.0 Hz, 1H), 3.48 (d, J = 12.0 Hz, 1H), 2.44–2.72 (m, 5H), 2.30 (q, J = 6.4 Hz, 2H), 1.78 (s, 3H), 1.31 (t, J = 7.2 Hz, 1H), 0.80 (d, J = 6.8 Hz, 3H), 0.49 (d, J = 6.8 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 173.3, 171.2, 159.6, 150.1, 140.3, 130.8, 128.9, 121.0, 117.8, 84.8, 58.6, 47.9, 44.0, 40.0, 36.2, 31.2, 24.0, 23.9, 23.7, 19.5, 16.1; IR (neat) 2965, 2930, 1655, 1534, 1410, 1201, 1142, 1026; HRMS (ESI) m/z calcd for C23H32N5O3S2 [M+H]+ 490.1947, found 490.1952; [α]D = +129 (c 0.08, CHCl3).
Peptide isostere pyridyl “IN” (9)
To a solution of peptide pyridine “IN” thiol 10 (25.0 mg, 0.051 mmol) in THF (1.0 mL) at ambient temperature was treated with N-octanoyl–imidazole (19.8 mg, 0.102 mmol), imidazole (6.9 mg, 0.102 mmol), and DMAP (0.6 mg, 0.005 mmol). The reaction mixture was warmed to 50 °C and stirred for 6 h. The mixture was cooled and concentrated and purified using flash chromatography (0.5% to 6% MeOH in CH2Cl2) to obtain the desired octanoyl masked thiol (20.6 mg, 66%): 1H-NMR (CD3OD, 400 MHz) δ 8.08 (t, J = 7.6 Hz, 1H), 7.84 (d, J = 7.6 Hz, 1H), 7.66 (d, J = 7.6 Hz, 1H), 7.00 (d, J = 10.8 Hz, 1H), 5.52–5.64 (m, 2H), 4.92 (d, J = 17.6 Hz, 1H), 4.77–4.83 (m, 1H), 4.53–4.57 (m, 1H), 4.38 (d, J = 17.2 Hz, 1H), 3.93 (d, J = 11.6 Hz, 1H), 3.67 (d, J = 12.0 Hz, 1H), 2.89 (t, J = 7.2 Hz, 2H), 2.68 (dd, J = 14.0, 11.2 Hz, 1H), 2.53 (dd, J = 14.0, 4.0 Hz, 1H), 2.52 (t, J = 7.2 Hz, 2H), 2.34–2.42 (m, 1H), 2.23–2.30 (m, 2H), 1.89 (s, 3H), 1.59–1.63 (m, 2H), 1.26–1.29 (m, 10H), 0.87 (t, J = 6.8 Hz, 3H), 0.75 (d, J = 7.2 Hz, 3H), 0.35 (d, J = 6.8 Hz, 3H); 13C-NMR (CD3OD, 100 MHz) δ 203.4, 178.9, 177.9, 175.9, 174.4, 161.2, 151.4, 143.9, 143.8 135.0, 132.5, 129.7, 127.3, 87.3, 61.6, 47.5, 47.3, 47.1, 43.8, 35.8, 35.3, 34.9, 32.5, 32.4, 31.6, 29.3, 26.5, 26.1, 22.4, 18.5, 16.9; IR (neat) 2957, 2928, 2855, 1678, 1530, 1403, 965; HRMS (ESI) m/z calcd for C31H46N5O4S2 [M+H]+ 616.2986, found 616.2988; [α]D = +107 (c 0.23, CHCl3).
Peptide isostere pyridyl “OUT” (11)
To a solution of peptide pyridine “OUT” thiol 12 (8.0 mg, 0.016 mmol) in CH2Cl2 (0.2 mL) at ambient temperature was treated with octanoyl chloride (.005 mL, 0.032 mmol), DIPEA (0.011 mL, 0.064 mmol), and DMAP (0.1 mg, 0.008 mmol). The reaction mixture was stirred for 2 h before cooling to 0 °C and quenching with methanol (5.0 mL). The mixture was cooled and concentrated and purified using flash chromatography (0.5% to 6% MeOH in CH2Cl2) to obtain the desired octanoyl masked thiol (5.4 mg, 55%): 1H-NMR (CDCl3, 400 MHz) δ 8.65 (dd, J = 4.8, 0.8 Hz, 1H), 7.88 (d, J = 0.8 Hz, 1H), 7.33 (dd, J = 4.8, 1.6 Hz, 1H), 6.27–6.33 (m, 2H), 6.12 (d, J = 10.0 Hz, 1H), 5.61–5.72 (m, 2H), 4.90–4.95 (m, 1H), 4.75 (dd, J = 16.8, 6.8 Hz, 1H), 4.53 (dd, J = 17.2, 6.0 Hz, 1H), 4.46 (dd, J = 10.4, 4.0 Hz, 1H), 3.83 (d, J = 12.0, 1H), 3.48 (d, J = 11.6 Hz, 1H), 2.86 (t, J = 7.2 Hz, 2H), 2.71 (dd, J = 15.6, 4.4 Hz, 1H), 2.56 (dd, J = 15.6, 7.6 Hz, 1H), 2.43–2.51 (m, 2H), 2.26–2.33 (m, 1H), 1.77 (s, 3H), 1.56–1.64 (m, 2H), 1.24–1.29 (m, 10H), 0.85 (t, J = 6.8 Hz, 3H), 0.82 (d, J = 6.8 Hz, 3H), 0.52 (d, J = 6.8 Hz, 3H); 13C-NMR (CD3OD, 100 MHz) δ 199.5, 173.4, 171.7, 170.9, 170.4, 158.3, 148.5, 142.1, 130.9, 128.5, 121.3, 118.9, 84.5, 58.1, 43.7, 43.4, 42.9, 39.2, 32.0, 31.4, 31.1, 28.6, 28.5, 27.7, 25.4, 22.3, 22.2, 18.9, 15.8, 12.9; IR (neat) 2956, 2927, 2855, 1679, 1649, 1519, 1403, 1031, 965; HRMS (ESI) m/z calcd for C31H46N5O4S2 [M+H]+ 616.2981, found 616.2981; [α]D = +40.6 (c 0.35, CHCl3).
HDAC Biochemical Assay
Compounds 1-12 were tested against HDAC1-9 and the activity was determined with an optimized homogenous assay performed in a 384-well plate. Reactions were performed in assay buffer (50 mM HEPES, 100 mM KCl, 0.001% Tween-20, 0.05% BSA and pH 7.4. Additional 200 μM TCEP was added for HDAC6) and followed by fluorogenic release of 7-amino-4-methylcoumarin from substrate upon deacetylase and trypsin enzymatic activity. Fluorescence measurements were obtained every five minutes using a multilabel plate reader and plate-stacker (Envision; Perkin-Elmer). Each plate was analyzed by plate repeat, and the first derivative within the linear range was imported into analytical software (Spotfire DecisionSite). Replicate experimental data from incubations with inhibitor were normalized to DMSO controls ([DMSO] < 0.5%). IC50 is determined by logistic regression with unconstrained maximum and minimum values. The recombinant, full-length HDAC protein (BPS Biosciences) was incubated with fluorophore conjugates substrate, MAZ1600 and MAZ1675 at Km=[substrate].
797 and 10326 Cell Viability
For dose-response cellular viability assays, 797 and 10326 cells were seeded onto separate 384-well tissue culture-treated plates at a density of 2.0 × 104 cells/well in a volume of 50 μL/well. Addition of compound was performed with a JANUS Workstation (PerkinElmer Life and Analytical Sciences) using a 384-well pinhead tool that is calibrated to deliver 100 nL drug/well. Final DMSO concentration in the well was 0.02%. After 48 hours of incubation with compound, cells were analyzed for cell viability using the ATPLite (Perkin Elmer) luminescent assay kit per the manufacturer’s instructions. Luminescence was read on an EnVision 2104 Multilabel Plate Reader (PerkinElmer Life and Analytical Sciences). Replicate measurements were analyzed with respect to dose and estimates of IC50 were calculated by logistic regression (GraphPad Prism). Standard error of the LogIC50 values were performed using GraphPad according to the following equation:
where:
Pi: i-th adjustable(non-constant) parameter SS: sum of squared residuals
DF: degrees of freedom (the number of data points minus number of parameters fit by regression)
Cov(i,i): i-th diagonal element of covariance matrix
sqrt(): square root
Computational Details
An adapted strategy was used to investigate the conformational space of Largazole and the pyridyl derivatives described here in the free thiol form and bound to HDACs 1, 6, and 8.10 Monte Carlo conformational searches were used to generate the conformational ensemble of the ligands in solution. Then, molecular dynamics (MD) simulations were used to sample the available conformations of the bound ligands. The bound conformations were compared to the conformational ensemble in solution via heavy atom root-mean-squared distance (rmsd) and the energy difference between the closest conformation and the global minimum was recorded.
Molecular Dynamics
Initial coordinates for the protein structures were derived from previously developed homology models for HDACs 1113,21,22 and 615 and the recently published HDAC8-Largazole crystal structure14 (pdb code 3RQD). For MD simulations the ionizable residues were set to most likely ionization states at pH 7. The 6, 8, 10 and 12 were modeled with the pyridyl nitrogen protonated. The 15 inhibitor-enzyme complexes were solvated with a periodic box of TIP3P23 water molecules extending 10 Å from any protein atoms. The crystallographic potassium ions were kept for the HDAC8 complexes and sodium ions were added to neutralize the system.
The ff0324,25 version of the all-atom AMBER force field and GAFF26 were employed to model the protein structures and inhibitors, respectively. Atom-centered partial charges were fit using the RESP method.27 Energy minimizations were used first to relax only solvent molecules and counterions before relaxing the full complexes to remove any bad contacts in the initial geometries. The MD simulations were performed with the PMEMD module in the AMBER 12 suite of programs. All bonds containing hydrogen were constrained with the SHAKE28 algorithm and the timestep was chosen to be 2 fs. A nonbonded cutoff 10.0 Å was chosen, and the nonbonded pair list was updated every 25 steps. The temperature was kept constant at 300 K using Langevin dynamics with a collision frequency of 1.0 ps−1, and the pressure was kept constant at 1 atm with isotropic position scaling. Periodic boundary conditions were employed to simulation a continuous system with contributions from long range interactions included with the Particle-Mesh-Ewald (PME) method.
Supplementary Material
Scheme 7.

Synthesis of peptide containing inhibitors.
Reagents and conditions: (a) TFA, CH2Cl2; (b) 32, PyBOP, DIPEA, 84% over 2 steps; (c) 36, PyBOP, DIPEA, 92% over 2 steps; (d) LiOH, THF, H2O; (e) TFA, CH2Cl2; (f) HATU, HOBt, DIPEA, CH3CN, 33% over 3 steps; (g) HATU, HOBt, DIPEA, CH3CN, 32% over 3 steps; (h) TFA, iPr3SiH, CH2Cl2, 94%; (i) TFA, iPr3SiH, CH2Cl2, 89%; (j) N-octanoyl–imidazole, imidazole, DMAP, THF, 50 ° C, 66%; (k) octanoyl chloride, Et3N, 55%.
Acknowledgments
This work was supported by the National Institutes of Health (Grant 1RO1 CA152314 to RMW, JEB and OW). We also acknowledge financial support from the Colorado State University Cancer Supercluster. Mass Spectra were obtained on instruments supported by the NIH Shared Instrument Grant GM49631. We thank Ralph Mazitschek for the provision of acetylated substrate. JEB acknowledges support by grants from the National Cancer Institute (1K08CA128972) and the Burroughs-Wellcome Foundation (CAMS). We would also like to acknowledge Christine Dunne for her support with the manuscript.
Abbreviations
- HDACi
Histone Deacetylase Inhibitor
Footnotes
The authors declare no competing financial interest. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. 1H-NMR and 13C-NMR of all compounds reported in the experimental section, LC50 plots, sequence alignment and analysis of surface residues surrounding the active site, RMSD plots and pymol sessions files of all complexes of HDAC with Largazole analogs discussed. This material is available free of charge via the Internet at http://pubs.acs.org. Pymol sessions with additional snapshots from the MD simulations can be obtained from the authors upon request.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Taori K, Paul VJ, Luesch H. Structure and Activity of Largazole, a Potent Antiproliferative Agent from the Floridian Marine Cyanobacterium Symploca sp. J Am Chem Soc. 2008;130:1806–1807. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- 2.Taori K, Paul VJ, Luesch H. Structure and Activity of Largazole, a Potent Antiproliferative Agent from the Floridian Marine Cyanobacterium Symploca sp. J Am Chem Soc. 2008;130:13506. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- 3.Newkirk TL, Bowers AA, Williams RM. Discovery, Biological Activity, Synthesis and Potential Therapeutic Utility of Naturally Occurring Histone Deacetylase Inhibitors. Nat Prod Rep. 2009;26:1293–1320. doi: 10.1039/b817886k. [DOI] [PubMed] [Google Scholar]
- 4.Hong J, Luesch H. Largazole: From Discovery to Broad-Spectrum Therapy. Nat Prod Rep. 2012;29:449–456. doi: 10.1039/c2np00066k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li S, Yao H, Xu J, Jiang S. Synthetic Routes and Biological Evaluation of Largazole and Its Analogues as Potent Histone Deacetylase Inhibitors. Molecules. 2011;16:4681–4694. doi: 10.3390/molecules16064681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowers A, West N, Taunton J, Schreiber SL, Bradner JE, Williams RM. Total Synthesis and Biological Mode of Action of Largazole: A Potent Class I Histone Deacetylase Inhibitor. J Am Chem Soc. 2008;130:11219–11222. doi: 10.1021/ja8033763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gregoretti L, Lee Y, Goodson HV. Molecular Evolution of the Histone Deacetylase Family: Functional Implications of Phylogenetic Analysis. J Mol Biol. 2004:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Dokmanovic M, Clarke C, Marks PA. Histone Deacetylase Inhibitors: Overview and Perspectives. Mol Cancer Res. 2007;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 9.Minucci S, Pelicci PG. Histone Deacetylase Inhibitors and the Promise of Epigenetic (and more) Treatments for Cancer. Nature Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 10.Bowers AA, Greshock TJ, West N, Estiu G, Schreiber SL, Wiest O, Williams RM, Bradner JE. Synthesis and Conformation-Activity Relationships of the Peptide Isosteres of FK228 and Largazole. J Am Chem Soc. 2009;131:2900–2905. doi: 10.1021/ja807772w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bowers AA, West N, Newkirk TL, Troutman-Youngman AE, Schreiber SL, Wiest O, Bradner JE, Williams RM. Synthesis and Histone Deacetylase Inhibitory Activity of Largazole Analogs: Alteration of the Zinc-Binding Domain and Macrocyclic Scaffold. Org Lett. 2009;11:1301–1304. doi: 10.1021/ol900078k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guerra-Bubb JM, Bowers AA, Smith WB, Paranal R, Estiu G, Wiest O, Bradner JE, Williams RM. Synthesis and HDAC Inhibitory Activity of Isosteric Thiazoline-Oxazole Containing Largazole Analogs. Biorg Med Chem. 2013;23:6025–6028. doi: 10.1016/j.bmcl.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang DF, Helquist P, Wiech N, Wiest O. Toward selective Histone Deacetylase Inhibitor Design: Homology Modeling, Docking Studies and Molecular Dynamics Simulations of Human Clkass I Histone Deacetylases. J Med Chem. 2005;48:6936–6947. doi: 10.1021/jm0505011. [DOI] [PubMed] [Google Scholar]
- 14.Cole KE, Dowling DP, Boone MA, Phillips AJ, Christianson DW. Structural Basis of the Antiproliferative Activity of Largazole, a Depsipeptide Inhibitor of the Histone Deacetylases. J Am Chem Soc. 2011;133:12474. doi: 10.1021/ja205972n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Estiu G, Greenberg E, Harrison CB, Kwiatkowski NP, Mazitschek R, Bradner JE, Wiest O. J Med Chem. 2008;51:2898. doi: 10.1021/jm7015254. [DOI] [PubMed] [Google Scholar]
- 16.Crimmins MT, Chaudhary K. Titanium Enolates of Thiazolidinethione Chiral Auxiliaries: Versatile Tools for Asymmetric Aldol Additions. Org Lett. 2000;2:775–777. doi: 10.1021/ol9913901. [DOI] [PubMed] [Google Scholar]
- 17.Fürstner A. Olefin Metathesis and Beyond. Angew Chem Int Ed. 2000;39:3012–3043. [PubMed] [Google Scholar]
- 18.Mulqueen GC, Pattenden G, Whiting D. A Synthesis of the Thiazoline-Based Siderophore (S)-Desferrithiocin. Tetrahedron. 1993;49:5359–5364. [Google Scholar]
- 19.Somoza JR, Skene RJ, Katz BA, Moi C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang BC, Verner E, Wynands R, Leahy EM, Dougan D, Snell G, Navre M, Knuth MW, Swanson RV, McRee D, Tari LW. Structural Snapshots of Human HDAC8 Provide Insights into the Class I Histone Deacetylases. Structure. 2004;12:1325–1334. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 20.Estiu G, West N, Mazitschek R, Greenberg E, Bradner J, Wiest O. On the Inhibition of Histone Deacetylase 8. Bioorg Med Chem. 2010;18:4103–4110. doi: 10.1016/j.bmc.2010.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang DF, Wiest O, Helquist P, Lan-Hargest HY, Wiech NL. J Med Chem. 2004;47:3409. doi: 10.1021/jm0498497. [DOI] [PubMed] [Google Scholar]
- 22.Weerasinghe SVW, Estiu G, Wiest O, Pflum MKH. Residues in the 11 Å Channel of Histone Deacetylase 1 Promote Catalytic Activity: Implications for Designing Isoform-Selective Histone Deacetylase Inhibitors. J Med Chem. 2008;51:5542–5551. doi: 10.1021/jm800081j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 24.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong GM, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang JM, Kollman P. J Comp Chem. 2003;24:1999. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 25.Lee MC, Duan Y. Distinguish Protein Decoys by Using a Scoring Function Based on a New AMBER Force Field, Short Molecular Dynamics Simulations, and the Generalized Born Solvent Model. Proteins. 2004;55:620–634. doi: 10.1002/prot.10470. [DOI] [PubMed] [Google Scholar]
- 26.Wang JM, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and Testing of a General Amber Force Field. J Comp Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 27.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 28.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical Integration of the Cartesian Equations of Motions of a System with Constraints: Molecular Dynamics of n-Alkanes. J Comp Phys. 1977;23:327–341. [Google Scholar]
- 29.Pilon JL, Clausen DJ, Hansen RJ, Lunghofer PJ, Charles B, Rose BJ, Thamm DH, Gustafson DL, Bradner JE, Williams RM. Comparative pharmacokinetic properties and antitumor activity of the marine HDACi Largazole and Largazole peptide isostere. Cancer Chemother Pharmacol. 2015;75:671–682. doi: 10.1007/s00280-015-2675-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.