

SUMMARY
Upon antigen recognition and co-stimulation, T lymphocytes up-regulate the metabolic machinery necessary to proliferate and sustain effector function. This metabolic reprogramming in T cells regulates T cell activation and differentiation but is not just a consequence of antigen recognition. While such metabolic reprogramming promotes the differentiation and function of T effector cells, the differentiation of regulatory T cells employs different metabolic reprogramming. We therefore hypothesized that inhibition of glycolysis and glutamine metabolism might prevent graft rejection by inhibiting effector generation and function and promoting regulatory T cell generation. We devised an anti-rejection regimen involving a glycolytic inhibitor, 2-Deoxyglucose (2-DG), an anti-Type II diabetes drug (metformin) and an inhibitor of glutamine metabolism, 6-Diazo-5-oxo-L-norleucine (DON). Using this triple drug regimen we were able to prevent/delay graft rejection in fully mismatched skin and heart allograft transplantation models.
Graphical Abstract
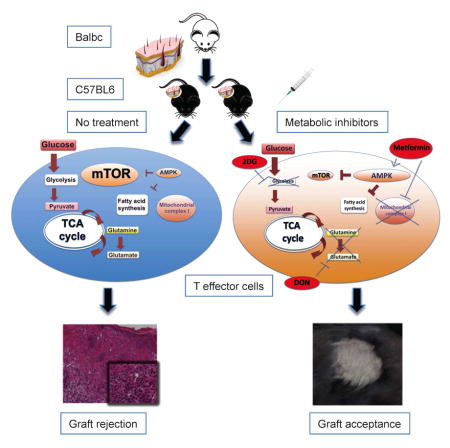
INTRODUCTION
Advances in immunosuppressive regimens have played an essential role in driving forward the field of organ transplantation (Sayegh and Carpenter, 2004). However, long-term use of immunosuppressants results in a broad range of co-morbidity. For example, calcineurin inhibitors are associated with hyperlipidemia, hyperglycemia, neuro- and nephro-toxicity, as well as an increased risk of malignancy (Arnold et al., 2013; Crutchlow and Bloom, 2007; Guba et al., 2004; Hoorn et al., 2012; Roodnat et al., 2014). In addition, such agents inhibit negative regulatory and tolerance-inducing responses (Wu et al., 2012). That is, the calcineurin inhibitors are truly immunosuppressive in that they inhibit both activating and inhibitory signaling pathways (Powell and Zheng, 2006). As such, whereas the ultimate goal of anti-rejection strategies is to induce immune tolerance in the absence of long-term immunosuppression, current treatment regimens thwart this goal by inhibiting the induction of tolerance. Therefore new approaches to preventing graft rejection are required.
Recently, metabolic signaling pathways have been shown to play critical roles in dictating the outcomes of T cell responses (Pollizzi and Powell, 2014; Waickman and Powell, 2012; Yang and Chi, 2012). The coordination of metabolism reprogramming and T cell function reflects the ability of how low-frequency antigen-specific naïve T cells rapidly expand in response to a pathogen (Powell et al., 2013a). In the presence of oxygen, naïve or resting T cells rely on mitochondrial oxidative phosphorylation (OXPHOS) to generate energy necessary for immune surveillance (Pearce et al., 2013). In contrast, both CD4+ and CD8+ effector T cells employ aerobic glycolysis to meet their biosynthetic demands (Jones and Thompson, 2007; Pearce et al., 2013). This use of glycolysis in the presence of oxygen was first described by Otto Warburg in cancer cells (Warburg, 1956) and was subsequently found to be important in activated T cells (Warburg et al., 1958). It has been proposed that aerobic glycolysis permits the generation of the substrates necessary for the generation of amino acids, nucleic acids and lipids, all of which are crucial for activation and proliferation (Vander Heiden et al., 2009). Essential for this activation-induced glycolytic response is glucose uptake (Cham et al., 2008; Cham and Gajewski, 2005). Indeed, the increased expression of the glucose transporter GLUT1 on the cell surface is a critical aspect of TCR-induced activation (Jacobs et al., 2008). Similarly, the uptake and metabolism of amino acids, especially glutamine, is essential for T cell activation (Carr et al., 2010). Glutamine deprivation blocks T cell proliferation and cytokine production (Carr et al., 2010). While the considerations depicted above reflect the metabolic needs of T cells during activation and explosive proliferation, what has also become apparent is that different T cell subsets require different metabolic programs. For example, Th1, Th2, and Th17 effector T cells have been found to depend on glucose uptake and glycolysis. Alternatively, regulatory T cells (Tregs) are not dependent upon glycolysis and appear to rely more on lipid oxidation to generate energy (Huynh et al., 2015; Michalek et al., 2011). Blocking glycolysis inhibits effector development but promotes Treg formation (Shi et al., 2011). Furthermore, CD4+ effector T cells with GLUT1 deficiency are impaired in proliferation and function in vivo, whereas Tregs are enriched and functionally unaffected (Macintyre et al., 2014). Likewise, glutamine is critically required by naïve CD4+ T cell to differentiate to Th1 and Th17 effector T cells but not to Tregs (Nakaya et al., 2014). Thus, strategies designed to inhibit metabolic reprogramming to prevent naïve T cells from activating and differentiating into effector T cells, might provide a means to inhibit transplant rejection. In this study, we demonstrate that combined inhibition of glycolysis and glutamine metabolism leads to a profound suppression in CD4+ and CD8+ effector T cell responses while preserving mechanisms of immune regulation. We propose that this anti-metabolic regimen represents a promising strategy to promote allograft survival.
RESULTS
Blocking glycolysis inhibits activation-induced T cell proliferation and cytokine production
To assess the effect of metabolic inhibitors on the metabolic changes of resting T cells in response to stimulation, we measured the two intrinsic cellular bioenergetic parameters, oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) with XF96 extracellular flux analyzer (Seahorse Bioscience). OCR is an indicator of mitochondrial OXPHOS, and ECAR is predominantly a measure of lactic acid formed during glycolytic energy metabolism, and thus reflects overall glycolytic flux. Naïve 5C.C7 CD4+ T cells were incubated with pigeon cytochrome c (PCC) peptide for 48 hrs and then expanded and rested in IL-2 for an additional 5 days to generate previously activated CD4+ cells. After measuring the baseline bioenergetics of these resting CD4+ cells, cells were re-stimulated with anti-CD3/CD28 (Figures 1A and 1B). Upon activation, the T cells exhibited a marked increase in ECAR and a modest increase in OCR in response to anti-CD3/CD28 (Figures 1A and 1B). Upon reaching the peak of glycolytic flux, metabolic inhibitors were administered to the cells. 2-DG is an ATP depleting agent and glucose analog that inhibits hexokinase, the first enzyme in the glycolytic pathway (Rowe et al., 2013). Adding 2-DG led to a sharp decrease of ECAR levels and an insignificant increase in OCR (Figures 1A and 1B), reflecting that 2-DG inhibited glycolysis but minimally affected OXPHOS (Cheng et al., 2012; Yamasaki et al., 2011).
Figure 1. 2-DG combined with Metformin inhibits T cell response through suppression of glycolysis.

(A and B) ECAR and OCR of resting CD4+ cells measured in real time under basal conditions and in response to anti-CD3/CD28/cross-linking IgG1 (anti-CD3, 2 μg/ml; anti-CD28, 2 μg/ml; cross-linking IgG1, 1 μg/ml) with or without the presence of individual or combination of drugs (2-DG, 10 mM; Metformin, 50 mM). Bar graphs display data of ECAR and OCR measured at the endpoint of the experiment (205 min). Data are shown as mean ± SEM of 5 measurements. (C and D) Naïve splenocytes labeled with cell proliferation dye eFluor 670 were stimulated with anti-CD3 in in the presence of media control, 2-DG alone, metformin alone or 2-DG + metformin (2-DG, 0.6mM; Metformin, 1mM). (C) 24-hour IFN-γ secretion to supernatants was interrogated by enzyme-linked immunosorbent assay (ELISA). Data are shown as mean ± SEM of three independent samples. (D) 72-hour eFluor dilution of CD4+ and CD8+ T cells. n.s., not significant, *p<0.05, **p < 0.01, ****p < 0.0001 (Student’s t test). Data are representative of at least two independent experiments.
Metformin is a commonly used oral hypoglycemic that activates AMPK, inhibits mitochondrial respiratory complex I and promotes fatty acid oxidation (Buzzai et al., 2007; El-Mir et al., 2000). Consistent with its ability to inhibit Complex I (El-Mir et al., 2000), the addition of metformin led to a marked suppression of OCR and an increase in ECAR (Figures 1A and 1B). It has been shown that organic cation transporters mediate uptake of metformin (Graham et al., 2011). Interestingly, we observed increased expression of organic cation transporter 1 (OCT1) upon T cell activation (Figure S1). These findings suggest that metformin will preferentially affect activated T cells which in turn express more OCT1.
The concomitant use of 2-DG and metformin together has been shown to potentiate the inhibition of glycolysis in tumor cells (Cheng et al., 2014; Cheong et al., 2011). Indeed, when combined with metformin, 2-DG inhibits ECAR in T cells more effectively than when employed alone (Figure 1A). In addition, the combination of 2-DG and metformin led to a lower OCR compared to that of the no treatment group but higher than that of metformin alone (Figure 1B). Thus, similar to observations in tumor cells, combining 2-DG and metformin leads to a profound decrease in glycolysis in activated T cells.
Given the critical role of glycolysis in promoting T cell effector function, we next sought to determine the effect of 2-DG and metformin on T cell proliferation and cytokine production. To this end, splenocytes from wild-type (WT) C57BL/6 mice were stimulated with anti-CD3 in the presence of media control, 2-DG alone, metformin alone or 2-DG + metformin for 72 hrs. Note, initial extensive titration studies demonstrated that 2-DG or metformin monotherapy inhibited T cell proliferation and cytokine production in a dose-dependent manner (data not shown). Importantly, consistent with their combined metabolic effects, the combination of 2-DG and metformin showed a more significant inhibition of cell proliferation and IFN-γ production of activated CD4+ and CD8+ T cells than either compound alone (Figures 1C and 1D). Together, these data suggest that 2-DG in combination with metformin potently inhibit both metabolic reprogramming and proliferation/cytokine production in activated T cells.
Combined inhibition of glycolysis and glutamine metabolism profoundly abrogates activation-induced T cell proliferation and cytokine production
Upon activation, glutamine oxidation increases as well as glycolysis to meet biosynthetic demand and allowing for continued tricarboxylic acid (TCA) flux (MacIver et al., 2013). Consequently, T cell activation, differentiation and function are critically dependent upon adequate glutamine availability (Carr et al., 2010; Nakaya et al., 2014). To this end, upon activation, T cells coordinately increase the expression of glutamine transporters and glutaminase activity required for glutamine oxidation (Carr et al., 2010; Nakaya et al., 2014).
6-Diazo-5-oxo-L-norleucine (DON) is a glutamine analog that inhibits multiple targets in the glutamine metabolic pathway, including glutaminase and glutamine transporters (Thomas et al., 2014b). To explore the role of glutaminase activity in T cell activation, naïve CD4+ T cells were stimulated with anti-CD3/CD28 in medium with or without metabolic inhibitors for 24 hrs. The glutaminase activity of these CD4+ T cells was determined using a radiolabeled assay as described (Thomas et al., 2013). We observed that activated T cells exhibited increased glutaminase activity compared to naïve T cells (Figure 2A), indicating metabolic reprogramming toward glutaminolysis. As expected, glutaminase activity of activated T cells was significantly abrogated in the presence of DON (Figure 2A), and this inhibition was not affected by the presence of the glycolytic inhibitors (Figure 2A). Furthermore, when cultured with DON, activated CD4+ and CD8+ T cells had decreased IFN-γ production and proliferation (Figures 2B and 2C). Having confirmed previous studies showing that blocking glutamine metabolic pathway was also capable of suppressing T cell responses (Carr et al., 2010), we next wanted to investigate whether dual inhibition of glycolysis and glutamine metabolism had an additional effect on inhibiting T cell proliferation and function. To this end, we combined 2-DG, metformin and DON as a triple therapy regimen. Indeed, triple therapy had a more profound effect on suppressing T cell IFN-γ production and proliferation than only 2-DG and metformin or DON alone (Figures 2B and 2C). Of note, the decrease in function was not because of a decrease of cell viability (Figure S2). In addition, although glutamate can be further catabolized to fuel TCA cycle and ATP production (Pearce et al., 2013), in extracellular flux analysis we did not find that adding DON (individually or combined with 2-DG and metformin) had an obvious impact on either OXPHOS or glycolytic flux of activated cells (Figure S3). Similarly, while 2-DG, metformin and DON individually had minimal effects on de novo lipid synthesis, triple therapy resulted in marked suppression of de novo lipid synthesis (Figure 2D). When glycolysis is blocked in conjunction with glutamine-dependent biosynthetic pathways, the decreased acetyl-coA level results in decreased lipid production necessary for increased membrane formation essential to facilitate proliferation. Moreover, triple therapy was associated with a decrease in mTORC1 activation as measured by S6 ribosomal protein phosphorylation, which correlates with previous findings (Powell et al., 2013b) that suppression of mTORC1 results in decreased T cell effector function (Figure 2E). Therefore, by interrogating the inhibition of metabolic pathways known to play a critical role in T cell activation and function, we have determined that the inhibition of activation-induced T cell proliferation and function was most effective by simultaneously blocking glycolysis (with 2-DG and metformin) and glutamine metabolism (with DON).
Figure 2. Combined inhibition of glycolysis and glutaminolysis profoundly suppresses T cell responses.

(A) Glutaminase activity of CD4+ T cells cultured for 24 hrs in different conditions (anti-CD3, 2μg/ml; anti-CD28, 2μg/ml; DON, 5 μM; 2-DG, 0.6 mM; Metformin, 1 mM). Data are shown as mean ± SEM of three independent experiments. (B and C) Naïve WT C57BL/6 splenocytes labeled with eFluor 670 and stimulated with anti-CD3 in medium containing indicated metabolic inhibitors (DON, 5μM; 2-DG, 0.6mM; Metformin, 1mM). (B) 24-hour IFN-γ secretion to supernatants as interrogated by ELISA. Data are shown as mean ± SEM of three independent samples. (C) Proliferation of CD4+ and CD8+ T cells at 72h measured by dilution of eFluor 670. n.s., not significant, **p < 0.01, ***p < 0.001, ****p < 0.0001 (Student’s t test). Data are representative of at least two independent experiments. (D) 3H-acetate incorporation into lipids in preactivated and stimulated CD4+ T cells (anti-CD3, 1μg/ml; anti-CD28, 2μg/ml; cross-linking 0.75 μg/ml) with the presence of 2DG, Metformin, DON or in combination (2DG, 5mM; Metformin, 30mM; DON, 60μM). Data are shown as mean ± SEM of three independent samples. ***p < 0.001 (ANOVA). Data are representative of two independent experiments. (E) The phosphorylation state of the S6 ribosomal protein was measured in CD4+ T cells after 30 minutes stimulation (anti-CD3, 1μg/ml; anti-CD28, 2μg/ml; cross-linking 0.75 μg/ml) with the presence of Rapamycin, PP242 and metabolic inhibitors (Rapamycin, 1μM; pp242, 1μM; 2DG, 5mM, Metformin, 30mM; DON, 60μM). Data are representative of two independent experiments.
Inhibition of glycolysis and glutamine metabolism preferentially suppresses the proliferation and function of antigen-specific CD4+ effector T cells compared to Tregs in vivo
Given the observation that inhibition of glycolysis and glutamine metabolism suppresses the activation of T cells in vitro, we next wanted to examine this paradigm in an antigen-specific manner in vivo. To test the ability of metabolic inhibitors to mitigate CD4+ effector T cell responses, Thy1.1+ CD4+ OVA-specific (OT-II) TCR transgenic T cells were adoptively transferred into Thy1.2+ WT mice that were concomitantly infected with vaccinia-OVA, a recombinant vaccinia virus carrying chicken ovalbumin (OVA) on day 0. The mice were then treated with PBS (vehicle control), 2-DG+metformin or 2-DG+metformin+DON (triple therapy) for 3 days and the expansion of the OVA-specific Thy1.1+ CD4+ T cells was interrogated on day 4. Mice treated with 2-DG+metformin demonstrated a marked decrease in the expansion of the antigen-specific Thy1.1+ CD4+ T cells, which was even further reduced with the triple therapy (Figure 3A). In addition, the triple therapy regimen in vivo strongly inhibited antigen-specific IFN-γ production of splenocytes upon re-challenge with peptide ex vivo (Figure 3B). Triple therapy also led to a significantly higher frequency of Foxp3+ CD4+ T cells (Figure 3C), indicating a preservation of this mechanism of immune regulation. Of note, the expansion and absolute number of Thy1.1+ Foxp3+ T cells was decreased in the triple therapy treated mice when compared to the untreated mice. This indicates that as is the case for effector cells, the expansion of Foxp3+ T cells in response to antigen requires glycolysis and glutamine. However, the ratio of OVA-specific Thy1.1+ regulatory T cells:effector cells was increased in the treated mice (Figure 3D). Together, these results demonstrate that triple therapy had the most robust effect on inhibiting CD4+ T cell proliferation and cytokine production upon antigen recognition, while the differentiation into antigen-specific regulatory T cells was relatively preserved.
Figure 3. Metabolic Inhibitors suppress the proliferation and function of antigen-specific T cells while increase the relative frequency of Tregs in vivo.

(A to D) OT-II (Thy1.1+) CD4+ T cells were adoptively transferred into WT (Thy1.2+) recipient mice. The recipients were infected with OVA-expressing vaccinia virus and treated with vehicle, 2-DG+Metformin or 2-DG+metformin+DON (2-DG, 500 mg/kg once daily; metformin, 150 mg/kg once daily; DON, 1.6 mg/kg once every other day) for 3 days. Splenocytes from the recipients were harvested at day 4 to interrogate the frequency of antigen-specific T cells and regulatory T cells. (A) Percentage of Thy1.1+ cells relative to CD4+ cells were analyzed by flow cytometry (left) and plotted as cumulative data (right). (B) OT-II cells were rechallenged with OVA peptide class II (10 μg/ml) ex vivo for 24hrs. The supernatants were interrogated for IFN-γ production by ELISA. Data are mean ± SEM (n=6). (C) Percentage of Foxp3+ cells relative to CD4+ cells (left) and plotted as cumulative data (right). (D) The ratio of OVA-specific Foxp3+ T cells:Effector cells. (E to G) OT-I (Thy1.1+) CD8+ T cells were adoptively transferred into WT (Thy1.2+) recipient mice. The hosts were infected with vaccinia-OVA and treated with vehicle, 2-DG+metformin or triple therapy for 5 days. Host splenocytes were harvested at day 6 to interrogate the proliferation and function of antigen-specific CD8+ T cells. (E) Percent Thy1.1+ cells relative to CD8+ cells were analyzed by flow cytometry (left) and plotted as cumulative data (right) (F) Percent IFN-γ-producing cells relative to CD8+ cells (left) and plotted as cumulative data (right). (G) OT-I cells were rechallenged with OVA peptide class I (10μg/ml) ex vivo for 24hrs. The supernatants were interrogated for IFN-γ production by ELISA. Data are mean ± SEM (n=5 ~6) (H) The ability of metabolic inhibitors to suppress endogenous effector CD8+T cells development was assessed with in vivo CTL assay. Percent of specific killing was determined at 10 hrs after transferring target cells. Data are mean ± SEM (n=3mice/group). Each symbol represents an individual mouse. Horizontal lines indicate mean± SEM. n.s., not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (Student’s t test). Data are representative of more than three independent experiments.
Inhibition of glycolysis and glutamine metabolism suppresses the proliferation and function of antigen-specific CD8+ T cells
Next we examined the ability of the metabolic inhibitors to block antigen-specific CD8+ T cell responses. To this end, Thy1.1+ CD8+ OVA-specific (OT-I) TCR transgenic T cells were adoptively transferred into Thy1.2+ WT mice and the mice were infected with vaccinia-OVA and treated with metabolic inhibitors (the same regimen used in OT-II experiments). As was the case of CD4+ T cells, treatment of the mice with triple therapy led to a more profound inhibition of the expansion of the antigen-specific Thy1.1+ CD8+ T cells than 2DG + metformin (Figure 3E) or any compound alone (data not shown). Additionally, this combination of anti-metabolic therapy strongly inhibited the generation of IFN-γ-secreting CD8+ T cells (Figure 3F). The decreased frequency and function of Thy1.1+ CD8+ T cells was also reflected by the amount of IFN-γ secreted by splenocytes re-stimulated ex vivo with class I OVA peptide (Figure 3G). Having demonstrated the ability of inhibitors of glycolysis and glutamine metabolism to mitigate the expansion and function of exogenous effector T cells, we next examined the effect of anti-metabolic regimen on endogenous effector CD8+ T cells with an in vivo cytotoxicity (CTL) assay. Mice were immunized with vaccinia-OVA and treated with PBS (vehicle control), 2DG + metformin or triple therapy for 7 days, then CFSE-labeled target cells were injected on day 8 and percent of specific killing was evaluated 10 hrs later. We observed that most (96% ±2%) of the OVA peptide-pulsed target cells were preserved in the mice receiving triple therapy, whereas 78±5% and 45±10% of the target cells were eliminated in mice receiving PBS and 2-DG+metformin treatment, respectively (Figure 3H). These data suggest that the development of effective cytotoxic T lymphocytes is suppressed by the inhibition of glycolysis and glutamine metabolism.
Metabolic inhibitors promote allograft acceptance in mouse models of skin and heart transplantation
Having demonstrated the ability of combined anti-metabolic therapy to inhibit anti-viral responses in vivo, we next investigated whether anti-metabolic therapy could prolong allograft survival by using a skin transplantation model involving C57BL/6 mice receiving allogenic skin grafts from Balb/c mice. We found a significantly prolonged graft median survival time (MST) for mice that received 2-DG+metformin or DON alone, compared to those that received no treatment (13 vs. 11 days, P = 0.0048 and 22 vs. 11 days, P=0.0019, respectively) (Figure 4A). However, blocking both glycolysis and glutamine metabolism with triple therapy resulted in a marked increase of skin graft survival compared to no treatment (MST 40 vs. 11 days, P < 0.0001) (Figure 4A). In addition, the appearance and histology of skin grafts from the triple therapy group exhibited no evidence of tissue destruction, more intact tissue alignment, and less infiltration of inflammatory cells (Figures 4B). For comparison, the MST of this fully allogeneic skin allograft under the treatment of FK506 or cyclosporine has previously been reported to be less than 20 days (Lagodzinski et al., 1990). Likewise, in our own hands, median graft survival is approximately 16 days using rapamycin mono-therapy for immunosuppression (data not shown).
Figure 4. Metabolic inhibitors promote allograft survival.

(A) Balb/c to C57BL/6 skin graft survival, as monitored daily by assessment of macroscopic signs of rejection. (B) Representative photomicrographs of skin graft histology (hematoxylin and eosion (H&E) staining) on post-transplant days 7 and 40 under an optical microscope (outlet, X100; inlet: X200). (C) Balb/c to C57BL/6 heart graft survival, as monitored daily by palpation of heart beating. (D) Representative photomicrographs of cardiac graft histology (H&E staining) on indicated post-transplant day under an optical microscope (outlet, X200; inlet, X400). The anti-metabolic treatment was started from the day of graft (day 0) until rejection in both models. The dosages of all drugs were the same as described in Figure 3. **p < 0.01, ****p < 0.001 (Log-rank analysis). Data are representative of two independent experiments.
Finally, we employed this triple therapy regimen in a Balb/c to C57BL/6 heart transplantation, which is a vascularized solid organ transplantation model. Strikingly, when recipient mice were treated with anti-metabolic triple therapy, the grafted hearts continued beating for more than 100 days post-transplantation without rejection (Figure 4C). Likewise, the cardiac grafts from treatment group exhibited much healthier microscopic morphology with no evidence of necrosis, fibrosis, or lymphocyte infiltration (Figures 4D). Together, these data support the notion that the inhibition of glycolysis and glutamine metabolic pathways represents a potent means to prevent acute rejection and promote long-term graft acceptance.
DISCUSSION
In this study, we have defined a approach to prevent transplantation graft rejection by inhibiting metabolic pathways necessary for effector T cell function. This approach is based upon the emerging understanding that metabolic reprogramming in T cells is not simply the result of T cell activation, but rather plays a critical role in regulating T cell differentiation and function (Jones and Thompson, 2007; MacIver et al., 2013). The therapeutic index of our approach relies on the extraordinary metabolic requirements of effector T cell response that is distinct from the more flexible and adaptive ordinary T cell metabolic network. As such, even though the inhibitors employed are relatively non-specific, we achieve relative cellular selectivity based upon the extraordinary metabolic demands of the effector T cells. That is, while glycolysis and glutamine are clearly important for all cells, the markedly enhanced reliance of effector T cells on these pathways makes them relatively selective to treatment with these relatively non-selective inhibitors. Along these lines, T regulatory cells were more resistant to inhibition of glycolysis and glutamine. Such an observation is consistent with reports that unlike effector T cells, T regulatory cells rely more on oxidative phosphorylation and lipid oxidation and in fact glycolysis inhibits their functionality (Huynh et al., 2015).
Previous studies by our lab and others have demonstrated the importance of mTOR in regulating T cell activation, differentiation and function (Powell et al., 2012). Further, we and others have demonstrated that the ability of mTOR to regulate T cells is in part mediated by the role of mTOR signaling on metabolic reprogramming (Pollizzi et al., 2015; Pollizzi and Powell, 2014). Consequently, in this current report we sought to regulate T cell function by directly targeting metabolic pathways. Indeed, our triple therapy led to a marked decrease in the biosynthetic capabilities of the effector T cells. This in turn inhibited both expansion of effector T cells as well as their function. Interestingly, in as much as nutrient availability promotes mTOR activity, this approach also led to a decrease in mTOR activity. Thus, our approach not only inhibited metabolic programs but also potentially the ability of mTOR to promote “immunologic reprogramming” independent of metabolism.
In our study, inhibiting glycolysis by the combination of 2-DG and metformin was not sufficient to prevent acute rejection or induce long-term tolerance in a fully MHC-mismatched transplantation model. Inhibition of glutamine metabolism also blocks effector T cell differentiation, proliferation and function (Carr et al., 2010; Nakaya et al., 2014). Thus, we integrated DON, an inhibitor of glutamine metabolism, into our protocol. Ultimately, the combination of inhibiting glycolysis and glutamine pathways provided the most robust protection against graft rejection. Overall, we found that including metformin (which inhibits Complex I of oxidative phosphorylation and also has the ability to potentiate the effects of 2-DG in terms of inhibiting glycolysis) further improved our regimen. The doses and combinations of these agents were determined initially in in vitro titration experiments (data not shown). We hypothesize that targeting multiple metabolic pathways not only enhanced the efficacy of our regimen but also enabled us to use lower doses of each compound thus minimizing potential side effects. Additionally, we propose that the relative cellular selectivity based on metabolic demand was not only observed for cells of the immune system but also contributed to the relative lack of side effects observed. By tracking body weight and fur ruffling as markers of overall health of transplant recipient mice, in some cases for more than 100 days, we did not observe any systemic sickness or morbidity associated with the treatment regimen. Notably, the compounds employed in our study have all been studied in patients (Franciosi et al., 2013; Raez et al., 2013; Sullivan et al., 1988). Thus, we believe that our combination treatment strategy might be readily translated into the clinic. In previous trials using 2-DG or DON or metformin for the treatment of cancer, the efficacy was not as striking as what we have observed in using these agents for transplant rejection (Raez et al., 2013; Sullivan et al., 1988; Tsilidis et al., 2014). We believe that this is because while T effector cells and cancer cells both employ Warburg metabolism, mutations in tumors enable them to more effectively become resistant to these agents.
Steroids and calcineurin inhibitors remain the backbone of most current post-transplantation regimens. Steroids have multiple adverse effects, such as increased risk of infection, hyperglycemia, accelerated atherosclerosis, and gastrointestinal bleeding, while calcineurin inhibitors are associated with neurotoxicity and nephrotoxicity, as well as risk of infection and an increased risk of cancer (Arnold et al., 2013; Crutchlow and Bloom, 2007; Guba et al., 2004; Hoorn et al., 2012; Roodnat et al., 2014). In this regard, we propose that our anti-metabolic regimen has certain advantages over conventional treatments in terms of avoiding some of these side effects. First, one potential advantage of anti-metabolic therapy is that it will promote a favorable systemic metabolic profile. Indeed, the metabolic inhibitors employed counteract some of the metabolic abnormalities associated with current immunosuppression, such as increased blood glucose and triglycerides. Second, a major infectious complication of immunosuppression for transplantation is CMV infection. Interestingly, the agents employed in our anti-metabolic approach have been shown to inhibit viral replication (Chambers et al., 2010). Third, while calcineurin inhibitors are associated with an increased risk of developing neoplasms (Guba et al., 2004), targeting both glycolysis and glutamine pathways inhibits the growth of tumor cells (Cheng et al., 2014; Cheong et al., 2011; Lim et al., 2014; Willems et al., 2013). Also, calcineurin inhibitors can block the induction of immunologic tolerance in part by preventing T cell anergy and inhibiting the generation of regulatory T cells (Powell and Zheng, 2006). The metabolic inhibitors employed in our study both promote anergy and T regulatory cell generation (Figure 3C and (Zheng et al., 2009)). However, metabolic therapy alone was not sufficient to completely prevent skin allograft rejection. Further, even though we observed 100% heart graft survival (Figure 4D) while the mice were being treated, preliminary studies indicate that stopping therapy resulted in the eventual rejection of the hearts approximately 80 days later (data not shown). To this end, we are actively pursuing integrating tolerance inducing therapy such as costimulatory blockade with our anti-metabolic regimen (Bestard et al., 2011; Lo et al., 2014; Oderup et al., 2006; Pilon et al., 2014).
In conclusion, we have defined an approach to prevention of graft rejection by inhibiting metabolic pathways necessary for effector T cell function. The differential metabolic requirements of effector and regulatory T cells reveal a therapeutic window to simultaneously inhibit rejection and promote tolerance. Future studies combining anti-metabolic therapy with tolerance-inducing regimens such as co-stimulatory blockade and Treg therapy have the potential to promote long-term graft acceptance in the absence of long-term immunosuppression.
EXPERIMENTAL PROCEDURES
Mice
Mice were kept in accordance with guidelines of the Johns Hopkins University Institutional Animal Care and Use Committee. 5C.C7 mice (RAG2−/− CD4+ TCR-transgenic mice which recognize Pigeon cytochrome c (PCC)) were from Taconic Farms. OVA-specific OT-I and OT-II TCR transgenic mice from The Jackson Laboratory were bred to Thy1.1+ backgrounds. C57BL/6 (Thy1.2+, H-2b) and Balb/c (H-2d) mice were obtained from The Jackson Laboratory.
Antibodies and reagents
The following antibodies for flow cytometry were from BD Biosciences: anti-CD4 (RM4-5), anti-CD8 (Ly-3), anti-IFN-γ (XMG1.2), and anti-Thy1.1 (OX-7). Anti-Foxp3 (FJK-16s) was from eBioscience. Class I OVA peptide and class II OVA peptide were obtained from AnaSpec. PCC peptide 81–104 was synthesized at Johns Hopkins University. Rabbit polyclonal antibody against mouse OCT1 (SLC22A1, ab55916) was obtained from Abcam. Rabbit polyclonal antibody to pS6 Ser 235/236 was obtained from Cell Signaling. Alexa Fluor 488-conjugated donkey anti-rabbit-IgG F(Ab′)2 was from Life Technologies. 3H-acetate was obtained from Perkin Elmer.
Rapamycin was purchased from LC laboratories and PP242 was from Chemdea. 2-DG was purchased from Carbosynth. Metformin and DON were purchased from Sigma-Aldrich. For all in vivo experiments, individual metabolic inhibitors were dissolved in PBS and administrated intraperitoneally.
Cell culture
Splenocytes or T cells were cultured in 45% RPMI 1640 and 45% EHAA media supplemented with 10% FBS, penicillin/streptomycin, glutamine and BME. For proliferation studies, T cells were labeled with 5 μM eFluor 670 cell proliferation dye (eBioscience). Splenocytes were stimulated with anti-CD3 (1μg/ml). For isolated T cells stimulation, flat-bottomed plates were coated with anti-CD3 (5 μg/ml) diluted in PBS, and soluble anti-CD28 (2 μg/ml). For preparation of pre-activated CD4+ T cells, splenocytes from 5C.C7 mice were stimulated with 5 μM PCC peptide in complete medium for 48 hrs. Mouse recombinant IL-2 (1 ng/ml; peprotech) was then added to the culture. After 5–7 days, live cells were isolated by a Ficoll gradient (GE healthcare).
Extracellular Flux Analysis
Cells were initially plated to XF assay medium, modified DMEM (Seahorse Bioscience) containing 25 mM glucose, 2 mM L-glutamine, and 1 mM sodium pyruvate, and incubated in a non-CO2 incubator at 37°C for 30 minutes. OCR and ECAR were measured at 37°C in XF96 Extracellular Flux Analyzer (Seahorse Bioscience) using manufacturer-recommended protocols. After baseline measurements, OCR and ECAR were measured after sequentially adding of anti-CD3 (2 μg/ml)/CD28 (2 μg/ml)/cross-linking IgG1 (1 μg/ml) and metabolic inhibitors into each well to the indicated final concentrations using the included ports on the XF96 cartridges. All data were collected using the XF Reader software from Seahorse Bioscience.
Adoptive transfers
CD8+ (or CD4+) T cells were harvested from OT-I (or OT-II) mice and purified by negative selection with CD8+ (or CD4+) MACS cell isolation protocol (Miltenyi Biotec). 1 × 106 purified CD8+ (or CD4+) T cells were then injected intravenously into Thy1.2+ C57BL/6 recipients. Vaccinia-OVA (1 × 106 plaque-forming units) was simultaneously administered by injection into the retro-orbital venous sinus.
Glutaminase activity analysis
Glutaminase activity measurement was adapted from previously described protocols (Thomas et al., 2014a). Briefly, T cells were lysed in ice-cold potassium phosphate buffer (45 mM, pH 8.2) containing protease inhibitors (Roche, Complete Protease Inhibitor Cocktail, 1 tablet in 50 ml) and incubated with 3H-glutamine (0.03 μM, 0.91 μCi) for 90 min at room temperature. The reactions were carried out in 50 μl reaction volumes in a 96-well microplate. At the end of the reaction period, the assay was terminated upon the addition of imidazole buffer (20 mM, pH 7). 96-Well spin columns packed with strong anion ion-exchange resin (Bio-Rad, AG® 1-X2 Resin, 200–400 mesh, chloride form) were used to separate the substrate and the reaction product. Un-reacted 3H-glutamine was removed by washing with imidazole buffer. 3H-Glutamate, the reaction product, was then eluted with 0.1 N HCl and analyzed for radioactivity using Perkin Elmer’s TopCount instrument in conjunction with their 96-well LumaPlates.
Flow cytometry, intracellular cytokine staining, and ELISA
Flow cytometry data were acquired with a FACS Calibur (BD Biosciences) and were analyzed with FlowJo7.6 software (TreeStar). For intracellular staining, cells were stimulated at 37°C for 4 hrs in the presence of Monensin (GolgiStop; BD Biosciences), phorbol 12-myristate 13- acetate (PMA; Sigma), and ionomycin (Sigma). Cells were surface stained and underwent fixation/permeabilization with either a Cytofix/Cytoperm kit (BD Biosciences) or a Fixation/Permeabilization kit (eBioscience), followed by staining for intracellular cytokines. Gates were determined appropriately through the use of un-stimulated control cells. Voltages were determined from unstained controls. IFN-γ concentration in the supernatant of cell cultures was analyzed by ELISA as recommended by the manufacturer (eBioscience). Cell viability was determined by 7-aminoactinomycin D (7-AAD; BD Biosciences) exclusion.
In vivo cytotoxicity assay
Splenocytes from WT mice were labeled with either 20 μM or 2 μM of carboxyfluorescein diacetate succinimidyl (CFSE, Invitrogen). The CSFEhigh cells were pulsed with class I OVA peptide (10 μM). Equal numbers (5 × 106 cells) of peptide-pulsed CFSEhigh cells and un-pulsed CFSElow cells were co-administered i.v. into syngeneic host mice that had been previously (day -7) immunized with vaccinia-OVA (1 × 106 plaque-forming units). After 10 hrs, splenocytes were isolated from host spleens. The two target populations are distinguished based on the differences in their CFSE intensity by flow cytometry. The loss of peptide-pulsed target cells is indicative of cytotoxicity. The ratio of pulsed to un-pulsed target cells in the indicated groups of mice was calculated and the percent killing was determined by using the formula: [1− (ratio in experimental mouse/ratio in naïve mouse)] × 100%.
Lipid biosynthesis assay
Naïve and preactivated T cells from 5C.C7 mice were incubated with 3H-acetate (0.6 uCi per 1 million cells) in culture media containing anti-CD3/anti-CD28/cross-linking, vehicle (PBS), 2-DG, Metformin, DON or triple therapy. After 4hr incubation at 37°C, total cellular lipids were extracted with chloroform/methanol by the Folch method (Folch et al., 1957) and radioactivity was counted by liquid scintillation and normalized to μg protein.
S6 ribosomal protein phosphorylation assay
Splenocytes from wild-type C57BL/6 mice were stimulated with anti-CD3 (1 μg/ml), anti-CD28 (2 μg/ml), cross-linked with antibody to hamster IgG1 (0.75 μg/ml) and combined with vehicle, Rapamycin, PP242 and triple therapy for 30 minutes. The cells were stained using rabbit polyclonal antibody to S6 ribosomal protein Ser 235/236 and donkey anti-rabbit labeled secondary antibody.
Murine skin transplantation
BALB/c mice served as skin donors and C57BL/6 mice serve as allograft recipients. Full-thickness skin grafts were harvested from the back and then fixed on the thoracic flank of recipient mice with simple separate stitches. The size of transplanted grafts was 1 × 1 cm2. Grafts were observed every day after the removal of the bandage at day 7 and considered rejected when ≥ 90% of the graft tissue became necrotic. We observed the MST of skin grafts. Seven days after surgery, skin tissue from the transplantation site was collected for optical microscopy after hematoxylin and eosion staining.
Murine heterotopic heart transplantation
BALB/c mice served as heart donors and C57BL/6 mice serve as allograft recipients. Either abdominal or cervical heterotopic heart transplantation was performed according to previously published methods (Corry et al., 1973; Oberhuber et al., 2014). Functionality of the transplanted heart was monitored daily by palpation. Clinical rejection was defined by cessation of palpable heartbeats and confirmed by autopsy. Loss of graft function within 48 hrs of transplantation was considered as a technical failure, and animals in which this occurred were omitted from the analysis.
Statistical analysis
Prism software version 5.0 (GraphPad Software) was used for statistical analyses, including unpaired Student’s t-test, two-way analysis of variance (ANOVA) and log-rank analysis. A P value less than 0.05 was considered statistically significant.
Acknowledgments
We thank members of the Powell Lab for discussions and critical review of this manuscript. This work was supported by the US National Institutes of Health grant R01AI077610.
Footnotes
AUTHOR CONTRIBUTIONS
G. B., B.S. S., M.J.W. and J. D. P. designed research; C.-F. L., Y.-C. L., G. J. F., B. O., V. A.-O., C.E.B., C-C. H. and A. G. T. performed research; C.-F. L. and Y.-C. L. analyzed data; C.-F. L., Y.-C. L. and J. D. P. wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arnold R, Pussell BA, Pianta TJ, Lin CS, Kiernan MC, Krishnan AV. Association between calcineurin inhibitor treatment and peripheral nerve dysfunction in renal transplant recipients. Am J Transplant. 2013;13:2426–2432. doi: 10.1111/ajt.12324. [DOI] [PubMed] [Google Scholar]
- Bestard O, Cassis L, Cruzado JM, Torras J, Franquesa M, Gil-Vernet S, Lucia M, Grinyo JM. Costimulatory blockade with mTor inhibition abrogates effector T-cell responses allowing regulatory T-cell survival in renal transplantation. Transpl Int. 2011;24:451–460. doi: 10.1111/j.1432-2277.2011.01223.x. [DOI] [PubMed] [Google Scholar]
- Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–6752. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, Turay AM, Frauwirth KA. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol. 2010;185:1037–1044. doi: 10.4049/jimmunol.0903586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cham CM, Driessens G, O’Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8(+) T cells. Eur J Immunol. 2008;38:2438–2450. doi: 10.1002/eji.200838289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cham CM, Gajewski TF. Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J Immunol. 2005;174:4670–4677. doi: 10.4049/jimmunol.174.8.4670. [DOI] [PubMed] [Google Scholar]
- Chambers JW, Maguire TG, Alwine JC. Glutamine metabolism is essential for human cytomegalovirus infection. J Virol. 2010;84:1867–1873. doi: 10.1128/JVI.02123-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G, Zielonka J, Dranka BP, McAllister D, Mackinnon AC, Jr, Joseph J, Kalyanaraman B. Mitochondria-targeted drugs synergize with 2-deoxyglucose to trigger breast cancer cell death. Cancer Res. 2012;72:2634–2644. doi: 10.1158/0008-5472.CAN-11-3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G, Zielonka J, McAllister D, Tsai S, Dwinell MB, Kalyanaraman B. Profiling and targeting of cellular bioenergetics: inhibition of pancreatic cancer cell proliferation. Br J Cancer. 2014;111:85–93. doi: 10.1038/bjc.2014.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong JH, Park ES, Liang J, Dennison JB, Tsavachidou D, Nguyen-Charles C, Wa Cheng K, Hall H, Zhang D, Lu Y, et al. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol Cancer Ther. 2011;10:2350–2362. doi: 10.1158/1535-7163.MCT-11-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corry RJ, Winn HJ, Russell PS. Primarily vascularized allografts of hearts in mice. The role of H-2D, H-2K, and non-H-2 antigens in rejection. Transplantation. 1973;16:343–350. doi: 10.1097/00007890-197310000-00010. [DOI] [PubMed] [Google Scholar]
- Crutchlow MF, Bloom RD. Transplant-associated hyperglycemia: a new look at an old problem. Clinical journal of the American Society of Nephrology : CJASN. 2007;2:343–355. doi: 10.2215/CJN.03671106. [DOI] [PubMed] [Google Scholar]
- El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–228. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. The Journal of biological chemistry. 1957;226:497–509. [PubMed] [Google Scholar]
- Franciosi M, Lucisano G, Lapice E, Strippoli GF, Pellegrini F, Nicolucci A. Metformin therapy and risk of cancer in patients with type 2 diabetes: systematic review. PloS one. 2013;8:e71583. doi: 10.1371/journal.pone.0071583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham GG, Punt J, Arora M, Day RO, Doogue MP, Duong JK, Furlong TJ, Greenfield JR, Greenup LC, Kirkpatrick CM, et al. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 2011;50:81–98. doi: 10.2165/11534750-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Guba M, Graeb C, Jauch KW, Geissler EK. Pro- and Anti-Cancer Effects of Immunosuppressive Agents Used in Organ Transplantation. Transplantation. 2004;77:1777–1782. doi: 10.1097/01.tp.0000120181.89206.54. [DOI] [PubMed] [Google Scholar]
- Hoorn EJ, Walsh SB, McCormick JA, Zietse R, Unwin RJ, Ellison DH. Pathogenesis of calcineurin inhibitor-induced hypertension. Journal of nephrology. 2012;25:269–275. doi: 10.5301/jn.5000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh A, DuPage M, Priyadharshini B, Sage PT, Quiros J, Borges CM, Townamchai N, Gerriets VA, Rathmell JC, Sharpe AH, et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol. 2015;16:188–196. doi: 10.1038/ni.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, Rathmell JC. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. 2008;180:4476–4486. doi: 10.4049/jimmunol.180.7.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity. 2007;27:173–178. doi: 10.1016/j.immuni.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Lagodzinski Z, Gorski A, Wasik M. Effect of FK506 and cyclosporine on primary and secondary skin allograft survival in mice. Immunology. 1990;71:148–150. [PMC free article] [PubMed] [Google Scholar]
- Lim JH, Luo C, Vazquez F, Puigserver P. Targeting Mitochondrial Oxidative Metabolism in Melanoma Causes Metabolic Compensation through Glucose and Glutamine Utilization. Cancer Res. 2014;74:3535–3545. doi: 10.1158/0008-5472.CAN-13-2893-T. [DOI] [PubMed] [Google Scholar]
- Lo YC, Lee CF, Powell JD. Insight into the role of mTOR and metabolism in T cells reveals new potential approaches to preventing graft rejection. Curr Opin Organ Transplant. 2014;19:363–371. doi: 10.1097/MOT.0000000000000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, et al. The Glucose Transporter Glut1 Is Selectively Essential for CD4 T Cell Activation and Effector Function. Cell Metab. 2014;20:61–72. doi: 10.1016/j.cmet.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31:259–283. doi: 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, Blonska M, Lin X, Sun SC. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. 2014;40:692–705. doi: 10.1016/j.immuni.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberhuber R, Cardini B, Kofler M, Ritschl P, Oellinger R, Aigner F, Sucher R, Schneeberger S, Pratschke J, Brandacher G, et al. Murine cervical heart transplantation model using a modified cuff technique. J Vis Exp. 2014:e50753. doi: 10.3791/50753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oderup C, Malm H, Ekberg H, Qi Z, Veress B, Ivars F, Corbascio M. Costimulation blockade-induced cardiac allograft tolerance: inhibition of T cell expansion and accumulation of intragraft cD4(+)Foxp3(+) T cells. Transplantation. 2006;82:1493–1500. doi: 10.1097/01.tp.0000244064.66136.04. [DOI] [PubMed] [Google Scholar]
- Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342:1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilon CB, Petillon S, Naserian S, Martin GH, Badoual C, Lang P, Azoulay D, Piaggio E, Grimbert P, Cohen JL. Administration of low doses of IL-2 combined to rapamycin promotes allogeneic skin graft survival in mice. Am J Transplant. 2014;14:2874–2882. doi: 10.1111/ajt.12944. [DOI] [PubMed] [Google Scholar]
- Pollizzi KN, Patel CH, Sun IH, Oh MH, Waickman AT, Wen J, Delgoffe GM, Powell JD. mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. The Journal of clinical investigation. 2015;125:2090–2108. doi: 10.1172/JCI77746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nature reviews Immunology. 2014;14:435–446. doi: 10.1038/nri3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JD, Heikamp EB, Pollizzi KN, Waickman AT. A modified model of T-cell differentiation based on mTOR activity and metabolism. Cold Spring Harb Symp Quant Biol. 2013a;78:125–130. doi: 10.1101/sqb.2013.78.020214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JD, Heikamp EB, Pollizzi KN, Waickman AT. A Modified Model of T-Cell Differentiation Based on mTOR Activity and Metabolism. Cold Spring Harb Symp Quant Biol. 2013b doi: 10.1101/sqb.2013.78.020214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol. 2012;30:39–68. doi: 10.1146/annurev-immunol-020711-075024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JD, Zheng Y. Dissecting the mechanism of T-cell anergy with immunophilin ligands. Curr Opin Investig Drugs. 2006;7:1002–1007. [PubMed] [Google Scholar]
- Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN, Rocha Lima CM, Schlesselman JJ, Tolba K, Langmuir VK, et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;71:523–530. doi: 10.1007/s00280-012-2045-1. [DOI] [PubMed] [Google Scholar]
- Roodnat JI, Hilbrands LB, Hene RJ, de Sevaux RG, Smak Gregoor PJ, Kal-van Gestel JA, Konijn C, van Zuilen A, van Gelder T, Hoitsma AJ, et al. 15-Year Follow-up of a Multicenter, Randomized, Calcineurin Inhibitor Withdrawal Study in Kidney Transplantation. Transplantation. 2014 doi: 10.1097/01.TP.0000442774.46133.71. [DOI] [PubMed] [Google Scholar]
- Rowe I, Chiaravalli M, Mannella V, Ulisse V, Quilici G, Pema M, Song XW, Xu H, Mari S, Qian F, et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med. 2013;19:488–493. doi: 10.1038/nm.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayegh MH, Carpenter CB. Transplantation 50 years later--progress, challenges, and promises. N Engl J Med. 2004;351:2761–2766. doi: 10.1056/NEJMon043418. [DOI] [PubMed] [Google Scholar]
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MP, Nelson JA, Feldman S, Van Nguyen B. Pharmacokinetic and phase I study of intravenous DON (6-diazo-5-oxo-L-norleucine) in children. Cancer Chemother Pharmacol. 1988;21:78–84. doi: 10.1007/BF00262746. [DOI] [PubMed] [Google Scholar]
- Thomas AG, O’Driscoll CM, Bressler J, Kaufmann W, Rojas CJ, Slusher BS. Small molecule glutaminase inhibitors block glutamate release from stimulated microglia. Biochem Biophys Res Commun. 2014a;443:32–36. doi: 10.1016/j.bbrc.2013.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AG, O’Driscoll CM, Bressler J, Kaufmann WE, Rojas CJ, Slusher BS. Small molecule glutaminase inhibitors block glutamate release from stimulated microglia. Biochem Bioph Res Co. 2014b;443:32–36. doi: 10.1016/j.bbrc.2013.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AG, Rojas C, Tanega C, Shen M, Simeonov A, Boxer MB, Auld DS, Ferraris DV, Tsukamoto T, Slusher BS. Kinetic characterization of ebselen, chelerythrine and apomorphine as glutaminase inhibitors. Biochem Biophys Res Commun. 2013 doi: 10.1016/j.bbrc.2013.06.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsilidis KK, Capothanassi D, Allen NE, Rizos EC, Lopez DS, van Veldhoven K, Sacerdote C, Ashby D, Vineis P, Tzoulaki I, et al. Metformin does not affect cancer risk: a cohort study in the U.K. Clinical Practice Research Datalink analyzed like an intention-to-treat trial. Diabetes Care. 2014;37:2522–2532. doi: 10.2337/dc14-0584. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev. 2012;249:43–58. doi: 10.1111/j.1600-065X.2012.01152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- Warburg O, Gawehn K, Geissler AW. Metabolism of leukocytes. Zeitschrift fur Naturforschung Teil B: Chemie, Biochemie, Biophysik, Biologie. 1958;13B:515–516. [PubMed] [Google Scholar]
- Willems L, Jacque N, Jacquel A, Neveux N, Maciel TT, Lambert M, Schmitt A, Poulain L, Green AS, Uzunov M, et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood. 2013;122:3521–3532. doi: 10.1182/blood-2013-03-493163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TT, Zhang LJ, Xu KU, Sun CM, Lei T, Peng JX, Liu GW, Wang RY, Zhao Y. Immunosuppressive drugs on inducing Ag-specific CD4(+)CD25(+)Foxp3(+) Treg cells during immune response in vivo. Transplant Immunology. 2012;27:30–38. doi: 10.1016/j.trim.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Yamasaki T, Tran TA, Oz OK, Raj GV, Schwarz RE, Deberardinis RJ, Zhang X, Brugarolas J. Exploring a glycolytic inhibitor for the treatment of an FH-deficient type-2 papillary RCC. Nature reviews Urology. 2011;8:165–171. doi: 10.1038/nrurol.2010.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Chi H. mTOR and metabolic pathways in T cell quiescence and functional activation. Semin Immunol. 2012;24:421–428. doi: 10.1016/j.smim.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T cells are metabolically anergic. J Immunol. 2009;183:6095–6101. doi: 10.4049/jimmunol.0803510. [DOI] [PMC free article] [PubMed] [Google Scholar]