

Abstract
Background: 1-methylpropyl 2-imidazolyl disulfide (PX-12), a thioredoxin 1 (Trx1) inhibitor, has been investigated in a number of ancers, but its effectiveness in the treatment of hepatocellular carcinoma (HCC) has not been reported. PX-12 has generated considerable interest in its use in a variety of solid tumors, yet most studies have confined their interests to using PX-12 as a single agent. The aim of this study is to investigate whether PX-12 inhibits cell growth and has a synergistic anti-tumor effect in combination with 5-fluorouracil (5-FU) in HCC. Methods: Cells were treated with different concentrations of PX-12 and 5-FU. Cell viability assays, colony formation assay, cell cycle assay, reactive oxygen species (ROS) assay, apoptosis analysis, western blot assay, immunohistochemistry and xenograft tumorigenicity assay were performed. Results: Treatment with PX-12 inhibited cell growth, induced S-phase arrest, and increased ROS levels. PX-12-induced apoptosis and inhibition of colony formation were associated with the generation of ROS, and inhibition of ROS attenuated PX-12-induced apoptosis and inhibition of colony formation. Treatment with PX-12 increased the expression of bax and reduced the expression of bcl-2, indicating that PX-12-mediated apoptosis is mitochondria-dependent. PX-12 also exerted a synergistic effect with 5-FU tosignificantly suppress tumorigenicity both in vitro and in vivo. Inhibition of ROS accumulation reduced the synergistic effect of PX-12 and 5-FU. Conclusions: PX-12 has anti-tumor activity and a synergistic effect in combination with 5-FU in HCC. Treatment with PX-12 alone or in combination with 5-FU may have clinical use in the treatment of HCC and other cancers.
Keywords: Hepatocellular carcinoma, thioredoxin 1, PX-12, 5-FU, ROS
Introduction
1-methylpropyl 2-imidazolyl disulfide (PX-12), an inhibitor of thioredoxin 1 (Trx1) is currently being used as a therapy for advanced cancers in phase II/IB clinical trials. Trx1 is an important protein because of its antioxidant activity. Modification of thiols in thioredoxin interrupts signaling mechanisms involved in cell growth, proliferation, and apoptosis. Trx1 is upregulated in a wide variety of carcinomas [1-4]. Increased Trx1 levels have been correlated with increased proliferation and decreased apoptosis of human gastric tumors [5] and with decreased patient survival in non-small cell lung cancer [6]. Inactivation of Trx-1 increases reactive oxygen species (ROS) levels. ROS mediates cell mitochondrial dysfunction [7], leads to autophagic cell death of hepatocellular carcinoma (HCC) cells [8], and promotes apoptosis via activation of JNK and p38 [9]. At low levels, ROS act as signaling molecules to activate proliferation and survival pathways. However, high ROS levels induce cell senescenceor death. Therefore, Trx-1inhibitors have been regarded as potential anti-tumor drugs.
PX-12 causes rapid reversible thioalkylation of the catalytic site Cys32 and Cys35 residues ofTrx1, and slower irreversible thioalkylation of Cys73 that is outside the catalytic site, and prevents the reduction of Trx1 by thioredoxin reductase 1 [10]. PX-12 has been shown to have antitumor activities in several types of cancer cells [11-13]. PX-12 inhibits HIF-1α and VEGF protein levels in MCF-7 tumor xenografts in vivo [14]. Although PX-12 has been investigated in many cancers, its effectiveness in the treatment of HCC has not been reported.
The compound 5-fluorouracil (5-FU) is one of the most commonly used chemotherapeutic drugs for HCC [15], colorectal and gastric cancers [16]. It acts by blocking nucleoside metabolism that leads to cell cycle arrest and subsequent apoptosis. Increased ROS levels elevate the toxicity of 5-FUin MCF-7 cells [17]. PX-12 enhances the generation of ROS [12-14], whether PX-12 increases the inhibitory effect of 5-FU on HCC remains unknown.
In this study, we have investigated the roles of PX-12 in HCC and the effects of the combination of PX-12 and 5-FU on HCC in vitro and in vivo. We found that PX-12 induced S-phase arrest and ROS-dependent apoptosis and inhibition of colony formation. PX-12 sensitized HCC to 5-FU both in vitro and in vivo. Inhibition of ROS reduced the synergistic effect of PX-12 and 5-FU. Collectively, our data suggests that treatment with PX-12 alone or in combination with 5-FU has significant potential as an anti-tumor agent for HCC and other cancers.
Materials and methods
Cell culture and reagents
HCC cell lines HepG2 and SMMC7721 were obtained from China Center for Type Culture Collection (CCTCC, Wuhan, China). Cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco, USA) with 100 U/mL penicillin and 100 μg/mL streptomycin (Invitrogen Life Technologies, USA) at 37°C in a 5% CO2 humidified incubator. PX-12 (Santa Cruz, USA) was diluted to 100 mM. 5-FU was purchased from Sigma-Aldrich (St. Louis, MO, USA). N-Acetyl Cysteine (NAC) (Beyotime Institute of Biotechnology, China) was diluted to 200 mM.
Cell viability assays
For Cell Counting Kit-8 assays, indicated cells (2000 cells/well) were cultured in 96-well plates for the indicated time periods. Cell Counting Kit-8 (CCK-8, Dojindo, Japan) was added in the plates for 2 h to test the optical density (OD) value at 450 nm. IC50 was then calculated using SPSS software.
Cell cycle assay
For cell cycle analysis, HepG2 or SMMC-7721 cells were seeded in 6-well plates (1×105 cells/well) and serum starved for 24 h. The cells were treated with PX-12 for 48 h. The cells were harvested and washed with PBS. The cells were then fixed with 70% ethanol. Immediately prior to the analysis, the cells were incubated with fresh propidium iodide containing RNase A for 30 min at 37°C. A total of 1×104 cells were analyzed from each sample on a fluorescence-activated cell sorting Calibur flow cytometer (Becton Dickinson).
Assay of the intracellular ROS level
The intracellular ROS level was measured by using a dichlorofluorescein assay (Beyotime Institute of Biotechnology, China). 2, 7-dichlorodihydrofluorescein diacetate (DCFH-DA) was used to evaluate the generation of ROS in oxidative damage. Cells (1×105 cells/well) were seeded in 6-well plates in a humidified atmosphere containing 5% CO2 at 37°C and serum starved for 24 h. The cells were treated with the indicated concentrations of PX-12 in the presence or absence of NAC (5 mM) or 5-FU and further incubated for 48 h. Thereafter, the cells were harvested and incubated with 100 µM DCFH-DA for 20 min in a 5% CO2 humidified incubator. Finally, the cells were washed three times with phosphate-buffered saline (PBS, pH 7.4). Fluorescence was measured using flow cytometry (Becton Dickinson).
Apoptosis detection
For apoptosis analysis, HepG2cells were seeded in 6-well plates (1×105 cells/well) and serum starved for 24 h. The cells were treated with PX-12 or 5-FU for 48 h. The cells were harvested and stained by FITC-labeled Annexin V/PI apoptosis assay kit (BD Biosciences, San Jose, CA, USA). Cells (1×104) were analyzed from each sample on flow cytometry. Three independent assays were performed with at least 3 replicates.
Colony formation assay
HepG2 cells (500 cells/well) were plated in 6-well plates and treated with PX-12 and 5-FU for 48 h. Then, fresh culture media were replaced every two days. 12 days later, the plates were stained with 1% crystal violet (Sigma-Aldrich, USA) and photographed. Colonies were counted and analyzed using Alpha Innotech Imaging system (Alphatron Asia Pte Ltd, Singapore).
Western blot analysis
Cells were lysed in RIPA lysis buffer (50 mM Tris-HCl at pH 8.0, 1% NP40, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 0.02% sodium azide and 150 mM NaCl) containing Protease Inhibitor cocktail (Roche, Switzerland) at 4°C. 40 μg proteins were separated on precasted 10% sodium dodecyl sulfate-polyacrylamide gels and then transferred onto PVDF membranes (Millipore). The blots were blocked in 5% non-fat milk and incubated overnight at 4°C with primary antibodies (anti-bcl-2, bax, PARP, cleaved PARP antibodies at 1:1000 dilution; other antibodies at 1:2000 dilution). All the antibodies were bought from Cell Signaling Technology, USA. The blots were then incubated with horseradish peroxidase-conjugated secondary antibody at 1:2000 dilution for 1 h at 37°C. The signals were visualized using the enhanced chemiluminescence system (Bio-Rad, USA). Protein expression was quantified by densitometry and normalized to β-actin expression using Image Lab software.
Drug combination effects
To determine the drug combination effects, the commercially available program CalcuSyn (Biosoft Ferguson, MO, USA) was used to calculate combination index (CI) values and dose-reduction index (DRI) values for combination of drugs. CI: A quantitative measure of the degree of drug interaction in terms of additive effect (CI=1), synergism (CI 1), or antagonism (CI>1) for a given endpoint of the effect measurement. DRI: A measure of how much the dose of each drug in a synergistic combination may be reduced at a given effect level compared with the doses of each drug alone. Drug combinations that acted synergistically can be identified as those that exhibited significant dose reduction values.
Xenograft tumorigenicity assay
All of the in vivo studies satisfied the National Institutes of Health guidelines (NIH publication 86-23 revised 1985) and the protocol was approved by the Committee on the Ethics of Animal Experiments of the Tongji Medical College, Huazhong University of Science and Technology. 5×106 HepG2 cells were subcutaneously injected into the mice at day 0. Drug administration started when tumor xenograft grew up to approximately 100 mm3 in size. Mice were treated with vehicle control (0.9% NaCl solution), 25 mg/kg PX-12 (2.5 mg/mL in vehicle) i.v. injection, 30 mg/kg 5-FU (3.0 mg/mLin vehicle) i.p. injection, or combination of PX-12 with 5-FU three times per week till the end of the study. Subcutaneous tumors were removed, fixed and sectioned for proliferation and apoptosis analysis.
Immunohistochemistry analysis
Immunohistochemistry was carried out as described previously [18,19]. The Ki-67 primary antibody was purchased from Dako (Golstrup, Denmark). The apoptosis of paraffin-embedded sections of the tumors was detected by a TUNEL assay kit (R&D Systems, Minneapolis, MN, USA).
Statistical analysis
Data analyses were performed by SPSS 13.0 (Chicago, IL, USA) or GraphPad Prism 5.0 (La Jolla, CA, USA). All experiments were at least three independent times and the results were presented as mean ± SEM. Comparisons between the different groups were evaluated using one-way ANOVA, and p<0.05 was considered statistically significant.
Results
PX-12 inhibits cell growth
We started by investigating the effect of PX-12 on HepG2 and SMMC-7721HCC cell lines in vitro. HCC cells were incubated for 24 or 48 hours with increasing concentrations of PX-12. We found a progressive reduction in HCC cell numbers that was proportional to both the concentration of PX-12 and the duration of culture (Figure 1). The IC50 values of PX-12 on HepG2 and SMMC-7721 cells for the 24 h time point were 30.30 and 25.15 μM. The IC50 values of PX-12 on HepG2 and SMMC-7721 cells for the 48 h time point were 6.32 and 13.38 μM.
Figure 1.
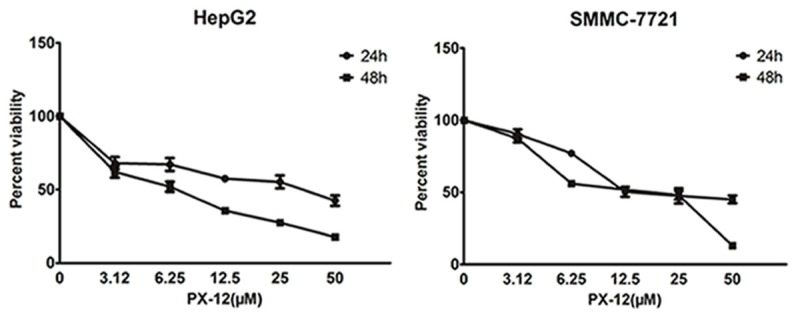
PX-12 inhibits cell growth. Cell growth inhibition was evaluated by CCK8. HepG2 and SMMC-7721 cells were treated with escalated doses of PX-12 (0, 3.12, 6.25, 12.5, 25, and 50 μM, respectively) and incubated for 24 h or 48 h. CCK8 assay was used to calculate the number of cells. All experiments were performed in triplicate.
PX-12 induces S-phase arrest and increases the accumulation of ROS
To further examine the mechanism by which PX-12 inhibited cell growth, we studied the effects of PX-12 on cell cycle. PX-12 treatment led to a dose-dependent induction of S-phase arrest in HepG2 and SMMC-7721 (Figure 2A, 2B). The fluorescent probe DCFH-DA was used to monitor intracellular ROS levels in different concentrations of PX-12. PX-12-treated cells had significantly higher ROS levels compared with untreated cells (Figure 2C, 2D).
Figure 2.

PX-12 induces S-phase arrest and increases the accumulation of ROS. A, B. HepG2 and SMMC-7721 cells were treated with various concentrations of PX-12 for 48 h, and the DNA content was analyzed by flow cytometry. The percentages of cells in the G1, S and G2/M phases of the cell cycle were shown. C, D. DCFH-DA was used to detect ROS in HepG2 and SMMC-7721 cells. Relative ratio of mean fluorescence intensity was normalized to control. All the results are represented as the mean ± SEM from three independent trials. *P<0.05, **P<0.01.
NAC inhibits ROS accumulation and activation of the mitochondria-dependent apoptosis induced by PX-12
We next investigated whether ROS accumulation induced by PX-12 was responsible for apoptosis in HCC cells. Cells were pre-treated with the antioxidant NAC (5 mM) 1 h before being incubated with PX-12 for a further 48 h. Pretreatment with NAC caused a significant decrease in ROS levels induced by PX-12 (Figure 3A, 3B). NAC prevented cell from apoptosis induced by PX-12 (Figure 3C, 3E). Addition of NAC significantly reduced the inhibitive effect of PX-12 on colony formation (Figure 3D, 3F). Moreover, the presence of NAC resulted in a reversal in the ability of PX-12 to inhibit HCC cell growth (Figure 3G). Bcl-2 is a protein of the anti-apoptotic family, and bax is a protein of the pro-apoptotic family. PX-12 induced an increase in bcl-2 and PARP cleavage and a decrease in bax (Figure 3H). These data shows that PX-12-induced apoptosis is mitochondria-dependent. Treatment of NAC reversed the changes of bcl-2, bax, and PARP cleavage. To conclude, these results suggest that PX-12 induces mitochondria-dependent apoptosis via ROS accumulation.
Figure 3.

NAC inhibits ROS accumulation and activation of the mitochondria-dependent apoptosis induced by PX-12. A, B. HepG2 and SMMC-7721 cells were treated with the antioxidant NAC at 1 h before adding PX-12 and incubation for an additional 48 h. ROS level was measured using DCFH-DA. Relative ratio of mean fluorescence Intensity was normalized to control. C, E. Annexin V/PI staining was used to identify apoptosis of HepG2 cells induced by PX-12 or in the presence or absence of NAC. The percentage of apoptosis was shown as the mean ± SEM from three independent experiments. D, F. HepG2 cells were treated with PX-12 in the presence or absence of NAC for 48 h. The plates were stained and photographed 12 days later. The percentage of the colonies was calculated. G. HepG2 and SMMC-7721 cells were treated with NAC at 1 h before adding PX-12 and incubation for an additional 24 and 48 h. CCK8 assay was used to monitor the cell growth. H. HepG2 cells were treated with or without NAC at 1 h before adding PX-12 and incubation for an additional 48 h. Western blot was used to analyze the expression of apoptosis-related protein. All the results are represented as the mean ± SEM from three independent trials. *P<0.05, **P<0.01.
PX-12 potentiates anti-tumor effect of 5-FU in vitro
5-FU remains the principle cytotoxic chemotherapy in the treatment of HCC. We next explored the effects of treatment with PX-12 and 5-FUon HCC. We found that the combination of PX-12 with 5-FU had a synergistic effect in inhibiting cell growth (Figure 4A). This was demonstrated by CI<1 and DRI (Figure 4B, Table 1). The dose of each drug used in the combination to achieve a specific measurable effect level was substantially reduced when compared with the dose needed to achieve the same inhibition rate when the drugs were given alone (Table 1). Incubation of HCC cells with either 5-FU or PX-12 alone caused a slight increase in apoptosis, while combination of the two agents strongly increased apoptosis (Figure 4C, 4D). Western blot analysis revealed a stable expression of bax and cleaved-PARP after incubation with PX-12 and 5-FU, while antiapoptotic opponent bcl-2 was diminished (Figure 4E). All these results demonstrated that PX-12 acts synergistically with5-FU to inhibit the growth of HCC.
Figure 4.

PX-12 potentiates anti-tumor effect of 5-FU in vitro. A. 2000 cells/well were plated overnight followed by treatment with increasing doses of PX-12and 5-FU in combination at a fixed ratio (PX-12/5-FU=1.25). CCK8 assay was used to calculate the inhibition rate of PX-12 or 5-FU on cells. B. CI was obtained using CalcuSyn2.0 for synergy analyses. C. Annexin V/PI staining was used to identify apoptosis induced by PX-12 and/or 5-FU. D. The percentage of cell apoptosis was shown as the mean ± SEM from three independent experiments. E. Western blot analysis of the expression levels of apoptosis-related protein in HepG2 cells which incubated for 48 h. All the results are represented as the mean ± SEM from three independent trials. *P<0.05, **P<0.01.
Table 1.
Combination of PX-12 with 5-FU in HepG2 cells
| Combination | Drug alone | Fa | CI | DRI | |||
|---|---|---|---|---|---|---|---|
|
|
|
|
|||||
| PX-12 | 5-FU | PX-12 | 5-FU | PX-12 | 5-FU | ||
| 0 | 0 | 0 | 0 | ||||
| 3.12 | 2.5 | 7.63±2.74 | 7.58±0.55 | 0.53±0.02 | 0.78±0.17 | 2.45±0.88 | 3.03±0.22 |
| 6.25 | 5 | 27.8±2.23 | 20.00±6.16 | 0.71±0.05 | 0.57±0.18 | 3.57±0.96 | 4.00±1.23 |
| 12.5 | 10 | 42.18±0.25 | 37.01±4.23 | 0.80±0.01 | 0.57±0.03 | 3.37±0.02 | 3.70±0.42 |
| 25 | 20 | 135.56±42.19 | 102.76±12.90 | 0.90±0.00 | 0.39±0.03 | 5.42±1.69 | 5.14±0.65 |
| 50 | 40 | 613.72±298.45 | 408.58±191.72 | 0.96±0.01 | 0.22±0.08 | 12.27±5.97 | 10.21±4.79 |
Fa the fraction affected by the dose.
Combination of PX-12 with 5-FU induces apoptosis, reduces colony formation of HCC cells via ROS-dependent mechanism.
Combination of PX-12 with 5-FU increased ROS levels compared with single agents, while pre-treatment of NAC down-regulated ROS accumulation (Figure 5A, 5D). Combination of both agents significantly reduced colony formation and increased apoptosis, addition of NAC decreased the combined effects of the two agents (Figure 5B, 5C, 5E, 5F). Western blot assay showed that combination of the two agents decreased bax and increased bcl-2 and PARP cleavage, while NAC significantly reversed the changes of apoptosis-related proteins induced by the two agents (Figure 5G). These data indicated that combination of PX-12 with 5-FU reduces colony formation and induces apoptosis of HCC cells via ROS-dependent mechanism in vitro.
Figure 5.
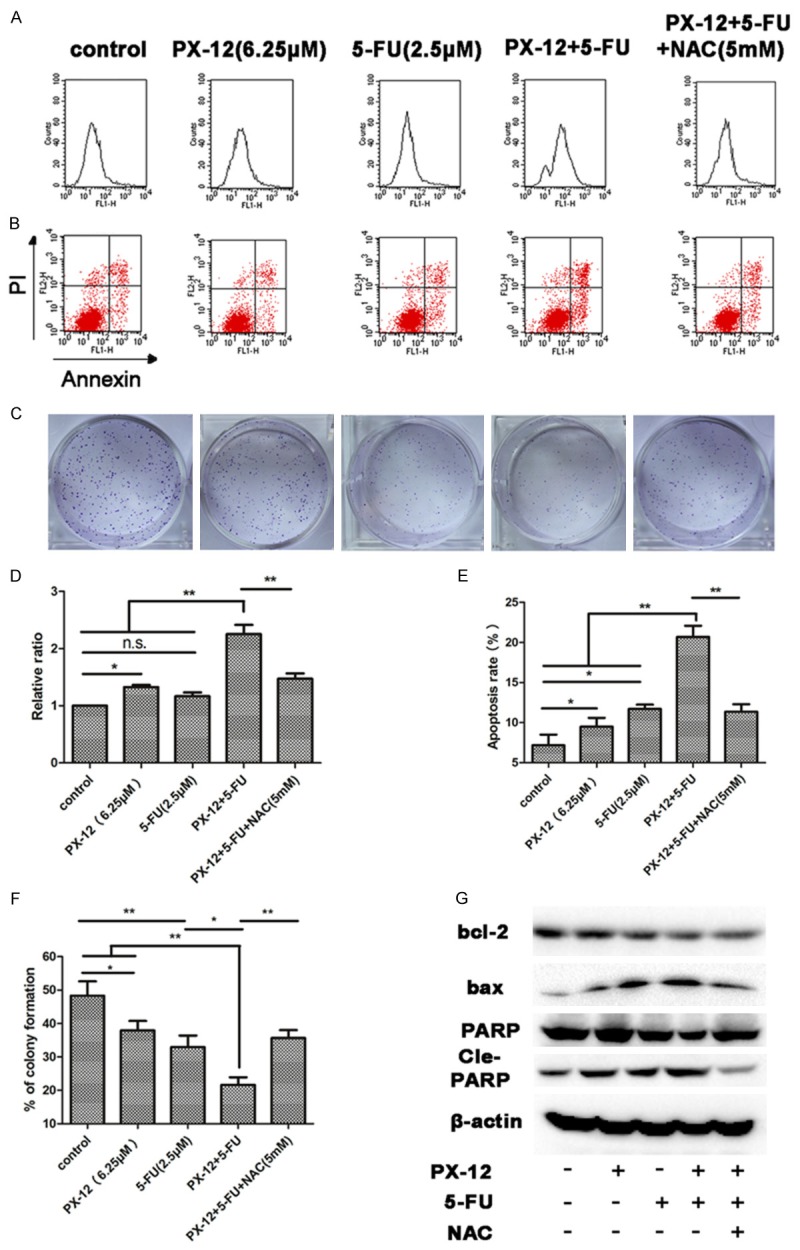
Combination of PX-12 with 5-FU induces apoptosis, reduces colony formation of HCC cells via ROS-dependent mechanism. A, D. HepG2 cells were treated with PX-12 and/or 5-FU in the presence or absence of NAC for 48 h. After incubation for 48 h, DCFH-DA was used to detect ROS level. Relative ratio of mean fluorescence intensity was normalized to control. B, E. Annexin V/PI staining was used to identify apoptosis induced by PX-12 and/or 5-FU in the presence or absence of NAC. C, F. HepG2 cells were treated with PX-12 and/or 5-FUin the presence or absence of NAC for 48 h. The plates were stained and photographed 10 days later. The percentage of the colonies was calculated. G. Western blot analysis of the expression levels of apoptosis-related protein in HepG2 cells which incubated for 48 h. All the results are represented as the mean ± SEM from three independent experiments. *P<0.05, **P<0.01, n.s. means no significance.
PX-12 enhances the anti-tumor effect of 5-FU in vivo
To further verify PX-12 and 5-FU have synergistic antitumor effect, we next explored the effect of both agents on HCC cell growth in vivo. Mice were subcutameously injected with HCC cell line HepG2. Drug treatment was initiated as soon as the tumor volume reached approximately 100 mm3 at day 9. At day 24, mice were sacrificed. Treatment with PX-12 and 5-FU had no influence on body weight (Figure 6A). Although treatment with either PX-12 (25 mg/kg) or 5-FU (30 mg/kg) alone slightly reduced HCC cell growth compared with vehicle, co-treatment resulted in a significant reduction in HCC tumor size and weight (Figure 6B-D). TUNEL staining and Ki-67 of the tumor sections were used to detect the apoptosis and proliferation in vivo. After treatment with either PX-12 or 5-FU alone, the apoptotic index determined by the percentage of TUNEL stained nuclei was slightly increased and the proliferation index determined by the percentage of Ki-67 positive cells was also slightly decreased, while co-treatment significantly increased apoptosis and reduced proliferation (Figure 6E-G).
Figure 6.

PX-12 enhances the anti-tumor effect of 5-FU in vivo. A. The body weight curve of mice in all groups. B. The tumor growth curve. At day 9 after implant, drug administration started and the tumor volume was calculated every three days. C. The general view of the subcutaneous tumors in the four groups. D. The tumor weights in different groups. E. Hematoxylin and eosin (H&E) staining. E, F. TUNEL staining was performed for apoptosis analysis. The apoptotic index was calculated by the percentage of TUNEL stained nuclei. E, G. Ki-67 staining was used for growth analysis. The proliferation index as determined by the percentage of Ki-67 stained nuclei was calculated in different groups. All the results are represented as the mean ± SEM from three independent trials. **p<0.01, n.s. means no significance.
Discussion
PX-12, a Trx1 inhibitor, is currently being assessed in phase II trials in the treatment of advanced pancreatic cancer [20] and in a phase IB trials in treatment of advanced gastrointestinal cancers [21]. Its ability to inhibit the ability of Trx1 has generated considerable interest in its use in a variety of solid tumors, yet most studies have confined their interests to using PX-12 as a single agent. In this study, we testify whether PX-12 inhibits cell growth and enhances the chemosensitivity to 5-FU in HCC cells both in vitro and in vivo. PX-12 greatly inhibited the anchorage-dependent growth of COLO-357 cells, and this inhibitory effect was markedly attenuated in CS7 clones that expressed high levels of Trx [22]. Human Trx1 belongs to a family of small redox proteins that undergo NADPH-dependent reduction by thioredoxin reductase (TrxR), and reduce oxidized cysteine groups on proteins. Trx1 plays an important role in regulating cell redox homeostasis and cell growth, differentiation, and apoptosis. The expression of Trx1 is high in a wide variety of carcinomas such as hepatocellular carcinoma [1,2], colorectal cancer [3], and thyroid cancer [4]. Increased serum levels of Trx1 in patients secreted by cancer cells are associated with aggressive tumor growth and decreased survival [23-26]. Recombinant human Trx attenuated ethanol-induced increases in markers of oxidative stress, inflammatory cytokine expression, and apoptosis [27]. Overexpressed hTrx is responsible for the development of cellular resistance to CDDP, possibly by scavenging intracellular toxic oxidants generated by this anticancer agent [28].
In this study, PX-12 reduced cell number by cell cycle arrest and inducing activation of the mitochondria-dependent apoptosis. Furthermore, apoptosis was associated with the enhanced generation of ROS and was inhibited in the presence of NAC, a potent antioxidant agent. Antioxidant can reduce ROS level and ameliorate oxidative stress [29]. PX-12 promotes ROS-dependent apoptosis in several cell lines, such as HeLa [12], A549 [13], and Calu-6 [30]. NAC, an antioxidant, can prevent dissociation of Trx-ASK1 and inactivates p38 MAPK pathway [31]. ROS induces mitochondrial dysfunction [7], promotes apoptosis via activation of JNK and p38 [9], and leads to death of HCC cells through autophagy [8]. We found that PX-12 increased the expression of PARP cleavage and bax and reduced the expression of bcl-2. It suggests that PX-12 induced apoptosis via mitochondria-dependent mechanism. NAC reduced the effect of PX-12 on the expression of PARP cleavage, bax, and bcl-2. It suggests that ROS plays an important role in PX-12-induced cell activation of the mitochondria-dependent apoptosis pathway. In addition to inducing apoptosis, PX-12 also induced S-phase arrest. Therefore, the S-phase arrest and apoptosis in PX-12-treated cells are underlying mechanisms to inhibit cell growth.
PX-12 increased generation of ROS in our study, we therefore predicted that it may augment the cytotoxicity of 5-FU via ROS-dependent mechanism in HCC cells. The compound 5-FU has been used against cancer for about 40 years [32]. 5-FUis one of the most commonly used chemotherapeutic drugs for hepatocellular carcinoma [15], colorectal and gastric cancers in clinical practice [16]. There is compelling evidence that cellular adaptation to ROS stress has a part in maintaining a cellular cancer phenotype and chemotherapy resistance [33]. Increased ROS levels increased toxicity of 5-FU in MCF-7 cells [17]. We deduce that PX-12 may increase cytotoxicity of 5-FU in HCC cells. To test this hypothesis, we measured CI and DRI of PX-12 and 5-FU. If drug combinations interact in a manner that result in synergy, then the dose of each drug used in the combination to achieve a specific measurable effect level will be substantially reduced when compared with the dose needed to achieve the same effect level when the drugs are given alone. We found that PX-12 and 5-FU had a synergistic effect on inhibition of cell growth (CI<1 and DRI). Combination of PX-12 with 5-FU significantly increased ROS level, induced apoptosis, and inhibited colony formation.NAC reduced the effects of PX-12 and 5-FU on ROS accumulation, apoptosis, and inhibition of colony formation. Combined treatment with PX-12 and 5-FU increased the expression of bax and cleaved-PARP and diminished the expression of bcl-2 compared with each agent used alone. NAC reversed the changes of apoptosis-related proteins caused by the combined treatment. These results suggest that PX-12 increase cytotoxicity of 5-FU via ROS-dependent mechanism.
Finally, Xenograft tumorigenicity assay was used to verify PX-12 and 5-FU synergistically exert the antitumor effect in vivo. We found combination of PX-12 with 5-FU significantly reduced tumor burden compared with single agents that matched with the results in vitro.
Conclusions
In summary, PX-12 inhibits the growth of HCC and the ability of PX-12 and 5-FU to synergistically inhibit growth in HCC. Combination therapy may yield success when applied in the clinical setting.PX-12 is currently being assessed in phase II/IB trials. This underscores the realistic and potentially rapid clinical application of these findings.
Acknowledgements
This work was supported by the State Key Project on Infection Diseases of China (Grant No. 2012ZX10002016-004, 2012ZX10002010-001-004), the National Natural Science Foundation of China (No. 81372495 and 81372327), and the Hepatic surgery clinical study center of Hubei Province, China (No. 2014BKB089, 2013BCB026, and 2012DCA13003). The authors thank Laurence Arian for his assistance in revising the draft.
Disclosure of conflict of interest
None.
Abbreviations
- PX-12
1-methylpropyl 2-imidazolyl disulfide
- 5-FU
5-fluorouracil
- HCC
Hepatocellularcarcinoma
- Trx1
thioredoxin 1
- HDAC
Histone deacetylase
- CI
Combination index
- DRI
Dose-reduction index
- TUNEL
TdT-mediated dUTP Nick-End Labeling
- DCFH-DA
2, 7-dichlorodihydrofluorescein diacetate
- PARP
Poly ADP-ribose polymerase
References
- 1.Ding SJ, Li Y, Shao XX, Zhou H, Zeng R, Tang ZY, Xia QC. Proteome analysis of hepatocellular carcinoma cell strains, MHCC97-H and MHCC97-L, with different metastasis potentials. Proteomics. 2004;4:982–994. doi: 10.1002/pmic.200300653. [DOI] [PubMed] [Google Scholar]
- 2.Cunnea P, Fernandes AP, Capitanio A, Eken S, Spyrou G, Bjornstedt M. Increased expression of specific thioredoxin family proteins; a pilot immunohistochemical study on human hepatocellular carcinoma. Int J Immunopathol Pharmacol. 2007;20:17–24. doi: 10.1177/039463200702000103. [DOI] [PubMed] [Google Scholar]
- 3.Raffel J, Bhattacharyya AK, Gallegos A, Cui H, Einspahr JG, Alberts DS, Powis G. Increased expression of thioredoxin-1 in human colorectal cancer is associated with decreased patient survival. J Lab Clin Med. 2003;142:46–51. doi: 10.1016/S0022-2143(03)00068-4. [DOI] [PubMed] [Google Scholar]
- 4.Lincoln DT, Al-Yatama F, Mohammed FM, Al-Banaw AG, Al-Bader M, Burge M, Sinowatz F, Singal PK. Thioredoxin and thioredoxin reductase expression in thyroid cancer depends on tumour aggressiveness. Anticancer Res. 2010;30:767–775. [PubMed] [Google Scholar]
- 5.Grogan TM, Fenoglio-Prieser C, Zeheb R, Bellamy W, Frutiger Y, Vela E, Stemmerman G, Macdonald J, Richter L, Gallegos A, Powis G. Thioredoxin, a putative oncogene product, is overexpressed in gastric carcinoma and associated with increased proliferation and increased cell survival. Hum Pathol. 2000;31:475–481. doi: 10.1053/hp.2000.6546. [DOI] [PubMed] [Google Scholar]
- 6.Kakolyris S, Giatromanolaki A, Koukourakis M, Powis G, Souglakos J, Sivridis E, Georgoulias V, Gatter KC, Harris AL. Thioredoxin expression is associated with lymph node status and prognosis in early operable non-small cell lung cancer. Clin Cancer Res. 2001;7:3087–3091. [PubMed] [Google Scholar]
- 7.Guo Y, Zhang W, Yan YY, Ma CG, Wang X, Wang C, Zhao JL. Triterpenoid pristimerin induced HepG2 cells apoptosis through ROSmediated mitochondrial dysfunction. J BUON. 2013;18:477–485. [PubMed] [Google Scholar]
- 8.Guo W, Zhao Y, Zhang Z, Tan N, Zhao F, Ge C, Liang L, Jia D, Chen T, Yao M, Li J, He X. Disruption of xCT inhibits cell growth via the ROS/autophagy pathway in hepatocellular carcinoma. Cancer Lett. 2011;312:55–61. doi: 10.1016/j.canlet.2011.07.024. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Z, Zhang C, Ding Y, Zhao Q, Yang L, Ling J, Liu L, Ji H, Zhang Y. The activation of p38 and JNK by ROS, contribute to OLO-2-mediated intrinsic apoptosis in human hepatocellular carcinoma cells. Food Chem Toxicol. 2014;63:38–47. doi: 10.1016/j.fct.2013.10.043. [DOI] [PubMed] [Google Scholar]
- 10.Kirkpatrick DL, Kuperus M, Dowdeswell M, Potier N, Donald LJ, Kunkel M, Berggren M, Angulo M, Powis G. Mechanisms of inhibition of the thioredoxin growth factor system by antitumor 2-imidazolyl disulfides. Biochem Pharmacol. 1998;55:987–994. doi: 10.1016/s0006-2952(97)00597-2. [DOI] [PubMed] [Google Scholar]
- 11.Li C, Thompson MA, Tamayo AT, Zuo Z, Lee J, Vega F, Ford RJ, Pham LV. Over-expression of Thioredoxin-1 mediates growth, survival, and chemoresistance and is a druggable target in diffuse large B-cell lymphoma. Oncotarget. 2012;3:314–326. doi: 10.18632/oncotarget.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin HR, You BR, Park WH. PX-12-induced HeLa cell death is associated with oxidative stress and GSH depletion. Oncol Lett. 2013;6:1804–1810. doi: 10.3892/ol.2013.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.You BR, Shin HR, Park WH. PX-12 inhibits the growth of A549 lung cancer cells via G2/M phase arrest and ROS-dependent apoptosis. Int J Oncol. 2014;44:301–308. doi: 10.3892/ijo.2013.2152. [DOI] [PubMed] [Google Scholar]
- 14.Welsh SJ, Williams RR, Birmingham A, Newman DJ, Kirkpatrick DL, Powis G. The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1alpha and vascular endothelial growth factor formation. Mol Cancer Ther. 2003;2:235–243. [PubMed] [Google Scholar]
- 15.de Lima Lopes G Jr, Dicksey JS, Peters WP, Palalay M, Chang AY. Final results of a prematurely discontinued Phase 1/2 study of eniluracil with escalating doses of 5-fluorouracil administered orally in patients with advanced hepatocellular carcinoma. Cancer Chemother Pharmacol. 2011;68:1067–1073. doi: 10.1007/s00280-011-1588-x. [DOI] [PubMed] [Google Scholar]
- 16.Diasio RB, Johnson MR. The role of pharmacogenetics and pharmacogenomics in cancer chemotherapy with 5-fluorouracil. Pharmacology. 2000;61:199–203. doi: 10.1159/000028401. [DOI] [PubMed] [Google Scholar]
- 17.Pandey V, Chaube B, Bhat MK. Hyperglycemia regulates MDR-1, drug accumulation and ROS levels causing increased toxicity of carboplatin and 5-fluorouracil in MCF-7 cells. J Cell Biochem. 2011;112:2942–2952. doi: 10.1002/jcb.23210. [DOI] [PubMed] [Google Scholar]
- 18.Ding ZY, Jin GN, Wang W, Chen WX, Wu YH, Ai X, Chen L, Zhang WG, Liang HF, Laurence A, Zhang MZ, Datta PK, Zhang B, Chen XP. Reduced expression of transcriptional intermediary factor 1 gamma promotes metastasis and indicates poor prognosis of hepatocellular carcinoma. Hepatology. 2014;60:1620–1636. doi: 10.1002/hep.27273. [DOI] [PubMed] [Google Scholar]
- 19.Zhang B, Chen X, Bae S, Singh K, Washington MK, Datta PK. Loss of Smad4 in colorectal cancer induces resistance to 5-fluorouracil through activating Akt pathway. Br J Cancer. 2014;110:946–957. doi: 10.1038/bjc.2013.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramanathan RK, Abbruzzese J, Dragovich T, Kirkpatrick L, Guillen JM, Baker AF, Pestano LA, Green S, Von Hoff DD. A randomized phase II study of PX-12, an inhibitor of thioredoxin in patients with advanced cancer of the pancreas following progression after a gemcitabine-containing combination. Cancer Chemother Pharmacol. 2011;67:503–509. doi: 10.1007/s00280-010-1343-8. [DOI] [PubMed] [Google Scholar]
- 21.Baker AF, Adab KN, Raghunand N, Chow HH, Stratton SP, Squire SW, Boice M, Pestano LA, Kirkpatrick DL, Dragovich T. A phase IB trial of 24-hour intravenous PX-12, a thioredoxin-1 inhibitor, in patients with advanced gastrointestinal cancers. Invest New Drugs. 2013;31:631–641. doi: 10.1007/s10637-012-9846-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnold NB, Ketterer K, Kleeff J, Friess H, Buchler MW, Korc M. Thioredoxin is downstream of Smad7 in a pathway that promotes growth and suppresses cisplatin-induced apoptosis in pancreatic cancer. Cancer Res. 2004;64:3599–3606. doi: 10.1158/0008-5472.CAN-03-2999. [DOI] [PubMed] [Google Scholar]
- 23.Miyazaki K, Noda N, Okada S, Hagiwara Y, Miyata M, Sakurabayashi I, Yamaguchi N, Sugimura T, Terada M, Wakasugi H. Elevated serum level of thioredoxin in patients with hepatocellular carcinoma. Biotherapy. 1998;11:277–288. doi: 10.1023/a:1008032703468. [DOI] [PubMed] [Google Scholar]
- 24.Tabata C, Terada T, Tabata R, Yamada S, Eguchi R, Fujimori Y, Nakano T. Serum thioredoxin-1 as a diagnostic marker for malignant peritoneal mesothelioma. J Clin Gastroenterol. 2013;47:e7–11. doi: 10.1097/MCG.0b013e31824e901b. [DOI] [PubMed] [Google Scholar]
- 25.Park BJ, Cha MK, Kim IH. Thioredoxin 1 as a serum marker for breast cancer and its use in combination with CEA or CA15-3 for improving the sensitivity of breast cancer diagnoses. BMC Res Notes. 2014;7:7. doi: 10.1186/1756-0500-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park BJ, Cha MK, Kim IH. Thioredoxin 1 as a serum marker for ovarian cancer and its use in combination with CA125 for improving the sensitivity of ovarian cancer diagnoses. Biomarkers. 2014;19:604–610. doi: 10.3109/1354750X.2014.956793. [DOI] [PubMed] [Google Scholar]
- 27.Cohen JI, Roychowdhury S, DiBello PM, Jacobsen DW, Nagy LE. Exogenous thioredoxin prevents ethanol-induced oxidative damage and apoptosis in mouse liver. Hepatology. 2009;49:1709–1717. doi: 10.1002/hep.22837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sasada T, Iwata S, Sato N, Kitaoka Y, Hirota K, Nakamura K, Nishiyama A, Taniguchi Y, Takabayashi A, Yodoi J. Redox control of resistance to cis-diamminedichloroplatinum (II) (CDDP): protective effect of human thioredoxin against CDDP-induced cytotoxicity. J Clin Invest. 1996;97:2268–2276. doi: 10.1172/JCI118668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim SH, Smith AJ, Tan J, Shytle RD, Giunta B. MSM ameliorates HIV-1 Tat induced neuronal oxidative stress via rebalance of the glutathione cycle. Am J Transl Res. 2015;7:328–338. [PMC free article] [PubMed] [Google Scholar]
- 30.You BR, Shin HR, Han BR, Park WH. PX-12 induces apoptosis in Calu-6 cells in an oxidative stress-dependent manner. Tumour Biol. 2015;36:2087–95. doi: 10.1007/s13277-014-2816-x. [DOI] [PubMed] [Google Scholar]
- 31.Hsieh CC, Papaconstantinou J. Thioredoxin-ASK1 complex levels regulate ROS-mediated p38 MAPK pathway activity in livers of aged and long-lived Snell dwarf mice. FASEB J. 2006;20:259–268. doi: 10.1096/fj.05-4376com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen CT, Hung MC. Beyond anti-VEGF: dual-targeting antiangiogenic and antiproliferative therapy. Am J Transl Res. 2013;5:393–403. [PMC free article] [PubMed] [Google Scholar]
- 33.Pervaiz S, Clement MV. Tumor intracellular redox status and drug resistance--serendipity or a causal relationship? Curr Pharm Des. 2004;10:1969–1977. doi: 10.2174/1381612043384411. [DOI] [PubMed] [Google Scholar]