

Abstract
Excessive nitric oxide (NO) produced in inflammation may result in oxidative stress, which is closely related to the neurodegenerative diseases and brain damage. Massive NO production can enhance NF-κB activity in various neural cells, but the function of this activation by NO and the target genes transactivated by NF-κB are still largely unknown. In the present study, our results showed sodium nitropruside (SNP), a NO donor, triggered apoptotic cell death and NF-κB activation in human neuroblastoma SH-EP1 cells, and inhibition of NF-κB activation by its super endogenous inhibitor, I-κBαM, sensitized SH-EP1 cells to NO-induced apoptosis. Conversely, NF-κB activation induced by insulin-like growth factor (IGF)-1 antagonizes NO-induced apoptotic cell death in SH-EP1 cells. In addition, cDNA microarray analysis showed biglycan, an extracellular glycoprotein, was up-regulated by NF-κB, and recombinant biglycan protein conferred a protective effect on NF-κB mediated NO-induced apoptotic cell death in SH-EP1 cells. These findings suggest biglycan may serve as a potential target in preventing NO-induced neurodegenerative diseases.
Keywords: Nuclear factor-kappaB, neuroblastoma, nitric oxide, apoptosis, biglycan
Introduction
Nitric oxide (NO), a free radical, is a short-lived and multifaceted molecule that is produced from L-arginine by the members of NO synthase (NOS) family including neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS) [1]. In the central nervous system (CNS), NO plays an important role in the neurotransmission, synaptic plasticity, learning, and memory [2]. However, excessive NO production during inflammation may result in oxidative stress, which is closely related to the neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), and Huntington’s disease (HD), as well as brain damage following ischemia and stroke [3]. NO exerts its cytotoxic effects in diverse cell types by inducing redox reactions to produce reactive nitrogen species (RNS), especially peroxinitrite (ONOO-, PN), which is the most toxic derivative of NO and a source for nitrosative stress in neuronal cells via affecting mitochondrial function and damaging DNA, proteins, and lipids by oxidation [4-8]. Such damage in turn triggers downstream signal transduction pathways, leading to apoptosis or necrosis [9,10].
Nuclear factor-kappaB (NF-κB) is crucial for a variety of cellular processes, such as inflammation, immunity, cell proliferation, and apoptosis [11]. It consists of five members: RelA (p65), c-Rel, RelB, NF-κB1 (p50) and NF-κB2 (p52) [12,13]. All NF-κB proteins share a highly conserved Rel homology domain (RHD) responsible for DNA binding and dimerization [14]. NF-κB subunits form various homodimers or heterodimers with p65/p50 heterodimer (also known as NF-κB complex) being the most common one in most cell types [15,16]. Under physiological conditions, NF-κB dimers are sequestered in the cytoplasm due to the binding of inhibitor of κB (IκB) family which consists of IκBα, IκBβ, and IκBε [17]. Upon stimulation, the IκB kinase complex (IKK), which consists of two catalytically active kinases, IKKα and IKKβ, and the regulatory subunit IKKγ, becomes activated, leading to the proteasomal degradation of IκB proteins and the translocation of NF-κB dimers to the nucleus [17-19]. Once NF-κB dimers translocate to the nucleus, they bind to a consensus DNA motif (5’-GGGRNNYYCC-3’) to activate a set of target genes.
NF-κB has been found to be involved in neuroprotection against death-inducing stimuli such as β-amyloid protein, 6-OHDA, and oxidative stress [20-22]. In some cases, however, NF-κB also promotes neuronal death. For example, NF-κB was found essential to hydrogen peroxide- and dopamine-induced apoptosis in rat PC12 neural cells [23,24]. In addition, our previous study demonstrated that NF-κB promoted MPP+-induced apoptosis of SH-EP1 cells [25]. Recent studies also reveal that NF-κB can be activated in neurons in response to massive NO production, but the function of this activation and the target genes regulated by NF-κB and NO in various types of neural cells are largely unknown [26,27]. The present study aimed to investigate the potential role of NF-κB in the NO toxicity in human neuroblastoma SH-EP1 cells and explore the target genes of NF-κB involved in this process.
Materials and methods
Cell culture
Human neuroblastoma SH-EP1 cells obtained from Evelyne Goillot (Laboratoire d'Immunologie, Centre Leon Berard, Lyon, France) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum (FCS), 100 U/ml penicillin, and 100 µg/ml streptomycin at 37°C in humidified 5% CO2. pCMV-IκBα-M provided by Dr Douglas R. Green [28] was transfected into SH-EP1 cells in the presence of Lipofectamine 2000 (Invitrogen, USA) following the manufacturer’s instructions. The transfected cells were selected with G418 (500 µg/ml) (Sigma, USA), and the expression of IκBα-M in selected cell clones was determined by Western blot assay with IκBα antibody. Cell clones stably expressing IκBα-M were designated as I-κBαM cells.
Total, cytoplasmic and nuclear protein preparation
Cells were lysed in lysis buffer (50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 5 mM MgCl2, 10% glycerol, 1% Triton X-100 and complete protease inhibitor mixture [Roche]) and kept on ice for 30 min for the extraction of total protein. After centrifugation at 14,000 g for 15 min at 4°C, the supernatant was collected as total protein. For cytoplasmic protein extraction, cells were suspended in ice-cold lysis buffer (100 mM NaCl, 20 mM Tris-HCl, pH 8.0, 1% Nonidet P-40, complete protease inhibitor mixture), homogenized and centrifuged at 16,000 g at 4°C for 60 min, and the supernatant was collected as cytoplasmic protein. To extract nuclear proteins, cells were homogenized in buffer A (20 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol, and complete protease inhibitor complex). After centrifugation at 600 g, the pellets containing nuclei were re-suspended in buffer C (20 mM HEPES, pH 7.9, 1.5 mM MgCl2, 420 mM KCl, 1 mM EDTA, 25% glycerol, 0.5 mM dithiothreitol, and complete protease inhibitor complex) and centrifuged at 100,000 g for 20 min at 4°C. The supernatant was collected as nuclear protein.
Electrophoretic mobility shift assay (EMSA)
For EMSA, reactions were conducted in a total volume of 20 μl. In brief, 5 μg of nuclear extract was mixed with the reaction buffer (100 mM KCl, 10% glycerol, 0.2 mM EDTA, 0.5 mM dithiothreitol, 1 mM PMSF, 1 μg/ml elupeptin, 2 µg/ml poly [dI-dC], 20 mM HEPES, pH 7.9). The mixture was incubated on ice for 20 min in the presence of 105 cpm (3,000 Ci/mmol) of [g-32P] ATP-conjugated oligonucleotide probe with an NF-κB binding site (5’-AGT TGA GGG GAC TTT CCC AGG C-3’) (Santa Cruz Biotechnology, USA). In the supershift assay, 1 μg of p65 antibody was added together with nuclear protein, followed by incubation for another 20 min at 4°C. Samples were separated in 5% native polyacrylamide gels. Then the gels were dried and autoradiographed with intensifying screens at -88(80?)°C.
Transcription factor reporter assay
The transcription factor reporter assay was performed using the TransAM Assay kit (Active Motif, USA) according to the manufacturer’s instructions. In brief, after treatment with 1 mM SNP, 2 µg of nuclear extract was incubated in oligonucleotide-coated 96-well plate for 1 h. After washing, the bound complexes were incubated with antibodies against NF-κB p65 for 1 h. The plates were then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for another 1 h. Finally, the developing solution was added and incubated for 2-10 min, followed by measurement of absorbance at 450 nm with the Tecan reader (Männedorf, Switzerland).
Determination of biglycan concentration in cell culture medium
Cell culture media were collected and filtered through 0.2-µm filter to remove floating cells. The biglycan from the cell culture media was prepared by incubating with 100 mU of chondroitinase ABC (Sigma, USA) for 16 h. Proteins were then concentrated by incubating with Strataclean resin (Agilent Technologies, USA) for 30 min on ice. After centrifugation at 14,000 g for 5 min, the pellet was suspended in 2× SDS sample buffer and boiled for 5 min at 100°C. Then, the samples were centrifuged at 14,000 g for 5 min at 4°C and the supernatant wascollected.
Western blot assay
Proteins were separated by SDS-PAGE and transferred onto polyvinyl difluoride (PVDF) membrane (Millipore, USA). The membrane was blocked in 5% non-fat milk in phosphate-buffered saline with 0.1% Tween 20 (PBST) for 1 h and probed with different primary antibodies (anti-IκBα from Santa Cruz Biotechnology, anti-β-actin from Sigma, anti-cleaved caspase-9 and cleaved caspase-3 from Cell Signaling Technology). After washing with PBST, the membrane was probed with corresponding HRP-conjugated secondary antibodies (Sigma, USA). Thereafter, the membrane was rinsed in PBST and visualized with the enhanced chemiluminescence (Pierce, USA).
Immunohistochemistry
Immunohistochemistry for p65 of NF-κB was performed by using the coverslips where cells were grown. In brief, cells were washed with PBS and fixed in 2% paraformaldehye for 10 min, followed by washing with PBST 3 times. Cells were permeated with 0.2% Triton X-100 in PBS for 15 min. After incubation with blocking buffer (4% bovine serum albumin in PBS) for 1 h, cells were incubated overnight at 4°C with rabbit polyclonal antibody against p65 (1:40; Santa Cruz Biotechnology) in blocking buffer. Cells were washed thrice (5 min for each) with PBS. Cells were incubated with FITC-conjugate anti-rabbit polyclonal antibodies (1:50) for 1 h in dark. Finally, cells were examined under a fluorescence microscope (Carl Zeiss, USA).
Cell viability assay
Neural cell viability was assessed by crystal violet staining as described previously [29]. In brief, cells (2×104/well) were seeded in 96-well plates in triplicate. Following overnight incubation, cells were washed with fresh medium and treated with SNP for up to 24 h. The plate was stained with 0.5% crystal violet in 20% methanol for 20 min and washed with tap water. Crystal violet in stained cells was dissolved with 20% acidic acid, and measured at 570 nm with Tecan Reader. Absorption of samples was normalized by the mean of control SH-EP1 cells grown in DMEM alone (defined as 100%). At least three independent experiments were performed in triplicate.
PI staining
Cells were seeded on cover-slips coated with 0.1% poly-L-lysine (PLL) in 12-well plates. After treatment with SNP for 24 h, cells were washed with PBS and fixed with 4% cold paraformaldehyde for 15 min. Cells were washed with PBS thrice, and then permeabilized with 70% ethanol for 30 min followed by propidium iodide (PI) staining (0.1% Triton X-100, 0.1 mM EDTA, 0.5 mg/ml RNaseA, and 50 µg/ml PI). Cells were observed under a fluorescent microscope (Carl Zeiss, USA).
Fluorescence-activated cell sorting (FACS)
Cells undergoing apoptosis were analyzed by FACS assay. In brief, cells were plated in 6-well plates until 80% confluence was achieved. Upon treatment, cells were trypsinized and neutralized with 10% FCS in DMEM and centrifuged at 800 g for 5 min. The supernatant was removed, and cells were stained with PI (50 µg/mL) in PBST prior to flow cytometry (R&D Systems, USA). Finally, the numbers of living and apoptotic cells were expressed as percentages of total stained cells.
Real time PCR
Total RNA was isolated from cells using Trizol reagent (Invitrogen) following the manufacturer’s instructions. RT-PCR of total RNA was performed as described in first strand cDNA synthesis (Invitrogen). The real-time PCR was performed on ABI Prism 7500 Sequence detection system (Applied Biosystems, CA) with the KAPA SYBR qPCR Kit (KAPA Biosystems, USA) according to the manufacturer’s instructions. The primers for PCR were as follows: Biglycan, 5’-GGA CTC TGT CAC ACC CAC CT-3’ (forward) and 5’-AGC TCG GAG ATG TCG TTG TT-3’ (reverse). PCR was performed under the following conditions: 2 min at 50°C, 1 min at 95°C, 40 cycles at 95°C for 3 s, 60°C for 30 s, followed by melting curve analysis at the end of each run from 60°C to 95°C.
Statistical analysis
Statistical analysis was performed with SPSS version 17.0. Data are expressed as mean ± standard error (S.E.). Comparisons were done with analysis of variance followed by Bonferroni test. A value of P < 0.05 was considered statistically significant.
Results
SNP induces apoptosis and activation of NF-κB in SH-EP1 cells
NO donors including SNP have been used widely to study oxidative stress and cellular responses by mimicking endogenous NO generation [30]. To verify the effect of NO on human neuroblastoma SH-EP1 cells, cell viability assay was conducted following treatment with SNP at 0.5, 1 and 1.5 mM. As shown in Figure 1A, SNP induced SH-EP1 cell death in a concentration-dependent manner. The viability of SH-EP1 cells decreased by about 28% after treatment with 0.5 mM SNP for 24 h, whereas 90% reduction was observed when the SNP concentration was increased up to 1.5 mM. Meanwhile, PI staining was performed to assess the cytotoxicity induced by SNP. As shown in Figure 1B, SNP induced a relatively higher death rate. Consistently, the SNP cytotoxicity analyzed by FACS demonstrated similar cell toxicity of SNP under the same conditions (Figure 1C). Western blot was performed to detect the expressions of caspase-9 and caspase-3, two typical markers of apoptosis. As expected, SNP elicited a significant activation of caspase-9 and caspase-3 at 16 and 24 h post-treatment (Figure 1D), suggesting that SNP induces cell apoptosis.
Figure 1.
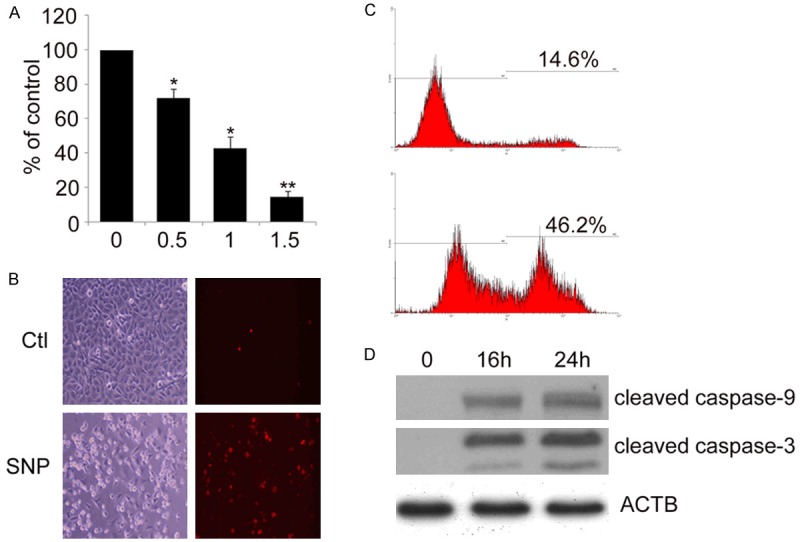
SNP induces apoptotic cell death of SH-EP1 cells. (A) SH-EP1 cells were treated with SNP (0.5, 1 and 1.5 mM) for 24 h. Cell viability was determined by crystal violet staining. Survival rate is represented as the percentage of viable control cells. Data are expressed as mean ± S.E. from at least experiments performed in triplicate. *P < 0.05 and **P < 0.01, vs untreated SH-EP1 cells. SH-EP1 cells were subjected to 1 mM SNP for 24 h, and SNP cytotoxicity was examined by PI staining (B) and FACS (C). (D) SH-EP1 cells were treated with 1 mM SNP for 16 and 24 h. Samples were assessed by Western blot assay with antibodies against cleaved caspase-9 and cleaved caspase-3. β-actin (ACTB) served as a loading control.
NF-κB activation has been shown to be involved in NO-induced apoptosis [31]. To determine whether NF-κB activation is involved in SNP-induced apoptosis of SH-EP1 cells, the activation of NF-κB was determined by EMSA. Nuclear extract of SH-PE1 cells treated with SNP was probed with a NF-κB-specific binding oligonucleotide. DNA binding activity of NF-κB increased at 1 and 2 h but reduced at 4 h (Figure 2A). In addition, DNA binding activity of NF-κB reduced in the presence of p65 (RelA) antibody at 2 h implied that the DNA complex consists of p65 subunit of NF-κB (Figure 2A). The activation of NF-κB by SNP was further demonstrated by immunocytochemistry. After exposure to SNP for 30 min, NF-κB p65 (green) translocated from cytoplasm to nucleus, suggesting the NF-κB activation (Figure 2B). These results clearly demonstrate that SNP induces the activation of p65 NF-κB in SH-EP1 cells.
Figure 2.
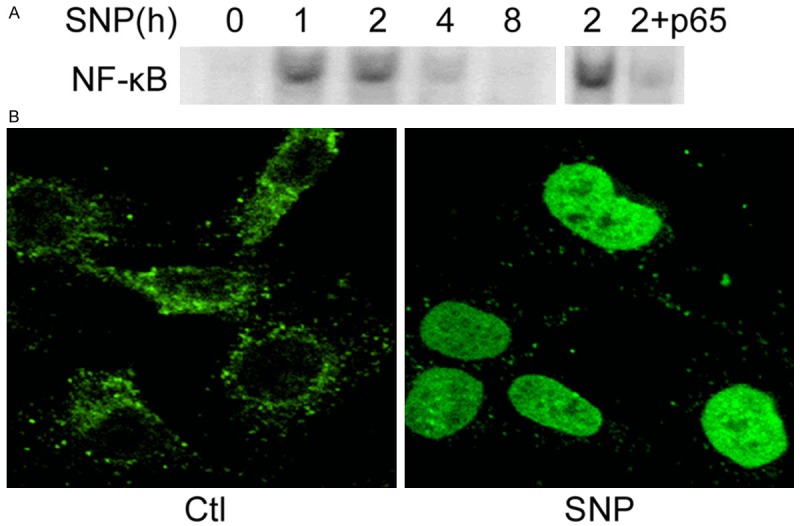
SNP induces NF-κB activation in SH-EP1 cells. A. Left: SH-EP1 cells and IκBα-M cells were treated with SNP (1 mM) for 1, 2, 4, and 8 h. The nuclear extract was prepared after SNP treatment, and EMSA was performed using an oligonucleotide containing NF-κB binding sites. Right: supershift assay with nuclear extract of SH-EP1 cells treated with SNP for 2 h using p65 antibody. B. SH-EP1 cells were incubated with SNP (1 mM) for 1 h and immunocytochemistry was performed for NF-κB p65 (green).
Dominant negative I-κBα sensitizes SH-EP1 cells to NO-induced apoptosis
To clarify the role of NF-κB in NO-induced apoptosis of SH-EP1 cells, we prepared SH-EP1 cells stably expressing I-κBαM that deviates from I-κBα and can bind and inhibit NF-κB. I-κBαM acts as an effective dominant negative inhibitor of NF-κB activation as the mutation of two key phosphorylation sites in I-κBαM prevents its phosphorylation and subsequent degradation. Western blot assay confirmed that I-κBαM was over-expressed in SH-EP1 cells (Figure 3A). To confirm the inhibitory effects of I-κBαM on NF-κB activation, EMSA was performed. As stated earlier, NF-κB activation was observed in SH-EP1 cells treated with SNP, but it remarkably decreased in SNP-treated I-κBαM cells (Figure 3B). These results indicate that I-κBαM over-expression effectively blocks NF-κB activation. Thus, the effect of NF-κB activation on SNP-induced cytotoxicity was examined. It was clearly shown in Figure 3C that I-κBαM cells were more sensitive to SNP-induced cell death as compared to control SH-EP1 cells. Besides, the cell viability assay and detection of cleaved caspase-9 were also performed in IκBα-M cells. As shown in Figure 3D, cleaved caspase-9 was also observed in IκBα-M cells after SNP treatment for 24 h, but the cleaved caspase-9 in IκBα-M cells increased significant significantly as compared to SNP-treated SH-EP1 cells. These results suggest that the blockage of NF-κB activity sensitizes SH-EP1 cells to NO-induced apoptosis.
Figure 3.
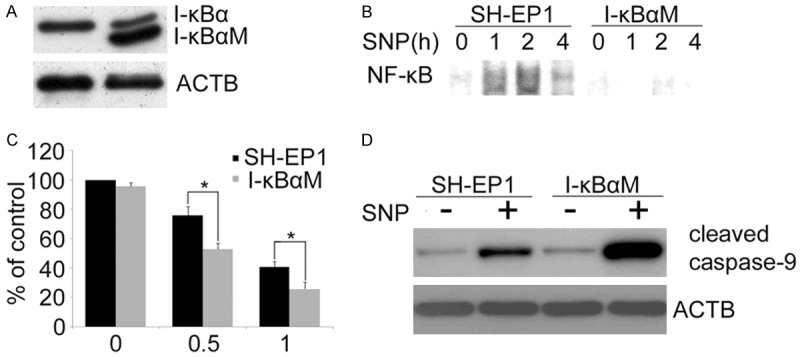
I-κBαM sensitizes SH-EP1 to NO-induced apoptosis. A. Western blot assay of I-κBα expression in control SH-EP1 cells transfected with empty plasmid vector and SH-EP1 cells transfected with I-κBαM, a dominant negative inhibitor of NF-κB. B. Nuclear extract of SH-EP1 cells and I-κBαM cells was prepared after SNP (1 mM) treatment. EMSA was performed using an oligonucleotide containing NF-κB binding site. C. SH-EP1 cells and I-κBαM cells were treated for 24 h with SNP at different concentrations. Cell viability was quantified by crystal violet staining. Data are expression as mean ± S.E. from at least three experiments performed in triplicate. D. Western blot assay of cell extract was performed with an antibody against the cleaved caspase-9. β-actin (ACTB) served as a loading control.
NF-κB activation induced by IGF-1 protects SH-EP1 cells from NO-induced apoptosis
IGF-1 had been reported to induce NF-κB activation [32,33]. To further confirm the protective effect of NF-κB activation against NO-induced apoptosis in SH-EP1 cells, SH-EP1 cells were treated with IGF-1 to mimic NF-κB activation. The effect of IGF-1 on NF-κB activation was examined by transcription factor reporter assay. As expected (Figure 4A), NF-κB activity increased at 30 min, reached a peak at 1 h and decreased substantially at 2 h after IGF-1 (100 ng/ml) treatment. However, IGF-1-induced NF-κB activation was significantly attenuated in I-κBαM cells.
Figure 4.
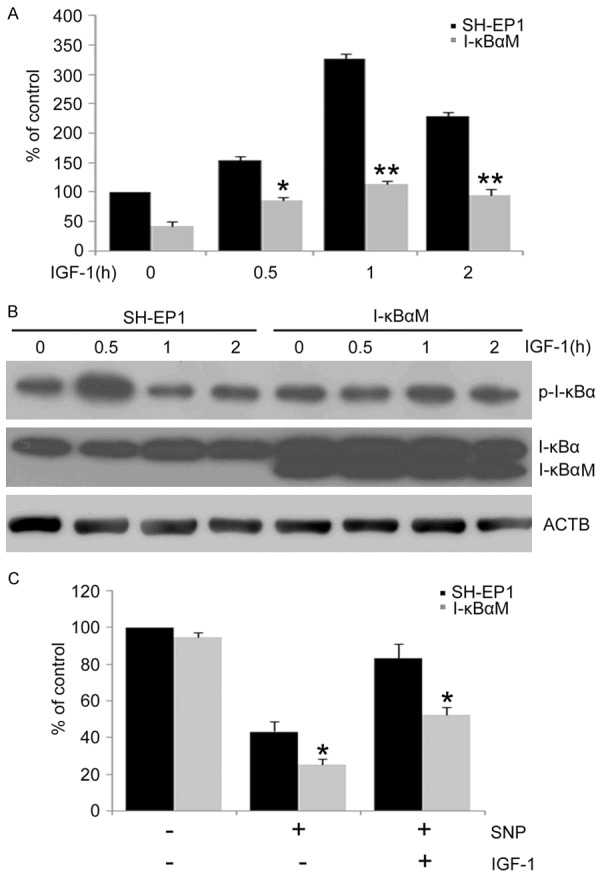
NF-κB activation protects SH-EP1 cells from NO-induced apoptosis. A. SH-EP1 and I-κBαM cells were treated with IGF-1 (100 ng/ml) for 0.5, 1, and 2 h. NF-κB activation was determined using NF-κB reporter assay. Data are from three independent experiments and expressed as mean ± S.E. *P < 0.05 and **P < 0.01, vs IGF-1 treated SH-EP1 cells. B. SH-EP1 cells and I-κBαM cells were treated with IGF-1. Phospho-I-κBα and I-κBα in SH-EP1 cells and I-κBαM cells were determined by Western blot assay. β-actin (ACTB) served as a loading control. C. SH-EP1 cells and I-κBαM cells were incubated with SNP (1 mM) in the presence or absence of IGF-1 for 24 h. Data are expressed as mean ± S.E. from at least three experiments performed in triplicate. *P < 0.05, vs SH-EP1 cells.
It has been reported that the phosphorylation of IκBα results in the degradation of IκBα, the release of NF-κB and subsequent NF-κB activation [34]. To further confirm the effect of IGF-1 on the NF-κB activation, the phosphorylation of I-κBα was detected in SH-EP1 and I-κBαM cells by Western blot assay. As shown in Figure 4B, the phosphorylated I-κBα significantly increased at 0.5 h after IGF-1 treatment and decreased rapidly afterwards, but the phosphorylated I-κBα in IGF-1-treated I-κBαM cells remained relatively stable, suggesting that I-κBαM inhibits the NF-κB activation induced by IGF-1. Interestingly, no degradation of I-κBα was observed in IGF-1-treated SH-EP1 cells and I-κBαM cells during the IGF-1 treatment, indicating that the phosphorylated I-κBα is sufficient to activate NF-κB in this scenario.
Then, the effects of IGF-1 induced NF-κB activation on NO-induced cell death were further examined in SH-EP1 cells and I-κBαM cells. IGF-1 at 100 ng/ml increased the survival of NO-treated SH-EP1 cells. In contrast, IGF-1 had no significant effects on the survival of NO-treated I-κBαM cells (Figure 4C), suggesting a pro-survival role of IGF-1 induced NF-κB. Taken together, these findings further confirm that NF-κB activation protects SH-EP1 cells from NO-induced cell death.
NF-κB activation upregulates biglycan expression in SH-EP1 cells
To identify the genes involved in the protection of NF-κB against NO-induced apoptosis in SH-EP1 cells, the RNA of I-κBαM cells and IGF-1-treated SH-EP1 cells were harvested for cDNA microarray assay. Only genes with more than 4-fold change in their expression were categorized. Among these genes, biglycan was found to be up-regulated by 194 fold. Considering that biglycan has been shown to play a pro-survival role in other cell types, such as mesangial cells and cardiomyocytes [35,36], biglycan was selected for further investigation. Our results confirmed the up-regulation of biglycan by real-time PCR. As shown in Figure 5A, IGF-1 up-regulated biglycan mRNA expression in SH-EP1 cells, but not in I-κBαM cells, indicating that NF-κB contributes to the induction of biglycan expression.
Figure 5.
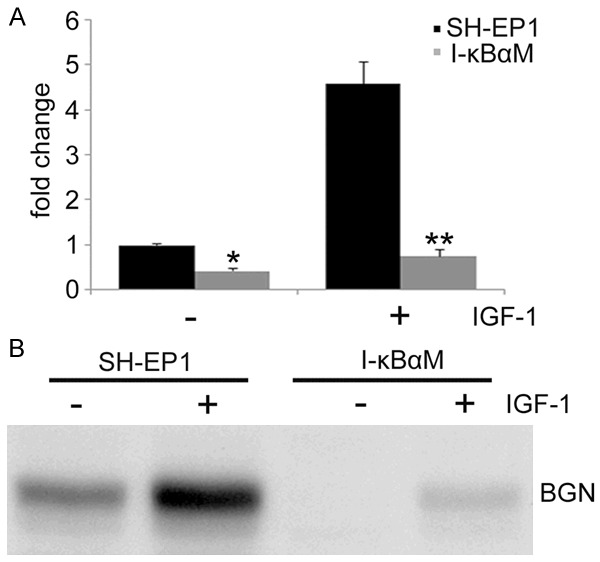
NF-κB activation up-regulates biglycan expression. A. Real-time PCR was carried out to detect the mRNA expression of biglycan. Data are expressed as mean ± S.E. from at least three experiments performed in triplicate. *P < 0.05 and **P < 0.01, vs SH-EP1 cells. B. SH-EP1 cells and I-κBαM cells were treated with 100 ng/ml IGF-1, and the culture medium was digested with chondroitinase ABCase. Total protein was then concentrated using StrataClean resin and subjected to western blot assay with antibody against biglycan.
In order to further confirm that biglycan expression is NF-κB-dependent, the baseline biglycan expression was compared between control SH-EP1 and I-κBαM cells. Since biglycan functions as a secretory protein, total protein in the supernatant was concentrated for Western blot assay. As expected, the baseline biglycan in I-κBαM cells was significantly lower than that in control SH-EP1 cells, suggesting the biglycan expression is dependent on NF-κB activity (Figure 5B). More convincingly, upon IGF-1 induced NF-κB activation, biglycan expression in both SH-EP1 cells and I-κBαM cells substantially increased, although the biglycan expression in SH-EP1 cells was still markedly higher than in I-κBαM cells. Taken together, these findings indicate that biglycan may function as a target gene of NF-κB.
Biglycan protects I-κBαM cells from NO-induced apoptosis
To confirm that biglycan is involved in NF-κB-mediated cell protection against NO-induced apoptosis, recombinant biglycan was introduced to cells and cell viability assay was performed. SH-EP1 and I-κBαM cells were pretreated with biglycan prior to SNP treatment and cell survival was determined 24 h later. As expected, biglycan confered cell protection against SNP cytotoxicity in both SH-EP1 cells and I-κBαM cells and the protection was comparable between SH-EP1 cells and I-κBαM cells (Figure 6A). Consistently, SNP-induced caspase-9 cleavage was clearly inhibited by biglycan in both SH-EP1 cells and I-κBαM cells (Figure 6B). These results suggest that biglycan is a NF-κB downstream effector, which is responsible for NF-κB-mediated protection against NO-induced apoptosis.
Figure 6.
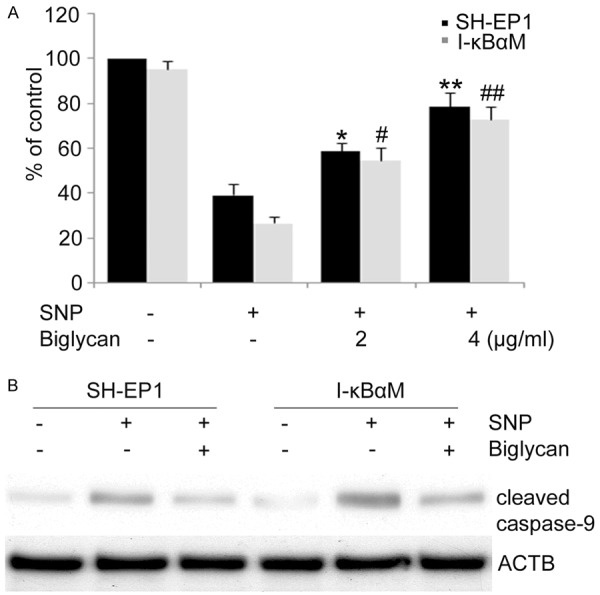
Biglycan protects SH-EP1 cells and I-κBαM cells from SNP-induced apoptosis. A. SH-EP1 cells and I-κBαM cells were treated with 1 mM NO in the presence of biglycan for 24 h and then subjected to crystal violet staining for the determination of cell viability. Data are expressed as mean ± S.E. from at least three experiments performed in triplicate. *P < 0.05 and **P < 0.01, vs SNP-treated SH-EP1 cells. #P < 0.05 and ##P < 0.01, vs SNP-treated I-κBαM cells. B. In the presence of biglycan (4 μg/ml), cells were incubated with SNP for 24 h, samples were examined by western blot assay with an antibody against cleaved caspase-9. β-actin (ACTB) served as a loading control.
Discussion
NF-κB activation, which is induced by neuroinflammation and associated with many neurodegenerative diseases [37], usually plays an important role in cell survival, yet the role of NF-κB in cell viability is still controversial. A pro-survival role of NF-κB was seen in neurotoxin kainate-induced damage to hippocampal pyramidal neurons of mice lacking the p50 subunit of NF-κB as compared to wild-type mice [38]. Similarly, NF-κB activation is essential for the erythropoietin-mediated neuroprotection against apoptosis induced by N-methyl-Daspartate (NMDA) or NO in cerebrocortical neurons in vitro [39]. Consistently, our findings suggest that NF-κB plays a protective role against NO-induced apoptosis in SH-EP1 cells in three observations: (1) NO increases DNA binding activity and promotes the nuclear translocation of p65 in human neuroblatoma SH-EP1 cells, resulting in NF-κB activation; (2) introduction of a NF-κB dominant negative mutant (IκBα-M), which effectively inhibits NF-κB activity, sensitizes SNP-induced cytotoxicity, resulting in decreased cell viability and increased caspase-9 cleavage in SNP treated IκBα-M cells as compared to SH-EP1 cells; (3) IGF-1 induced NF-κB activation significantly reduced SNP-induced cell death and caspase-9 cleavage, which were significantly reversed in IκBα-M cells. In contrast to these findings, other studies also show that NF-κB activation promotes the death of neurons under certain conditions. For example, in ischemia models, NF-κB activation appears to contribute to the brain damage as demonstrated by the fact that mice lacking the IKK2 or the p50 subunit of NF-κB exhibit decreased infarct volume [40,41]. Additionally, NF-κB activation potentiates the seizure-induced neuronal damage in the rat hippocampus [42]. Taken together, these findings suggest that whether NF-κB inhibits or promotes apoptosis is dependent upon the cell types and the nature of the apoptosis-inducing stimulus.
NF-κB was found to be activated by IGF-1 in many cell types, such as porcine vascular smooth muscle cells, human umbilical vein endothelial cells, rat immortalized GT1-7 hypothalamic neuronal cells and mature primary cerebellar granule neurons [43-45]. Consis-tently, our NF-κB p65 reporter assay showed a time-dependent activation of NF-κB in response to IGF-1 treatment in SH-EP1 cells, and a transient but potent phosphorylation of IκBα was observed in SH-EP1 cells upon IGF-1 treatment. IGF-1 induced NF-κB activation protected SH-EP1 cells against oxidative stress, which was reversed in IκBα-M cells. However, dominant-negative IκBα failed to completely block IGF-1-mediated cell survival, which might be due to the fact that the dominant-negative IκBα is not sufficient to block IGF-1-induced NF-κB activation. Another explanation is that additional factors, such as AKT which was previously reported to be involved in IGF-1-mediated protection against MPP+-induced apoptosis of SH-EP1 cells [29], together with NF-κB, are involved in the protection of neuronal cells against oxidative stress. Collectively, these results suggest that it is plausible to use IGF-1 to mimic NF-κB activation.
It is not clear precisely how NF-κB regulates cell survival, although it has been reported that NF-κB is able to induce the expression of some genes such as anti-apoptotic bcl-2 homolog A1, and some members of the inhibitor of apoptosis proteins (IAPs) [46,47]. To identify the genes involved in the NF-κB-mediated protection against NO-induced apoptosis in SH-EP1 cells, cDNA microarray was em-ployed. As a result, the mRNA expression of biglycan was significantly up-regulated by IGF-1 in SH-EP1 cells but not in IκBα-M cells. Biglycan belongs to the family of small leucine-rich proteoglycans and consists of a core protein with 331 amino acids covalently bound to two chondroitin sulphate- or dermatan sulphate-containing glycosaminoglycan side chains [48]. The up-regulated biglycan expression was further proven by real-time PCR and Western blot assay, but such up-regulation was not observed in IκBα-M cells. The delayed presence between NF-κB activation and biglycan up-regulation strongly suggest biglycan as a direct target of NF-κB in SH-EP1 cells. Consistently, our results showed recombinant biglycan protected SH-EP1 cells and IκBα-M cells from NO-induced apoptotic cell death. Our findings for the first time demonstrated that biglycan served as a direct target gene involved in NF-κB-mediated cell protection against NO insults.
In summary, our findings show that NF-κB provides a strong protection against NO-mediated SH-EP1 cells death through up-regulating the biglycan expression. Our results provide a novel target for the treatment of stroke and neurodegenerative diseases.
Disclosure of conflict of interest
None.
References
- 1.Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 2007;8:766–775. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- 2.Boulton CL, Southam E, Garthwaite J. Nitric oxide-dependent long-term potentiation is blocked by a specific inhibitor of soluble guanylyl cyclase. Neuroscience. 1995;69:699–703. doi: 10.1016/0306-4522(95)00349-n. [DOI] [PubMed] [Google Scholar]
- 3.Bennett S, Grant MM, Aldred S. Oxidative stress in vascular dementia and Alzheimer’s disease: a common pathology. J Alzheimers Dis. 2009;17:245–257. doi: 10.3233/JAD-2009-1041. [DOI] [PubMed] [Google Scholar]
- 4.Lipton SA. Neuronal protection and destruction by NO. Cell Death Differ. 1999;6:943–951. doi: 10.1038/sj.cdd.4400580. [DOI] [PubMed] [Google Scholar]
- 5.Kroncke KD, Fehsel K, Kolb-Bachofen V. Nitric oxide: cytotoxicity versus cytoprotection--how, why, when, and where? Nitric Oxide. 1997;1:107–120. doi: 10.1006/niox.1997.0118. [DOI] [PubMed] [Google Scholar]
- 6.Schulz JB, Matthews RT, Beal MF. Role of nitric oxide in neurodegenerative diseases. Curr Opin Neurol. 1995;8:480–486. doi: 10.1097/00019052-199512000-00016. [DOI] [PubMed] [Google Scholar]
- 7.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghasemi M, Fatemi A. Pathologic role of glial nitric oxide in adult and pediatric neuroinflammatory diseases. Neurosci Biobehav Rev. 2014;45C:168–182. doi: 10.1016/j.neubiorev.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 9.Brown GC. Nitric oxide and neuronal death. Nitric Oxide. 2010;23:153–165. doi: 10.1016/j.niox.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 10.Drechsel DA, Estevez AG, Barbeito L, Beckman JS. Nitric oxide-mediated oxidative damage and the progressive demise of motor neurons in ALS. Neurotox Res. 2012;22:251–264. doi: 10.1007/s12640-012-9322-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gloire G, Legrand-Poels S, Piette J. NFkappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72:1493–1505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 12.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 13.Radhakrishnan SK, Kamalakaran S. Proapoptotic role of NF-kappaB: implications for cancer therapy. Biochim Biophys Acta. 2006;1766:53–62. doi: 10.1016/j.bbcan.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 14.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 15.Ruland J. Return to homeostasis: downregulation of NF-kappaB responses. Nat Immunol. 2011;12:709–714. doi: 10.1038/ni.2055. [DOI] [PubMed] [Google Scholar]
- 16.Prasad S, Ravindran J, Aggarwal BB. NFkappaB and cancer: how intimate is this relationship. Mol Cell Biochem. 2010;336:25–37. doi: 10.1007/s11010-009-0267-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skaug B, Jiang X, Chen ZJ. The role of ubiquitin in NF-kappaB regulatory pathways. Annu Rev Biochem. 2009;78:769–796. doi: 10.1146/annurev.biochem.78.070907.102750. [DOI] [PubMed] [Google Scholar]
- 18.Karin M, Cao Y, Greten FR, Li ZW. NFkappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 19.O’Dea E, Hoffmann A. The regulatory logic of the NF-kappaB signaling system. Cold Spring Harb Perspect Biol. 2010;2:a000216. doi: 10.1101/cshperspect.a000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barger SW, Horster D, Furukawa K, Goodman Y, Krieglstein J, Mattson MP. Tumor necrosis factors alpha and beta protect neurons against amyloid beta-peptide toxicity: evidence for involvement of a kappa B-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci U S A. 1995;92:9328–9332. doi: 10.1073/pnas.92.20.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park SH, Choi WS, Yoon SY, Ahn YS, Oh YJ. Activation of NF-kappaB is involved in 6-hydroxydopamine-but not MPP+ -induced dopaminergic neuronal cell death: its potential role as a survival determinant. Biochem Biophys Res Commun. 2004;322:727–733. doi: 10.1016/j.bbrc.2004.07.193. [DOI] [PubMed] [Google Scholar]
- 22.Mattson MP, Goodman Y, Luo H, Fu W, Furukawa K. Activation of NF-kappaB protects hippocampal neurons against oxidative stressinduced apoptosis: evidence for induction of manganese superoxide dismutase and suppression of peroxynitrite production and protein tyrosine nitration. J Neurosci Res. 1997;49:681–697. doi: 10.1002/(SICI)1097-4547(19970915)49:6<681::AID-JNR3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 23.Panet H, Barzilai A, Daily D, Melamed E, Offen D. Activation of nuclear transcription factor kappa B (NF-kappaB) is essential for dopamine-induced apoptosis in PC12 cells. J Neurochem. 2001;77:391–398. doi: 10.1046/j.1471-4159.2001.00213.x. [DOI] [PubMed] [Google Scholar]
- 24.Liu Q, Kou JP, Yu BY. Ginsenoside Rg1 protects against hydrogen peroxide-induced cell death in PC12 cells via inhibiting NF-kappaB activation. Neurochem Int. 2011;58:119–125. doi: 10.1016/j.neuint.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 25.Yang HJ, Wang L, Xia YY, Chang PN, Feng ZW. NF-kappaB mediates MPP+-induced apoptotic cell death in neuroblastoma cells SH-EP1 through JNK and c-Jun/AP-1. Neurochem Int. 2010;56:128–134. doi: 10.1016/j.neuint.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 26.Contestabile A. Regulation of transcription factors by nitric oxide in neurons and in neural-derived tumor cells. Prog Neurobiol. 2008;84:317–328. doi: 10.1016/j.pneurobio.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 27.Socodato R, Brito R, Portugal CC, de Oliveira NA, Calaza KC, Paes-de-Carvalho R. The nitric oxide-cGKII system relays death and survival signals during embryonic retinal development via AKT-induced CREB1 activation. Cell Death Differ. 2014;21:915–928. doi: 10.1038/cdd.2014.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alphainduced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 29.Wang L, Yang HJ, Xia YY, Feng ZW. Insulinlike growth factor 1 protects human neuroblastoma cells SH-EP1 against MPP+-induced apoptosis by AKT/GSK-3beta/JNK signaling. Apoptosis. 2010;15:1470–1479. doi: 10.1007/s10495-010-0547-z. [DOI] [PubMed] [Google Scholar]
- 30.Li L, Feng Z, Porter AG. JNK-dependent phosphorylation of c-Jun on serine 63 mediates nitric oxide-induced apoptosis of neuroblastoma cells. J Biol Chem. 2004;279:4058–4065. doi: 10.1074/jbc.M310415200. [DOI] [PubMed] [Google Scholar]
- 31.D’Acquisto F, de Cristofaro F, Maiuri MC, Tajana G, Carnuccio R. Protective role of nuclear factor kappa B against nitric oxide-induced apoptosis in J774 macrophages. Cell Death Differ. 2001;8:144–151. doi: 10.1038/sj.cdd.4400784. [DOI] [PubMed] [Google Scholar]
- 32.Jimenez Del Rio M, Velez-Pardo C. Insulinlike growth factor-1 prevents Abeta[25-35] /(H2O2)- induced apoptosis in lymphocytes by reciprocal NF-kappaB activation and p53 inhibition via PI3K-dependent pathway. Growth Factors. 2006;24:67–78. doi: 10.1080/08977190500361788. [DOI] [PubMed] [Google Scholar]
- 33.Harvey AE, Lashinger LM, Hays D, Harrison LM, Lewis K, Fischer SM, Hursting SD. Calorie restriction decreases murine and human pancreatic tumor cell growth, nuclear factor-kappaB activation, and inflammation-related gene expression in an insulin-like growth factor-1-dependent manner. PLoS One. 2014;9:e94151. doi: 10.1371/journal.pone.0094151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Espinosa L, Margalef P, Bigas A. Nonconventional functions for NF-kappaB members: the dark side of NF-kappaB. Oncogene. 2015;34:2279–2287. doi: 10.1038/onc.2014.188. [DOI] [PubMed] [Google Scholar]
- 35.Schaefer L, Beck KF, Raslik I, Walpen S, Mihalik D, Micegova M, Macakova K, Schonherr E, Seidler DG, Varga G, Schaefer RM, Kresse H, Pfeilschifter J. Biglycan, a nitric oxide-regulated gene, affects adhesion, growth, and survival of mesangial cells. J Biol Chem. 2003;278:26227–26237. doi: 10.1074/jbc.M210574200. [DOI] [PubMed] [Google Scholar]
- 36.Csont T, Gorbe A, Bereczki E, Szunyog A, Aypar E, Toth ME, Varga ZV, Csonka C, Fulop F, Santha M, Ferdinandy P. Biglycan protects cardiomyocytes against hypoxia/reoxygenation injury: role of nitric oxide. J Mol Cell Cardiol. 2010;48:649–652. doi: 10.1016/j.yjmcc.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 37.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu Z, Zhou D, Bruce-Keller AJ, Kindy MS, Mattson MP. Lack of the p50 subunit of nuclear factor-kappaB increases the vulnerability of hippocampal neurons to excitotoxic injury. J Neurosci. 1999;19:8856–8865. doi: 10.1523/JNEUROSCI.19-20-08856.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Digicaylioglu M, Lipton SA. Erythropoietinmediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature. 2001;412:641–647. doi: 10.1038/35088074. [DOI] [PubMed] [Google Scholar]
- 40.Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. doi: 10.1038/8432. [DOI] [PubMed] [Google Scholar]
- 41.Herrmann O, Baumann B, de Lorenzi R, Muhammad S, Zhang W, Kleesiek J, Malfertheiner M, Kohrmann M, Potrovita I, Maegele I, Beyer C, Burke JR, Hasan MT, Bujard H, Wirth T, Pasparakis M, Schwaninger M. IKK mediates ischemia-induced neuronal death. Nat Med. 2005;11:1322–1329. doi: 10.1038/nm1323. [DOI] [PubMed] [Google Scholar]
- 42.Chang CC, Chen SD, Lin TK, Chang WN, Liou CW, Chang AY, Chan SH, Chuang YC. Heat shock protein 70 protects against seizure-induced neuronal cell death in the hippocampus following experimental status epilepticus via inhibition of nuclear factor-kappaB activationinduced nitric oxide synthase II expression. Neurobiol Dis. 2014;62:241–249. doi: 10.1016/j.nbd.2013.10.012. [DOI] [PubMed] [Google Scholar]
- 43.Balaram SK, Agrawal DK, Edwards JD. Insulin like growth factor-1 activates nuclear factor-kappaB and increases transcription of the intercellular adhesion molecule-1 gene in endothelial cells. Cardiovasc Surg. 1999;7:91–97. doi: 10.1177/096721099900700118. [DOI] [PubMed] [Google Scholar]
- 44.Yerneni KK, Bai W, Khan BV, Medford RM, Natarajan R. Hyperglycemia-induced activation of nuclear transcription factor kappaB in vascular smooth muscle cells. Diabetes. 1999;48:855–864. doi: 10.2337/diabetes.48.4.855. [DOI] [PubMed] [Google Scholar]
- 45.Heck S, Lezoualc’h F, Engert S, Behl C. Insulin-like growth factor-1-mediated neuroprotection against oxidative stress is associated with activation of nuclear factor kappaB. J Biol Chem. 1999;274:9828–9835. doi: 10.1074/jbc.274.14.9828. [DOI] [PubMed] [Google Scholar]
- 46.Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaB control. Proc Natl Acad Sci U S A. 1997;94:10057–10062. doi: 10.1073/pnas.94.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang CY, Guttridge DC, Mayo MW, Baldwin AS Jr. NF-kappaB induces expression of the Bcl-2 homologue A1/Bfl-1 to preferentially suppress chemotherapy-induced apoptosis. Mol Cell Biol. 1999;19:5923–5929. doi: 10.1128/mcb.19.9.5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamamoto K, Ohga N, Hida Y, Maishi N, Kawamoto T, Kitayama K, Akiyama K, Osawa T, Kondoh M, Matsuda K, Onodera Y, Fujie M, Kaga K, Hirano S, Shinohara N, Shindoh M, Hida K. Biglycan is a specific marker and an autocrine angiogenic factor of tumour endothelial cells. Br J Cancer. 2012;106:1214–1223. doi: 10.1038/bjc.2012.59. [DOI] [PMC free article] [PubMed] [Google Scholar]