

Abstract
Autophagy is an intracellular degradation process that clears away aggregated proteins or aged and damaged organelles. Abnormalities in autophagy result in defects in clearance of these misfolded and aggregate proteins, which have been associated with neurodegenerative disorders. A key neuropathological hallmark of amyotrophic lateral sclerosis (ALS) that contributes to the progressive loss of motor neurons is abnormal protein aggregation of mutant Cu/Zn superoxide dismutase1 (SOD1). TFEB is a recently described gene that regulates autophagy. Several studies have reported that autophagy is altered in ALS, but little is known about the role and mechanisms of TFEB-mediated autophagy during the progression of ALS. In this study, altered expression of TFEB and Beclin-1 were detected in the spinal cords of ALS transgenic mice at different stages and in an NSC-34 cell model with the SOD1-G93A mutation using RT-PCR, western blot, and immunohistochemistry. The majority of cells positive for TFEB and Beclin-1 are β-tubulin III-labeled neurons, especially in the anterior horn of the gray matter. Overexpression of TFEB in NSC-34 cells with the SOD1-G93A mutation increased the mRNA and protein levels of Beclin-1, accompanied by increased levels of LC3-II protein. MTS assay revealed that TFEB overexpression increased proliferation and survival of NSC-34 cells with the SOD1-G93A mutation. Our findings suggest that TFEB promotes autophagy by enhancing the expression of Beclin-1. The altered autophagy mediated by TFEB is a key element in the pathogenesis of ALS, making TFEB a very promising target for the development of novel drugs and new gene therapeutics for ALS.
Keywords: Amyotrophic lateral sclerosis, autophagy, TFEB, Beclin-1, SOD1-G93A transgenic mice, NSC-34 cells
Introduction
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a neurodegenerative disorder characterized by the progressive death of motor neurons. It clinically manifests in progressive weakness, muscle atrophy, and paralysis [1,2]. Despite substantial efforts to decipher the etiology of the disease, the primary events triggering its pathology remain to be elucidated. Glutamate-induced excitotoxicity, inflammation, mitochondrial dysfunction, oxidative stress, protein aggregation, transcription deregulation, apoptosis, and epigenetic modifications are associated with the pathogenesis of ALS [3,4]. Numerous distinct therapeutic interventions have been explored, but there is still no efficacious treatment for ALS. The disease exists most frequently in its sporadic form, but also occurs as familial ALS. A key neuropathological hallmark of ALS that contributes to the progressive loss of motor neurons is the abnormal protein aggregation of mutate Cu/Zn superoxide dismutase1 (SOD1) [5]. Familial ALS is strongly associated with dominant mutations in the SOD1 gene. SOD1 protein alterations have also been reported in sporadic ALS patients [6,7]. The transgenic mouse that overexpresses the human mutant form of the SOD1 gene with glycine-to-alanine conversion at the 93rd codon is the mostly studied model and recapitulates the main features of both familial and sporadic forms of ALS [8,9].
Autophagy is an intracellular degradation process that clears away aggregated proteins or aged and damaged organelles [10]. Autophagy prevents the accumulation of disease-associated proteins, including the polyQ-expanded Htt in Huntington’s disease (HD), -syn in Parkinson’s disease (PD), amyloid-β in Alzheimer’s disease (AD), and SOD1 in ALS. Abnormalities in autophagy result in defects in clearance of these misfolded and aggregate proteins, which have been associated with these neurodegenerative disorders [11-13].
Transcription factor EB (TFEB), a recently described basic helix-loop-helix transcriptional activator, which is known to control lysosomal biogenesis, also regulates autophagy [14,15]. Because TFEB is a transcription factor, it probably regulates autophagy by controlling gene expression. Autophagy is a complex and important homeostatic process. A family of genes whose products are involved in autophagy are called autophagy-related genes (ATGs). Among the core ATG genes, Beclin-1, the orthologue of Atg6, encodes a coiled-coil protein that binds to the Class III phosphatidylinositol-3-kinase Vps34, which generates the phosphatidylinositol-3-phosphate (PtdIns (3) P) required for autophagosome formation [16]. LC3 (also known as MAP1LC3) is an intracellular protein, the accumulation of which is often used to identify shifts in autophagic flux. Upon initiation of autophagy, the cytosolic LC3-I form of the protein is cleaved and conjugated to phosphatidylethanolamine to form LC3-II, which associates with autophagosomal membranes. LC3-II is commonly used as a marker of autophagy [17]. Several studies have reported that the LC3-II levels increase in the spinal cord of G93A-SOD1 mice during the early stage (90 d) of ALS [18], and the changes become prominent at the end stage [18-20], indicating that autophagy is altered. However, little is known about changes and mechanisms underlying TFEB-mediated autophagy during ALS progression.
In this study, we found altered expression of TFEB and Beclin-1 in the spinal cords of ALS transgenic mice at different stages and a NSC-34 cell model with a mutated form of the SOD1 gene. Overexpression of TFEB in NSC-34 cells with the SOD1-G93A mutation increased the mRNA and protein levels of Beclin-1, accompanied by increased levels of LC3-II protein. Collectively, these findings suggest that altered expression of TFEB is a key element in the pathogenesis of ALS, making TFEB a very promising target to counteract autophagy dysfunction and neurodegeneration. A deeper understanding of the biology of TFEB-mediated autophagy will help identify novel drug targets and hopefully provide new therapeutics for treating ALS.
Material and methods
Animals and tissue preparations
Transgenic mice with the G93A mutation in the human SOD1 gene, originally obtained from Jackson Laboratories (Bar Harbor, ME, USA), were described previously [21,22]. ALS and age-matched wild-type (WT) mice were killed at 70 days (asymptomatic stage), 95 days (early stage), 108 days (middle stage), or 122 days (end stage), and tissues were preserved as previously described for further analysis [21,22]. All animal care and surgical procedures were approved by the Animal Studies Committee of Weifang Medical University in accordance with the National Institutes of Health Guidelines for animal experimentation.
Cell culture
Mouse motor neuron-like hybrid (NSC-34) cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 2 mM L-glutamine, 10% fatal bovine serum, and 100 mg/mL each of penicillin and streptomycin. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2. When they reached 80-90% confluence in tissue-culture flasks, cells were divided into subsequent culture plates as required.
Human SOD1 plasmid and cell transfection
The plasmids pEGFP-wt-SOD1 and pEGFP-G93A-SOD1 expressing the Green FP (GFP)-tagged human SOD1 [23] were the kind gift of Professor Angelo Poletti (University of Milan, Italy). NSC-34 cells were seeded in six-well plates at 300,000 cells/ml density. Transfection was carried out when the cells reached 70-80% confluence using Lipofectamine™ 2000 (Life Technologies) according to the manufacturer’s protocol. Plasmid and lipofectamine™ 2000 were diluted in Opti-MEM (Invitrogen) separately and incubated for 5 min at room temperature. The diluted solutions were then mixed and incubated for 20 min at room temperature. Subsequently, the mixtures were added to each well containing the cells and medium. The cell culture plates were then incubated for 6 h at 37°C in a CO2 incubator. The cell medium was then replaced with a complete culture medium. At 24 h, 48 h, and 72 h after transfection, cells were harvested for further analysis. Total RNA or proteins were extracted from transfected NSC-34 cells at different time points for RT-PCR and western blot analysis.
TFEB cloning and cell transfection
Mouse TFEB cDNA was cloned into the XhoI/AgeI sites of the GV316-DsRed2 vector (Jikai, Shanghai, China) to generate DsRed2-TFEB. Before transfection NSC-34 cells were seeded in six-well plates at 70% confluence, which was performed according to the manufacturer’s protocols. The following cells were analyzed: co-transfected control group with DsRed2 vector and pEGFP-G93A-SOD1; and co-transfected test group with DsRed2-TFEB and pEGFP-G93A-SOD1. At 24 h, 48 h, and 72 h after transfection, cells were harvested for further analysis.
MTS assay
The viability of NSC-34 cells was measured by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay (Promega). Briefly, 1×104 transfected cells were plated into each well of a 96-well plate. At 24, 48, 72, 96, and 120 h after transfection, 20μl MTS reagent (5 mg/ml) was added to each well and incubated for one additional hour at 37°C. The optical density (OD) of the wells was measured at a wavelength of 490 nm. All experiments were performed in triplicate. The cell proliferation curve was plotted at each time point.
RNA extraction and RT-PCR
Total RNA was extracted from spinal cord tissue and NSC-34 cells using TRIzol Reagent (Invitrogen) following the manufacturer’s instructions. The synthesis of cDNA and procedures for PCR amplification were described previously [21]. The primers used in this study are as follows: TFEB (sense, 5’-CAGCAGGTGGTGAAGCAAGAGT-3’; antisense, 5’-TCCAGGTGATGGAACGGAGACT-3’); Beclin-1 (sense, 5’-TTTCAGACTGGGTCGCTTGC-3’; antisense, 5’-CTTTTGTCCACTGCTCCTCCG-3’); and β-actin (sense, 5’-CGTTGACATCCGTAAAGACC-3’; antisense, 5’-ACAGTCCGCCTAGAAGCAC-3’).
Protein extraction and western blotting
Samples of spinal cord tissue and the transfected NSC34 cells were homogenized in RIPA buffer containing 1 mM PMSF and 10 μl/ml of protease inhibitor cocktail (Sigma-Aldrich). Other protocols for protein extraction and western blotting were carried out as described previously [21,24]. We used the following primary antibodies: rabbit anti-TFEB (1:800, Anbo), rabbit anti-Beclin-1 (1:2000, Sigma-Aldrich), mouse anti-GAPDH (1:2000; Proteintech Group), and rabbit anti-LC3II (1:1000, Abcam). Secondary antibodies against the appropriate primary species were used at 1:20,000 dilution.
Immunohistochemistry and immunofluorescence
The method of immunohistochemistry used for frozen sections was previously described [27]. The primary antibodies used include rabbit anti-TFEB (1:100; Anbo) and rabbit anti-Beclin-1 (1:200; Sigma). Specific reactivity was detected using a DAB kit (ZsBio, Beijing, China). For double immunofluorescence labeling, a mixture of the following antibodies was used at the dilutions indicated: rabbit anti-TFEB (1:100; Anbo), mouse anti-GFAP (1:300; Cell signaling technology), and chicken anti-β-III tubulin (1:600; Abcam). Secondary antibodies against the appropriate primary species were used: goat anti-rabbit Cy3 (1:400; Jackson ImmunoResearch), goat anti-chicken AlexaFluor 488 (1:400; Jackson ImmunoResearch), and donkey anti-mouse AlexaFluor 488 (1:400; Jackson ImmunoResearch). The images were obtained by confocal microscopy (Olympus FV500, Japan).
For cell immunofluorescence, NSC-34 cells were plated onto coverslips in complete culture medium for 24 h. The cells were then transfected with the plasmids. At 24, 48, and 72 h after transfection, the cells were fixed with 4% paraformaldehyde for 20 min, incubated in 0.3% Triton X-100-PBS for 10 min at room temperature, followed by blocking with 5% goat serum at 37°C for 30 min. The cells were then incubated with the following primary antibodies at 4°C overnight: rabbit anti-TFEB (1:100; Anbo), rabbit anti-Beclin-1 (1:200; Sigma-Aldrich). After being soaked in goat anti-rabbit IgG conjugated to Cy3 secondary antibody (1:400; Jackson ImmunoResearch) at 37°C for 1 h, the sample was stained with Hoechst33258 (1:1000; Sigma-Aldrich). Images were obtained using an inverted fluorescence microscope (Olympus, Japan).
Statistical analysis
All data are shown as mean ± SD. SPSS 13.0 was used for statistical evaluation. The two data sets were compared using the two-tailed Student’s t-test. A probability level of 0.05 or less was considered statistically significant.
Results
The altered expression of TFEB and Beclin-1 in ALS transgenic mice
To explore the role of TFEB in the pathogenesis of ALS, we detected its expression in SOD1-G93A ALS transgenic mice at different stages using RT-PCR and western blot. TFEB mRNA and protein remained unchanged during the asymptomatic stage (70 d, data not shown) but were up-regulated in the early stage (95 d) and down-regulated in the middle and end stages (108 d and 122 d) in the spinal cords of ALS mice, compared with WT mice. The differences found in the symptomatic stages were all significant (*P < 0.05, **P < 0.01, ***P < 0.001) (Figure 1A-D).
Figure 1.
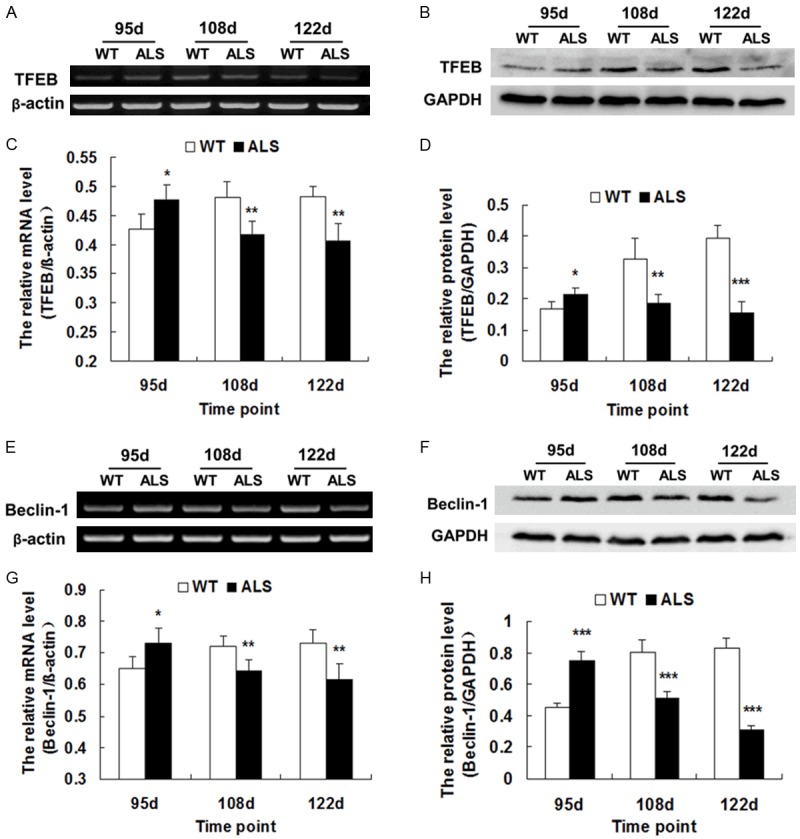
The expression of TFEB and Beclin-1 in the spinal cords of ALS mice and WT mice. A. Representative RT-PCR of TFEB. β-actin was used as an internal control. B. Representative western blot of TFEB protein. GAPDH was used as an internal control. C. The relative mRNA levels of TFEB as analyzed by RT-PCR (n = 5). D. The relative intensity of TFEB protein as analyzed by western blot (n = 5). E. Representative RT-PCR of Beclin-1. β-actin was used as an internal control. F. Representative western blot of Beclin-1 protein. GAPDH was used as an internal control. G. The relative mRNA level of Beclin-1 as analyzed by RT-PCR (n = 5). H. The relative intensity of Beclin-1 protein as analyzed by western blot (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001, vs. WT.
Similarly, the expression of Beclin-1, an important autophagy-regulatory gene, was markedly altered, similar to the expression of TFEB in ALS transgenic mice (*P < 0.05, **P < 0.01, ***P < 0.001) (Figure 1E-H). This pattern of changes indicates that the function of TFEB and Beclin-1 in the early stage of disease may be different from the middle and end stages.
The distribution and localization of TFEB and Beclin-1 in ALS transgenic mice
To investigate the distribution and localization of TFEB and Beclin-1, we analyzed their expression by immunohistochemistry labeling. TFEB-positive cells were detected in the gray and white matter of the spinal cord, but none were found in the central canal. The majority of the TFEB-positive cells were located in the ventral horn within the gray matter. Within the white matter, nerve fibers were also immunopositive. At 70 d, the immunoreactivity of TFEB was unchanged in the spinal cord of ALS mice compared to age-matched WT mice. At 95 d, the immunoreactivity of TFEB was strong in the ALS mice but weak in WT mice, in both gray and white matter. At 108 d and 122 d, the immunoreactivity of TFEB was weak in ALS mice but strong in WT mice (Figure 2). At 108 d and 122 d, the nerve fibers in the white matter of ALS mice became irregular and also decreased because of serious degeneration of motor neurons. The distribution of Beclin-1 was similar to that of TFEB (Figure 2).
Figure 2.
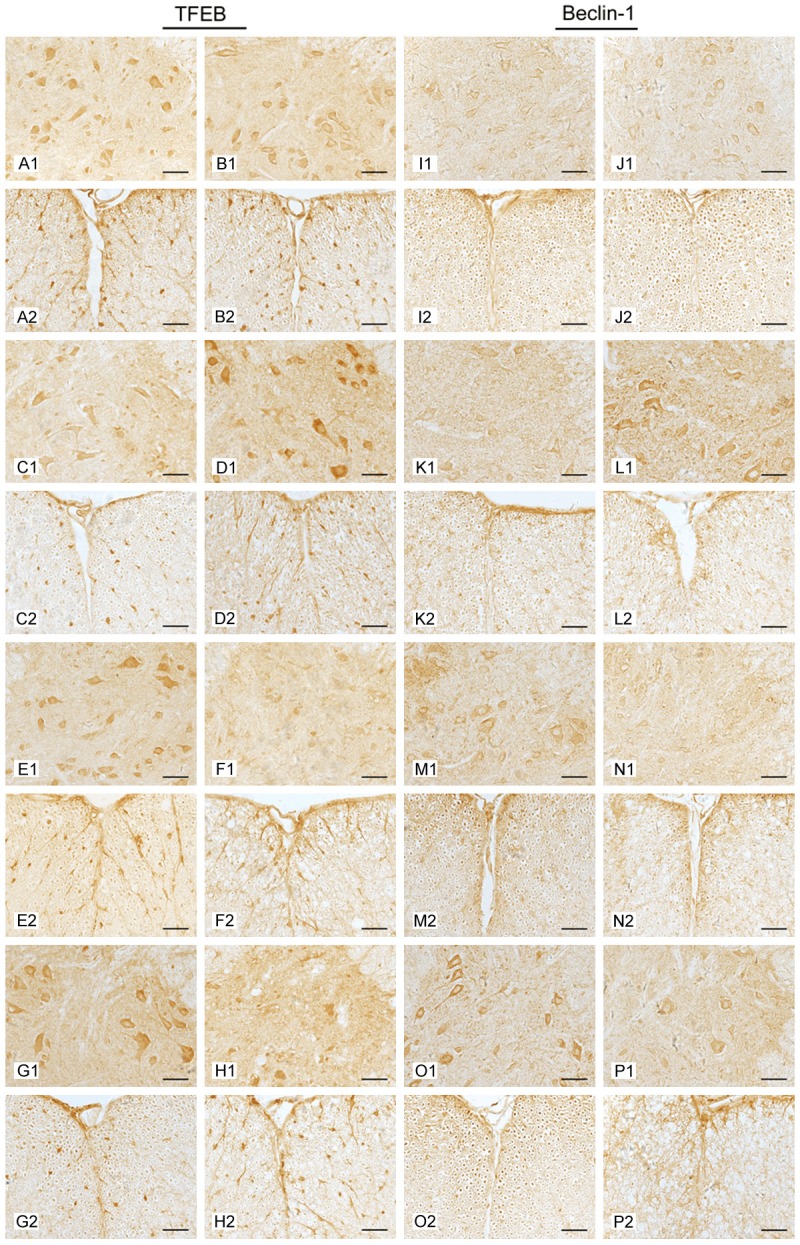
The distribution of TFEB and Beclin-1 in the spinal cords of ALS mice and WT mice detected by immunohistochemical staining. Immunohistochemistry images were taken of gray matter (GM) and white matter (WM) of the spinal cords at different stages. 70 d (A, B, I, J); 95 d (C, D, K, L); 108 d (E, F, M, N); 122 d (G, H, O, P); GM (1); WM (2); WT (A, C, E, G, I, K, M, O); ALS (B, D, F, H, J, L, N, P). Scale bar = 50 μm.
To determine whether TFEB or Beclin-1 was colocalized with β-tubulin III for neurons or GFAP for mature astrocytes, we performed double-immunofluorescence staining. The result revealed that TFEB/β-tubulin III and Beclin-1/β-tubulin III double-positive cells were detected in both ALS and WT mice, and most of the double-positive cells were located in the ventral horn (Figure 3). However, TFEB/GFAP and Beclin-1/GFAP double-positive cells were not found in either ALS or WT mice (Figure 3), suggesting that TFEB and Beclin-1 were expressed in neurons but not in mature astrocytes. The majority of the positive cells were expressed mainly in neurons.
Figure 3.
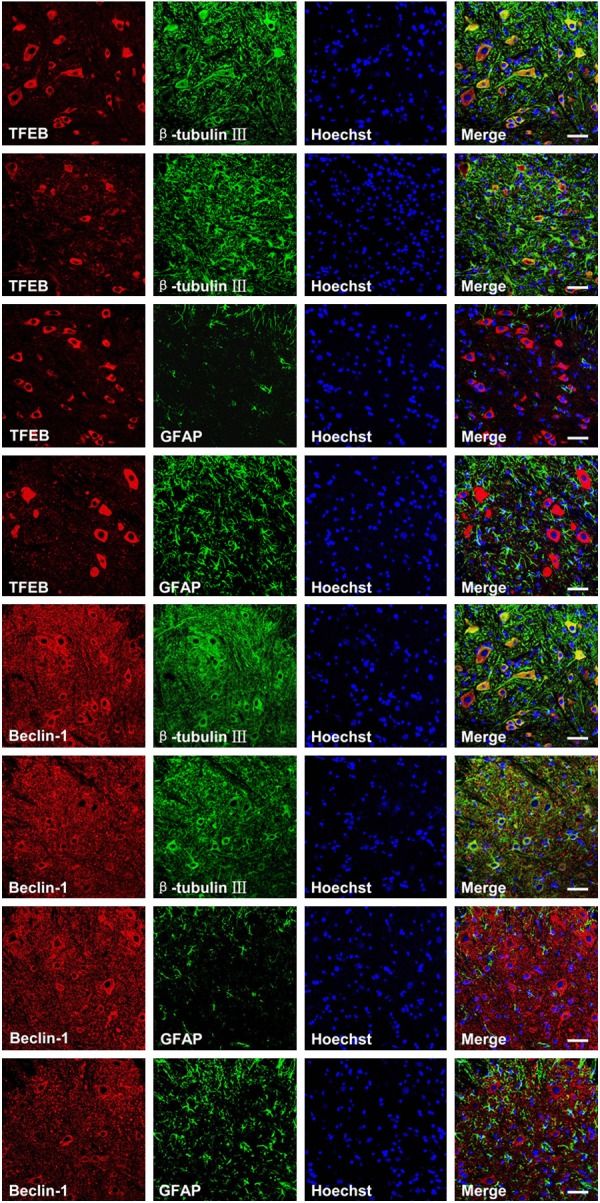
Double immunofluorescence staining results showing the colocalization of TFEB or Beclin-1 with β-tubulin III or GFAP. Representative confocal images were taken of gray matter (GM) of 108-day-old mice. WT (panel 1, 3, 5, and 7) and ALS (panel 2, 4, 6, and 8). Scale bar = 50 μm.
Altered expression of TFEB and Beclin-1 in the NSC-34 cell model
We transfected NSC-34 cells using the pEGFP-G93A-SOD1 and pEGFP-WT-SOD1 plasmids to explore the expression of TFEB and Beclin 1 in the NSC-34 cell model. At 24, 48, and 72 h after transfection, TFEB and Beclin-1 mRNA and protein were detected by RT-PCR and western blot. At 24 h, there was no significant difference in the expression of TFEB or Beclin-1 of NSC-34 cells transfected with the pEGFP-G93A-SOD1 and pEGFP-wt-SOD1 plasmids. The NSC-34 cells that carried the pEGFP-G93A-SOD1 plasmid had lower levels of TFEB or Beclin-1 mRNA and protein at both 48 h and 72 h than NSC-34 cells transfected with the wild-type plasmid (**P < 0.01, ***P < 0.001) (Figure 4A-H), suggesting that the expression of TFEB and Beclin-1 were altered in the ALS cell model, in agreement with our results using the animal model. Immunofluorescence staining showed that the immunoreactivity of TFEB was weak in the NSC-34 cells transfected with pEGFP-G93A-SOD1 but strong in the NSC-34 cells transfected with pEGFP-wt-SOD1 at 48 h and 72 h (Figure 4I).
Figure 4.
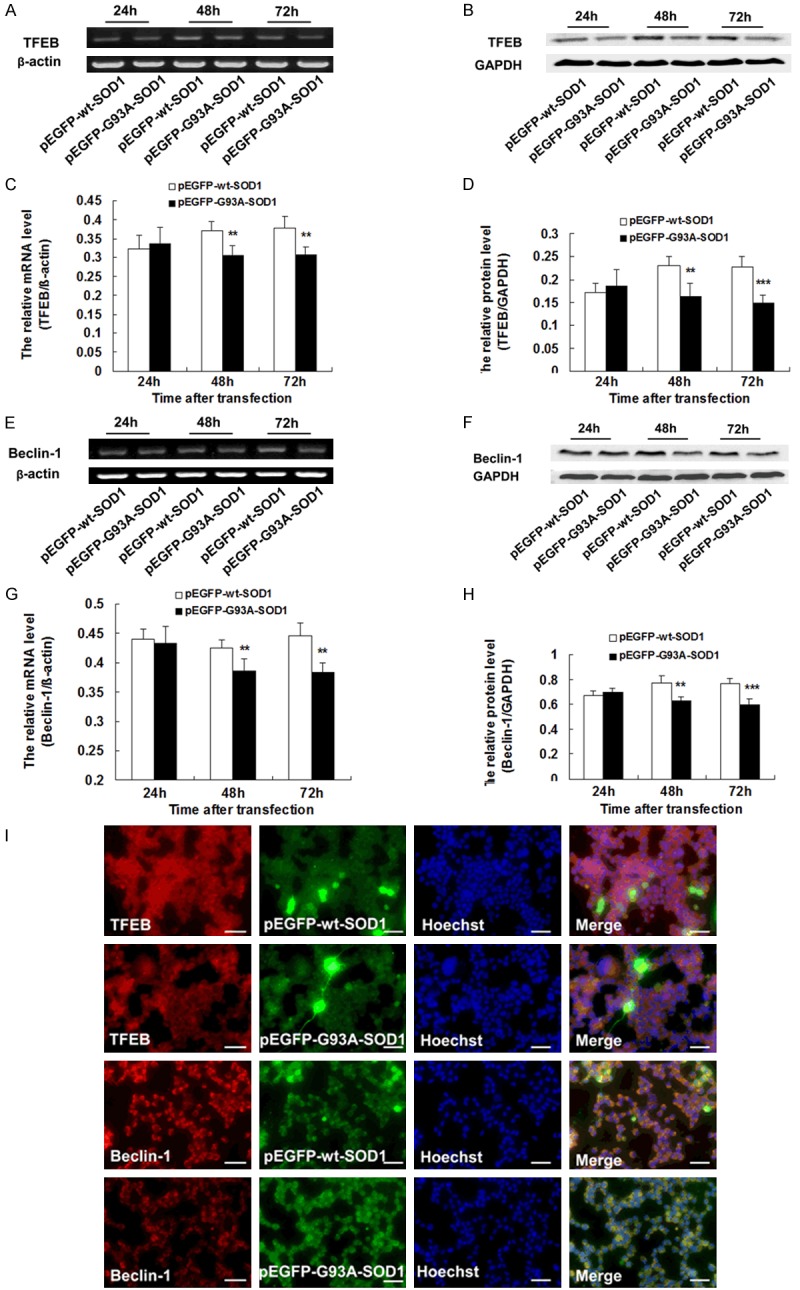
Altered expression of TFEB and Beclin 1 in NSC-34 cells transfected with the pEGFP-SOD1-G93A or pEGFP-SOD1-WT plasmids. A. Representative RT-PCR of TFEB. β-actin was used as an internal control. B. Representative western blot of TFEB protein. GAPDH was used as an internal control. C. The relative mRNA levels of TFEB as analyzed by RT-PCR (n = 5). D. The relative intensity of TFEB protein as analyzed by western blot (n = 5). E. Representative RT-PCR of Beclin-1. β-actin was used as an internal control. F. Representative western blot of Beclin-1 protein. GAPDH was used as an internal control. G. The relative mRNA level of Beclin-1 as analyzed by RT-PCR (n = 5). H. The relative intensity of Beclin-1 protein as analyzed by western blot (n = 5). I. Immunofluorescence staining results showing the expression of TFEB and Beclin-1 in NSC-34 cells 72 h after transfection. Scale bar = 50 μm. **P < 0.01, ***P < 0.001, vs. pEGFP-wt-SOD1.
Overexpression of TFEB promotes autophagy in vitro
To determine whether TFEB was involved in autophagy in the pathogenesis of ALS, we constructed a plasmid with TFEB overexpression. At 24 h, 48 h, and 72 h after co-transfection with pEGFP-G93A-SOD1 and DsRed2-TFEB plasmids, we examined the efficiency of TFEB overexpression using RT-PCR and western blot. The result showed that at 24 h, 48 h, and 72 h after transfection, the TFEB mRNA of NSC-34 cells co-transfected with pEGFP-G93A-SOD1 and DsRed2-TFEB plasmids were all up-regulated significantly compared with the NSC-34 cells co-transfected with pEGFP-G93A-SOD1 and DsRed2 empty vectors (*P < 0.05, **P < 0.01, ***P < 0.001) (Figure 5A-D). The TFEB protein levels in NSC34 cells co-transfected with pEGFP-G93A-SOD1 and DsRed2-TFEB plasmids were up-regulated significantly compared to NSC-34 cells co-transfected with pEGFP-G93A-SOD1 and DsRed2 empty vectors.
Figure 5.
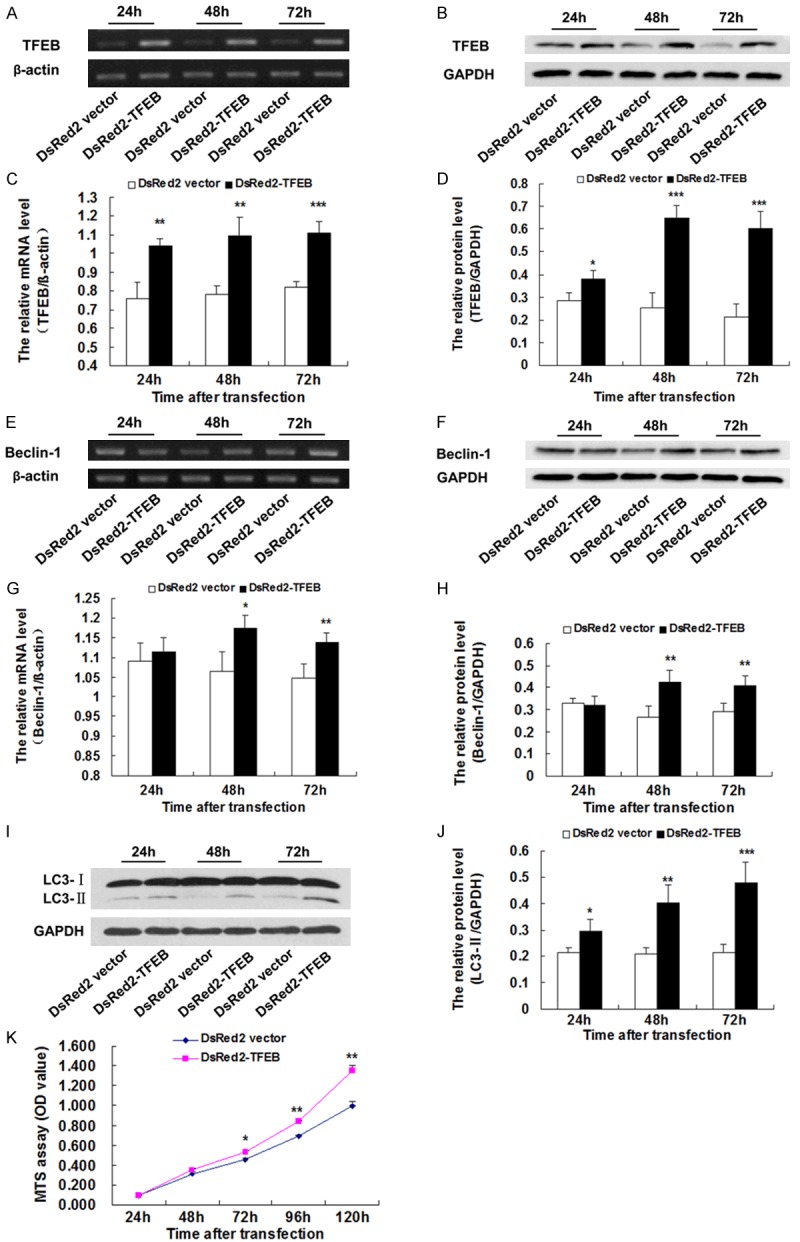
TFEB overexpression increased the expression of both Beclin-1 and LC3-II and cell survival in NSC-34 cells with a SOD1-G93A mutation. A. Representative RT-PCR of TFEB. β-actin was used as an internal control. B. Representative western blot of TFEB protein. GAPDH was used as an internal control. C. RT-PCR analysis illustrating the overexpression of TFEB (n = 4). D. Western blot analysis illustrating the overexpression of TFEB. The amount of TFEB was quantified and normalized against GAPDH (n = 4). E. Representative RT-PCR of Beclin-1. β-actin was used as an internal control. F. Representative western blot of Beclin-1 protein. GAPDH was used as an internal control. G. RT-PCR analysis illustrating up-regulation of Beclin-1 (n = 4). H. Western blot analysis illustrating the up-regulation of Beclin-1. The amount of Beclin-1was quantified and normalized against GAPDH (n = 4). I. Representative western blot of LC3-II protein. GAPDH was used as an internal control. J. Western blot analysis illustrating up-regulation of LC3-II. The amount of LC3-II was quantified and normalized against GAPDH (n = 4). K. TFEB overexpression increases cell survival of NSC-34 cells with the SOD1-G93A mutation evaluated by MTS assay. *P < 0.05, **P < 0.01, ***P < 0.001, vs. DsRed2 vector.
To elucidate how TFEB overexpression alters autophagy, the protein expression of LC3-II, a marker of autophagy, was measured in NSC34 cells with the SOD1-G93A mutation. Western blot analysis revealed that, compared with the group transfected with empty vector. LC3-II protein in the NSC-34 cells with DsRed2-TFEB was significantly up-regulated at 24 h, 48 h and 72 h after transfection (*P < 0.05, **P < 0.01, ***P < 0.001) (Figure 5I, 5J), suggesting that the overexpression of TFEB promotes autophagy.
TFEB overexpression results in the increased expression of Beclin-1 in vitro
To explore the molecular mechanisms underlying TFEB’s regulation of autophagy, we measured the expression of Beclin-1, an important factor in autophagy, using RT-PCR and western blot. Compared with the group transfected with empty vector, Beclin-1 mRNA levels were up-regulated at 48 h and 72 h after transfection. No significant difference was found at 24 h. The protein levels of Beclin-1 were significantly up-regulated at 48 h and 72 h after transfection (*P < 0.05, **P < 0.01) (Figure 5E-H), suggesting that the overexpression of TFEB promotes autophagy by up-regulating the expression of Beclin-1.
TFEB overexpression increased cell proliferation and survival of NSC-34 cells with the SOD1-G93A mutation
To examine the role of TFEB in cell proliferation and survival of NSC-34 cells with the SOD1-G93A mutation, we examined the effect of TFEB overexpression on cell growth. As shown in Figure 5K, the OD value of NSC-34 cells co-transfected pEGFP-G93A-SOD1 and DsRed2-TFEB was lower than that of NSC-34 cells co-transfected with pEGFP-G93A-SOD1 and DsRed2 empty vector, which suggests that TFEB overexpression increases cell proliferation and survival of NSC-34 cells with the SOD1-G93A mutation.
Discussion
ALS is a motor neuron disease characterized by the aggregation of misfolded proteins. Autophagy is a fundamental intracellular degradative pathway that plays a central role in clearing away aggregated proteins; defects in clearance of these misfolded proteins have been associated with several neurodegenerative disorders. LC3-II, a well-known autophagic marker, was increased in SOD1-G93A transgenic mice in the symptomatic stage compared with WT mice [18,19]. An et al. [25] also reported increased LC3-II expression in the brainstem motor neurons of SOD1-G93A mice. Crippa et al. [26] found that LC3 mRNA was significantly up-regulated in the skeletal muscle of tgG93A-SOD1 mice at symptomatic (16 weeks) stages. Accumulating evidence indicates that altered autophagy is involved in ALS motor neuron degeneration.
Recently, a new transcription-dependent mechanism regulating autophagy was identified. TFEB, a member of the MITF/TFE subfamily of basic helix-loop-helix leucinezipper (bHLH-Zip) transcription factors, not only controls lysosomal biogenesis, but also regulates autophagy. TFEB overexpression in HeLa cells increases the expression of LC3, and depletion of TFEB results in decreased levels of LC3-II [18,19,27,28]. However, changes in the expression of TFEB in the spinal cord of ALS have not yet been detected, and the role and mechanisms of TFEB in pathogenesis remain uncertain.
Here we found that TFEB mRNA and protein were up-regulated in the early stage (95 d) but down-regulated in the middle and end stages (108 d and 122 d) of disease in the spinal cords of ALS mice, compared with wild-type mice. We also measured the expression of TFEB in a cell model. The NSC-34 cells that carried the pEGFP-G93A-SOD1 plasmid had lower levels of TFEB mRNA and proteins at both 48 h and 72 h than those in NSC-34 cells transfected with the pEGFP-wt-SOD1 plasmid, suggesting that the SOD1 with a mutation affected the expression of TFEB. The expression of TFEB was altered in both our cell model and our animal model, suggesting that TFEB may be closely related to the pathogenesis of ALS. The majority of the TFEB-positive cells are expressed mainly in β-tubulin III-labeled neurons, indicating that the altered expression of TFEB in ALS pathogenesis mainly affects neurons.
To determine whether TFEB was involved in autophagy in the pathogenesis of ALS, NSC34 motor neuron cells were transfected with the TFEB plasmid. The efficiency test showed that at 24, 48, and 72 h after transfection, TFEB mRNA were all up-regulated significantly. TFEB protein levels of NSC-34 cells carrying the pEGFP-G93A-SOD1 and DsRed2-TFEB plasmids were also up-regulated significantly compared to NSC-34 cells carrying the pEGFP-G93A-SOD1 and empty vectors. To determine how TFEB overexpression alters autophagy, we measured the protein expression of LC3-II by western blot analysis. LC3-II protein levels were significantly up-regulated at 24 h, 48 h and 72 h after transfection in response to the overexpression of TFEB, suggesting that TFEB overexpression promotes autophagy.
Furthermore, several recent studies demonstrated that TFEB is also involved in other neurodegenerative diseases. For example, the presence of α-synuclein is characteristic of PD. TFEB is a key player in the induction of α-synuclein-induced toxicity and PD pathogenesis. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity, thus identifying TFEB as a promising target for therapies aimed at neuroprotection and disease modification in PD [29,30]. Enhancement of TFEB function has been shown to stimulate the autophagy, promote protein clearance, and protect neurons in cellular models of protein misfolding and oxidative stress, as well as in a mouse model of HD [27,31]. La et al. [32] found decreased expression of TFEB in HD cells and mice and noted that PPARGC1A is upstream of TFEB in promoting proteostasis. TFEB does appear to be a key regulator of cellular protein and organelle homeostasis, suggesting that further investigation into its biology and regulation is warranted.
To test whether TFEB regulates the expression of autophagy genes, we measured the expression of Beclin-1in both our animal and cell models. Beclin-1 mRNA and protein were up-regulated at 95 d but down-regulated at 108 d and 122 d in the spinal cords of ALS mice compared to wild-type mice. Recently, Nassif et al. [33] reported that BECN1 mRNA and protein levels were upregulated in symptomatic SOD1-G86R transgenic mice. We used another ALS transgenic mouse with a SOD1-G93A mutation to examine the expression changes of Beclin-1 in the early, middle, and end stages of disease. Our results showed more detailed changes in Beclin-1 in the pathogenesis of ALS. We found a high degree of coexpression of Beclin-1 and β-tubulin III, a marker for neurons, but no Beclin-1/GFAP double-positive cells were found, in agreement with the results of Nassif et al. Studies in models of PD, HD, and AD indicate that abnormal interaction of disease-related proteins with Beclin-1 may alter its biological function, contributing to neurodegeneration [34-36].
Beclin-1 mRNA and protein were also decreased in NSC-34 cells carrying the pEGFP-G93A-SOD1 plasmid compared to those carrying the pEGFP-WT-SOD1 plasmid. The expression of Beclin-1 is consistent with that of TFEB; perhaps they are linked in function. Overexpression of TFEB in NSC-34 cells increased Beclin-1 expression (both mRNA and protein) with a concordant increase in cellular autophagy measured by LC3-II. Our results indicate that TFEB is associated with altered autophagy via regulation of Beclin-1 expression. Many researchers have focused on the role and mechanisms of TFEB in autophagy and attempted to identify the targets of TFEB. Palmieri et al. [37] found that Beclin-1 is one of the direct targets of TFEB. They used a combination of genomic approaches, including ChIP-seq (sequencing of chromatin immunoprecipitate) analysis, profiling of TFEB-mediated transcriptional induction, genome-wide mapping of TFEB target sites, and recursive expression meta-analysis of TFEB targets, and identified 471 TFEB direct targets that represent essential components of the CLEAR (Coordinated Lysosomal Expression and Regulation, CLEAR) network. Settembre et al. [14] reported that TFEB overexpression significantly affected the expression of several genes in different steps of autophagy, including MAPLC3B, ATG9B, SQSTM1, UVRAG, and VPS11. These genes carry at least one TFEB target site in their promoters and appear to be targets of TFEB.
Emerging studies have demonstrated that up-regulation of autophagy is beneficial in various models of neurodegenerative diseases [38,39]. Kabuta et al. [40] found that the activation of autophagy decreases the expression of mutant SOD1 and reduces its neurotoxicity. Lithium, one of the autophagy inducers, can protect motor neurons from damage and prolong the lifespan of ALS transgenic mice by clearing the aggregation of abnormal mutant SOD1 protein. In fact, lithium delays progression of ALS [41]. Therefore, understanding the molecular mechanisms underlying the regulation of autophagy is crucial to the development of strategies for therapy. Targeting autophagy may serve as an effective therapeutic strategy against neurodegenerative diseases. Song et al. [42] reported that 2-hydroxypropylbeta-cyclodextrin, a small molecule, promotes transcription factor EB-mediated activation of autophagy, which may suggest possible therapeutic approaches.
Conclusions
TFEB plays a critical role in the pathogenesis of ALS by regulating cellular autophagy. TFEB promotes autophagy by enhancing the expression of Beclin-1, increasing cell proliferation and survival. The altered autophagy mediated by TFEB is a key element in the pathogenesis of ALS, making TFEB a very promising target for the development of novel drugs and new gene therapeutics for ALS.
Acknowledgements
We thank Professor Angelo Poletti (Inter-University Center on Neurodegenerative Diseases of the Universities of Milan, Florence, and Rome, Italy) for SOD1 plasmids. This work was supported by grants from the National Natural Science Foundation of China (81401066; 81271413), the Shandong Province Natural Science Foundation of China (ZR2012HQ021), the Shandong Province Science and Technology Development Program of China (2012GSF11827), the Shandong Provincial Education Department of China (J11LF16; J12LK51; J12LK05; J13LK05), the Science and Technology Development Program of Weifang in China (201301074), the Muscular Dystrophy Association (254530), the ALS Therapy Alliance (2013D001622), the Bill and Melinda Gates Foundation, the International Cooperation Program for international lecturers of Shandong Provincial Education Department, and the Shandong Province Taishan Scholar Project of China.
Disclosure of conflict of interest
None.
References
- 1.Ryu HH, Jun MH, Min KJ, Jang DJ, Lee YS, Kim HK, Lee JA. Autophagy regulates amyotrophic lateral sclerosis-linked fused in sarcoma-positive stress granules in neurons. Neurobiol Aging. 2014;35:2822–2831. doi: 10.1016/j.neurobiolaging.2014.07.026. [DOI] [PubMed] [Google Scholar]
- 2.Zhu Y, Fotinos A, Mao LL, Atassi N, Zhou EW, Ahmad S, Guan Y, Berry JD, Cudkowicz ME, Wang X. Neuroprotective agents target molecular mechanisms of disease in ALS. Drug Discov Today. 2015;20:65–75. doi: 10.1016/j.drudis.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 3.Pasquali L, Ruffoli R, Fulceri F, Pietracupa S, Siciliano G, Paparelli A, Fornai F. The role of autophagy: what can be learned from the genetic forms of amyotrophic lateral sclerosis. CNS Neurol Disord Drug Targets. 2010;9:268–278. doi: 10.2174/187152710791292594. [DOI] [PubMed] [Google Scholar]
- 4.Deng M, Wei L, Zuo X, Tian Y, Xie F, Hu P, Zhu C, Yu F, Meng Y, Wang H, Zhang F, Ma H, Ye R, Cheng H, Du J, Dong W, Zhou S, Wang C, Wang Y, Wang J, Chen X, Sun Z, Zhou N, Jiang Y, Liu X, Li X, Zhang N, Liu N, Guan Y, Han Y, Lv X, Fu Y, Yu H, Xi C, Xie D, Zhao Q, Xie P, Wang X, Zhang Z, Shen L, Cui Y, Yin X, Liang B, Zheng X, Lee TM, Chen G, Zhou F, Veldink JH, Robberecht W, Landers JE, Andersen PM, Al-Chalabi A, Shaw C, Liu C, Tang B, Xiao S, Robertson J, van den Berg LH, Sun L, Liu J, Yang S, Ju X, Wang K, Zhang X. Genome-wide association analyses in Han Chinese identify two new susceptibility loci for amyotrophic lateral sclerosis. Nat Genet. 2013;45:697–700. doi: 10.1038/ng.2627. [DOI] [PubMed] [Google Scholar]
- 5.Chattopadhyay M, Valentine JS. Aggregation of copper-zinc superoxide dismutase in familial and sporadic ALS. Antioxid Redox Signal. 2009;11:1603–1614. doi: 10.1089/ars.2009.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D, Frosch MP, Agar JN, Julien JP, Brady ST, Brown RH Jr. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13:1396–1403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rotunno MS, Bosco DA. An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front Cell Neurosci. 2013;7:253. doi: 10.3389/fncel.2013.00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 9.Guareschi S, Cova E, Cereda C, Ceroni M, Donetti E, Bosco DA, Trotti D, Pasinelli P. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc Natl Acad Sci U S A. 2012;109:5074–5079. doi: 10.1073/pnas.1115402109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 12.Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13:805–811. doi: 10.1038/nn.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gal J, Chen J, Barnett KR, Yang L, Brumley E, Zhu H. HDAC6 regulates mutant SOD1 aggregation through two SMIR motifs and tubulin acetylation. J Biol Chem. 2013;288:15035–15045. doi: 10.1074/jbc.M112.431957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Settembre C, Ballabio A. TFEB regulates autophagy: an integrated coordination of cellular degradation and recycling processes. Autophagy. 2011;7:1379–1381. doi: 10.4161/auto.7.11.17166. [DOI] [PubMed] [Google Scholar]
- 16.Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19:5360–5372. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- 18.Morimoto N, Nagai M, Ohta Y, Miyazaki K, Kurata T, Morimoto M, Murakami T, Takehisa Y, Ikeda Y, Kamiya T, Abe K. Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res. 2007;1167:112–117. doi: 10.1016/j.brainres.2007.06.045. [DOI] [PubMed] [Google Scholar]
- 19.Li L, Zhang X, Le W. Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy. 2008;4:290–293. doi: 10.4161/auto.5524. [DOI] [PubMed] [Google Scholar]
- 20.Tan CC, Yu JT, Tan MS, Jiang T, Zhu XC, Tan L. Autophagy in aging and neurodegenerative diseases: implications for pathogenesis and therapy. Neurobiol Aging. 2014;35:941–957. doi: 10.1016/j.neurobiolaging.2013.11.019. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Guan Y, Liu H, Wu X, Yu L, Wang S, Zhao C, Du H, Wang X. Activation of the Wnt/beta-catenin signaling pathway is associated with glial proliferation in the adult spinal cord of ALS transgenic mice. Biochem Biophys Res Commun. 2012;420:397–403. doi: 10.1016/j.bbrc.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 22.Chen Y, Guan Y, Zhang Z, Liu H, Wang S, Yu L, Wu X, Wang X. Wnt signaling pathway is involved in the pathogenesis of amyotrophic lateral sclerosis in adult transgenic mice. Neurol Res. 2012;34:390–399. doi: 10.1179/1743132812Y.0000000027. [DOI] [PubMed] [Google Scholar]
- 23.Poletti A, Cattaneo E, Taroni F. The neurotoxicity of mutant proteins 20 years after the discovery of the first mutant gene involved in neurodegeneration. Foreword. Prog Neurobiol. 2012;97:53. doi: 10.1016/j.pneurobio.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 24.Li X, Guan Y, Chen Y, Zhang C, Shi C, Zhou F, Yu L, Juan J, Wang X. Expression of Wnt5a and its receptor Fzd2 is changed in the spinal cord of adult amyotrophic lateral sclerosis transgenic mice. Int J Clin Exp Pathol. 2013;6:1245–1260. [PMC free article] [PubMed] [Google Scholar]
- 25.An T, Shi P, Duan W, Zhang S, Yuan P, Li Z, Wu D, Xu Z, Li C, Guo Y. Oxidative stress and autophagic alteration in brainstem of SOD1-G93A mouse model of ALS. Mol Neurobiol. 2014;49:1435–1448. doi: 10.1007/s12035-013-8623-3. [DOI] [PubMed] [Google Scholar]
- 26.Crippa V, Boncoraglio A, Galbiati M, Aggarwal T, Rusmini P, Giorgetti E, Cristofani R, Carra S, Pennuto M, Poletti A. Differential autophagy power in the spinal cord and muscle of transgenic ALS mice. Front Cell Neurosci. 2013;7:234. doi: 10.3389/fncel.2013.00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 28.Pastore N, Ballabio A, Brunetti-Pierri N. Autophagy master regulator TFEB induces clearance of toxic SERPINA1/alpha-1-antitrypsin polymers. Autophagy. 2013;9:1094–1096. doi: 10.4161/auto.24469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A. TFEBmediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci U S A. 2013;110:E1817–1826. doi: 10.1073/pnas.1305623110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ebrahimi-Fakhari D, Wahlster L. Restoring impaired protein metabolism in Parkinson’s disease--TFEB-mediated autophagy as a novel therapeutic target. Mov Disord. 2013;28:1346. doi: 10.1002/mds.25601. [DOI] [PubMed] [Google Scholar]
- 31.Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, Lazarowski ER, Damian VA, Masliah E, La Spada AR. PGC-1alpha rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012;4:142ra97. doi: 10.1126/scitranslmed.3003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.La Spada AR. PPARGC1A/PGC-1alpha, TFEB and enhanced proteostasis in Huntington disease: defining regulatory linkages between energy production and protein-organelle quality control. Autophagy. 2012;8:1845–1847. doi: 10.4161/auto.21862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nassif M, Valenzuela V, Rojas-Rivera D, Vidal R, Matus S, Castillo K, Fuentealba Y, Kroemer G, Levine B, Hetz C. Pathogenic role of BECN1/Beclin 1 in the development of amyotrophic lateral sclerosis. Autophagy. 2014;10:1256–1271. doi: 10.4161/auto.28784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu JC, Qi L, Wang Y, Kegel KB, Yoder J, Difiglia M, Qin ZH, Lin F. The regulation of N-terminal Huntingtin (Htt552) accumulation by Beclin1. Acta Pharmacol Sin. 2012;33:743–751. doi: 10.1038/aps.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mealer RG, Murray AJ, Shahani N, Subramaniam S, Snyder SH. Rhes, a striatal-selective protein implicated in Huntington disease, binds beclin-1 and activates autophagy. J Biol Chem. 2014;289:3547–3554. doi: 10.1074/jbc.M113.536912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khandelwal PJ, Herman AM, Hoe HS, Rebeck GW, Moussa CE. Parkin mediates beclindependent autophagic clearance of defective mitochondria and ubiquitinated Abeta in AD models. Hum Mol Genet. 2011;20:2091–2102. doi: 10.1093/hmg/ddr091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20:3852–3866. doi: 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- 38.Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, Webster JA, Lewis TA, O’Kane CJ, Schreiber SL, Rubinsztein DC. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol. 2007;3:331–338. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 40.Kabuta T, Suzuki Y, Wada K. Degradation of amyotrophic lateral sclerosis-linked mutant Cu,Zn-superoxide dismutase proteins by macroautophagy and the proteasome. J Biol Chem. 2006;281:30524–30533. doi: 10.1074/jbc.M603337200. [DOI] [PubMed] [Google Scholar]
- 41.Fornai F, Longone P, Cafaro L, Kastsiuchenka O, Ferrucci M, Manca ML, Lazzeri G, Spalloni A, Bellio N, Lenzi P, Modugno N, Siciliano G, Isidoro C, Murri L, Ruggieri S, Paparelli A. Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2008;105:2052–2057. doi: 10.1073/pnas.0708022105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song W, Wang F, Lotfi P, Sardiello M, Segatori L. 2-Hydroxypropyl-beta-cyclodextrin promotes transcription factor EB-mediated activation of autophagy: implications for therapy. J Biol Chem. 2014;289:10211–10222. doi: 10.1074/jbc.M113.506246. [DOI] [PMC free article] [PubMed] [Google Scholar]