

Abstract
Heart failure remains a leading cause of death and it is a major cause of morbidity and mortality affecting tens of millions of people worldwide. Despite decades of extensive research conducted at enormous expense, only a handful of interventions have significantly impacted survival in heart failure. Even the most widely prescribed treatments act primarily to slow disease progression, do not provide sustained survival advantage, and have adverse side effects. Since mortality remains about 50% within five years of diagnosis, the need to increase our understanding of heart failure disease mechanisms and development of preventive and reparative therapies remains critical. Currently, the vast majority of basic science heart failure research is conducted using animal models ranging from fruit flies to primates; however, insights gleaned from decades of animal-based research efforts have not been proportional to research success in terms of deciphering human heart failure and developing effective therapeutics for human patients. Here we discuss the reasons for this translational discrepancy which can be equally attributed to the use of erroneous animal models and the lack of widespread use of human-based research methodologies and address why and how we must position our own species at center stage as the quintessential animal model for 21st century heart failure research. If the ultimate goal of the scientific community is to tackle the epidemic status of heart failure, the best way to achieve that goal is through prioritizing human-based, human-relevant research.
Keywords: Heart failure, animal models, translatability, human relevance, human-based research
Introduction
Heart failure is the leading cause of hospitalization in the United States and a major cause of morbidity and mortality, affecting more than five million Americans and tens of millions worldwide [1]. Human heart failure is a complex, multifactorial, heterogeneous disease primarily caused by coronary artery disease, hypertension, and diabetes mellitus. Other common causes include valvular heart disease and myocarditis, and influencing risk factors include age, gender, ethnicity, family history, and lifestyle issues such as obesity, dietary content, and cardiovascular fitness [2]. Few interventions have significantly impacted survival in heart failure. Even the most widely prescribed treatments act primarily to slow disease progression, do not provide sustained survival advantage, and have important adverse side effects. Despite advances in the management of heart failure, prognosis is poor because mortality has persisted at about 50% within five years of diagnosis [1], and therefore the need to develop novel preventive and reparative therapies remains a high priority in the field.
Decades of research have provided an understanding of mechanisms underlying various aspects of heart failure, but many molecular and physiological aspects of human heart failure remain unclear. Many basic science research studies have been devoted to studying heart failure in various animal models in species ranging from fruit flies to rodents to nonhuman primates [3,4]. Tens of millions of dollars of research funding are spent on these animal models of heart failure every year, but insights gleaned from animal-based research efforts have shown poor translation in terms of deciphering human heart failure and developing effective therapies. Examples of translational failure are plentiful within this field. Failed drugs such as endothelin antagonists, cytokine inhibitors, and vasopeptide blockers illustrate how developing novel therapies for heart disease has been a difficult task to achieve [5], and heart failure with preserved left ventricular ejection fraction presents a considerable problem for which there is no proven therapy that can improve clinical outcome [6]. While the reasons for such translational failure can be multifactorial, lack of concordance between animal models and human disease state has been acknowledged as a major contributing factor. Over the years, various researchers across the field have tried to address this translational discrepancy. For example, in 2013, the former director of the National Institutes of Health (NIH), Dr. Elias Zerhouni, emphasized the need to expand research in humans in order to decipher human disease biology and facilitate better therapies. In 2014, the question of how to make the human a better experimental model for heart, lung, blood, and sleep disorders was at the center of discussions at a ‘Strategic Visioning Retreat’ held by the NIH’s National Heart, Lung and Blood Institute. Following decades-long research with limited translational success which can be equally attributed to the use of erroneous animal models and the lack of widespread use of human-based research methodologies here we discuss why and how we must use the human subject as the quintessential animal model for 21st century heart failure research.
Heart failure research: the inadequacy of conventional models
The most prevalent animal models of heart failure may be broadly categorized as either small animal models (primarily mouse, rat, rabbit, and cat models) or large animal models (primarily dog, pig, cow, and nonhuman primate models). Several techniques are widely used to create heart failure in these animals: rapid cardiac pacing, coronary artery embolization, coronary artery ligation, transverse aortic constriction, volume overload, pressure overload, mitral valve avulsion, transmyocardial direct current shock, toxic agents, dysrhythmias, and hypertension [4]. In addition, various genetic manipulation techniques (e.g., knock-out, knock-in, and transgenesis) have been used to generate human heart failure-like conditions in model organisms such as rodents and fruit flies. However, developing animal models that can sufficiently replicate key features of human heart failure has been challenging, given that human heart failure is a complex multifactorial disease significantly influenced by genetic, environmental, and lifestyle factors.
Our analysis of the literature indicates that the lack of translational success for heart failure animal research is primarily attributable to immutable species differences in genetic determinants, cardiovascular physiology and pathophysiology, and the inability to accurately replicate human heart failure causes, natural history, manifestations, comorbidities, complications, and responses to treatments in animals. Given the scope of this manuscript, it is not possible to provide herein a comprehensive analysis of animal models of heart failure. Rather, we provide representative examples for an overview of the common animal models and their primary limitations.
Heart failure models: from fruit flies to nonhuman primates
For more than a century, scientists have been creating models of heart failure in animals ranging from fruit flies to nonhuman primates. For example, Drosophila melanogaster, Danio renio, and Caenorhabditis elegans are widely used to study inherited forms of cardiomyopathies due to the ease of genetic manipulation with these species. While disease models involving species like Drosophila may have provided insight into genetic and molecular pathways, with a single-chamber heart (linear tube of a single layer of cardiomyocytes) lacking coronary arteries [3], these models are unable to approximate any of the major causes or consequences of human heart failure. Accurate extrapolations cannot be made from primitive heart-like structures present in zebrafish that differ developmentally and physiologically from all mammalian hearts. Similarly, mechanical and electrical dysfunction associated with human cardiomyopathies cannot be recreated in the striated muscle present in the body wall of C. elegans, a nematode lacking a heart and circulatory system.
Research from the most popular model organism for heart failure, the mouse, has been equally disappointing. Although they are mammalian 4-chambered hearts, small murine hearts that beat rapidly (300-600 beats per min) differ significantly from human hearts and therefore cannot accurately replicate human cardiac physiology under normal or pathological conditions. From sarcomeric gene expression to excitation-contraction coupling, and from natural history to drug responses, mouse hearts differ to the extent that even with thousands of cardiac gene modification studies, very few results have made it from murine models to the human clinic. For example, genes known to cause heart failure in the mouse may not cause the same disease phenotype in humans [7] or cause irrelevant undesired effects [8,9], and drugs shown to have cardio-therapeutic effects in mice [10] can give rise to fatal consequences in humans [11]. Furthermore, cardiovascular data from mice often do not correlate with data from large animal models [12], and important species differences exist even between more closely related species, such as mice and rats [13], and even among different strains of the same species [14]. All of these effects are confounded by factors such as sex and age [15,16] as well as laboratory conditions [17] and even animal husbandry practices [18].
While most researchers acknowledge the limitations of small animal models, large animal models of heart failure are often thought to be more applicable. However, the relevance of such experiments to human health, especially regarding translation of research results to advances for the prevention and treatment of human heart failure, appears to be quite minimal. For example, one of the most commonly used pacing-induced canine heart failure models produces cardiac dysfunction that is etiologically and temporally unrelated to human heart failure [19]. The differences between the etiology, natural history, mechanisms, and complications of heart failure in humans and experimentally induced heart failure in large animal models complicate translation of results from animals to humans. Balke and Shorofsky stated the complexity of heart failure animal modelling succinctly: “The characteristics of cardiac hypertrophy and heart failure are model-dependent. Even in the same species, the experimental results are profoundly influenced by the method used to create hypertrophy and/or heart failure (e.g., volume-overload, pressure-overload, genetically altered, rapidly paced, coronary artery ligation, etc.)” [19].
In addition, much of this animal research is outdated or irrelevant, because there is an abundance of information from similar studies in humans, which contradict the findings from animal studies in important ways. For example, while data from a rapid-pacing canine model showed that muscle metaboreflex restores blood flow to contracting muscle during exercise, similar studies in humans showed that the pressor response to ischemic exercise does not improve blood flow to contracting muscle [20]. Furthermore, since a wealth of data is available through human research efforts, the less-relevant animal research is at best redundant to the human studies. The reality is that no animal model can replicate the clinical pathophysiology of human heart failure due to complex, species-specific genetic, biochemical, physiological and environmental factors. Even nonhuman primates including our closest relative, the chimpanzee, exhibit differential heart disease in terms of etiology, pathogenesis and manifestations [21].
Heart failure models: rising concerns
Limitations of the conventional research paradigm are clearly reflected by the limited number of areas of relative success in heart failure treatment, such as angiotensin converting enzyme inhibitors, β-blockers, aldosterone antagonists, coronary revascularization, heart transplantation, and mechanical devices. While it is true that description of remodelling after myocardial infarction, for example, was first deciphered from experimental work in rodents by Pfeffer and Braunwald [22] (showing that there was a dynamic process post-myocardial infarction that was amenable to being altered in rodents) and was subsequently confirmed in human patients [23] such studies are the exception, not the norm. In reality, the vast majority of basic science experiments have not provided valuable insight into the limited success in heart failure treatment seen to date. Such limited translatability of animal data is well-described by the noted cardiovascular researcher Igor Efimov, Ph.D. Dr. Efimov states in an NIH-funded grant proposal that there is translational failure despite extensive knowledge acquired over decades of research conducted at enormous expense: the complexity of human cardiac physiology cannot be accurately modelled in animals due to “significant genetic, molecular, cellular, anatomical, and systemic differences among species,” and Dr. Efimov proposes ways to study cardiac physiology in our own species. Therefore, it is time to re-evaluate whether in the second decade of the 21st century there is benefit to focusing primarily on animals to develop the substrate for subsequent studies or to understand the underlying physiology that may identify important principles that would lead to successful clinical interventions for heart failure.
Heart failure is one of the most challenging chronic diseases in humans. Treatments are limited and the prognosis remains poor and not much improved overall. In 2002, this “impossible task of developing a new treatment for heart failure” [5] was discussed by the former president of the Heart Failure Society of America, Milton Packer, M.D. Referring to a number of large-scale clinical trials involving endothelin, cytokine, and angiotensin II receptor antagonists, Packer discussed how clearly defined and scientifically valid benefits of new drugs which produced marked improvements in experimental heart failure models did not lead to favourable outcomes in humans, and even appeared to have been harmful in some cases [5]. Just as Packer asked in 2002, the question to ask again is “given the almost insurmountable challenges that we face in developing a new treatment for heart failure, what should we do?” [5]. The study of cardiovascular diseases in humans using in vitro, ex vivo, in vivo, and in silico methods more readily transferable to humans must be the primary approach to improve understanding, prevention, and treatment of heart failure.
Heart failure research: incorporating the human subject
There are many examples of how genetic, biochemical, physiological, anatomical, and technical limitations inherent in animal models can be overcome with similar studies in humans. Since it is not possible to provide herein a comprehensive list of such strategies, the following representative examples serve to illustrate how - from nucleic acid level to organism level to human populations - increased utilization of existing human-based methods and the development of novel human-based methods can dramatically improve translatability of heart failure research. As shown in Figure 1, an integrative human-based research framework incorporating every level of human biological complexity can serve as the primary platform from which basic science discoveries are launched and more effective treatment strategies are developed and tested.
Figure 1.
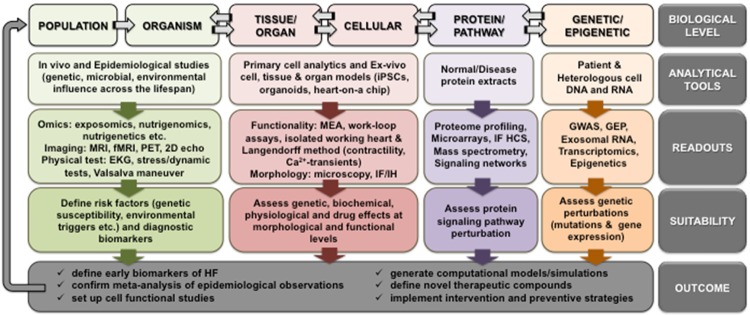
An integrative human-based framework for heart failure research. This is an overview of the human-based biological tools (and their associated readouts, suitability and outcomes) available to study human heart failure at every level of biological complexity from nucleic acid level to population level. GWAS, genome-wide association studies; GEP, gene expression profiling; IF HCS, immunofluorescence-high content screening; MS, mass spectrometry; MRS, magnetic resonance spectroscopy; MEA, microelectrode array; iPSCs, induced pluripotent stem cells; IF/IH, immunofluorescence/immunohistochemistry; MRI, magnetic resonance imaging; fMRI, functional magnetic resonance imaging; PET, positron emission tomography; EKG, electrocardiogram.
The human subject: genetic, epigenetic, protein and pathway levels
In spite of recent advances in genomics technologies, our knowledge of all genes and variants contributing to the genetic susceptibility of human heart failure is incomplete. Therefore, the elucidation of human transcriptional regulation, functional genetic variants (e.g., expression quantitative loci), epigenetic regulation, signal transduction, and a plethora of other genetic and biochemical information using human-based low- and high-throughput analyses is highly desirable. For example, blood transcriptome signature and putative marker genes can be identified in human white blood cell (and in other cell types) gene expression profiling with genomic microarrays [24]. Researchers can readily capture exosomal RNA, including miRNA, mRNA, and long non-coding RNA from human plasma, cerebrospinal fluid, and cell culture supernatants. When combined with deep sequencing-based transcriptome profiling, it is possible to ascertain important information regarding the pathogenesis of human heart failure (for example, in advanced heart failure and remodelling after left ventricular assist device support) [25]. New insights into cardiac transcriptome complexity and involvement of novel components of the transcriptome (at individual exon and transcript level) must be identified in the normal and failing human myocardium, not in the murine myocardium [26]. Novel candidate genes or mechanisms of drug action can be identified with human tissue samples rather than with tissue from animal models like the canine microembolic heart failure model [27] that do not accurately replicate the etiopathogenesis of human heart failure. The generation of anatomically precise, genome-wide transcript maps of the human heart - as has been done for the human brain [28] - will help correlate genomic data to cardiac function and eliminate the need for a priori hypothesis generation in the cardiac transcriptomics arena. Elaborate networks of gene regulation must be characterized in human heart tissue to avoid species-specific differences.
Another form of gene regulation that has recently emerged as a key player in cardiovascular diseases is epigenetics. Epigenetic studies in humans have revealed a significant number of modifications affecting cardiovascular disease development, progression, pathophysiology, risk factors, and therapeutics [29]. As researchers probe this area in search of environmental and lifestyle factors modulating cardiac pathophysiology, it is important to conduct these studies using human subjects and samples since differential epigenomic regulation is associated with genetic changes, transcriptional divergence, and disease genes even in our closest relative, the chimpanzee [30].
The next level is to capture the functional information on the genome that resides in the proteome. The complex pathophysiology of human heart failure can be unravelled at the protein level by incorporating proteomics strategies such as protein microarrays, multidimensional liquid chromatography, tandem mass spectrometry, immunofluorescence high-content screening, multiple reaction monitoring (ng/ml or sub-ng/ml high-sensitivity detection of proteins in serum or plasma), and magnetic resonance spectroscopy to elucidate human proteome-profiling, comparative expression analysis, post-translational modifications, protein-protein interactions, and signaling pathway aberrations, among others [31]. The study of signaling pathways using human samples is critical, as there are numerous species differences in many cardiac signaling networks such as Ca2+ signaling, ion channels involved in excitation-contraction coupling, and protein kinase signaling, all of which are subject to an array of interactions with other proteins and further levels of regulation [32]. The integration of information derived from human-based molecular methods will undoubtedly aid in teasing apart the contributions of the human genome, epigenome, proteome, and signaling networks to the etiology, pathogenesis, and treatment of human cardiovascular diseases such as heart failure.
The human subject: cellular, tissue, and organ levels
The underlying molecular mechanisms for major cardiovascular diseases such as coronary artery disease, myocardial infarction, hypertrophy, and heart failure are still largely unknown. In discovery and preclinical sciences, a plethora of genetic, biochemical, and physiological information can be delineated from human cardiac cells under normal and disease conditions. Freshly isolated primary adult human cardiac myocytes can be used for many acute assays (e.g., contractility and Ca2+ transient measurements) [33] and can easily be cultured for more than a week for long-term assays (e.g., gene silencing) [34]. Even autopsy-derived tissue (up to 24 hours of autolysis) can be a practical source, as this can be utilized for biologically relevant gene expression experiments [35]. Heterologous myocyte cell lines are not currently available to researchers (only rodent-derived cell lines like HL-1 are available); however, as has been shown for human pancreatic β-cells [36], robust novel technology involving targeted oncogenesis in human fetal tissue should be adopted for making human cardiac cell lines using cardiac-specific promoters. Meanwhile, patient and disease-specific human induced pluripotent stem cell-derived cardiomyocytes are proving to be invaluable for disease modelling, high throughput drug discovery, preclinical cardiotoxicity assessment, and personalized medicine [37]. However, it is critical to utilize human-derived stem cells rather than murine stem cells since fundamental species differences in molecular signatures, cellular signalling mechanisms, colony shape, growth rate, surface markers, developmental potential, and other factors will hamper data extrapolation from mouse stem cells to humans [38]. With the aid of easy-to-use co-culture (2D or 3D) systems like the Quasi-Vivo system [39], researchers can now study complex cardiac biochemistry and physiology in a more advanced, physiologically relevant setting.
Novel in vitro techniques such as the work loop technique (utilizing cardiac tissue samples to assess force-length work loop contractions mimicking those of the pressure-volume work loops seen in the heart in vivo) provide a rapid and easy platform to assess detailed biomechanics of drug-induced cardiac dysfunction [40]. Although the original work was done with murine myocardium, it has already been tested with human cardiac tissue and will be published by the same research group in the near future (personal communication). Another versatile tool for basic and preclinical research is functional engineered human cardiac tissue: with > 90% troponin-positive cells expressing many cardiac-specific genes and functionally intact electrophysiology with reconstituted Frank-Starling mechanism, these tissues amenable to gene silencing can serve as a powerful model for discovery research and preclinical toxicology [41]. Biochemical, physiological, and drug responses can also be evaluated in human hearts ex-vivo by isolated working heart and/or the Langendorff method [42,43], especially before proceeding to human clinical trials. Increased reliance on the use of such human-based preclinical testing methods rather than poorly predictive animal models will greatly enhance the predictive validity for humans since sequence homology or functional equivalence does not guarantee pharmacological equivalence among different species. Microfluidic technologies such as heart-on-a-chip [44] (and multiple-organs-on-a chip) are available to researchers in some instances, and in the near to intermediate future, it will be feasible to evaluate integrated multi-organ drug responses in a human-relevant manner.
The human subject: organism and population levels and beyond
For decades, human subjects have been used to study various aspects of cardiac function in vivo. In the modern clinic, sophisticated techniques are used for anatomical imaging, functional imaging, perfusion, and delayed enhancement to assess cardiac morphology and function. Techniques such as echocardiography in all its variations and applications still serve as the fundamental tool for heart failure diagnosis. With the advent of non-invasive techniques such as magnetic resonance spectroscopy (with 31P, 1H 13C, and 23Na probes), researchers can gain detailed insight into relationships among cardiac structure, function, metabolism, and perfusion in human subjects under normal and pathological conditions as well as during therapeutic interventions [45]. However, since the use of magnetic resonance spectroscopy is currently primarily limited to scientific investigation due to low temporal and spatial resolution and low reproducibility [45], isolated perfused human hearts- instead of animal hearts [46] - should be employed for the development, validation, and translation of novel magnetic resonance techniques. On the other hand, magnetic resonance imaging (MRI) and computed tomography (CT) are commonly used to characterize human cardiac structure and performance, while myocardial perfusion imaging methods allow monitoring of disease extent and progression as well as responses to lifestyle modifications and therapeutic interventions [47]. The ability to characterize myocardial fiber architecture ex vivo and in vivo using diffusion tensor MRI tractography continues to provide new insights into a range of conditions affecting human cardiovascular health [48]. Advanced myocardial imaging techniques will be a rapid, powerful and relevant way to translate bench science into bedside therapeutics for human patients.
In addition to individual in vivo studies, epidemiological studies will continue to form a cornerstone of modern cardiovascular research since we can gain valuable hypothesis-generating information regarding the contributions of genetic, epigenetic, gut microbiome, geographical, ethnic, and lifestyle factors to the development, etiopathogenesis, and treatment of human heart failure [49,50]. In particular, as emerging evidence indicates that epigenetic regulation and the gut microbiome play an important role in the pathogenesis of heart failure [51,52], it is important to conduct these studies using human subjects and samples due to species specificity of these biological processes. Since it is well known that heart failure has a strong lifestyle component, it is equally important to dedicate more time and resources to conduct research on preventive strategies in human populations.
Identification of reliable biomarkers to diagnose and phenotype heart failure using human-based methods and integration of human data through a systems biology approach will greatly enhance the translational potential [53]. The diverse pathophysiology of human heart failure at the systems level can be deduced by integrating information from every level of biological complexity (Figure 1). The information fostered by mapping the human genome can be assessed through different sources to define various aspects of heart failure pathology. For example, environmental risk factors that contribute to disease etiology can be determined by examining the human exposome (which encompasses the totality of human environmental exposures from conception onward) through blood specimens [54]. Novel biomarkers can be identified using metabolomics (lipids, amino acid, hormones and other metabolites) in plasma samples from human patients [55], and nutrigenomics can identify how dietary components may modulate risk conferred by genetic susceptibility [56], while metabonomics serve as a platform to study drug toxicity and gene function. In this era of ‘big data’, as we enter the dawn of personalized medicine (feasible with human data from a multitude of sources such as omics technologies, single cell analytics, nanofluidics, imaging, etc.) [57], it is important to implement robust systems biology platforms to promote basic and clinical research integration. In addition to basic science applications, these experiments offer clear potential for improved diagnosis and treatment when performed in the clinical setting, especially in human patients at risk for or in the early phase of heart failure.
Heart failure research: an integrative human framework
Heart failure remains a leading cause of death and hospitalization worldwide. Despite decades of research, treatment options remain limited. In the two decades leading up to 2002, nearly 1,000 new drugs and devices had been developed in preclinical studies to treat heart failure, but only nine received regulatory approval and are currently used in the clinical arena [5]. Today, more than a decade later, the scientific community is not much further along. Given the extensive history of translational failure, the time has come for researchers to acknowledge and accept that no amount of molecular or physiological tinkering can lead to accurate recapitulation of human heart failure and drug responses in model organisms, even in the presence of conserved genomes and molecular pathways and adherence to rigorous research methods.
It is time to objectively evaluate the scientific evidence and realize that the development of preventive methods and definitive therapies has been hindered by animal-based preclinical studies that are not fit for purpose. We should transition from the quest to generate ‘better animal models’ to the prioritized use of human-based methods in order to advance preventive and reparative approaches for human heart failure. This requires investment in both clinical research and fundamental discovery studies using human-based methods. It requires improved teamwork from both basic science and clinical investigators, and a better integration from clinical observation to basic discovery and back to clinical validations. With the establishment of new collaborative interactions and initiatives among academic, industry, and government agencies, it will only be a matter of time until the human subject becomes the quintessential animal model for heart failure research.
Fortunately, current efforts consistent with these goals are advancing at the National Institutes of Health and academic centers, with aims to develop and validate better translational capacities for human diseases [58]. Researchers must rebalance review systems for grants and resource distribution at national and institutional levels by encouraging clinical translation and basic/clinical integration. It is imperative for our review systems to prioritize our limited resources toward solving difficult and longstanding patient dilemmas, and heart failure is just such a dilemma. For example, with the advent and the widespread use of gene editing technologies like CRISPR/Cas9, researchers must stop and ask again whether transgenic mice are the ‘alkahest’ to understanding heart failure [59]. Instead of funding the generation of triple-knockout mice purely based on observations in rodents, funding agencies should reward forward-looking basic science that taps into the bounty of information available from humans, including naturally occurring ‘human knockouts’ using human-relevant methods [60].
It is imperative that emphasis be placed on the development of novel ‘next generation’ technologies for use with the human subject. It is through such efforts that we now have technologies like organ-on-a-chip at our disposal. In the meantime, it is equally critical to widely incorporate existing human-based methods that are currently vastly underutilized. For example, explanted human hearts not suitable for transplantation should be used for experimentation, and all surgical remnants from consenting patients should be used freshly or archived frozen for research use. In this regard, research institutions affiliated with hospitals should not have difficulty facilitating such collaborations - it is an enormous waste that the vast majority of human surgical remains are sent to the incinerator when they can be utilized for research purposes. Instead of resorting to animal tissue because it is sometimes difficult to obtain human heart samples, researchers must actively seek to form collaborative partnerships with hospitals and the NIH to share resources, in particular through the establishment of biobanks. In addition, NIH and other suitable institutions should establish and expand human cell and tissue banks to facilitate cost-effective distribution of human samples. For hypothesis-driven discovery science grants, it should be mandatory to demonstrate public health relevance up front with human-based preliminary data (whether cells, tissues, in vivo, or in silico studies). Positive results obtained from such human models should form the fundamental basis for these investigations, rather than exaggerated claims of future translational potential derived from unreliable animal models. Just as current NIH director Francis Collins acknowledged in 2011 [57], the time is indeed right - as it has been for years - for reengineering translational science.
Researchers in the field are emphasizing that in the second decade of the 21st century, the scientific community must reflect on how it is paradoxical that we know so much more about mouse, rat, rabbit, and canine cardiac physiology than our own species, and that assuming a clinically robust flawless translation between species will only hinder our research progress and delay the transition into the clinic [38]. If the ultimate goal is to tackle the epidemic status of chronic debilitating diseases such as heart failure, future research success should be measured not by the number of publications or citations, nor by journal impact factors, but by important advances in human medicine. Following the footsteps of other exemplary researchers in the field, we must “modify and enhance the currently dominant translational paradigm and provide new important directions of research, which will stimulate and reinvigorate a biomedical research community that has ignored human physiology and thus delayed effective translation of needed therapies for heart failure”. The best way to achieve that goal is through prioritizing human-based heart failure research.
Disclosure of conflict of interest
None.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER 3rd, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics--2014 update: a report from the American Heart Association. Circulation. 2015;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wexler RK, Elton T, Pleister A, Feldman D. Cardiomyopathy: an overview. Am Fam Physician. 2009;79:778–784. [PMC free article] [PubMed] [Google Scholar]
- 3.Piazza N, Wessells RJ. Drosophila models of cardiac disease. Prog Mol Biol Transl Sci. 2011;100:155–210. doi: 10.1016/B978-0-12-384878-9.00005-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Houser SR, Margulies KB, Murphy AM, Spinale FG, Francis GS, Prabhu SD, Rockman HA, Kass DA, Molkentin JD, Sussman MA, Koch WJ. Animal models of heart failure: a scientific statement from the American Heart Association. Circ Res. 2012;111:131–150. doi: 10.1161/RES.0b013e3182582523. [DOI] [PubMed] [Google Scholar]
- 5.Packer M. The impossible task of developing a new treatment for heart failure. J Card Fail. 2002;8:193–196. doi: 10.1054/jcaf.2002.128001. [DOI] [PubMed] [Google Scholar]
- 6.Butler J, Fonarow GC, Zile MR, Lam CS, Roessig L, Schelbert EB, Shah SJ, Ahmed A, Bonow RO, Cleland JG, Cody RJ, Chioncel O, Collisn SP, Dunnmon P, Filippatos G, Lefkowitz MP, Marti CN, McMurray JJ, Misselwitz F, Nodari S, O’Connor C, Pfeffer MA, Pieske B, Pitt B, Rosano G, Sabbah HN, Senni M, Solomon SD, Stockbridge N, Teerlink JR, Georgiopoulos VV, Gehorghiade M. Developing therapies for heart failure with preserved ejection fraction: current state and future directions. JACC Heart Failure. 2014;2:97–112. doi: 10.1016/j.jchf.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mayosi BM, Kardos A, Davies CH, Gumedze F, Hovnanian A, Burge S, Watkins H. Heterozygous disruption of SERCA2a is not associated with impairment of cardiac performance in humans: implications for SERCA2a as a therapeutic target in heart failure. Heart. 2006;92:105–109. doi: 10.1136/hrt.2004.051037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang WY, Aramburu J, Douglas PS, Izumo S. Transgenic expression of green fluorescence protein can cause dilated cardiomyopathy. Nat Med. 2000;6:482–483. doi: 10.1038/74914. [DOI] [PubMed] [Google Scholar]
- 9.Buerger A, Rozhitskaya O, Sherwood MC, Dorfman AL, Bisping E, Abel ED, Pu WT, Izumo S, Jay PY. Dilated cardiomyopathy resulting from high level myocardial expression of Cre recombinase. J Card Fail. 2006;12:392–398. doi: 10.1016/j.cardfail.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 10.LaPointe MC, Mendez M, Leung A, Tao Z, Yang XP. Inhibition of cyclooxygenase-2 improves cardiac function after myocardial infarction in the mouse. Am J Physiol Heart Circ Physiol. 2004;286:H1416–1424. doi: 10.1152/ajpheart.00136.2003. [DOI] [PubMed] [Google Scholar]
- 11.Harirforoosh S, Asghar W, Jamali F. Adverse effects of nonsteroidal anti-inflammatory drugs: an update of gastrointestinal, cardiovascular and renal complications. J Pharm Pharm Sci. 2013;16:821–847. doi: 10.18433/j3vw2f. [DOI] [PubMed] [Google Scholar]
- 12.Kaiser RA, Lyons JM, Duffy JY, Wagner CJ, Mclean KM, O’neill TP, Pearl JM, Molkentin JD. Inhibition of p38 reduces myocardial infarction injury in the mouse but not pig after ischemiareperfusion. Am J Physiol Heart Circ Physiol. 2005;289:H2747–H2751. doi: 10.1152/ajpheart.01280.2004. [DOI] [PubMed] [Google Scholar]
- 13.Hintz KK, Norby FL, Duan J, Cinnamon MA, Doze VA, Ren J. Comparison of cardiac excitation-contraction coupling in isolated ventricular myocytes between rat and mouse. Comp Biochem Physiol A Mol Integr Physiol. 2002;133:191–198. doi: 10.1016/s1095-6433(02)00177-0. [DOI] [PubMed] [Google Scholar]
- 14.van den Borne SW, van de Schans VA, Strzelecka AE, Vervoort-Peters HT, Lijnen PM, Cleutjens JP, Smits JF, Daemen MJ, Janssen BJ, Blankesteijn WM. Mouse strain determines the outcome of wound healing after myocardial infarction. Cardiovascular Research. 2009;84:273–282. doi: 10.1093/cvr/cvp207. [DOI] [PubMed] [Google Scholar]
- 15.Leinwand LA. Sex is a potent modifier of the cardiovascular system. J Clin Invest. 2003;112:302–307. doi: 10.1172/JCI19429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rice KM, Fannin JC, Gillette C, Blough ER. Efficacy of female rat models in translational cardiovascular aging research. J Aging Res. 2014;2014:1–14. doi: 10.1155/2014/153127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin B, Ji S, Maudsley S, Mattson MP. Control laboratory rodents are metabolically morbid: why it matters. Proc Natl Acad Sci U S A. 2010;107:6127–6133. doi: 10.1073/pnas.0912955107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duke JL, Zammit TG, Lawson DM. The effects of routine cage-changing on cardiovascular and behavioral parameters in male Sprague-Dawley rats. Contemp Top Lab Anim Sci, 2001;40:17–20. [PubMed] [Google Scholar]
- 19.Balke CW, Shorofsky SR. Alterations in calcium handling in cardiac hypertrophy and heart failure. Cardiovasc Res. 1998;37:290–299. doi: 10.1016/s0008-6363(97)00272-1. [DOI] [PubMed] [Google Scholar]
- 20.O’Leary DS, Joyner MJ. Point-Counterpoint: The muscle metaboreflex does/does not restore blood flow to contracting muscles. J Appl Physiol. 2006;100:357–361. [PubMed] [Google Scholar]
- 21.Varki N, Anderson D, Herndon JG, Pham T, Gregg CJ, Cheriyan M, Murphy J, Strobert E, Fritz J, Else JG, Varki A. Heart disease is common in humans and chimpanzees, but is caused by different pathological processes. Evol Appl. 2009;2:101–112. doi: 10.1111/j.1752-4571.2008.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81:1161–1172. doi: 10.1161/01.cir.81.4.1161. [DOI] [PubMed] [Google Scholar]
- 23.Konstam MA, Kramer DG, Patel AR, Maron MS, Udelson JE. Left Ventricular Remodeling in heart failure: current concepts in clinical significance and assessment. J Am Coll Cardiol Img. 2011;4:98–108. doi: 10.1016/j.jcmg.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 24.Smih F, Desmoulin F, Berry M, Turkieh A, Harmancey R, Iacovoni J, Trouillet C, Delmas C, Pathak A, Lairez O, Koukoui F, Massabuau P, Ferrieres J, Galinier M, Rouet P. Blood signature of pre-heart failure: A microarrays study. PLoS One. 2011;6:e20414. doi: 10.1371/journal.pone.0020414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang KC, Yamada KA, Patel AY, Topkara V, George I, Cheema FH, Ewald GA, Mann DL, Nerbonne JM. Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNA in failing human heart and remodeling with mechanical circulatory support. Circulation. 2014;129:1009–1021. doi: 10.1161/CIRCULATIONAHA.113.003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee JH, Gao C, Peng G, Greer C, Ren S, Wang Y, Xiao X. Analysis of transcriptome complexity via RNA sequencing in normal and failing murine hearts. Circ Res. 2011;109:1332–1341. doi: 10.1161/CIRCRESAHA.111.249433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lanfear DE, Yan JJ, Mishra S, Sabbah HN. Genome-wide approach to identify novel candidate genes for beta blocker response in heart failure using an experimental model. Discov Med. 2011;11:369–366. [PMC free article] [PubMed] [Google Scholar]
- 28.Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, van de Lagemaat LN, Smith KA, Ebbert A, Riley ZL, Abajian C, Beckmann CF, Bernard A, Bertagnolli D, Boe AF, Cartagena PM, Chakravarty MM, Chapin M, Chong J, Dalley RA, Daly BD, Dang C, Datta S, Dee N, Dolbeare TA, Faber V, Feng D, Fowler DR, Goldy J, Gregor BW, Haradon Z, Haynor DR, Hohmann JG, Horvath S, Howard RE, Jeromin A, JOchim JM, Kinnunen M, Lau C, Lazarz ET, Lee C, Lemon TA, Li L, Li Y, Morris JA, Overly CC, Parker PD, Parry SE, Reding M, Royall JJ, Schulkin J, Sequeira PA, Slaughterbeck CR, Smith SC, Sodt AJ, Sunkin SM, Swanson BE, Vawter MP, Williams D, Wohnoutka P, Zielke HR, Geschwind DH, Hof PR, Smith SM, Koch C, Grant SG, Jones AR. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489:391–399. doi: 10.1038/nature11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abi Khalil C. The emerging role of epigenetics in cardiovascular disease. Ther Adv Chronic Dis. 2014;5:178–187. doi: 10.1177/2040622314529325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukuda K, Ichiyanagi K, Yamada Y, Go Y, Udono T, Wada S, Maeda T, Soejima H, Saitou N, Ito T, Sasaki H. Regional DNA methylation differences between humans and chimpanzees are associated with genetic changes, transcriptional divergence and disease genes. J Hum Genet. 2013;58:446–454. doi: 10.1038/jhg.2013.55. [DOI] [PubMed] [Google Scholar]
- 31.Gregorich ZR, Chang YH, Ge Y. Proteomics in heart failure: top-down or bottom-up? Pflugers Arch. 2014;466:1199–1209. doi: 10.1007/s00424-014-1471-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fearnley CJ, Roderick HL, Bootman MD. Calcium signaling in cardiac myocytes. Cold Spring Harb Perspect Biol. 2011;3:a004242. doi: 10.1101/cshperspect.a004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piacentino V, Weber CR, Chen X, Weisser-Thomas J, Margulies KB, Bers DM, Houser SR. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res. 2003;92:651–658. doi: 10.1161/01.RES.0000062469.83985.9B. [DOI] [PubMed] [Google Scholar]
- 34.Hsieh YP, Huang CH, Lee CY, Lin CY, Chang CC. Silencing of hepcidin enforces the apoptosis in iron-induced human cardiomyocytes. J Occup Med Toxicol. 2014;9:11–18. doi: 10.1186/1745-6673-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gupta S, Halushka MK, Hilton GM, Arking DE. Postmortem cardiac tissue maintains gene expression profile even after late harvesting. BMC Genomics. 2012;13:26–37. doi: 10.1186/1471-2164-13-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ravassard P, Hazhouz Y, Pechberty S, Bricout-Neveu E, Armanet M, Czernichow P, Scharfmann R. A genetically engineered human pancreatic beta cell line exhibiting glucose-induced insulin secretion. J Clin Invest. 2011;121:3589–3597. doi: 10.1172/JCI58447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chow M, Boheler KR, Li RA. Human pluripotent stem cell-derived cardiomyocytes for heart regeneration, drug discovery and disease modeling: from the genetic, epigenetic, and tissue modeling perspectives. Stem Cell Res Ther. 2013;14:97–109. doi: 10.1186/scrt308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schnerch A, Cerdan C, Bhatia M. Distinguishing between mouse and human pluripotent stem cell regulation: the best laid plans of mice and men. Stem Cells. 2010;28:419–430. doi: 10.1002/stem.298. [DOI] [PubMed] [Google Scholar]
- 39.Sbrana T, Ahluwalia A. Engineering Quasi-Vivo in vitro organ models. Adv Exp Med Biol. 2012;745:138–153. doi: 10.1007/978-1-4614-3055-1_9. [DOI] [PubMed] [Google Scholar]
- 40.Gharanei M, Hussain A, James RS, Janneh O, Maddock H. Investigation into the cardiotoxiceffects of doxorubicin on contractile function and the protection afforded by cyclosporin A using the work-loop assay. Toxicol In Vitro. 2014;28:722–731. doi: 10.1016/j.tiv.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 41.Turnbull IC, Karakikes I, Serrao GW, Backeris P, Lee JJ, Xie C, Senyei G, Gorden RE, Li RA, Akar FG, Hajjar RJ, Hulot JS, Costa KD. Advancing functional engineered cardiac tissues toward a preclinical model of human myocardium. FASEB J. 2014;28:644–654. doi: 10.1096/fj.13-228007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Federov VV, Glukhov AV, Ambrosi CM, Kostecki G, Chang R, Janks D, Schuessler RB, Moazami N, Nichols CG, Efimov IR. Effects of KATP channel openers diazoxide and pinacidil in coronary-perfused atria and ventricles from failing and non-failing human hearts. J Mol Cell Cardiol. 2011;51:215–225. doi: 10.1016/j.yjmcc.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holzem KM, Gomez JF, Glukhov AV, Madden EJ, Koppel AC, Ewald GA, Trenor B, Efimov IR. Reduced response to IKr blockade and altered hERG1a/1b stoichiometry in human heart failure. J Mol Cell Cardiol. 2015 doi: 10.1016/j.yjmcc.2015.06.008. pii: S0022-2828(15)00194-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Agarwal A, Goss JA, Cho A, McCain ML, Parker KK. Microfluidic heart on a chip for higher throughput pharmacological studies. Lab Chip. 2013;13:3599–3608. doi: 10.1039/c3lc50350j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hudsmith LE, Neubauer S. Magnetic resonance spectroscopy in myocardial disease. JACC Cardiovasc Imaging. 2009;2:87–96. doi: 10.1016/j.jcmg.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 46.Schuster A, Grünwald I, Chiribiri A, Southworth R, Ishida M, Hay G, Neumann N, Morton G, Perera D, Schaeffter T, Nagel E. An isolated perfused pig heart model for the development, validation and translation of novel cardiovascular magnetic resonance techniques. J Cardiovasc Magn Reson. 2010;12:53–61. doi: 10.1186/1532-429X-12-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferreira F, Kilner PJ, McGill L, Nielles-Vallespin S, Scott AD, Ho SY, McCarthy KP, Haba MM, Ismail TF, Gatehouse PD, de Silva R, Lyon AR, Prasad SK, Firmin DN, Pennell DJ. In vivo cardiovascular magnetic resonance diffusion tensor imaging shows evidence of abnormal myocardial laminar orientations and mobility in hypertrophic cardiomyopathy. J Cardiovasc Magn Reson. 2014;16:87. doi: 10.1186/s12968-014-0087-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mekkaoui C, Reese TG, Jackowski MP, Bhat H, Kostis WJ, Sosnovik DE. In vivo fiber tractography of the right and left ventricles using diffusion tensor MRI of the entire human heart. J Cardiovasc Magn Reson. 2014;16(Suppl 1):17. [Google Scholar]
- 49.Mahmood S, Levy D, Vasan RS, Wang TJ. The Framingham Heart Study and the epidemiology of cardiovascular disease: a historical perspective. Lancet. 2014;383:999–1008. doi: 10.1016/S0140-6736(13)61752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cox AJ, Azeem A, Yeboah J, Soliman EZ, Aggarwal SR, Bertoni AG, Car JJ, Freedman BI, Herrington DM, Bowden DW. Heart rate-corrected QT interval is an independent predictor of allcause and cardiovascular mortality in individuals with type 2 diabetes: the Diabetes Heart Study. Diabetes Care. 2014;37:1454–1461. doi: 10.2337/dc13-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cao DJ. Epigenetic regulation and heart failure. Expert Rev Cardiovasc Ther. 2014;12:1087–1098. doi: 10.1586/14779072.2014.942285. [DOI] [PubMed] [Google Scholar]
- 52.Loscalzo J. Lipid metabolism by gut microbes and atherosclerosis. Circ Res. 2011;109:127–129. doi: 10.1161/RES.0b013e3182290620. [DOI] [PubMed] [Google Scholar]
- 53.Hood L, Tian Q. Systems approaches to biology and disease enable translational systems medicine. Genomics Proteomics Bioinformatics. 2012;10:181–185. doi: 10.1016/j.gpb.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rappaport SM, Barupal DK, Wishart D, Vineis P, Scalbert A. The blood exposome and its role in discovering causes of disease. Environ Health Perspect. 2014;122:769–774. doi: 10.1289/ehp.1308015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Senn T, Hazen SL, Tang WH. Translating metabolomics to cardiovascular biomarkers. Prog Cardiovasc Dis. 2012;55:70–76. doi: 10.1016/j.pcad.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Corella D, Ordovas JM. Nutrigenomics in cardiovascular medicine. Circ Cardiovasc Genet. 2009;2:637–651. doi: 10.1161/CIRCGENETICS.109.891366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pah AR, Rasmussen-Torvik LJ, Goel S, Greenland P, Kho AN. Big data: what is it and what does it mean for cardiovascular research and prevention policy. Curr Cardiovasc Risk Rep. 2015;9:424. [Google Scholar]
- 58.Collins FS. Reengineering translational science: the time is right. Sci Transl Med. 2011;3:90cm17. doi: 10.1126/scitranslmed.3002747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cook SA, Clerk A, Sugden PH. Are transgenic mice the ‘alkahest’ to understanding myocardial hypertrophy and failure? J Mol Cell Cardiol. 2009;46:118–129. doi: 10.1016/j.yjmcc.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 60.Sakuma T, Nishikawa A, Kume S, Chavama K, Yamamotao T. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci Rep. 2014;4:5400. doi: 10.1038/srep05400. [DOI] [PMC free article] [PubMed] [Google Scholar]