

Abstract
Advances in the treatment of acute leukemia have resulted in significantly improved remission rates, although disease relapse poses a significant risk. By utilizing sensitive, non-invasive imaging guidance, detection of early leukemic infiltration and the extent of residual tumor burden after targeted therapy can be expedited, leading to more efficient treatment planning. We demonstrated marked survival benefit and therapeutic efficacy of a new-generation vascular disrupting agent, combretastatin-A1-diphosphate (OXi4503), using reporter gene-imaging technologies and mice systemically administered luc+ and GFP+ human leukemic cells (LCs). Before treatment, homing of double-transduced cells was serially monitored and whole-body cellular distributions were mapped using bioluminescence imaging (BLI). Imaging findings strongly correlated with quantitative GFP expression levels in solid organs/tissues, suggesting that the measured BLI signal provides a highly sensitive and reliable biomarker of tumor tissue burden in systemic leukemic models. Such optical technologies can thereby serve as robust non-invasive imaging tools for preclinical drug discovery and for rapidly screening promising therapeutic agents to establish potency, treatment efficacy and survival advantage. We further show that GFP+ HL-60 cells reside in close proximity to VE-cadherin- and CD31-expressing endothelial cells, suggesting that the perivascular niche may have a critical role in the maintenance and survival of LCs.
Keywords: HL-60, GFP, luciferase, OXi4503
INTRODUCTION
Chemotherapy induces remission in the majority of adult patients with acute myeloid leukemia, but is curative in only a small percentage of patients treated.1 Currently, the majority of adults with acute leukemias inevitably relapse, succumbing to this fatal disease.2,3 The capacity of leukemic cells (LCs) to circulate and potentially infiltrate the vascular beds of various organs is a differentiating feature of these solid tumors. Relapse of leukemias may be partially ascribed to the persistence of small populations of residual LCs residing within specialized perivascular niches. It is possible that LCs home to such specialized niches, which protect them from cytotoxic agents. The ability to sensitively and non-invasively monitor early time-dependent changes in homing patterns, tumor growth and the distribution/extent of residual tumor burden within solid organs after targeted therapy would offer insights into mechanisms underlying disease relapse, promote the development of new targeted therapies aimed at disrupting niches and reducing relapse rates, and expedite treatment planning and management.
LCs, like CD34+ hematopoietic stem cells, are thought to initially localize to the epiphyseal region of bones (osteoblastic niche), followed by expansion in the inner vascular and diaphyseal compartments.4 Certain subsets of residual chemotherapy-resistant LCs appear to adhere to blood vessel walls and possibly within the microvascular beds of other organs, leading to resistance to conventional chemotherapies.3,5,6 Previous studies have shown that LCs expressing integrin adhesion molecules, such as very late antigen-4 (VLA4)7 and beta-1 integrin fibronectin receptor,8 adhere to vascular endothelial cell adhesion molecule-1 (VCAM-1), a cognate receptor for VLA4, as well as the extracellular ligand fibronectin, respectively. Indeed, VLA4-expressing LCs are associated with much higher relapse than that seen with VLA4−LCs. Chemoresistance is attributed to the affinity of these VLA4 + LCs to attach to vascular endothelial cell adhesion molecule-1 + activated endothelial cells. Thus, this cell adhesion-mediated drug resistance has a major role in leukemic relapse and mortality, and has led to the use of vascular disrupting agents (VDAs) designed to directly target tumor vasculature and to promote cellular necrosis.9
One of the most commonly used and well-characterized VDAs is combretastatin-A4-phosphate (CA4P, Zybrestat).9,10 CA4P binds to tubulin, resulting in microtubule depolymerization, collapse of the endothelial cell cytoskeleton and obstruction of the tumor vasculature by enlarged endothelial cells.11 Recently, a new-generation, dual-mechanism VDA, combretastatin-A1-diphosphate (OXi4503, OXiGENE, Waltham, MA, USA), has been synthesized, which, like its structural analog CA4P, obstructs and obliterates tumor vasculature, resulting in extensive tumor necrosis.12,13 Preclinical studies have shown that OXi4503 has single-agent activity against a number of xenograft models,14–16 and demonstrates synergistic/additive effects when combined with chemotherapeutics17 or molecular-targeted therapies.18–20 OXi4503 is currently being evaluated as a monotherapy in clinical trials.
Most therapeutic efficacy evaluations with VDAs have utilized cellular and histological assays to assess acute vascular perfusion and/or tissue necrotic changes relative to controls.10,14–16,21–23 By contrast, more chronic assessments, typically at discrete time intervals of longer than 1 month,3,17,24 have monitored human LC engraftment24 or xenografts or chloromas in cohorts of treated mice. Relatively few studies have evaluated shorter-term post-treatment effects (that is, days-weeks)13,18,25 or employed systemic models more accurately reflecting human disease. Although several studies utilized real-time noninvasive imaging methods to examine acute vascular flow changes after administration of OXi450323 or other VDA,26–28 the impact of such changes on cell viability or other biological correlate was not explored. One such approach, bioluminescence imaging (BLI), has been used to sensitively monitor small populations of luc+ cells in vivo.29–31 To the best of our knowledge, the use of this noninvasive imaging tool to guide OXi4503 treatment of LC infiltrates and serially track their growth and regression over relatively short time intervals (that is, weeks) has not been previously reported in systemic models and confirmed with quantitative histological assays.
In the present study, we utilize reporter gene-imaging technology to serially monitor in vivo distributions of systemically administered luc+ and GFP+ HL-60 cells before and after administration of OXi4503 monotherapy over short-term intervals. Correlation of BLI data with quantitative GFP expression levels in reticuloendothelial organs and other tissues, as well as with co-labeling assays, has been performed.
MATERIALS AND METHODS
Animal xenograft and systemic models
Systemic HL-60 experiments were done in accordance with protocols approved by the Institutional Animal Care and Use Committee of Memorial Sloan-Kettering Cancer Center and followed National Institutes of Health guidelines for animal welfare. SCID/Beige mice (8–12 weeks old, Taconic Farms, Hudson, NY, USA) were irradiated (250 rad, 2.3 min) by a Gammacell 40 Exactor (Best Theratronics, Ottawa, ON, Canada)20,32 to enhance angiogenic potential. For systemic models, mice were inoculated with luc+/GFP+ HL-60 cells (1.0 × 107 cells in 200 μl saline vehicle) 5h post irradiation and serially monitored with BLI.
Xenograft models were also generated using non-transduced HL-60 (TB) cells (1.0 × 107) propagated in an in vivo ascites passage, and subsequently implanted subcutaneously in the right flank using a 23-gauge trocar needle. To maintain as homogeneous a population of tumor-bearing mice as possible, tumors within a given size range (78–156 mm3) were selected for treatment at 11 days post-inoculation (p.i.).
Treatment of leukemic xenografts
Ten million HL-60 cells, propagated in an in vivo passage, were implanted subcutaneously into the right flank of mice using a 23-gauge trocar needle to establish leukemic xenografts. A minimum of 10 animals was used in each treatment group in order to assess response. In an initial set of studies, three treatment groups were evaluated. CA4P (75 mg/kg) and OXi4503 (75 mg/kg) were administered once a week for 2 weeks, while Ara-C (20 mg/kg) was administered three times per day every 4 days. In a subsequent study, mice were assigned to one of four different treatment groups to evaluate the response of HL-60 tumors to a range of OXi4503 doses (2.5, 10, 25 and 100 mg/kg) administered once a week for 2 weeks. Control mice were administered 0.9% saline vehicle for both sets of experiments. Tumor sizes were measured using calipers over the treatment interval. Survival time was recorded as the number of days between tumor cell inoculation and euthanasia.
Serial BLI imaging in a systemic leukemic model
BLI acquisition and analysis were performed in irradiated SCID/Beige mice (n = 14) inoculated with luc+/GFP+. HL-60 cells using the IVIS 200 to monitor tumor growth. Firefly D-luciferin was diluted to 30mg/ml stock in phosphate-buffered saline and filtered before use. Groups of mice were placed in the specimen chamber and injected with D-luciferin via a retro-orbital approach using 50 μl of D-luciferin (75 mg/kg body weight). One-minute images were acquired until maximum whole-body signal was detected. BLI flux values were serially monitored over a 13–15-day period, with initial scans acquired 5 h after irradiation. At the end of the study, all mice were euthanized, and tissues (lung, liver, spleen, femur, spine, sternum and brain) harvested for ex vivo BLI imaging and correlative histology. IGOR image analysis software (V.4.02A, WaveMetrics, Portland, OR, USA) was used to quantify BLI average total flux values (photons/second, p/s) following the manual construction of regions of interest over major organs and tissues.
Additional cohorts of SCID/Beige mice were assigned to either treatment (OXi4503) or control (phosphate-buffered saline vehicle) arms for serial response monitoring every one to three days over a 13-day p.i. interval of luc+/GFP+ HL-60 tumor cells. Initial baseline BLI scans were acquired before therapy to monitor tumor growth following inoculation. One cohort of mice (n = 8) then received two doses of i.v.-administered OXi4503 (0.5 mg in 100 μl phosphate-buffered saline) on days 7 and 10 p.i., while a second cohort of mice (n = 6) received phosphate-buffered saline vehicle, and BLI flux monitored post treatment. Post- to pre-treatment signal changes (ΔS) were computed by dividing the mean BLI flux value 3 days after the first and second treatments by their respective pre-treatment baseline values (that is, days 7 and 10).
Differences in overall survival were examined for cohorts of irradiated mice inoculated with luc+/GFP + (n = 9) or non-transduced (n = 10) HL-60 cells and subsequently treated with double-dose OXi-4503 at 7 and 10 days p.i. Survival time evaluations were based on the number of days between the termination of treatment and euthanasia.
RESULTS
Efficient transduction and in vitro characterization of luc+/GFP+ HL-60 cells
Fluorescence microscopy (Supplementary Figure 1a) of double-transduced HL-60 cells confirmed GFP+ expression. No alteration in survival, proliferative capacity or morphology was found for GFP+ compared with non-GFP+ cells via bright-field microscopy. To determine the sensitivity of BLI, mean total flux values were measured for serial dilutions of luc+ cells (1.0 × 104−5.0 × 105), as against non-transduced (luc−) cell preparations, following addition of D-luciferin (150mg/ml). A strong linear correlation (R =0.993) was found between the BLI flux (maximum ~4× 106 p/s) and cell number (Supplementary Figure 1b). Flow cytometry showed ~99% GFP+ cells 24h after transduction3 (Supplementary Figure 1c), and the percentage of GFP+ -expressing cells was found to correlate strongly with total BLI flux values (R = 0.93) (inset, Supplementary Figure 1b). These results demonstrate high levels of reporter gene expression in HL-60 cells using this lentiviral-mediated delivery approach. Cell viability and cell growth over time were similar for double-transduced and non-transduced cells using a viability analyzer (Vi-CELL Cell Viability Analyzer, Beckman Coulter, Brea, CA, USA) (Supplementary Figure 1d).
Serial BLI monitoring of luc+/GFP+HL-60 cell migration in vivo
To probe whether spatial and temporal changes in HL-60 cellular migration could be sensitively monitored using BLI, SCID/Beige mice (n = 4) were systemically inoculated with 1.0 × 107 luc + /GFP+ HL-60 cells 5 h after low-dose whole-body irradiation. Serial BLI acquisition was performed over a 13-day period every 2–3 days, on average, with mice imaged in both the ventral and dorsal positions (Figure 1a). For all imaged mice, BLI flux and s.d. values were determined over the lung, liver, spine and femur regions as a function of post-injection time, and plots were constructed for each of the animals. Whole-body BLI flux values ranged from 4.5 × 105 to 1.5 × 107 p/s over the first 8 days p.i. Signal initially appeared in the lungs at 5 h p.i. (7.3 × 104 p/s), followed by dramatic increases in signal throughout the body, particularly in the femur, spine, lung and liver regions over days 6–13 p.i., which neared/exceeded values of 107 p/s (Figure 1b). Time-varying whole-body BLI flux ranged from 6.8 × 105 to 2.3 × 108 p/s ventrally and 4.6 × 105 to 1.4 × 108 p/s dorsally (inset, Figure 1b).
Figure 1.
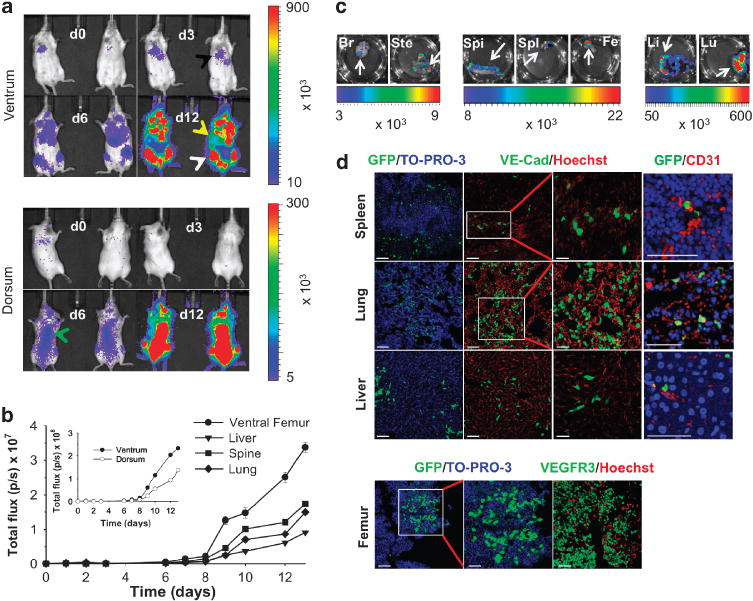
Serial BLI monitoring of human leukemic cell migration in untreated animals with correlative histology. (a) Serial in vivo monitoring of luc + /GFP + HL-60 cells in SCID/Beige mice over a 12-day interval p.i. Representative in vivo ventral and dorsal BLI images acquired on days 3, 6 and 12 p.i. (lung, black arrow; liver, yellow arrow; femur, white arrow; spine, green arrow). Data indicated as mean flux values (p/s ± s.d.). (b) Time series of average BLI flux values for femur, liver, spine and lungs in individual mice (mean p/s ± s.d.). Inset shows average whole-body flux values for the dorsum and ventrum. (c) BLI of harvested ex vivo tissues. (d) Immunofluorescence characterization (Zeiss LSM 510 Meta Confocal Microscope with EC-Plan Neofluor 10×/0.3 objective, AIM software) reveals GFP+ cells and endothelial cells expressing VE-cadherin, and VEGFR3 within harvested spleen, lung, liver and femur tissue with nuclear counterstaining (TO-PRO-3, Hoechst). High-resolution images of spleen, lung and liver demonstrate close proximity of GFP+ cells to CD31-expressing endothelial cells (col 4). Scale bars = 50 μm (spleen, lung, liver: cols 1, 2; femur: cols 1,3); higher magnification GFP/TO-PRO-3 (femur: 25 μm, col 2) and VE-Cadherin/Hoechst (spleen, lung, liver: 25 μm, col 3); high magnification GFP/CD31 (spleen, lung, liver: 100 μm, col 4). Br, brain; Fe, femur; Li, liver/bowel; Lu, lung; Spi, spine; Spl, spleen; Ste, sternum.
All animals were euthanized on p.i. day 13, and the major organs/tissues were harvested for correlative ex vivo BLI. The corresponding signal was observed in the brain, sternum, spine, femur, spleen, liver and lung specimens (Figure 1c), with flux values ranging from ~103 p/s (brain) to 107 p/s (femur). The in vivo signal observed over the brain region, greater than that observed in ex vivo specimens, was attributed to partial volume effects with overlying tissue. BLI signal was confirmed histologically on the basis of GFP+ expression levels in selected tissues using immunofluorescence. Co-localization of GFP+ -expressing cells with pan-endothelial cell markers, platelet endothelial cell adhesion molecule (CD31) and/or VE-cadherin and VEGFR3 was observed in the spleen, lung, liver and femur (Figure 1d), raising the possibility of LC interactions with endothelial cells in perivascular niches.
In vitro OXi4503 mechanism of action and evaluation of dose–response
Treatment response of luc + /GFP + HL-60 cells to the metabolically active form of OXi4503 was evaluated in vitro using BLI and viability assays. OXi4503 is known to be dephosphorylated by tissue phosphatases to the active tubulin-binding compound, combretastatin A133 (Supplementary Figure 2a), which rapidly inhibits tubulin polymerization and accelerates necrosis by functionally disrupting tumor neovasculature. Combretastatin A1 is further metabolized, in the presence of oxidative enzymes (tyrosinase, peroxidases), to a reactive orthoquinone intermediate, leading to the generation of reactive oxygen species,34,35 apoptotic changes and to enhanced antitumor activity.34,35 Significant decreases in cell viability and BLI flux values, roughly 70–80%, were observed for the order of magnitude changes in combretastatin A1 concentrations (that is, 50–500 nM) (Supplementary Figure 2b). At concentrations of 200 nM, mean BLI flux decreased to about one-third of baseline values.
Treatment efficacy in HL-60 xenograft models
To establish the best candidate drug for achieving maximum therapeutic efficacy, cohorts of mice (n = 10 each) bearing hindlimb HL-60 xenografts were treated with OXi4503, CA4P or Ara-C, and the appropriate dosing parameters were established. Mice treated with OXi4503 showed statistically significant differences in tumor volumes on days 16 and 23 (P<0.001) relative to tumors treated with CA4P, Ara-C or administered 0.9% saline vehicle (controls) (Figure 2a). These latter cohorts demonstrated increases in tumor volumes, while essentially constant volumes were observed in mice treated with OXi4503, resulting in at least factor of 200 decreases relative to controls at day 23. Weights of animals treated with OXi4503 remained relatively constant over the treatment interval (Figure 2b). A statistically significant survival benefit for OXi4503-treated mice was found (inset, P<0.001) relative to other agents. Tumor volumes measured over a range of OXi4503 concentrations (10–75 mg/kg/injection, 2 injections) revealed significant growth delays over an ~15-day period (that is, day 12-day 26) relative to that observed in cohorts receiving saline vehicle or 2.5 mg/kg OXi4503 (days 19, 26; P<0.001) (Figure 2c). Weights of mice treated with concentrations of at least 10 mg/kg/injection were essentially stable compared with controls and those treated with 2.5 mg/kg/injection (Figure 2d). Statistically significant improvements in survival were observed in cohorts administered 10, 25, or 75 mg/kg/injection (inset, P<0.001), suggesting a distinct survival benefit over those given 2.5 mg/kg/injection or no treatment.
Figure 2.
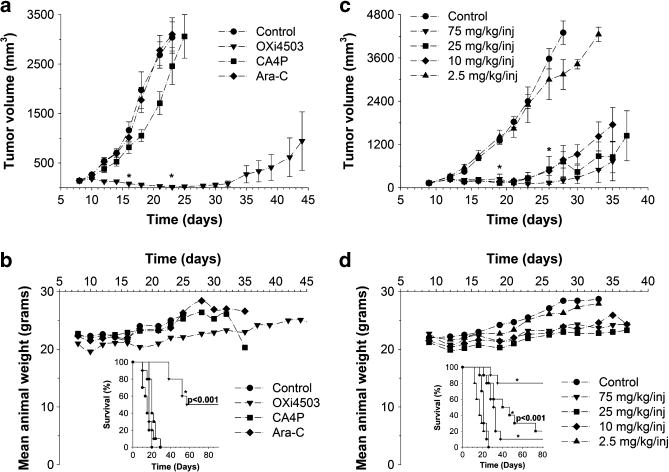
Therapeutic efficacy in HL-60 xenografts. (a) Time-dependent changes in xenograft volume following administration of OXi4503, CA4P, Ara-C or saline vehicle. *P<0.001 for OXi4503 versus CA4P or Ara-C. (b) Corresponding mean weight fluctuations and survival data (inset) demonstrate a statistically significant survival benefit of OXi4503 relative to other monotherapies and saline vehicle (*P<0.001). (c) Time-dependent changes in xenograft volumes as a function of administered OXi4503 (2.5–75mg/kg/inj) or saline vehicle. *P<0.001 for doses of at least 10 mg/kg/inj versus vehicle. (d) Corresponding animal weights and survival data (inset) show a statistically significant survival benefit of OXi4503 versus vehicle for doses of at least 10 mg/kg (*P<0.001).
Serial non-invasive BLI monitoring of treatment response in systemic models
Based on the xenograft data, a dose of 25 mg/kg/injection OXi4503 was selected for treatment of the disease in systemic models, and serial response monitoring was performed using BLI. Measured xenograft volumes using this dose were not significantly different from those found at higher doses, and offered an added benefit of reducing any potential systemic toxicity. Following systemic inoculations of mice (n = 14) with luc + / GFP+ HL-60 cells, BLI was conducted over a nearly 2-week period (Figure 3a). All untreated mice and mice treated with double-dose OXi4503 served as their own controls. Treatments were administered on the basis of observed increases in whole-body signal (that is, 7 and 10 days p.i.).
Figure 3.
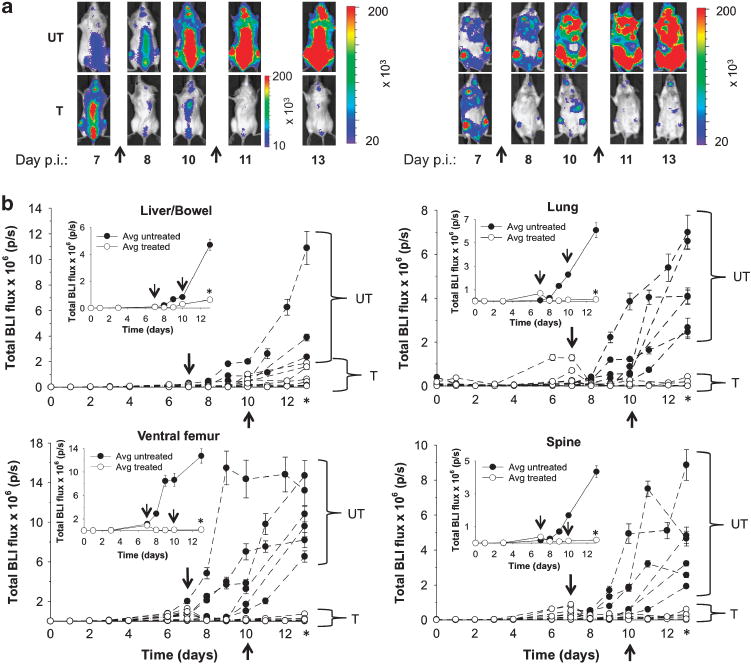
In vivo response monitoring in systemic HL-60 models with BLI. (a) Representative in vivo BLI (IVIS 200, Xenogen) before (day 7) and after (days 8, 10, 11, 13) OXi4503 (25 mg/kg) or saline vehicle administration (arrows) in mice systemically inoculated with luc+ /GFP + HL-60 cells (n = 14). Ventral (right-hand side) and dorsal views (left-hand side) are shown. Color scales represent total flux values (p/s±s.d.). (b) Serial monitoring of BLI flux changes in individual animals over a 13-day period following systemic inoculation. The inset shows average flux changes for treated and control mice. Color scales represent total flux values (p/s±s.d.). Arrows represent times of drug administration. One-tailed Mann–Whitney U-tests demonstrate statistical significance (*) between cohorts of untreated (UT, n = 6) and treated (T, n = 8) mice on day 13 at the P≤0.001 level for liver/bowel (P = 0.0015), lung (P = 0.001), ventral femur (P = 0.0005) and spine (P = 0.0005).
Up to a factor of 10 signal reductions were observed over the femur, lungs, liver and spine on ventral and dorsal views following treatment on days 7 and 10 p.i. (Figure 3a), as compared with the findings on ventral and dorsal views in untreated mice (controls). The corresponding BLI time-course data for the liver, lung, femur and spine in untreated mice and mice treated with double-dose OXi4503 are graphically represented for each individual animal (Figure 3b), along with the mean values for each cohort (inset). Statistically significant differences in BLI flux values were found between the untreated and treated groups of mice (P≤0.001) at day 13 p.i. for all analyzed tissues. Supplementary Table 1 summarizes corresponding mean tissue-specific signal changes (ΔS) for individual and overall treatment intervals. For the femur, spine and lung significantly greater reductions in ΔS were found after the initial, rather than the second, administered dose of OXi4503, whereas smaller differences (that is, factor of 2) were observed over the liver. Overall changes in DS (that is, day 7–13) were significantly larger for treated mice than controls.
No statistically significant difference (P>0.05) in overall survival was found for mice receiving double-dose OXi4503 after inoculation with either transduced or non-transduced HL-60 cells (Supplementary Figure 3), suggesting that viral transduction does not alter overall survival.
Correlative BLI-tissue analyses: tissue GFP expression and co-labeling studies
To confirm the presence and extent of tumor cell infiltration in harvested tissues from the liver, lung, spine, femur and spleen, GFP expression levels were evaluated by quantitative analyses of immunohistochemical preparations (that is, automated and/or manual cell-counting procedures, Supplementary Figures 4a and b) or by hematoxylin and eosin staining of adjacent tissue sections (Supplementary Figures 4c and d). Before treatment, the largest % GFP + HL-60 cells was found in the spine (n = 4), femur (n = 4) and the splenic specimens (n = 4), while only scattered GFP + HL-60 cells were identified in the brain tissue (data not shown). The % GFP+ expression values for untreated and treated tissues are graphically depicted in Figure 4a. Statistically significant differences in these values were found for all tissues, but were greatest for lung and femur (P = 0.015). In addition, strong correlations were found between computed % GFP+ cells and image-derived flux values (Figure 4b) for treated liver (n = 3, R2 = 0.973), lung (n = 3, R2 = 0.950), femur (n = 4, R2 = 0.861) and spine (n = 4, R2 = 0.929), as well as for untreated liver (n = 3, R2 = 0.973), lung (n = 4, R2 = 0.950) and femur (n = 3, R2 = 0.861).
Figure 4.
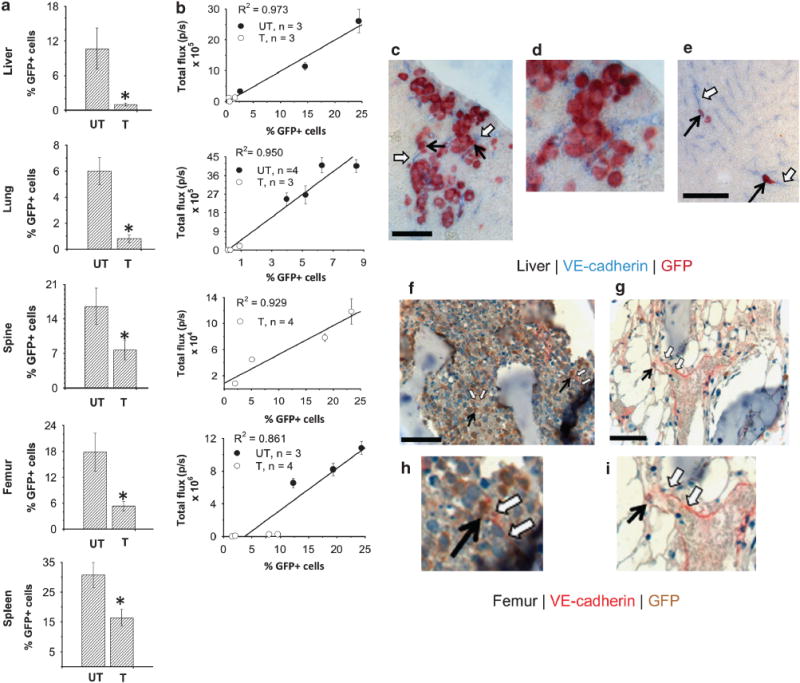
Quantitative GFP+ expression differences and co-labeling studies in treated and untreated mice. (a) Tissue-specific % GFP + -expressing cells for treated (T) and untreated (UT) mice (mean±s.e., n = 4 per group). One-tailed Mann-Whitney U-tests demonstrate statistical significance (*) at the P≤0.05 level for lung (P = 0.014) and femur (P = 0.014), and borderline significance for liver, spleen and spine (P = 0.057). (b) In vivo BLI signal versus % GFP+ expression in ex-vivo specimens using regression analysis. (c–i) Co-labeling of GFP and endothelial cells expressing VE-cadherin (Zeiss LSM 510 Meta Confocal Microscope (Carl Zeiss, Thornwood, NY, USA) with EC-Plan Neofluor 10 × /0.3 objective and AIM software (Carl Zeiss)) in untreated liver (c, d), untreated femur (f, h), and treated liver (e) and femur (g, i) sections. Endothelial cell marker: femur (red, open arrows); liver (blue, open arrows). GFP+ cells: femur (brown, solid arrows); liver (red, solid arrows). Scale bar = 25μm (c, e) and 50 μm (f, g). Representative higher-magnification images (d, h, i) reveal GFP+ cells located close to VE-cadherin-expressing endothelial cells.
Adjacent hematoxylin and eosin-stained tissue sections demonstrated necrotic and morphological changes in a number of treated (Supplementary Figure 4d) tissues relative to untreated specimens (Supplementary Figure 4c). Treated marrow tissues of the spine (Supplementary Figure 4d, row 3) and femur (row 4) revealed a preponderance of adipocytes, along with markedly diminished numbers of bone marrow-derived cells. Reductions in the fractions of splenic cell populations were also observed in treated spleen, along with extensive hemorrhagic changes (row 5) and grossly contracted morphological features. Treated lung specimens revealed a greater volume of air-filled sacs/cavities relative to untreated samples (row 2). Tissue morphology was found to be similar in untreated and treated liver (row 1) and brain specimens (data not shown); however, these tissues revealed corresponding reductions in % GFP+ cells. No apoptotic changes in any harvested tissue specimens were detected at the termination of the study.
Co-localization of GFP+ HL-60 cells with VE-cadherin-expressing endothelial cells was observed in both untreated and treated liver (Figures 4c–e) and femur sections (Figures 4f–i). Representative high-power microscopy images revealed greater numbers of GFP+ cells in untreated tissues, relative to treated tissues, with the latter showing co-localization of these cell populations. As HL-60 cells are seen to reside in close proximity to endothelial cells, the importance of perivascular niches to the maintenance and survival of LCs is implicated.
DISCUSSION
We report on the use of reporter gene-imaging technologies for sensitively and non-invasively guiding OXi4503 treatment of LC infiltrates in solid organs and tissues, as well as serially tracking their migration, growth and regression over relatively short time intervals (that is, 2 weeks). Our results highlight the distinct advantages offered by such an approach. As a surrogate tool for monitoring viability, serial BLI evaluations in mice before and after treatment obviate the need for killing large numbers of mice at arbitrary time points to detect maximal response. Rather, the use of BLI identifies times at which maximum treatment-induced signal losses occur, thus confining histological confirmation to those points. Such optical approaches thus serve as powerful imaging tools for use in preclinical drug discovery and response-monitoring applications in order to assess therapeutic efficacy, potency and survival advantage at early post-treatment time points.36,37 Further, they will be crucial for identifying tissue sites that potentially harbor leukemic niches in an effort to stem relapse and obliterate disease, as suggested by the results of our co-localization studies. Additional advantages include the low costs of such imaging strategies, their relative ease of use, and the practical advantages offered by real-time data acquisition over discrete tissue sampling methods for optimizing therapeutic protocols and/or altering treatment management.
Following systemic administration of luc + /GFP + HL-60 cells, we found that flux values (that is, luc + cells) measured over the liver, lung, spine and femur provided highly sensitive and reliable non-invasive biomarkers of tumor burden and distribution on the basis of correlative tissue-specific GFP expression levels (Figure 1). BLI signal was initially identified over the lung regions; subsequent migration to and expansion of luc + cells within reticuloendothelial organs was then observed over a 2-week interval, notably involving the liver and marrow compartments of prominent osseous structures. At the termination of the imaging study, in vivo signal overlying the liver, femur and lung regions was confirmed in corresponding harvested organ samples using BLI (Figure 1c), as well as by clusters of GFP+ cells on tissue immunofluorescence (Figure 1d). Although the presence of luc + signal overlying the region of the spleen was not reliably identified given its relatively small size and depth within the body, ex-vivo imaging of splenic tissue showed luc+ signal, and scattered GFP+ cells were present within corresponding tissue specimens.
We demonstrated that administration of double-dose OXi4503 as a monotherapy to cohorts of HL-60 xenograft mice resulted in statistically significant improvements in survival, as well as significant tumor growth delays at higher VDA concentrations, as compared with the findings for CA4P, Ara-C or saline vehicle. Petit3 previously showed treatment efficacy and prolonged survival following intraperitoneal administration of low-dose CA4P (<50 mg/kg), as against a control vehicle, using histological methods at longer time intervals (that is, 1 month). Further, acute tissue perfusion changes (approximately minutes to hours) were observed in colorectal and lucerifase-expressing (luc +) human mammary MDA-MB-23110 xenografts following CA4P administration, the latter study utilizing BLI to monitor changes.
The collective results of our OXi4503 studies confirm many of the observations noted in more acute13,25 and chronic xenograft models,18,38 namely that multi-dose treatment leads to tumor growth delays and/or vascular disruption with widespread necrosis. As shown by us and a recent prior human LC investigation,20 OXi4503 offers potential therapeutic efficacy. However, in the latter study, treatment response was assessed in more chronic human LC models (that is, chloroma, engraftment), necessitating longer time intervals of observation (that is, 8 weeks). Although we confirmed the resulting vascular disruption and cytotoxic effects, we did not observe the discrete peripheral tumor layer that survived VDA treatment in this study, presumably given our shorter post-treatment time course. We have further extended the methods of others by assessing the feasibility of using measured BLI signal changes as sensitive and reliable biomarkers of tumor tissue burden, correlating these findings with the % GFP + -expressing cells and their proximity to panendothelial markers.
Applied to systemic models, we further showed that double doses of OXi4503 significantly suppressed increases in BLI flux values at early time points, which led to enhanced DS values over the entire treatment interval in the spine, femur, lung and liver, relative to untreated cohorts. For individual mice, the largest variation in untreated and treated BLI flux values was found over the liver region. In a single control animal, for instance, a minimum flux value of 1.62 × 106 p/s was found to overlap the values determined for the treated cohort. This result was felt to be the consequence of partial volume effects involving adjacent abdominal cavity structures (that is, bowel). However, differences in time-varying average liver pre- and post-treatment flux values (Figure 3b, inset) were statistically significant at the termination of the imaging period.
Measured pre- and post-treatment flux values in the liver, lung, spine and femur were confirmed by the presence of GFP + cells in corresponding histological sections, and flux values were found to strongly correlate with % GFP + expression levels in all but untreated tissues from the spine (Figure 4b). In this latter case (data not shown), the lack of a strong correlation between the measured flux values and % GFP+ expression was thought to be due to limitations in the ability to derive accurate manual cell counts under conditions of very dense marrow cellularity coupled with excess background staining. By contrast, manual counting of treated spine tissue was not limited by these conditions, as significant losses in bone marrow cellularity and attenuation of tissue background staining were observed as a result of tissue necrotic changes.
In several treated tissue specimens, % GFP+ cells served as a highly sensitive and quantitative indicator of therapeutic response, complementing necrotic and morphological changes observed by hematoxylin and eosin staining (Supplementary Figure 4d). Relative to untreated specimens (Supplementary Figure 4c), disrupted tissue architecture and loss of the normal structural landmarks were observed. Necrotic changes were most prominent in the spine, femur and spleen. The marrow compartments of these bony specimens revealed significant losses in % GFP+ cells; these were accompanied by partial replacement of marrow-derived populations with adipocytes. In treated spleens, decreased % GFP+ cells corresponded to marked reductions in splenic cell populations on increasingly hemorrhagic backgrounds, in conjunction with grossly contracted morphological features. In other tissues, the % GFP+ cells served as a more sensitive indicator of response than corresponding structural alterations, particularly where histopathological changes were inconclusive and/or not readily identified. For instance, although no significant morphological changes were visually evident within treated liver specimens, statistically significant losses in % GFP+ cells were seen. No apoptotic changes were found in any tissues at the termination of the imaging study, suggesting that such changes occurred much earlier than our pathological assessment.
The ability to accurately track individual and/or clusters of cells within histological sections over time in order to determine whether such cells stay within perivascular niches is challenging following vascular disruption. Heterogeneous tissue responses to one or more therapies, manifested as spatially dependent microstructural alterations, make this process difficult to monitor. Given our findings that surviving GFP+ cells are identified within perivascular niches in all tissues evaluated, both before and after treatment, it can be concluded that these niches have a largely protective role, one which facilitates conditions favorable to the selective maintenance and expansion of residual human LCs. It is these small, persisting cell populations that are suspected to promote resistance to conventional therapies and subsequent disease relapse.
In summary, we showed statistically significant survival benefit and therapeutic efficacy of OXi4503 in human leukemic models using non-invasive reporter gene imaging. Mapping of cellular distributions across spatial scales enabled earlier detection of whole-body leukemic infiltration and residual tumor burden over a period of days to weeks following targeted therapy. Confirmed by cellular necrosis in multiple tissues and quantitative estimates of tissue GFP + expression, the findings suggested that BLI signal can serve as an exquisitely sensitive biomarker of tumor tissue burden in systemic leukemic models, underscoring the use of such powerful non-invasive imaging approaches for optimizing treatment selection and response assessment. As GFP+ HL-60 cells reside in close proximity to VE-cadherin- and CD31-expressing endothelial cells before and after treatment in all tissues evaluated, it can be concluded that the perivascular niche may have a critical role in the maintenance and survival of LCs.
Supplementary Material
Acknowledgments
We acknowledge M Gönen for providing assistance with biostatistical analyses and L Johnson in the Laboratory of Comparative Pathology for assistance with interpretation of histological specimens. This study was supported in part by an ICMIC P50 CA86438 grant. Technical services provided by the MSKCC Small-Animal Imaging Core Facility, supported in part by NIH Small-Animal Imaging Research Program (SAIRP) Grant No R24 CA83084 and NIH Center Grant No P30 CA08748, are gratefully acknowledged.
Footnotes
CONFLICT OF INTEREST
DC is currently the Head of Research and Development at OXiGENE. BS previously held the position of Director of Research at OXiGENE, but is no longer at the company. The remaining authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Leukemia website (http://www.nature.com/leu)
References
- 1.Pui CH, Jeha S. New therapeutic strategies for the treatment of acute lymphoblastic leukaemia. Nat Rev Drug Discov. 2007;6:149–165. doi: 10.1038/nrd2240. [DOI] [PubMed] [Google Scholar]
- 2.Jeha S. New therapeutic strategies in acute lymphoblastic leukemia. Semin Hematol. 2009;46:76–88. doi: 10.1053/j.seminhematol.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 3.Petit I, Karajannis MA, Vincent L, Young L, Butler J, Hooper AT, et al. The microtubule-targeting agent CA4P regresses leukemic xenografts by disrupting interaction with vascular cells and mitochondrial-dependent cell death. Blood. 2008;111:1951–1961. doi: 10.1182/blood-2007-05-089219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ninomiya M, Abe A, Katsumi A, Xu J, Ito M, Arai F, et al. Homing, proliferation and survival sites of human leukemia cells in vivo in immunodeficient mice. Leukemia. 2007;21:136–142. doi: 10.1038/sj.leu.2404432. [DOI] [PubMed] [Google Scholar]
- 5.Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood. 2002;100:59–66. doi: 10.1182/blood.v100.1.59. [DOI] [PubMed] [Google Scholar]
- 6.Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood. 2005;106:1154–1163. doi: 10.1182/blood-2005-01-0178. [DOI] [PubMed] [Google Scholar]
- 7.May AE, Neumann FJ, Schomig A, Preissner KT. VLA-4 (alpha(4)beta(1)) engagement defines a novel activation pathway for beta(2) integrin-dependent leukocyte adhesion involving the urokinase receptor. Blood. 2000;96:506–513. [PubMed] [Google Scholar]
- 8.Bohnsack JF, Chang J. Activation of beta 1 integrin fibronectin receptors on HL60 cells after granulocytic differentiation. Blood. 1994;83:543–552. [PubMed] [Google Scholar]
- 9.Pilat MJ, Lorusso PM. Vascular disrupting agents. J Cell Biochem. 2006;99:1021–1039. doi: 10.1002/jcb.20783. [DOI] [PubMed] [Google Scholar]
- 10.Zhao D, Richer E, Antich PP, Mason RP. Antivascular effects of combretastatin A4 phosphate in breast cancer xenograft assessed using dynamic bioluminescence imaging and confirmed by MRI. FASEB J. 2008;22:2445–2451. doi: 10.1096/fj.07-103713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaplin DJ, Hill SA. The development of combretastatin A4 phosphate as a vascular targeting agent. Int J Radiat Oncol Biol Phys. 2002;54:1491–1496. doi: 10.1016/s0360-3016(02)03924-x. [DOI] [PubMed] [Google Scholar]
- 12.Salmon HW, Mladinich C, Siemann DW. Evaluations of vascular disrupting agents CA4P and OXi4503 in renal cell carcinoma (Caki-1) using a silicon based microvascular casting technique. Eur J Cancer. 2006;42:3073–3078. doi: 10.1016/j.ejca.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 13.Holwell SE, Cooper PA, Grosios K, Lippert JW, III, Pettit GR, Shnyder SD, et al. Combretastatin A-1 phosphate a novel tubulin-binding agent with in vivo anti vascular effects in experimental tumours. Anticancer Res. 2002;22:707–711. [PubMed] [Google Scholar]
- 14.Wankhede M, Dedeugd C, Siemann DW, Sorg BS. In vivo functional differences in microvascular response of 4T1 and Caki-1 tumors after treatment with OXi4503. Oncol Rep. 2010;23:685–692. doi: 10.3892/or_00000685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan LS, Malcontenti-Wilson C, Muralidharan V, Christophi C. Effect of vascular targeting agent Oxi4503 on tumor cell kinetics in a mouse model of colorectal liver metastasis. Anticancer Res. 2007;27:2317–2323. [PubMed] [Google Scholar]
- 16.Chan LS, Malcontenti-Wilson C, Muralidharan V, Christophi C. Alterations in vascular architecture and permeability following OXi4503 treatment. Anticancer Drugs. 2008;19:17–22. doi: 10.1097/CAD.0b013e3282f077a1. [DOI] [PubMed] [Google Scholar]
- 17.Daenen LG, Shaked Y, Man S, Xu P, Voest EE, Hoffman RM, et al. Low-dose metronomic cyclophosphamide combined with vascular disrupting therapy induces potent antitumor activity in preclinical human tumor xenograft models. Mol Cancer Ther. 2009;8:2872–2881. doi: 10.1158/1535-7163.MCT-09-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siemann DW, Shi W. Dual targeting of tumor vasculature: combining Avastin and vascular disrupting agents (CA4P or OXi4503) Anticancer Res. 2008;28:2027–2031. [PMC free article] [PubMed] [Google Scholar]
- 19.Shaked Y, Ciarrocchi A, Franco M, Lee CR, Man S, Cheung AM, et al. Therapy-induced acute recruitment of circulating endothelial progenitor cells to tumors. Science. 2006;313:1785–1787. doi: 10.1126/science.1127592. [DOI] [PubMed] [Google Scholar]
- 20.Madlambayan GJ, Meacham AM, Hosaka K, Mir S, Jorgensen M, Scott EW, et al. Leukemia regression by vascular disruption and antiangiogenic therapy. Blood. 2010;116:1539–1547. doi: 10.1182/blood-2009-06-230474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hill SA, Toze GM, Pettit GR, Chaplin DJ. Preclinical evaluation of the antitumour activity of the novel vascular targeting agent Oxi 4503. Anticancer Res. 2002;22:1453–1458. [PubMed] [Google Scholar]
- 22.Holwell SE, Cooper PA, Thompson MJ, Pettit GR, Lippert LW, III, Martin SW, et al. Anti-tumor and anti-vascular effects of the novel tubulin-binding agent combretastatin A-1 phosphate. Anticancer Res. 2002;22:3933–3940. [PubMed] [Google Scholar]
- 23.Salmon HW, Siemann DW. Effect of the second-generation vascular disrupting agent OXi4503 on tumor vascularity. Clin Cancer Res. 2006;12:4090–4094. doi: 10.1158/1078-0432.CCR-06-0163. [DOI] [PubMed] [Google Scholar]
- 24.Madlambayan GJ, Meacham A, Hosaka K, Mir S, Jorgensen M, Scott EW, et al. Leukemia regression by vascular disruption and anti-angiogenic therapy. Blood. 2010;116:1539–1547. doi: 10.1182/blood-2009-06-230474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hua J, Sheng Y, Pinney KG, Garner CM, Kane RR, Prezioso JA, et al. Oxi4503, a novel vascular targeting agent: effects on blood flow and antitumor activity in comparison to combretastatin A-4 phosphate. Anticancer Res. 2003;23:1433–1440. [PubMed] [Google Scholar]
- 26.Lin CM, Singh SB, Chu PS, Dempcy RO, Schmidt JM, Pettit GR, et al. Interactions of tubulin with potent natural and synthetic analogs of the antimitotic agent combretastatin: a structure-activity study. Mol Pharmacol. 1988;34:200–208. [PubMed] [Google Scholar]
- 27.Seshadri M, Toth K. Acute vascular disruption by 5,6-dimethylxanthenone-4-acetic acid in an orthotopic model of human head and neck cancer. Transl Oncol. 2009;2:121–127. doi: 10.1593/tlo.09103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson HL, Yap JT, Miller MP, Robbins A, Jones T, Price PM. Assessment of pharmacodynamic vascular response in a phase I trial of combretastatin A4 phosphate. J Clin Oncol. 2003;21:2823–2830. doi: 10.1200/JCO.2003.05.186. [DOI] [PubMed] [Google Scholar]
- 29.Contag CH, Jenkins D, Contag PR, Negrin RS. Use of reporter genes for optical measurements of neoplastic disease in vivo. Neoplasia. 2000;2:41–52. doi: 10.1038/sj.neo.7900079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edinger M, Cao YA, Hornig YS, Jenkins DE, Verneris MR, Bachmann MH, et al. Advancing animal models of neoplasia through in vivo bioluminescence imaging. Eur J Cancer. 2002;38:2128–2136. doi: 10.1016/s0959-8049(02)00410-0. [DOI] [PubMed] [Google Scholar]
- 31.Bradbury MS, Panagiotakos G, Chan BK, Tomishima M, Zanzonico P, Vider J, et al. Optical bioluminescence imaging of human ES cell progeny in the rodent CNS. J Neurochem. 2007;102:2029–2039. doi: 10.1111/j.1471-4159.2007.04681.x. [DOI] [PubMed] [Google Scholar]
- 32.Agliano A, Martin-Padura I, Mancuso P, Marighetti P, Rabascio C, Pruneri G, et al. Human acute leukemia cells injected in NOD/LtSz-scid/IL-2Rgamma null mice generate a faster and more efficient disease compared to other NOD/scid-related strains. Int J Cancer. 2008;123:2222–2227. doi: 10.1002/ijc.23772. [DOI] [PubMed] [Google Scholar]
- 33.Pettit GR, Thornhill AJ, Moser BR, Hogan F. Antineoplastic agents. 552. Oxidation of combretastatin A-1: trapping the o-quinone intermediate considered the metabolic product of the corresponding phosphate prodrug. J Nat Prod. 2008;71:1561–1563. doi: 10.1021/np800179g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Folkes LK, Christlieb M, Madej E, Stratford MR, Wardman P. Oxidative metabolism of combretastatin A-1 produces quinone intermediates with the potential to bind to nucleophiles and to enhance oxidative stress via free radicals. Chem Res Toxicol. 2007;20:1885–1894. doi: 10.1021/tx7002195. [DOI] [PubMed] [Google Scholar]
- 35.Kirwan IG, Loadman PM, Swaine DJ, Anthoney DA, Pettit GR, Lippert JW, III, et al. Comparative preclinical pharmacokinetic and metabolic studies of the combretastatin prodrugs combretastatin A4 phosphate and A1 phosphate. Clin Cancer Res. 2004;10:1446–1453. doi: 10.1158/1078-0432.ccr-0518-03. [DOI] [PubMed] [Google Scholar]
- 36.Inoue Y, Izawa K, Kiryu S, Kobayashi S, Tojo A, Ohtomo K. Bioluminescent evaluation of the therapeutic effects of total body irradiation in a murine hematological malignancy model. Exp Hematol. 2008;36:1634–1641. doi: 10.1016/j.exphem.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 37.Jurczok A, Fornara P, Soling A. Bioluminescence imaging to monitor bladder cancer cell adhesion in vivo: a new approach to optimize a syngeneic, orthotopic, murine bladder cancer model. BJU Int. 2008;101:120–124. doi: 10.1111/j.1464-410X.2007.07193.x. [DOI] [PubMed] [Google Scholar]
- 38.Malcontenti-Wilson C, Chan L, Nikfarjam M, Muralidharan V, Christophi C. Vascular targeting agent Oxi4503 inhibits tumor growth in a colorectal liver metastases model. J Gastroenterol Hepatol. 2008;23(7 Part 2):e96–e104. doi: 10.1111/j.1440-1746.2007.04899.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.