

Abstract
Several mechanisms are associated with brain dysfunction during sepsis; one of the most important are activation of microglia and astrocytes. Activation of glial cells induces changes in permeability of the blood-brain barrier, secretion of inflammatory cytokines, and these alterations could induce neuronal dysfunction. Furthermore, blood-borne leukocytes can also reach the brain and participate in inflammatory response. Mechanisms involved in sepsis-associated brain dysfunction were revised here, focusing in neuroinflammation and involvement of blood-borne leukocytes and glial cells in this process.
Keywords: Sepsis, Glial cells, Blood-borne leukocytes, Neuroinflammation, Long-term cognitive dysfunction
Review
Introduction
Sepsis is referred as a systemic inflammatory response due to an infection [1] and presents a wide spectrum of severity: from severe sepsis to septic shock and multi-organ dysfunction syndrome [2]. It is a major cause of death in intensive care unit (ICU) and its incidence is increasing worldwide [3–5]. Sepsis-associated encephalopathy (SAE) is associated with an increased rate of morbidity and mortality. It is not fully understood the exact mechanism that drives brain dysfunction during sepsis development, but brain inflammation and oxidative stress are possible players [6].
Dysfunction of both innate and adaptive immune response plays a role in the induction of abnormal levels of immunoregulatory molecules that result in sepsis [7]. However, participation of effector immune system cells could also impair host response to the infective agents and induces tissue damage [8]. Thus, both an overwhelming inflammatory response and secondary tissue damage and cell dysfunction [9], and a decrease in the immune responses and inadequate infection control [8], could be associated with sepsis pathogenesis [9].
The clinical manifestations of sepsis are variable, depending on the initial site of infection, pathogenic organism, underlying health status of the patient, and time of treatment [10]. The majority of patients with sepsis develop reversible brain dysfunction, called sepsis-associated delirium or septic encephalopathy (SAE) [11, 12]. Moreover, septic patients are at risk for long-term cognitive impairment [12] that could be associated with cerebral atrophy [13–15]. Diagnosing brain dysfunction during sepsis implies a systematic approach of all potential factors. Furthermore, several mechanisms have been proposed to explain the pathophysiology of SAE [6, 7, 16–18]. SAE development involves cellular damage, mitochondrial and endothelial dysfunction, neurotransmission, and calcium homeostasis disturbances. Cerebral blood flow, integrity of blood-brain barrier (BBB) and cerebral water content may also be affected [6, 13, 19]. Apart from brain specific aspects, development of SAE could involve hypoxemia, hypotension, and electrolytes disturbances [20, 21]. Additionally, the long-term effects of sepsis in humans have been evaluated in some studies, all of which underscore the connection between SAE and subsequent cognitive decline [14, 15]. Mechanisms that link SAE and long-term cognitive dysfunction are not well understood, thus evidences that link neuroinflammation and both acute and long-term sepsis-associated brain dysfunction were here reviewed.
Neuroinflammation
In central nervous system (CNS) immune response to injury is initiated mainly by microglia and astroglia. Neuroinflammation is characterized by activation of microglial cells, followed by changes in permeability of BBB and infiltration of peripheral immune cells into CNS parenchyma. These alterations lead to secretion of inflammatory cytokines and neuronal dysfunction [22], and seem to be a common feature to all neuroinflammatory syndromes [23].
Besides acute symptoms, CNS dysfunction secondary to sepsis is characterized by long-term cognitive impairment. In this context, it has been demonstrated that cytokine levels, oxidative stress, and energetic metabolism alterations seen early after sepsis may persist for up to 30 days and this could be associated with cognitive damage [24–26]. Actually, several groups using different models of sepsis could reproduce cognitive impairment (from inhibitory avoidance to object recognition) that is similar to what is observed in humans [14, 27]. Thus, systemic inflammation is emerging as a significant driver of cognitive decline in the aged and vulnerable brain. A growing body of clinical and preclinical evidence demonstrates that various peripheral inflammatory insults can exacerbate CNS inflammation. In addition, sepsis itself is an independent risk factor for dementia [28]. In this context, severe systemic inflammation can produce a new neuropathology or accelerate cognitive decline previously installed [29]. Furthermore, both human and experimental studies suggest that cognitive impairment and dementia are important risk factors for delirium [30].
Among the far-reaching and sustained systemic effects of sepsis are hemodynamic alterations, which cause the changes in cerebral blood flow that are implicated in SAE. Although reduced cerebral blood flow and oxygenation occur in sepsis, they do not appear to drop low enough to threaten neuronal viability or cause electroencephalogram changes at least in preclinical models of SAE [31]. However, even when cerebral blood flow is sufficient to ensure neuronal integrity, slight reductions could still contribute to SAE when higher energy demands are present, as in the case of cognitive processing [31]. In addition, there are some evidences of alterations in the control of brain perfusion both in animal models and humans [32–35]. These alterations in auto regulation of brain perfusion could be associated with inflammation and the pathogenesis of SAE.
Abundant data have suggested adaptive immunity as a key regulator of brain cell renewal, behavior, learning, and memory. Immune deficiency was linked to impaired brain plasticity, and when adaptive immunity was boosted, brain function was restored or even enhanced [36–40]. Thus, neuroinflammation could be associated with brain (dys)function, inhibition of hippocampal neurogenesis, and disruption of cognitive ability [41]. Although this is clearly demonstrated in neurodegenerative disorders, its role in sepsis associated long-term cognitive impairment is less clear.
Microglia—do different phenotypes have different impact in brain dysfunction?
Microglia activation has a major role in the generation of oxidative damage and inflammation in the brain during several different CNS diseases. As demonstrated to peripheral immune cells, microglia are able to express several pathogens/damage recognition receptors, such as the toll-like receptors (TLRs). TLR4 and TLR2 expressed in microglial cells have been specifically associated with both neuroinflammation and clearance of aggregated proteins [42, 43]. Thus, it is plausible to suppose that microglia activation is a major determinant of SAE and long-term cognitive impairment after sepsis [44].
Morphology and density of microglia is region specific, being more common in the grey area of the CNS [45]. These differences may be associated with functional heterogeneity, but little is known about the nature of this heterogeneity among and within brain regions. Microglia share common features of cells of the myeloid lineage; they have the ability to secrete a plurality of immunomodulatory molecules, which coordinate signals to neighbor and circulating cells [46]. Thus, during inflammation, activated microglia modify the responses of supporting cells through the release of a diversity of factors [47].
Microglia show morphological and functional diversity in the brain, ranging from the ramified, “resting” phenotype associated with tissue surveillance in the healthy brain to amoeboid, cytokine-secreting, and phagocytic phenotypes in disease states [48]. Microglial activation, which plays a central role in neuroinflammation may be regulated by several intercellular interactions involving cell-surface molecules and soluble mediators, such as cytokines, reactive oxygen species (ROS), and neurotransmitters [22]. In healthy brain, microglial cells display a “homeostatic” phenotype, which monitor the surrounding environment [49]. In this phenotype, microglia express surface molecules and secrete soluble factors, which influence astrocytes and neuron function [50], promote the clearance of cellular debris and aggregated proteins [51]. Accordingly, microglia exhibit at least four functional behaviors: surveillance, neuroprotection, phagocytosis, and toxicity [23]. In addition, microglia could be “primed”, and in this context produces exaggerated levels of inflammatory cytokines in response to a stimulus [52]. Thus, it seems that microglia can be a double-edged sword [53].
Recent studies have demonstrated that microglia may simultaneously exhibit M1 and M2 phenotypes [54]. M1 cells can be neurotoxic, probably through the production of cytokines such as IL-6, IL-12, and TNF-α, reactive oxygen species, and deregulated release of glutamate [55]. Activated brain microglia can be neuroprotective by assuming an M2-like phenotype [56–58], a phenomena similar to what occur in macrophages. In response to lipopolysaccharide (LPS), TNF-α and/or IFN-γ, macrophages become classically activated and acquire an M1 phenotype, which express several pro-inflammatory cytokines and enzymes that promote a sustained tissue inflammation. In contrast, alternative activation in response to IL-4, IL-13, glucocorticoids, TGF-β, and/or IL-10, macrophages differentiate into the anti-inflammatory M2 phenotype, which is associated with the resolution of inflammation and tissue repair [59–61]. It seems that modulation of the M2 phenotype could represent a beneficial aspect of brain immune response during inflammatory injury [62–64], but to date, there is no sufficient information regarding M2 phenotype during the development of SAE.
Free radicals are a major causal factor of secondary insults, such as axonal damage or membrane lipid peroxidation, which result in evolving neurological deficits [65, 66]. Reactive nitrogen and oxygen species, as well as cytokines produced by M1-like microglia, such as TNF-α, can directly induce neuronal death [67–70]. Among the most frequently cited proinflammatory and damaging aspects of microglial activation is the production of nitric oxide (NO) via nitric oxide synthase (iNOS) [32]. iNOS expression in response to sepsis has been detected in neuronal and glial cells [71, 72]. In this context, neurons are highly sensitive to the toxic effects of NO [73]. NO produced by activated microglia is able to induce neuronal apoptosis [74] even when activated glial cells are present at relatively low numbers [75, 76]. In a model of systemic endotoxemia, it was observed an early upregulation of iNOS in several different brain regions, predominantly in microglial cells, and is associated with both neuronal and glial cell apoptosis [77]. Furthermore, iNOS activation can be found even late times after sepsis resolution [78]. NO might also include the stimulation of excitotoxicity, because the application of an acute low dose of the NMDA receptor antagonist MK-801 prevents memory deficits in the cecal ligation and puncture rat model [79].
In addition, microglia are able to generate oxidative burst involving the induction of multiple enzymes/complexes including NADPH oxidase and myeloperoxidase (MPO) [71]. Thus, a sort of strong oxidant agents can be produced by activated microglia, including superoxide, nitric oxide, hypochlorous acid, peroxynitrite, and hydroxyl radical that contributes to the progression of brain damage during SAE [80]. On the other hand, microglial can also help in clearance of free radicals (for example, by regulating ceruloplasmin levels), thus preventing free radical-mediated neuronal damage [70].
Activated microglia can also release large amounts of glutamate that can induce neuronal dysfunction [81]. High extracellular glutamate concentrations in the CNS promote excitotoxicity, which has been involved in various pathological conditions, including acute CNS trauma such as brain or axonal injury [82], ischemia [83], and epilepsy [84], as well as in chronic neurodegenerative disorders such as Parkinson’s disease (PD), AD and amyotrophic lateral sclerosis (ALS) [85]. When CNS is injured, glutamate buffering cells, astrocytes, are lost and the damaged site is repopulated by M1-like activated microglia [86]. Although neurons, astrocytes, and homeostatic microglia can release moderate levels of glutamate, M1-like activated microglia can release toxic amounts of glutamate through a mechanism that involves connexin channels and the cystine/glutamate antiporter system [81, 87]. Excessive stimulation of the ionotropic glutamate receptor N-methyl-d-aspartate (NMDA) in neurons promotes the deregulation of calcium influx, which leads to cellular death [88]. Thus, M1 phenotype exacerbates neuronal dysfunction by several different pathways, which include, at least, cytokine and ROS production and glutamate excitotoxicity [81, 88].
Furthermore, the concept of primed microglia is relevant to the understanding of its role in brain dysfunction during sepsis development. A primed microglia is able to make an exaggerated response to a typical stimulus [80]. Using the ME7 model of prion disease, Cunningham and colleagues demonstrated that the primed brain showed exaggerated response to several challenges, including LPS, poly I:C, IL-1β, and TNF-α [89–91]. This exaggerated response is phenotypically seen both in acute and chronic brain dysfunction after a systemic LPS challenge. This paradigm mimics what happens in the clinical setting. Both acute and chronic brain dysfunction is more severe/frequent in the aged or in patients wild mild cognitive impairment or clinical dementia; the clinical equivalent of the primed brain [92]. Little is known about the mechanisms of microglia activation during sepsis development. Our group recently demonstrated in an animal model of sepsis that the activation of microglia is crucial to acute brain inflammation and oxidative damage [44]. In this setting, microglia activation seems to be depend in part to the activation of CD40–CD40 ligand pathway. The activation of CD40 pathway drives microglia activation, brain inflammation, and oxidative damage and blood-brain barrier dysfunction. Using in vivo two-photon imaging in mice, Gyoneva et al., 2014 [93] showed that systemic inflammation affects the baseline morphology and dynamics of microglia 48 h after the initial stimuli. After systemic inflammation, microglia moved their processes at significantly higher mean speeds, which led to longer distances traveled, and this effect seems to be mediated by adenosine receptors.
Microglia activation is not only relevant to the acute phase of brain dysfunction after sepsis. In a model of endotoxemia, it was observed a long-lasting activation of microglia, but not astrocytes, and this was associated with decrease in expression of plasticity related genes and brain neurogenesis [94]. A single injection of LPS is able to induce microglia activation and inflammatory gene transcription, but not neuronal damage, as long as 2 months after the initial stimuli [78]. This was associated with disrupted synaptic structure and long-lasting behavioral deficits.
Is there a role for astrocytes in brain dysfunction after sepsis?
During CNS injury, astrocytes become reactive, migrate to the damaged site and form glial scar (reactive astrogliosis) [95]. In pathological conditions, a role for reactive astrogliosis as supportive or detrimental to neuronal survival remains undefined [96, 97]. Studies have shown that astrogliosis can have neuroprotective role by preserving bioenergetics [98] and trophic support [99]. In addition, astrocytes can prevent excitotoxicity [100, 101], decrease oxidative stress [102, 103], and apoptosis in neurons [104]. Astrocyte membranes contain numerous neurotransmitter receptors and transporters and can therefore sense and regulate formation, stability, and efficacy of synapses [105], and this was clearly demonstrated in dopaminergic neurons in vitro [106]. Studies suggest that by transforming from a basal to a reactive state, astrocytes neglect their supportive functions, thus rendering neurons vulnerable to neurotoxins, including proinflammatory cytokines and ROS [104].
Astrocytes seem to be also important to the maintenance of the BBB function [107–109]. They are suggested to regulate BBB permeability, water and ion exchange [110–112]. Presence of numerous astrocyte end-feet close to the BBB allows for a rapid regulation of BBB permeability [113]. Several recent studies have suggested a role for vascular factors in SAE-related injury of the brain vascular endothelium, changes in BBB permeability and microcirculatory dysfunction [114–116]. In this context, they can protect against neuroinflammation by invading T cells contributing to the immune privilege of the CNS [117]. Chapouly et al., 2015 [118] showed reactive astrocytes drive blood-brain barrier opening, via production of vascular endothelial growth factor A (VEGFA). In addition, thymidine phosphorylase (TYMP; previously known as endothelial cell growth factor 1, ECGF1) was identified as a second key astrocyte-derived permeability factor, which interacts with VEGFA to induce blood-brain barrier disruption [119]. Both are co-induced by NFκB1 in human astrocytes as a response to interleukin 1 beta (IL-1β), and inactivation of VEGFA in vivo potentiates TYMP induction. Nowadays, the BBB is understood as a complex regulated system. Terms such as neuro- or gliavascular unit (NVU, GVU) describe the strong influence of the microenvironment on the brain endothelium [120]. Neighboring cell types such as astrocytes, pericytes, microglia, or even neurons are known to influence the functionality of BBB in health as well as in disease, which is supported by their physical proximity and consequent small diffusion distances for signaling molecules [120, 121]. Astrocytes can also downregulate microglial activation by secretion of anti-inflammatory substances such as transforming growth factor (TGF) and prostaglandin E2 (PGE2) [122, 123], resembling a M2 phenotype, and may thereby limiting neuroinflammation.
Dysregulation of astroglial glutamate transporters has been implicated in neuroinflammation. Astrocytes are capable of modulating NMDAR activity through glutamate uptake transporters [121]. Dumont et al., 2014 [124] demonstrated the influence of inflammation on the control of glutamate transmission by astrocytes. Excitotoxic neuronal damage resulting from excessive glutamate is frequently associated with impaired handling of extracellular glutamate by astrocytes. Sepsis is associated with impaired glutamatergic transmission in brain, and inhibition of glutamate uptake by astrocytes through mechanisms that can be modulated by intracellular ascorbate [125]. Astrocytes take up DHAA (dehydroascorbic acid) and use it to synthesize ascorbate that is exported in response to increased glutamate concentrations [126].
Hernandes and colleagues (2014) [127] demonstrated that microglia and astrocytes were activated as many as 5 days after sepsis onset in the hippocampus and that this activation was dependent on the presence of functional Nox2. The results presented by Hernandes et al., 2014 [127] provide evidence that Nox2 is the main source of ROS involved in the oxidative damage to the hippocampus in SAE and that Nox2-derived ROS are determining factors for cognitive impairments after sepsis. They show the importance of Nox2-derived ROS as a central mechanism in glial cells activation and identify Nox2 as a potential target for future therapies to prevent SAE. Nox2 has been shown to regulate intracellular ROS levels in microglia and to result in both amplification of proinflammatory cytokines production and priming of microglia to additional stimuli [127]. Nox2 is essential for glial cell activation and emphasize the critical role of oxidative damage and Nox2-derived ROS as central factors contributing to acute and long-term brain dysfunction after sepsis [128].
In vitro experiments have recently shown that the stimulation of astrocytes with ligands for TLRs 2, 4, 5, or 6 enhances the production of ROS, IL-1β, IL-6, glutamate, and TNF-α, thereby favoring neuronal loss [129, 130]. Thus, activated astrocytes could display a neurotoxic behavior similar to that of activated microglia during inflammation of CNS [22]. An important and differential feature of activated astrocytes is the production of chemokines, including CCL2, CCL5, CCL20, CXCL10, CXCL12, CXCL1, CXCL2, and CX3CL1 [131]. These chemokines are involved in the recruitment of microglia, monocytes/macrophages, T cells and dendritic cells (DCs) into the inflamed sites of the CNS, thus favoring the formation of a more complex and long-lasting immune response during neuroinflammation [22]. Recently it was demonstrated that, as for microglia, astrocytes could also be primed [91]. Differently from microglia, primed astrocytes synthetize chemokines (CXCL1 and CCL2), resulting in markedly neutrophil, T cell and monocyte infiltration in the diseased brain. This suggests that primed astrocytes and microglia have different roles in brain inflammation during sepsis. These results point to a mixed constellation of inflammatory cells inducing brain damage during sepsis. There is an early role to activated/primed microglia that could be sustained for long periods of time after sepsis resolution. In addition, there is a second player that depends on the chemo attractive effects of activated astrocytes, and probably also depends on the breakdown of the BBB that are brain infiltration by peripheral inflammatory cells.
Blood-borne leukocytes and brain dysfunction
Monocyte-macrophage cells have the ability to phagocytize bacteria and interact with their products, resulting in the release of proinflammatory mediators, such as glutamate, free radicals, proteases, cytokines, leukotrienes, and nitric oxide [132–134] that could contribute to brain dysfunction [135, 136]. This can be further aggravated by the dysfunction of the BBB that is implicated in the pathogenesis of SAE [137]. Systemic-derived and brain-derived inflammatory mediators drive changes in the blood-brain barrier and help the influx of inflammatory cells and toxic mediators into the brain. We had previously demonstrated that activation of the brain microvasculature is an early event in an animal model of sepsis. There was an increase in leukocyte rolling and adhesion, as well as, cell migration into the brain that could contribute to brain inflammation after sepsis [138]. Activation of brain endothelial cells seems to be the main target of circulating inflammatory mediators to activate the brain circuits during systemic inflammation [139]. Recently, it was shown that after sepsis there was an increase in brain endothelial levels of CXCL1 and CX3CL1. This was dependent on leukocyte adhesion and purinergic signaling [140]. This signaling triggered microglia traffic to the injured site and could partially explain how systemic and brain inflammation communicates. In addition, endothelial cell activation seems to depend on estrogen signaling. Systemic LPS, TNF, or IFN administration affects endothelial cell function and BBB integrity only in males and reproductively senescent females but was not apparent in young females [141].
In addition, several immune components can actively cross the BBB via specific carrier inflammation during systemic inflammation [142]. Evidence from sepsis models suggests a role for C5a, and cytokines in the pathogenesis of the breakdown of the blood-brain barrier and the subsequent edema [143–146]. Thus, it seems that BBB dysfunction is a major fueling brain inflammation after sepsis. The mechanisms that drive BBB dysfunction are not fully understood, but include at least the activation of MMP-2, MM9, and MMP8 [147, 148]. These proteases are responsible to degrade key proteins that maintain the functionality of the BBB.
In addition to the innate immunity, lymphocytes could also be implicated in brain inflammation after systemic inflammation. In a neonate model of systemic inflammation, an important step of brain inflammation is the influx of the peripheral leukocytes through the choroid plexus. In this model, brain influx of leukocytes is dependent of an early onset of TH17-mediated immunity [149]. Actually, the choroid plexus has been considered as a possible route for cells to transfer to the central nervous system. Systemic inflammation stimulates expression of TLR and increases CSF cytokines and leukocytes levels, probably by affect blood-cerebrospinal fluid barrier (BCSFB) regulation [150]. This could be a double-edge sword, since deregulation of BCSFB was associated with worse signs of systemic inflammatory response syndrome in an animal model [148]. After systemic inflammation, the choroid plexus upregulated genes cluster into families implicated in immune-mediated cascades, in extracellular matrix remodeling, and in facilitating entry of cells into the cerebrospinal fluid, whereas those downregulated participate in maintenance of the barrier function [151, 152]. These findings reinforce the role for BCSFB in recruit blood-borne inflammatory cells, and in the amplification of brain.
Conclusions
Glial cells are important for the development of brain inflammation (Fig. 1). It seems reasonable to suppose that they play a central role in brain dysfunction during sepsis development. Furthermore, dysfunction of BBB and migration of blood-borne leukocytes take place in brain inflammation as well (Fig. 1). A more in depth understanding of the role of different subtypes of inflammatory cells and the specific role of each one is needed to further increase our understanding on the mechanisms of sepsis-associated brain dysfunction.
Fig. 1.
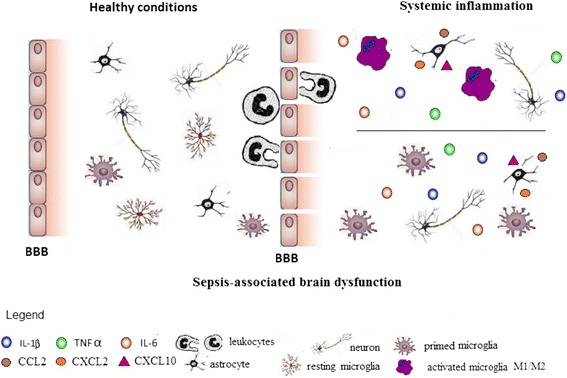
The transition from healthy to sepsis-associated brain dysfunction. In healthy conditions, resting microglia and astrocytes are predominant and are important to maintain several different brain functions. In addition, microglia could be primed, and in this context produces exaggerated levels of inflammatory cytokines in response to a stimulus. Systemic inflammation could activate both resting and primed microglia. Activated microglia could simultaneously exhibit M1 and M2 phenotypes. Despite the fact that classically M1 phenotype is associated with brain damage, during some conditions, the M2 phenotype could also be detrimental to brain function. In addition, astrocytes are also activated, lose the ability to maintain low the extracellular levels of glutamate, to maintain the integrity of the blood-brain barrier, and secrete chemokines. All these modifications are powerful stimuli to the chemotaxis of blood-borne leukocytes that also contribute to sepsis-associated brain dysfunction
Acknowledgements
This study was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Programa de Apoio aos Núcleos de Excelência (PRONEX–Project NENASC), Fundação de Apoio à Pesquisa do Estado de Santa Catarina (FAPESC) all from Brazil. M.M and A.V.S. received scholarships from CAPES and CNPq; T.B., J.Q., and F.D.P. are supported by research fellowships from CNPq.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MM and AVS drafted the manuscript, TB, JQ, and FDP have been involved in drafting the manuscript and revising it critically for important content. All authors read and approved the final version of the manuscript.
Contributor Information
Monique Michels, Email: moniquemichels@hotmail.com.
Amanda V. Steckert, Email: amandasteckert@yahoo.com.br
João Quevedo, Email: Joao.L.DeQuevedo@uth.tmc.edu.
Tatiana Barichello, Email: Tatiana.Barichello@uth.tmc.edu.
Felipe Dal-Pizzol, Email: fdpizzol@gmail.com.
References
- 1.Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644–1655. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 2.Vincent JL, Taccone F, Schmit X. Classification, incidence, and outcomes of sepsis and multiple organ failure. Contrib Nephrol. 2007;156:64–74. doi: 10.1159/000102071. [DOI] [PubMed] [Google Scholar]
- 3.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29(7):1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. New Eng J Med. 2003;348(16):1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 5.Dombrovskiy VY, Martin AA, Sunderram J, Paz HL. Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: a trend analysis from 1993 to 2003. Crit Care Med. 2007;35(5):1244–1250. doi: 10.1097/01.CCM.0000261890.41311.E9. [DOI] [PubMed] [Google Scholar]
- 6.Bozza FA, DÁvila JC, Ritter C, Sonneville R, Sharshar T, Dal-Pizzol F. Bioenergetics, mitochondrial dysfunction, and oxidative stress in the pathophysiology of septic encephalopathy. Shock. 2013;39(1):10–16. doi: 10.1097/SHK.0b013e31828fade1. [DOI] [PubMed] [Google Scholar]
- 7.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420(6917):885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 8.Mauri C, Bosma A. Immune regulatory function of B cells. Annual Rev Immunol. 2012;30:221–241. doi: 10.1146/annurev-immunol-020711-074934. [DOI] [PubMed] [Google Scholar]
- 9.Derek E, Dhanireddy K. Immunosuppression. Curr Opin Organ Transplant. 2012;17(6):616–618. doi: 10.1097/MOT.0b013e32835a7d3a. [DOI] [PubMed] [Google Scholar]
- 10.Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent JL, Ramsay G, SCCM/ESICM/ACCP/ATS/SIS 2001 International Sepsis Definitions Conference. Crit Care Med. 2003;31:1250–1256. doi: 10.1097/01.CCM.0000050454.01978.3B. [DOI] [PubMed] [Google Scholar]
- 11.Hopkins RO, Jackson JC. Long-term neurocognitive function after critical illness. Chest. 2006;130:869–878. doi: 10.1378/chest.130.3.869. [DOI] [PubMed] [Google Scholar]
- 12.Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H, Opal SM, Sevransky JE, Sprung CL, Douglas IS, Jaeschke R, Osborn TM, Nunnally ME, Towsend SR, Reinhart K, Kleinpell RM, Angus DC, Deutschman CS, Machado FR, Rubenfeld GD, Webb RJ, Beale RJ, Vicent JL, Moreno R. Surviving Sepsis Campaign Guidelines Committee including The Pediatric Subgroup. “Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock 2012. Crit Care Med. 2013;41:580–637. doi: 10.1097/CCM.0b013e31827e83af. [DOI] [PubMed] [Google Scholar]
- 13.Pytel P, Alexander JJ. Pathogenesis of septic encephalopathy. Curr Opin Neurol. 2009;22:283–287. doi: 10.1097/WCO.0b013e32832b3101. [DOI] [PubMed] [Google Scholar]
- 14.Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA. 2010;304:1787–1794. doi: 10.1001/jama.2010.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Semmler A, Widmann CN, Okulla T, Urbach H, Kaiser M, Widman G, Mormann F, Weide J, Fliessbach K, Hoeft A, Jessen F, Putensen C, Heneka MT. Persistent cognitive impairment, hippocampal atrophy and EEG changes in sepsis survivors. J Neurol Neurosurg Psychiatry. 2013;84:62–69. doi: 10.1136/jnnp-2012-302883. [DOI] [PubMed] [Google Scholar]
- 16.Gunther ML, Morandi A, Ely EW. Pathophysiology of delirium in the intensive care unit. Crit Care Clin. 2008;24:45–65. doi: 10.1016/j.ccc.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 17.Comim CM, Constantino LC, Barichello T, Streck EL, Quevedo J, Dal-Pizzol F. Cognitive impairment in the septic brain. Curr Neurovasc Res. 2009;6(3):194–203. doi: 10.2174/156720209788970045. [DOI] [PubMed] [Google Scholar]
- 18.Flierl MA, Rittirsch D, Huber-Lang MS, Stahel PF. Pathophysiology of septic encephalopathy—an unsolved puzzle. Crit Care. 2010;14:165. doi: 10.1186/cc9035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steckert AV, de Castro AA, Quevedo J, Dal-Pizzol F. Sepsis in the central nervous system and antioxidant strategies with N-acetylcysteine, vitamins and statins. Curr Neurovasc Res. 2014;11(1):83–90. doi: 10.2174/1567202610666131211111012. [DOI] [PubMed] [Google Scholar]
- 20.Kamel H, Iadecola C. Brain-immune interactions and ischemic stroke: clinical implications. Arch Neurol. 2012;69:576–581. doi: 10.1001/archneurol.2011.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quan N, Banks WA. Brain-immune communication pathways. Brain Behav Immun. 2007;21:727–735. doi: 10.1016/j.bbi.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 22.González H, Elgueta D, Montoya A, Pacheco R. Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J Neuroimmunol. 2014;274:1–13. doi: 10.1016/j.jneuroim.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 23.Chen Z, Trapp B. Microglia and neuroprotection. J Neurochem. 2015 doi: 10.1111/jnc.13062. [DOI] [PubMed] [Google Scholar]
- 24.Comim CM, Cassol-Jr OJ, Constantino LS, Felisberto F, Petronilho F, Rezin GT, Scaini G, Daufenbach JF, Streck EL, Quevedo J, Dal-Pizzol F. Alterations in inflammatory mediators, oxidative stress parameters and energetic metabolism in the brain of sepsis survivor rats. Neurochem Res. 2011;36(2):304–311. doi: 10.1007/s11064-010-0320-2. [DOI] [PubMed] [Google Scholar]
- 25.Tuon L, Comim CM, Petronilho F, Barichello T, Izquierdo I, Quevedo J, Dal-Pizzol F. Memory-enhancing treatments reverse the impairment of inhibitory avoidance retention in sepsis-surviving rats. Crit Care. 2008;12(5):R133. doi: 10.1186/cc7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steckert AV, Comim CM, Mina F, Mendonça BP, Dominguini D, Ferreira GK, Carvalho-Silva M, Vieira JS, Streck EL, Quevedo J, Dal-Pizzol F. Late brain alterations in sepsis-survivor rats. Synapse. 2013;67(11):786–793. doi: 10.1002/syn.21686. [DOI] [PubMed] [Google Scholar]
- 27.Götz T, Gunther A, Witte OW, Brunkhorst FM, Seidel G, Hamzei F. Long-term sequelae of severe sepsis: cognitive impairment and structural brain alterations—an MRI study (LossCog MRI) BMC Neurology. 2014;14:145. doi: 10.1186/1471-2377-14-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang HJ, Lynm C, Glass RM. JAMA patient page. Sepsis. JAMA. 2010;304(16):1856. doi: 10.1001/jama.304.16.1856. [DOI] [PubMed] [Google Scholar]
- 29.Cunningham C, Hennessy E. Co-morbidity and systemic inflammation as drivers of cognitive decline: new experimental models adopting a broader paradigm in dementia research. Alzheimers Res Ther. 2015;7:33. doi: 10.1186/s13195-015-0117-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fong TG, Davis D, Growdon ME, Albuquerque A, Inouye SK. The interface between delirium and dementia in elderly adults. Lancet Neurol. 2015;14:823–832. doi: 10.1016/S1474-4422(15)00101-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Widmann CN, Heneka MT. Long-term cerebral consequences of sepsis. Lancet Neurol. 2014;13:630–636. doi: 10.1016/S1474-4422(14)70017-1. [DOI] [PubMed] [Google Scholar]
- 32.Feng SY, Samarasinghe T, Phillips DJ, Alexiou T, Hollis JH, Yu VY, Walker AM. Acute and chronic effects of endotoxin on cerebral circulation in lambs. Am J Physiol Regul Integr Comp Physiol. 2010;298(3):760–766. doi: 10.1152/ajpregu.00398.2009. [DOI] [PubMed] [Google Scholar]
- 33.Taccone FS, Su F, De Deyne C, Abdellhai A, Pierrakos C, He X, Donadello K, Dewitte O, Vincent JL, De Backer D. Sepsis is associated with altered cerebral microcirculation and tissue hypoxia in experimental peritonitis. Crit Care Med. 2014;42(2):114–122. doi: 10.1097/CCM.0b013e3182a641b8. [DOI] [PubMed] [Google Scholar]
- 34.Taccone FS, Su F, Pierrakos C, He X, James S, Dewitte O, Vincent JL, De Backer D. Cerebral microcirculation is impaired during sepsis: an experimental study. Crit Care. 2010;14(4):R140. doi: 10.1186/cc9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berg RM, Plovsing RR, Ronit A, Bailey DM, Holstein-Rathlou NH, Møller K. Disassociation of static and dynamic cerebral autoregulatory performance in healthy volunteers after lipopolysaccharide infusion and in patients with sepsis. Am J Physiol Regul Integr Comp Physiol. 2012;303(11):R1127–R1135. doi: 10.1152/ajpregu.00242.2012. [DOI] [PubMed] [Google Scholar]
- 36.Kipnis J, Cohen H, Cardon M, Ziv Y, Schwartz M. T cell deficiency leads to cognitive dysfunction: implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc Natl Acad Sci U S A. 2004;101:8180–8185. doi: 10.1073/pnas.0402268101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kipnis J, Gadani S, Derecki NC. Pro-cognitive properties of T cells. Nat Rev Immunol. 2012;12:663–669. doi: 10.1038/nri3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolf SA, Steiner B, Akpinarli A, Kammertoens T, Nassenstein C, Braun A, Blankenstein T, Kempermann G. CD4-positive T lymphocytes provide a neuroimmunological link in the control of adult hippocampal neurogenesis. J Immunol. 2009;182:3979–3984. doi: 10.4049/jimmunol.0801218. [DOI] [PubMed] [Google Scholar]
- 39.Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 40.Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, Cohen H, Kipnis J, Schwartz M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosc. 2006;9:268–275. doi: 10.1038/nn1629. [DOI] [PubMed] [Google Scholar]
- 41.Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- 42.Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ, Lee SJ. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013;4:1562. doi: 10.1038/ncomms2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci. 2009;29:11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michels M, Vieira AS, Vuolo F, Zapelini HG, Mendonça BP, Mina F, Dominguini D, Steckert AV, Schuck PF, Quevedo J, Petronilho F, Dal-Pizzol F. The role of microglia activation in the development of sepsis-induced long-term cognitive impairment. Brain Behav Immun. 2015;43:54–59. doi: 10.1016/j.bbi.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 45.Lawson LJ, Perry V, Dri PH, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39(1):151–170. doi: 10.1016/0306-4522(90)90229-W. [DOI] [PubMed] [Google Scholar]
- 46.Garden GA, Moller T. Microglia biology in health and disease. J Neuroimmune Pharmacol. 2006;1(2):127–137. doi: 10.1007/s11481-006-9015-5. [DOI] [PubMed] [Google Scholar]
- 47.Michels M, Danielski LG, Dal-Pizzol F, Petronilho F. Neuroinflammation: microglial activation during sepsis. Curr Neurovasc Res. 2014;11(3):262–270. doi: 10.2174/1567202611666140520122744. [DOI] [PubMed] [Google Scholar]
- 48.Colton CA, Wilcock DM. Assessing Activation States in Microglia. CNS Neurol Disord Drug Targets. 2010;9:174–191. doi: 10.2174/187152710791012053. [DOI] [PubMed] [Google Scholar]
- 49.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 50.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 51.Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am J Pathol. 2010;177:2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25:9275–9284. doi: 10.1523/JNEUROSCI.2614-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4(4):399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fenn AM, Hall JC, Gensel JC, Popovich PG, Godbout JP. IL-4 signaling drives a unique arginase+/IL-1beta+ microglia phenotype and recruits macrophages to the inflammatory CNS: consequences of age-related deficits in IL-4Ralpha after traumatic spinal cord injury. J Neurosci. 2014;34:8904–8917. doi: 10.1523/JNEUROSCI.1146-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurotherapeutics. 2010;7:366–377. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen Z, Jalabi W, Shpargel KB, Farabaugh KT, Dutta R, Yin X, Kidd GJ, Bergmann CC, Stohlman SA, Trapp BD. Lipopolysaccharide induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci. 2012;32:11706–11715. doi: 10.1523/JNEUROSCI.0730-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJ, Ffrench-Constant C. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16:1211–1218. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shechter R, Miller O, Yovel G, Rosenzweig N, London A, Ruckh J, Kim KW, Klein E, Kalchenko V, Bendel P, Lira SA, Jung S, Schwartz M. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity. 2013;38:555–569. doi: 10.1016/j.immuni.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sandiego CM, Gallezot J, Pittman B, Nabulsi N, Lim K, Lin S, Matuskey D, Lee J, O’Connor KC, Huang Y, Carson RE, Hannestad J, Cosgrove KP. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. PNAS. 2015;112:12468–12473. doi: 10.1073/pnas.1511003112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mecha M, Feliú A, Carrillo-Salinas FJ, Rueda-Zubiaurre A, Ortega-Gutiérrez S, de Sola RG, Guaza C. Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav Immu. 2015;49:233–245. doi: 10.1016/j.bbi.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 63.Pan J, Jin JL, Ge HM, Yin KL, Chen X, Han LJ, Chen Y, Qian L, Li XX, Xu Y. Malibatol A regulates microglia M1/M2 polarization in experimental stroke in a PPARγ-dependent manner. J Neuroinflammation. 2015;14(12):51. doi: 10.1186/s12974-015-0270-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Won S, Lee JK, Stein DG. Recombinant tissue plasminogen activator promotes, and progesterone attenuates, microglia/macrophage M1 polarization and recruitment of microglia after MCAO stroke in rats. Brain Behav Immun. 2015;S0889–1591(15):00160–00169. doi: 10.1016/j.bbi.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 65.Siesjo BK, Agardh CD, Bengtsson F. Free radicals and brain damage. Cerebrovasc. Brain Metab Rev. 1989;1:165–211. [PubMed] [Google Scholar]
- 66.Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF. Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab. 2003;23:629–652. doi: 10.1097/01.WCB.0000073905.87928.6D. [DOI] [PubMed] [Google Scholar]
- 67.Ferrari CC, Pott Godoy MC, Tarelli R, Chertoff M, Depino AM, Pitossi FJ. Progressive neurodegeneration and motor disabilities induced by chronic expression of IL-1beta in the substantia nigra. Neurobiology Dis. 2006;24:183–193. doi: 10.1016/j.nbd.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 68.Gordon R, Anantharam V, Kanthasamy AG, Kanthasamy A. Proteolytic activation of proapoptotic kinase protein kinase Cdelta by tumor necrosis factor alpha death receptor signaling in dopaminergic neurons during neuroinflammation. J Neuroinflammation. 2012;9:82. doi: 10.1186/1742-2094-9-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reale M, Iarlori C, Thomas A, Gambi D, Perfetti B, Di Nicola M, Onofrj M. Peripheral cytokines profile in Parkinson's disease. Brain Beh Immunity. 2009;23:55–63. doi: 10.1016/j.bbi.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 70.Patel BN, Dunn RJ, Jeong SY, Zhu Q, Julien JP, David S. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J Neurosci. 2002;22:6578–6586. doi: 10.1523/JNEUROSCI.22-15-06578.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jacobs RA, Satta MA, Dahia PL, Chew SL, Grossman AB. Induction of nitric oxide synthase and interleukin-1beta, but not heme oxygenase, messenger rna in rat brain following peripheral administration of endotoxin. Brain Res Mol Brain Res. 1997;49(1–2):238–246. doi: 10.1016/S0169-328X(97)00150-2. [DOI] [PubMed] [Google Scholar]
- 72.Wong ML, Rettori V, al-Shekhlee A, Bongiorno PB, Canteros G, McCann SM, Gold PW, Licinio J. Inducible nitric oxide synthase gene expression in the brain during systemic inflammation. Nat Med. 1996;2(5):581–584. doi: 10.1038/nm0596-581. [DOI] [PubMed] [Google Scholar]
- 73.Heneka MT, Loschmann PA, Gleichmann M, Weller M, Schulz JB, Wullner U, Klockgether T. Induction of nitric oxide synthase and nitric oxide-mediated apoptosis in neuronal PC12 cells after stimulation with tumor necrosis factor-alpha/lipopolysaccharide. J Neurochem. 1998;71(1):88–94. doi: 10.1046/j.1471-4159.1998.71010088.x. [DOI] [PubMed] [Google Scholar]
- 74.Peterson PK, Hu S, Anderson WR, Chao CC. Nitric oxide production and neurotoxicity mediated by activated microglia from human versus mouse brain. J Infect Dis. 1994;170(2):457–460. doi: 10.1093/infdis/170.2.457. [DOI] [PubMed] [Google Scholar]
- 75.Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol. 1992;149(8):2736–2741. [PubMed] [Google Scholar]
- 76.Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci. 2001;21(17):6480–6491. doi: 10.1523/JNEUROSCI.21-17-06480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Semmler A, Okulla T, Sastre M, Dumitrescu-Ozimek L, Heneka MT. Systemic inflammation induces apoptosis with variable vulnerability of different brain regions. J Chem Neuroanat. 2005;30:144–157. doi: 10.1016/j.jchemneu.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 78.Weberpals M, Hermes M, Hermann S, Kummer MP, Terwel D, Semmler A, Berger M, Schäfers M, Heneka MT. NOS2 gene deficiency protects from sepsis-induced long-term cognitive deficits. J Neurosci. 2009;29(45):14177–14184. doi: 10.1523/JNEUROSCI.3238-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cassol-Jr OJ, Comim CM, Constantino LS, Rosa DV, Mango LA, Stertz L, Kapczinski F, Romano-Silva MA, Quevedo J, Dal-Pizzol F. Acute low dose of MK-801 prevents memory deficits without altering hippocampal DARPP-32 expression and BDNF levels in sepsis survivor rats. J Neuroimmunol. 2011;230(1–2):48–51. doi: 10.1016/j.jneuroim.2010.08.026. [DOI] [PubMed] [Google Scholar]
- 80.Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia. 2013;61:71–90. doi: 10.1002/glia.22350. [DOI] [PubMed] [Google Scholar]
- 81.Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, Sonobe Y, Mizuno T, Suzumura A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006;281:21362–21368. doi: 10.1074/jbc.M600504200. [DOI] [PubMed] [Google Scholar]
- 82.Alessandri B, Bullock R. Glutamate and its receptors in the pathophysiology of brain and spinal cord injuries. Prog Brain Res. 1998;116:303–330. doi: 10.1016/S0079-6123(08)60445-8. [DOI] [PubMed] [Google Scholar]
- 83.Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- 84.Fountain NB. Status epilepticus: risk factors and complications. Epilepsia. 2000;41(2):S23–S30. doi: 10.1111/j.1528-1157.2000.tb01521.x. [DOI] [PubMed] [Google Scholar]
- 85.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1998;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 86.Schwartz M, Shaked I, Fisher J, Mizrahi T, Schori H. Protective autoimmunity against the enemy within: fighting glutamate toxicity. Trends Neurosci. 2003;26:297–302. doi: 10.1016/S0166-2236(03)00126-7. [DOI] [PubMed] [Google Scholar]
- 87.Takeuchi K, Mizuno T, Zhang G, Wang J, Kawanokuchi J, Kuno R, Suzumura A. Neuritic beading induced by activated microglia is an early feature of neuronal dysfunction toward neuronal death by inhibition of mitochondrial respiration and axonal transport. J Biol Chem. 2005;280:10444–10454. doi: 10.1074/jbc.M413863200. [DOI] [PubMed] [Google Scholar]
- 88.Zou JY, Crews FT. TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res. 2005;1034:11–24. doi: 10.1016/j.brainres.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 89.Cunningham C, Campion S, Lunnon K, Murray CL, Woods JF, Deacon RM, Rawlins JN, Perry VH. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Pscychiatry. 2009;65:304–312. doi: 10.1016/j.biopsych.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Field R, Campion S, Warren C, Murray C, Cunningham C. Systemic challenge with the TLR3 agonist poly I:C induces amplified IFNalpha/beta and IL-1beta responses in the diseased brain and exacerbates chronic neurodegeneration. Brain Behac Immunol. 2010;24:996–1007. doi: 10.1016/j.bbi.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hennessy E, Griffin EW, Cunningham C. Astrocytes are primed by chronic neurodegeneration to produce exaggerated chemokine and cell infiltration responses to acute stimulation with the cytokines IL-1# and TNF-&UF061. J Neurosci. 2015;35:8411–8422. doi: 10.1523/JNEUROSCI.2745-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.van Gool WA, van de Beek D, Eikelenboom P. Systemic infection and delirium: when cytokines and acetylcholine collide. Lancet. 2010;375:773–775. doi: 10.1016/S0140-6736(09)61158-2. [DOI] [PubMed] [Google Scholar]
- 93.Gyoneva S, Davalos D, Biswas D, Swanger SA, Garnier-Amblard E, Loth F, Akassoglou K, Traynelis SF. Systemic inflammation regulates microglial responses to tissue damage in vivo. Glia. 2014;62(8):1345–1360. doi: 10.1002/glia.22686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Anderson TS, Commins S, Moynagh PN, Coogan AN. Lipopolysaccharide-induced sepsis induces long-lasting affective changes in the mouse. Brain Behav Immun. 2015;43:98–109. doi: 10.1016/j.bbi.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 95.Pekny M, Wilhelmsson U, Pekna M. The dual role of astrocyte activation and reactive gliosis. Neurosci Lett. 2014;565:30–38. doi: 10.1016/j.neulet.2013.12.071. [DOI] [PubMed] [Google Scholar]
- 96.O’Callaghan JP, Sriram K. Glial fibrillary acidic protein and related glial proteins as biomarkers of neurotoxicity. Exp Opin Drug Saf. 2005;4:433–442. doi: 10.1517/14740338.4.3.433. [DOI] [PubMed] [Google Scholar]
- 97.Zhang D, Hu X, Qian L, O’Callaghan JP, Hong JS. Astrogliosis in CNS pathologies: is there a role for microglia? Mol Neurobiol. 2010;41:232–241. doi: 10.1007/s12035-010-8098-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kajihara H, Tsutsumi E, Kinoshita A, Nakano J, Takagi K, Takeo S. Activated astrocytes with glycogen accumulation in ischemic penumbra during the early stage of brain infarction: immunohistochemical and electron microscopic studies. Brain Res. 2001;909:92–101. doi: 10.1016/S0006-8993(01)02640-3. [DOI] [PubMed] [Google Scholar]
- 99.Goss JR, O’Malley ME, Zou L, Styren SD, Kochanek PM, DeKosky ST. Astrocytes are the major source of nerve growth factor upregulation following traumatic brain injury in the rat. Exp Neurol. 1998;149:301–309. doi: 10.1006/exnr.1997.6712. [DOI] [PubMed] [Google Scholar]
- 100.Chorna NE, Santiago-Perez LI, Erb L, Seye CI, Neary JT, Sun GY, Weisman GA, Gonzalez FA. P2Y receptors activate neuroprotective mechanisms in astrocytic cells. J Neurochem. 2004;91:119–132. doi: 10.1111/j.1471-4159.2004.02699.x. [DOI] [PubMed] [Google Scholar]
- 101.Hansson E, Muyderman H, Leonova J, Allansson L, Sinclair J, Blomstrand F, Thorlin T, Nilsson M, Ronnback L. Astroglia and glutamate in physiology and pathology: aspects on glutamate transport, glutamateinduced cell swelling and gap-junction communication. Neurochem Int. 2000;7:317–329. doi: 10.1016/S0197-0186(00)00033-4. [DOI] [PubMed] [Google Scholar]
- 102.Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/S0301-0082(99)00060-X. [DOI] [PubMed] [Google Scholar]
- 103.Gegg ME, Beltran B, Salas-Pino S, Bolanos JP, Clark JB, Moncada S, Heales SJ. Differential effect of nitric oxide on glutathione metabolism and mitochondrial function in astrocytes and neurones: implications for neuroprotection/neurodegeneration? J Neurochem. 2003;86:228–237. doi: 10.1046/j.1471-4159.2003.01821.x. [DOI] [PubMed] [Google Scholar]
- 104.Nakase T, Sohl G, Theis M, Willecke K, Naus CC. Increased apoptosis and inflammation after focal brain ischemia in mice lacking connexin43 in astrocytes. Am J Pathol. 2004;164:2067–2075. doi: 10.1016/S0002-9440(10)63765-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fields RD, Stevens-Graham B. Neuroscience: new insights into neuron-glia communication. Science. 2002;298(5593):556–562. doi: 10.1126/science.298.5593.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mena MA, García de Yébenes J. Glial cells as players in parkinsonism: the “good”, the “bad”, and the “mysterious” glia. Neuroscientist. 2008;14(6):544–560. doi: 10.1177/1073858408322839. [DOI] [PubMed] [Google Scholar]
- 107.Fuller S, Münch G, Steele M. Activated astrocytes: a therapeutic target in Alzheimer's disease? Expert Rev Neurother. 2009;9(11):1585–1594. doi: 10.1586/ern.09.111. [DOI] [PubMed] [Google Scholar]
- 108.Janzer RC, Raff MC. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature. 1987;325:253–257. doi: 10.1038/325253a0. [DOI] [PubMed] [Google Scholar]
- 109.Neuhaus J, Risau W, Wolburg H. Induction of blood-brain barrier characteristics in bovine brain endothelial cells by rat astroglial cells in transfilter coculture. Ann NY Acad Sci. 1991;633:578–580. doi: 10.1111/j.1749-6632.1991.tb15667.x. [DOI] [PubMed] [Google Scholar]
- 110.Ronaldson PT, Davis TP. Blood-brain barrier integrity and glial support: mechanisms that can be targeted for novel therapeutic approaches in stroke. Curr Pharm Des. 2012;18:3624–3644. doi: 10.2174/138161212802002625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 112.Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;1(16):1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 113.Mathiisen TM, Lehre KP, Danbolt NC, Ottersen OP. The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia. 2010;1(58):1094–1103. doi: 10.1002/glia.20990. [DOI] [PubMed] [Google Scholar]
- 114.David S, Kumpers P, van Slyke P, Parikh SM. Mending leaky blood vessels: the angiopoietin-Tie2 pathway in sepsis. J Pharmacol Exp Ther. 2013;345:2–6. doi: 10.1124/jpet.112.201061. [DOI] [PubMed] [Google Scholar]
- 115.Ince C, Sinaasappel M. Microcirculatory oxygenation and shunting in sepsis and shock. Crit Care Med. 1999;27:1369–1377. doi: 10.1097/00003246-199907000-00031. [DOI] [PubMed] [Google Scholar]
- 116.Hernandez G, Bruhn A, Ince C. Microcirculation in sepsis: new perspectives. Curr Vasc Pharmacol. 2013;11:161–169. [PubMed] [Google Scholar]
- 117.Gimsa U, Mitchison NA, Brunner-Weinzierl MC. Immune privilege as an intrinsic CNS property: astrocytes protect the CNS against T-cell-mediated neuroinflammation. Mediators Inflamm. 2013;1(2013):320519. doi: 10.1155/2013/320519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chapouly C, Tadesse Argaw A, Horng S, Castro K, Zhang J, Asp L, Loo H, Laitman BM, Mariani JN, Straus Farber R, Zaslavsky E, Nudelman G, Raine CS, John GR. Astrocytic TYMP and VEGFA drive blood-brain barrier opening in inflammatory central nervous system lesions. Brain. 2015;1.138(Pt 6):1548–1567. doi: 10.1093/brain/awv077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Neuhaus W, Gaiser F, Mahringer A, Franz J, Riethmüller C, Förster C. The pivotal role of astrocytes in an in vitro stroke model of the blood-brain barrier. Front Cell Neurosci. 2014;28(8):352. doi: 10.3389/fncel.2014.00352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cecchelli R, Berezowski V, Lundquist S, Culot M, Renftel M, Dehouck MP, Fenart L. Modelling of the blood-brain barrier in drug discovery and development. Nat Rev Drug Discov. 2007;1(6):650–661. doi: 10.1038/nrd2368. [DOI] [PubMed] [Google Scholar]
- 121.Zimmer ER, Torrez VR, Kalinine E, Augustin MC, Zenki KC, Almeida RF, Hansel G, Muller AP, Souza DO, Machado-Vieira R, Portela LV. Long-term NMDAR antagonism correlates reduced astrocytic glutamate uptake with anxiety-like phenotype. Front Cell Neurosci. 2015;3(9):219. doi: 10.3389/fncel.2015.00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Font-Nieves M, Sans-Fons MG, Gorina R, Bonfill-Teixidor E, Salas-Pérdomo A, Márquez-Kisinousky L, Santalucia T, Planas AM. Induction of COX-2 enzyme and down-regulation of COX-1 expression by lipopolysaccharide (LPS) control prostaglandin E2 production in astrocytes. J Biol Chem. 2012;287(9):6454–6468. doi: 10.1074/jbc.M111.327874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vincent VA, Tilders FJ, van Dam AM. Inhibition of endotoxin-induced nitric oxide synthase production in microglial cells by the presence of astroglial cells: a role for transforming growth factor beta. Glia. 1997;1(19):190–198. doi: 10.1002/(SICI)1098-1136(199703)19:3<190::AID-GLIA2>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 124.Dumont AO, Goursaud S, Desmet N, Hermans E. Differential regulation of glutamate transporter subtypes by pro-inflammatory cytokine TNF-α in cortical astrocytes from a rat model of amyotrophic lateral sclerosis. PLoS One. 2014;9(5):e97649. doi: 10.1371/journal.pone.0097649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Korcok J, Wu F, Tyml K, Hammond RR, Wilson JX. Sepsis inhibits reduction of dehydroascorbic acid and accumulation of ascorbate in astroglial cultures: intracellular ascorbate depletion increases nitric oxide synthase induction and glutamate uptake inhibition. J Neurochem. 2002;1.81(1):185–193. doi: 10.1046/j.1471-4159.2002.00814.x. [DOI] [PubMed] [Google Scholar]
- 126.Wilson JX, Dragan M. Sepsis inhibits recycling and glutamate-stimulated export of ascorbate by astrocytes. Free Radic Biol Med. 2005;39(8):990–998. doi: 10.1016/j.freeradbiomed.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 127.Hernandes MS, D’Avila JC, Trevelin SC, Reis PA, Kinjo ER, Lopes LR, Castro-Faria-Neto HC, Cunha F, Britto LRG, Bozza FA. The role of Nox2-derived ROS in the development of cognitive impairment after sepsis. J Neuroinflammation. 2014;11:36. doi: 10.1186/1742-2094-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Surace MJ, Block ML. Targeting microglia-mediated neurotoxicity: the potential of NOX2 inhibitors. Cell Mol Life Sci. 2012;1.69(14):2409–2427. doi: 10.1007/s00018-012-1015-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ma D, Jin S, Li E, Doi Y, Parajuli B, Noda M, Sonoda Y, Mizuno T, Suzumura A. The neurotoxic effect of astrocytes activated with toll-like receptor ligands. J Neuroimmun. 2013;254:10–18. doi: 10.1016/j.jneuroim.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 130.Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer’s disease, role of cytokines. Sci World J. 2012;2012:756357. doi: 10.1100/2012/756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 132.De Pablo R, Monserrat J, Reyes E, Diaz-Martin D, Rodrigues Zapata M, Carballo F, de la Hera A, Pietro A, Alvarez-Mon M. Mortality in patients with septic shock correlates with anti-inflammatory but not proinflammatory immunomodulatory molecules. J Int Care Med. 2011;26(2):125–132. doi: 10.1177/0885066610384465. [DOI] [PubMed] [Google Scholar]
- 133.Salomao R, Brunialti MKC, Rapozo MM, Baggio-Zappia GL, Galanos C, Freudenberg M. Bacterial sensing, cell signaling, and modulation of the immune response during sepsis. Shock. 2012;38(3):227–242. doi: 10.1097/SHK.0b013e318262c4b0. [DOI] [PubMed] [Google Scholar]
- 134.Douglas JJ, Tsang JL, Walley KR. Sepsis and the innate-like response. Int Care Med. 2012;40(2):249–251. doi: 10.1007/s00134-013-3141-3. [DOI] [PubMed] [Google Scholar]
- 135.Kremlev SG, Roberts RL, Palmer C. Differential expression of chemokines and chemokine receptors during microglial activation and inhibition. J Neuroimmunol. 2004;149:1–9. doi: 10.1016/j.jneuroim.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 136.Sayyah M, Najafabadi IT, Beheshti S, Majzoob S. Lipopolysaccharide retards development of amygdala kindling but does not affect fully-kindled seizures in rats. Epilepsy Res. 2003;57:175–180. doi: 10.1016/j.eplepsyres.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 137.Deng YY, Fang M, Zhu GF, Zhou Y, Zeng HK. Role of microglia in the pathogenesis of sepsis-associated encephalopathy. CNS Neurol Disord Drug Targets. 2013;12:720–725. doi: 10.2174/18715273113126660178. [DOI] [PubMed] [Google Scholar]
- 138.Comim CM, Vilela MC, Constantino LS, Petronilho F, Vuolo F, Lacerda-Queiroz N, Rodrigues DH, da Rocha JL, Teixeira AL, Quevedo J, Dal-Pizzol F. Traffic of leukocytes and cytokine up-regulation in the central nervous system in sepsis. Intensive Care Med. 2011;37(4):711–718. doi: 10.1007/s00134-011-2151-2. [DOI] [PubMed] [Google Scholar]
- 139.Gosselin D, Rivest S. MyD88 signaling in brain endothelial cells is essential for the neuronal activity and glucocorticoid release during systemic inflammation. Mol Psychiatry. 2008;13(5):480–497. doi: 10.1038/sj.mp.4002122. [DOI] [PubMed] [Google Scholar]
- 140.Wang H, Hong LJ, Huang JY, Jiang Q, Tao RR, Tan C, Lu NN, Wang CK, Ahmed MM, Lu YM, Liu ZR, Shi WX, Lai EY, Wilcox CS, Han F. P2RX7 sensitizes Mac-1/ICAM-1-dependent leukocyte-endothelial adhesion and promotes neurovascular injury during septic encephalopathy. Cell Res. 2015;25(6):674–690. doi: 10.1038/cr.2015.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Maggioli E, McArthur S, Mauro C, Kieswich J, Kusters DH, Reutelingsperger CP, Yaqoob M, Solito E. Estrogen protects the blood-brain barrier from inflammation-induced disruption and increased lymphocyte trafficking. Brain Behav Immun. 2015 doi: 10.1016/j.bbi.2015.08.020. [DOI] [PubMed] [Google Scholar]
- 142.Pan W, Stone KP, Hsuchou H, Manda VK, Zhang Y, Kastin AJ. Cytokine signaling modulates blood-brain barrier function. Curr Pharm Des. 2011;17(33):3729–3740. doi: 10.2174/138161211798220918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mina F, Comim CM, Dominguini D, Cassol-Jr OJ, Dall’Igna DM, Ferreira GK, Silva MC, Galant LS, Streck EL, Quevedo J, Dal-Pizzol F. Il1-β involvement in cognitive impairment after sepsis. Mol Neurobiol. 2014;49(2):1069–1076. doi: 10.1007/s12035-013-8581-9. [DOI] [PubMed] [Google Scholar]
- 144.Lee EJ, Han JE, Woo MS, Shin JA, Park EM, Kang JL, Moon PG, Baek MC, Son WS, Ko YT, Choi JW, Kim HS. Matrix metalloproteinase-8 plays a pivotal role in neuroinflammation by modulating TNF-α activation. J Immunol. 2014;193(5):2384–2393. doi: 10.4049/jimmunol.1303240. [DOI] [PubMed] [Google Scholar]
- 145.Tsuge M, Yasui K, Ichiyawa T, Saito Y, Nagaoka Y, Yashiro M, Yamashita N, Morishima T. Increase of tumor necrosis factor-alpha in the blood induces early activation of matrix metalloproteinase-9 in the brain. Microbiol Immunol. 2010;54(7):417–424. doi: 10.1111/j.1348-0421.2010.00226.x. [DOI] [PubMed] [Google Scholar]
- 146.Flierl MA, Stahel PF, Rittirsch D, Huber-Lang M, Niederbichler AD, Hoesel LM, Touban BM, Morgan SJ, Smith WR, Ward PA, Ipaktchi K. Inhibition of complement C5a prevents breakdown of the blood-brain barrier and pituitary dysfunction in experimental sepsis. Crit Care. 2009;13(1):R12. doi: 10.1186/cc7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Dal-Pizzol F, Rojas HA, dos Santos EM, Vuolo F, Constantino L, Feier G, Pasquali M, Comim CM, Petronilho F, Gelain DP, Quevedo J, Moreira JC, Ritter C. Matrix metalloproteinase-2 and metalloproteinase-9 activities are associated with blood-brain barrier dysfunction in an animal model of severe sepsis. Mol Neurobiol. 2013;48(1):62–70. doi: 10.1007/s12035-013-8433-7. [DOI] [PubMed] [Google Scholar]
- 148.Vandenbroucke RE, Dejonckheere E, Van Lint P, Demeestere D, Van Wonterghem E, Vanlaere I, Puimège L, Van Hauwermeiren F, De Rycke R, Mc Guire C, Campestre C, López-Otin C, Matthys P, Leclercq G, Libert C. Matrix metalloprotease 8-dependent extracellular matrix cleavage at the blood-CSF barrier contributes to lethality during systemic inflammatory diseases. J Neurosci. 2012;32(29):9805–9816. doi: 10.1523/JNEUROSCI.0967-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yang D, Sun YY, Bhaumik SK, Li Y, Baumann JM, Lin X, Zhang Y, Lin SH, Dunn RS, Liu CY, Shie FS, Lee YH, Wills-Karp M, Chougnet CA, Kallapur SG, Lewkowich IP, Lindquist DM, Murali-Krishna K, Kuan CY. Blocking lymphocyte trafficking with FTY720 prevents inflammation-sensitized hypoxic-ischemic brain injury in newborns. J Neurosci. 2014;34(49):16467–16481. doi: 10.1523/JNEUROSCI.2582-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Stridh L, Ek CJ, Wang X, Nilsson H, Mallard C. Regulation of Toll-like receptors in the choroid plexus in the immature brain after systemic inflammatory stimuli. Transl Stroke Res. 2013;4(2):220–227. doi: 10.1007/s12975-012-0248-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Marques F, Sousa JC, Coppola G, Falcao AM, Rodrigues AJ, Geschwind DH, Sousa N, Correia-Neves M, Palha JÁ. Kinetic profile of the transcriptome changes induced in the choroid plexus by peripheral inflammation. J Cereb Blood Flow Metab. 2009;29(5):921–932. doi: 10.1038/jcbfm.2009.15. [DOI] [PubMed] [Google Scholar]
- 152.Marques F, Sousa JC, Coppola G, Geschwind DH, Sousa N, Palha JA, Correia-Neves M. The choroid plexus response to a repeated peripheral inflammatory stimulus. BMC Neurosci. 2009;10:135. doi: 10.1186/1471-2202-10-135. [DOI] [PMC free article] [PubMed] [Google Scholar]