

Abstract
Key points
Local regulation of vascular resistance adjusts coronary blood flow to metabolic demand, although the mechanisms involved are not comprehensively understood
We show that heart tissue surrounding rat coronary arteries releases diffusible factors that regulate vasoconstriction and relaxation
Perivascular tissue reduces rho‐kinase‐dependent smooth muscle Ca2+ sensitivity and constriction of coronary arteries to serotonin, the thromboxane analogue U46619 and the α1‐adrenergic agonist phenylephrine
Endothelium‐dependent relaxation of coronary arteries in response to cholinergic stimulation is inhibited by perivascular tissue as a result of reduced endothelial Ca2+ responses and attenuated H2S‐dependent signalling
These results establish cellular mechanisms by which perivascular heart tissue can modify local vascular tone and coronary blood flow
Abstract
Interactions between perivascular tissue (PVT) and the vascular wall modify artery tone and contribute to local blood flow regulation. Using isometric myography, fluorescence microscopy, membrane potential recordings and phosphospecific immunoblotting, we investigated the cellular mechanisms by which PVT affects constriction and relaxation of rat coronary septal arteries. PVT inhibited vasoconstriction to thromboxane, serotonin and α1‐adrenergic stimulation but not to depolarization with elevated extracellular [K+]. When PVT was wrapped around isolated arteries or placed at the bottom of the myograph chamber, a smaller yet significant inhibition of vasoconstriction was observed. Resting membrane potential, depolarization to serotonin or thromboxane stimulation, and resting and serotonin‐stimulated vascular smooth muscle [Ca2+]‐levels were unaffected by PVT. Serotonin‐induced vasoconstriction was almost abolished by rho‐kinase inhibitor Y‐27632 and modestly reduced by protein kinase C inhibitor bisindolylmaleimide X. PVT reduced phosphorylation of myosin phosphatase targeting subunit (MYPT) at Thr850 by ∼40% in serotonin‐stimulated arteries but had no effect on MYPT‐phosphorylation in arteries depolarized with elevated extracellular [K+]. The net anti‐contractile effect of PVT was accentuated after endothelial denudation. PVT also impaired vasorelaxation and endothelial Ca2+ responses to cholinergic stimulation. Methacholine‐induced vasorelaxation was mediated by NO and H2S, and particularly the H2S‐dependent (dl‐propargylglycine‐ and XE991‐sensitive) component was attenuated by PVT. Vasorelaxation to NO‐ and H2S‐donors was maintained in arteries with PVT. In conclusion, cardiomyocyte‐rich PVT surrounding coronary arteries releases diffusible factors that reduce rho‐kinase‐dependent smooth muscle Ca2+ sensitivity and endothelial Ca2+ responses. These mechanisms inhibit agonist‐induced vasoconstriction and endothelium‐dependent vasorelaxation and suggest new signalling pathways for metabolic regulation of blood flow.
Abbreviations
- 8‐SPT
8‐(p‐sulphophenyl)theophylline
- ACh
acetylcholine
- AUC
area under the curve
- Bis‐10
bisindolylmaleimide X
- CSE
cystathionine γ‐lyase
- EC
endothelial cell
- K‐PSS
physiological saline solution with elevated [K+]
- l‐NAME
N‐nitro‐l‐arginine methyl ester
- MYPT
myosin phosphatase targeting subunit
- PKC
protein kinase C
- PPG
dl‐propargylglycine
- PSS
physiological saline solution
- PVT
perivascular tissue
- SNAP
S‐nitroso‐N‐acetyl‐d,l‐penicillamine
- SNP
sodium nitroprusside
- VSMC
vascular smooth muscle cell
Introduction
Matching local blood flow to the metabolic demand is a critical physiological function in the heart that protects against ischaemia and consequent cardiac dysfunction. By contrast to heavily sympathetically innervated vascular beds (e.g. mesenteric arteries) where output from the cardiovascular centre in the brain stem controls vascular tone and resistance, the coronary vasculature is controlled mostly by local mechanisms. Regulation of coronary blood flow involves responses to physical forces, such as changes in transmural pressure (i.e. myogenic responses) and shear stress (i.e. flow‐mediated vasodilation), and responses to metabolites (e.g. build‐up of acidic waste products).
Regulation of vascular resistance is complex involving input to the vascular smooth muscle cells (VSMCs) from, for example, endothelial cells (ECs), nerve endings and the surrounding perivascular tissue (PVT). Despite the obvious physiological (e.g. during exercise) and pathophysiological (e.g. in ischaemic heart disease) importance of coronary blood flow regulation, the cellular and molecular background for cross‐talk between the perivascular cardiac tissue and the coronary resistance arteries remains incompletely understood. Previous studies focusing on the vasoactive effects of perivascular adipose tissue propose that the release of anti‐contractile factors from PVT is altered in obesity (Yudkin et al. 2005; Greenstein et al. 2009; Aghamohammadzadeh et al. 2013) and hypertension (Li et al. 2013), as well as in response to inflammation (Bhattacharya et al. 2013), suggesting that disturbed signalling between PVT and the arterial wall contributes to the development of cardiovascular disease.
A role for PVT in modifying artery tone has been suggested for several vascular beds, including the aorta (Lohn et al. 2002; Kohn et al. 2012), mesenteric (Lynch et al. 2013; Li et al. 2013; Verlohren et al. 2004; Weston et al. 2013), skeletal (Zavaritskaya et al. 2013), coronary (Payne et al. 2009; Reifenberger et al. 2007) and subcutaneous arteries (Aghamohammadzadeh et al. 2013; Greenstein et al. 2009). Although most of these studies describe an anti‐contractile influence of PVT, inhibition of endothelium‐dependent vasorelaxation (Payne et al. 2009) and potentiation of vasocontractile responses (Gao et al. 2006) have also been proposed. The effects of PVT surrounding coronary arteries are particularly debatable because previous studies reach opposing conclusions with respect to whether PVT modifies coronary artery tone: some investigators find that PVT inhibits acetylcholine (ACh)‐induced NO production (Payne et al. 2009), some describe inhibition of endothelin‐induced contractions but no effect on ACh‐induced relaxations (Reifenberger et al. 2007) and yet others report no vasoactive effects of PVT (Bunker & Laughlin, 2010). Adding to the controversy, PVT has been suggested to act as a diffusion hindrance to exogenously applied agonists under in vitro experimental conditions causing the magnitude of PVT‐mediated vasomotor effects to be overestimated (Li et al. 2013) and questioning its biological significance.
Although the cellular signalling mechanisms contributing to PVT‐dependent regulation of vascular tone have not been conclusively determined, H2S (Schleifenbaum et al. 2010), leptin (Dashwood et al. 2011), NO (Dashwood et al. 2007), angiotensin 1–7 (Lee et al. 2009), reactive oxygen species (Gao et al. 2006) and adiponectin (Greenstein et al. 2009) derived from adipocyte‐rich PVT have all been proposed to possess signalling potential. Pharmacological interventions and results from genetically modified mice, however, are in conflict with respect to the contribution of individual signalling molecules (Fesus et al. 2007; Greenstein et al. 2009). The cellular mechanisms responsible for the vasoactive influence of adipocyte‐rich PVT appears to involve the activation of K+ channels in VSMCs leading to cell hyperpolarization and vasorelaxation (Li et al. 2013; Lohn et al. 2002; Verlohren et al. 2004; Lynch et al. 2013; Zavaritskaya et al. 2013; Kohn et al. 2012; Weston et al. 2013), although altered release or bioavailability of NO from ECs has also been proposed (Payne et al. 2009; Greenstein et al. 2009). Although these disparate results may in part be explained by species differences (Kohn et al. 2012), the nature of the PVT‐derived vasoactive factors and their cellular effects also probably vary between different vascular beds. In particular, more evidence is required concerning the role of vasoactive factors and downstream cellular signalling mechanisms in arteries surrounded predominantly by non‐adipose tissue.
In the present study, we investigated the role of PVT‐derived vasoactive factors and their mechanism of action in rat coronary septal arteries surrounded primarily by myocardial tissue. We show that cardiomyocyte‐rich PVT releases diffusible factors that modify vasocontractile and relaxant responses and that, in their mode of action, are distinct from previously described factors from adipocyte‐rich PVT. PVT surrounding coronary septal arteries inhibits agonist‐induced vasoconstriction by lowering rho‐kinase‐dependent VSMC Ca2+ sensitivity. PVT also diminishes vasorelaxation in response to cholinergic stimulation by attenuating EC Ca2+ responses and inhibiting H2S‐dependent signalling.
Methods
Male Wistar rats (10–14 weeks old) were killed by CO2 inhalation followed by decapitation, their hearts placed in cold oxygenated buffer, and the coronary septal arteries isolated with or without surrounding PVT. PVT was either kept around the whole arterial circumference (‘1/1 PVT’) or left longitudinally around half of the arterial circumference (‘1/2 PVT’), see Fig. 1 for histology examples. In other experiments, PVT was wrapped and gently fixed with a loose suture around arteries dissected free of the surrounding tissue, or arteries without adherent PVT were investigated with PVT placed at the bottom of the myograph chamber. All animal handling was approved by the Danish Animal Experiments Inspectorate.
Figure 1. Coronary septal arteries are surrounded by myocardial tissue separated only by a thin layer of perivascular connective tissue .
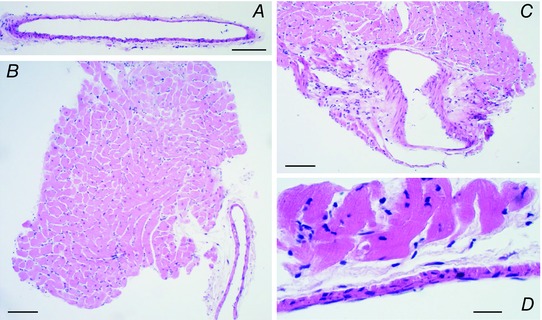
Low magnification micrographs of hematoxylin and eosin stained coronary septal arteries without PVT (A), with PVT around half of the circumference (B) and with PVT around the whole circumference (C). Scale bars in (A), (B) and (C) represent 100 μm. D,higher magnification micrograph of a haematoxylin and eosin stained coronary septal artery showing the close proximity between the arterial wall and the myocardial tissue separated only by a thin layer of connective tissue. Scale bar in (D) represents 20 μm.
Small vessel myography
Arteries were mounted in wire myographs (DMT, Aarhus, Denmark) and normalized to 90% of the internal diameter corresponding to a transmural pressure of 100 mmHg as described previously (Mulvany & Halpern, 1977). The proximal and distal parts of the septal artery were mounted in separate myograph chambers. Typically, one part of the artery was investigated with PVT, whereas the other part was investigated without PVT to allow for paired comparisons. PVT was kept alternately around the proximal or distal part of the artery to avoid any systematic error caused by structural or functional differences between the different artery segments. The normalized diameter was 242 ± 5 μm for arteries with PVT around half of the circumference compared to 269 ± 4 μm for matched arteries without PVT (n = 112, P < 0.001, paired two‐tailed Student's t test). When PVT was left around the whole arterial circumference, the normalized diameter was 212 ± 12 μm compared to 251 ± 12 μm for matched arteries without PVT (n = 13, P < 0.05, paired two‐tailed Student's t test). The relatively small difference in diameter estimates for arteries with and without PVT suggests that the presence of PVT does not interfere substantially with the passive mechanical properties of the arteries. Any potential effects of different passive wall stretch because of adherent PVT were controlled for by studying arteries wrapped in loose PVT.
Following normalization, and before data collection was initiated, the arteries were stimulated first with 80 mm extracellular K+ in combination with 300 nm thromboxane analogue U46619 or 1 μm serotonin (these maximal contractile levels are reported for each agonist in Fig. 2 and are summarized in Fig. 2 F), then with 1 μm serotonin and, finally, with half‐log cumulative additions of U46619 (from 1 nm to 3 μm). Arteries studied for contractile function, without PVT or with PVT around half the circumference, showed stable resting tension and produced more than 0.75 N/m active tension during (a) stimulation with 80 mm extracellular K+ combined with 300 nm U46619 or 1 μm serotonin and (b) subsequent cumulative exposure (1 nm to 3 μm) to U46619. Vasorelaxant responses were tested in arteries developing a stable level of preconstriction corresponding to ∼70% of maximal tension to U46619 by that artery, except for NaHS, which was tested at a level of preconstriction equivalent to ∼50% of maximal tension. In select experiments, arteries were endothelium‐denuded by passing air bobbles through the lumen. Successful denudation was confirmed by an abolished relaxant response to methacholine.
Figure 2. The presence of PVT inhibits active tone development in coronary septal arteries .
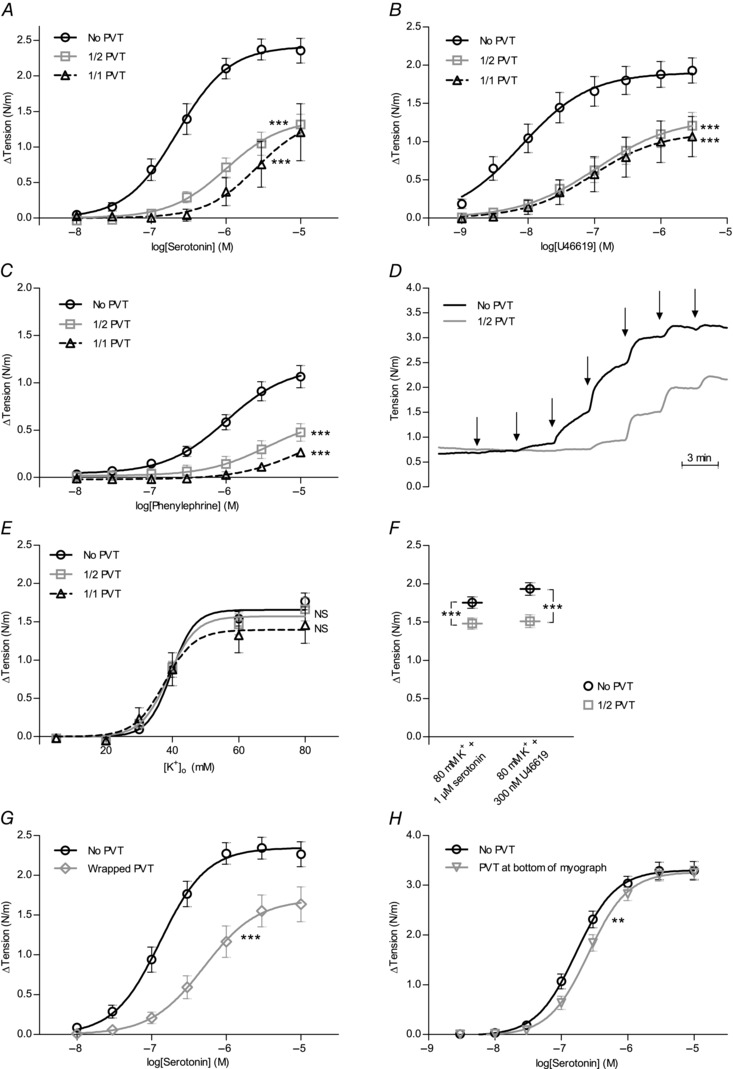
Average vasocontractile responses to serotonin (A) (n = 4–15), thromboxane analogue U46619 (B) (n = 6–17) and α1‐adrenergic agonist phenylephrine (C) (n = 4–10) were attenuated by the presence of PVT, whether left around the whole (1/1 PVT) or half (1/2 PVT) of the arterial circumference. Resting tension prior to the addition of agonist: (A) 0.62 ± 0.05 N/m (No PVT), 0.67 ± 0.05 N/m (1/2 PVT) and 0.78 ± 0.22 N/m (1/1 PVT); (B) 0.49 ± 0.04 N/m (No PVT), 0.43 ± 0.05 N/m (1/2 PVT) and 0.40 ± 0.10 N/m (1/1 PVT); and (C) 0.48 ± 0.04 N/m (No PVT), 0.42 ± 0.06 N/m (1/2 PVT) and 0.35 ± 0.05 N/m (1/1 PVT). Active tension developed in response to 80 mm K+ combined with 1 μm serotonin or 300 nm U46619 for the arteries: (A) 1.95 ± 0.21 N/m (No PVT) and 1.36 ± 0.17 N/m (1/2 PVT); (B) 1.54 ± 0.14 N/m (No PVT) and 1.40 ± 0.13 N/m (1/2 PVT); and (C) 1.45 ± 0.21 N/m (No PVT) and 1.40 ± 0.17 N/m (1/2 PVT). Summarized values for all arteries tested are provided in (F). D,original traces of contractile responses to cumulative application of serotonin. At each arrow, the concentration of serotonin was increased by a half‐log step (between 10 nm and 10 μm). E,average vasocontractile responses to depolarization induced by elevated extracellular [K+] were not affected by the presence of PVT (n = 6–14). Resting tension at 4 mm extracellular K+ was 0.47 ± 0.04 N/m (No PVT), 0.38 ± 0.06 N/m (1/2 PVT) and 0.35 ± 0.08 N/m (1/1 PVT). F,tension development to 80 mm extracellular K+ combined with 1 μm serotonin (n = 68) or 300 nm U46619 (n = 42) was moderately reduced by the presence of PVT. For arteries stimulated by 80 mm K+ and 1 μm serotonin, resting tension was 0.73 ± 0.03 N/m (No PVT) and 0.88 ± 0.04 N/m (1/2 PVT). For arteries stimulated with 80 mm K+ and 300 nm U46619, resting tension was 0.65 ± 0.02 N/m (No PVT) and 0.74 ± 0.03 N/m (1/2 PVT). G and H, reduced sensitivity to serotonin with respect to force production was also observed when loose PVT was wrapped around arteries (G) (n = 14) or placed at the bottom of the myograph chamber (H) (n = 4 rats). From each rat, two artery segments were tested both with and without PVT in the chamber and the order of the experiment was alternated to eliminate any potential effects of time. Resting tension prior to the addition of agonist: (G) 0.63 ± 0.03 N/m (No PVT) and 0.68 ± 0.04 N/m (Wrapped PVT) and (H) 0.63 ± 0.07 N/m (No PVT) and 0.54 ± 0.12 N/m (PVT at bottom). The data in (A, B, C, E, G and H) were fitted to sigmoidal curve fits and compared using extra sum‐of‐squares F tests. The data in (F) were compared using a paired two‐tailed Student's t test. **P < 0.01, ***P < 0.001. NS, not significantly different vs. arteries without PVT.
Experiments were performed in physiological saline solution containing (in mM): 140 Na+, 4 K+, 1.6 Ca2+, 1.2 Mg2+, 122 Cl−, 24 HCO3 −, 1.2 SO4 2−, 1.18 H2PO4 −, 10 Hepes, 5.5 glucose and 0.03 EDTA. Solutions containing increased [K+] were prepared by replacing the appropriate proportion of NaCl with equimolar amounts of KCl. All solutions were aerated with a gas mixture of 5% CO2 balance air and pH was adjusted to 7.4 at 37°C.
Intracellular [Ca2+] measurements
Intracellular Ca2+ dynamics in VSMCs and ECs were investigated by confocal fluorescence microscopy of intact isolated arteries loaded with the Ca2+‐sensitive fluorophores Fura Red and Calcium Green‐1 (Invitrogen, Naerum, Denmark) as described previously (Boedtkjer et al. 2011). When ECs were investigated, arteries were first mounted in a pressure myograph (110P; DMT) and perfused luminally with the acetoxymethyl ester derivatives of the fluorophores. Then, arteries were transferred to and mounted in a confocal wire myograph (120CW; DMT). When VSMCs were investigated, arteries were mounted directly in the confocal wire myograph and loaded by addition of the acetoxymethyl ester derivatives of the fluorophores directly to the myograph chamber. Fluorescence imaging was perfomed with a LSM 5 Pascal Axiovert 200 M confocal microscope (Carl Zeiss, Oberkochen, Germany). Images at 512 × 512 pixels were collected at 0.05 Hz (VSMCs) or 0.13 Hz (ECs). The arteries were excited at 488 nm and emission light collected in the range 505–600 nm (F 505–600) and at wavelengths longer than 615 nm (F > 615). The F 505–600/F > 615 fluorescence ratio was taken as a measure of intracellular [Ca2+]. In typical experiment, ∼10 cells were visible within the field of view and were included in the analyses.
Membrane potential measurements
Membrane potentials of VSMCs were measured as described previously (Boedtkjer et al. 2013). Arteries were mounted in a wire myograph (DMT) and normalized as described above. Membrane potential measurements were performed using aluminium silicate microelectrodes (WPI, Hitchin, UK) with a resistance of 40–120 MΩ when backfilled with 3 m KCl and recorded with an Intra‐767 amplifier (WPI), visualized on an oscilloscope (Gould‐Nicolet Technologies, Loughton, UK) and continuously stored with a PowerLab system (ADInstruments, Dunedin, New Zealand). Electrode entry into cells resulted in an abrupt drop in voltage followed by a sharp return to baseline upon retraction. In arteries with PVT around half of the circumference, impalements were performed on the side dissected free of the surrounding tissue.
Histological investigations
Coronary arteries with or without PVT were fixed in 4% neutral‐buffered formaldehyde (VWR, Herlev, Denmark) for 20 min, paraffin‐embedded and cut to 3 μm thick sections on a RM2165 microtome (Leica, Wetzlar, Germany). Tissue sections were deparaffinized and rehydrated through graded alcohol rinses, stained with haematoxylin and eosin, and imaged with a Moticam 2500 digital camera mounted on a BA310 microscope (Motic, Hong Kong, China).
Detection of phosphorylation levels by immunoblotting
We investigated the phosphorylation status at Thr850 of the myosin phosphatase targeting subunit (MYPT), which is a substrate for the rho‐kinase, using a protocol described in detail previously (Boedtkjer et al. 2011). Bound antibody was detected by enhanced chemiluminescence (ECL Plus; GE Healthcare, Brøndby, Denmark) using conventional photographic film or an ImageQuant LAS 4000 luminescent image analyser (GE Healthcare). Following densitometric analyses using ImageJ software (NIH, Bethesda, MD, USA), background intensities were subtracted from the analysed band intensities. Arteries without PVT were compared with arteries wrapped in PVT, rather than arteries with adherent PVT, to permit the quick removal of the PVT and isolation of proteins from the arterial wall only. Phosphorylation of MYPT was investigated in matched arteries (a) under resting conditions, (b) stimulated for 5 min with concentrations of serotonin giving half‐maximal tension in arteries without PVT or (c) depolarized with 40 mm extracellular K+.
Statistical analysis
Data are expressed as the mean ± SEM. Paired two‐tailed Student's t tests were used for comparison of single interventions between arteries from the same rats. More than two interventions on arteries from the same rats were compared by repeated measures one‐way ANOVA followed by Bonferroni post hoc tests. When measurements from arteries isolated from different rats were compared, unpaired statistical tests were employed. Concentration–response relationships were analysed by sigmoidal curve‐fits and the derived parameters (logEC50 and maximum values) compared by extra sum‐of‐squares F tests. The area under the curve (AUC) was calculated and compared to evaluate the effects of pharmacological inhibitors on concentration–response relationships. P < 0.05 was considered statistically significant. Statistical analyses were performed using Prism, version 5.03 (GraphPad Software Inc., San Diego, CA, USA).
Results
We investigated the effects of PVT on rat coronary septal arteries. The septal artery is located on the cardiac septum and is closely surrounded by myocardial tissue separated only by a thin layer of perivascular connective tissue containing limited amounts of fat (Fig. 1).
PVT marginally increases resting arterial tension
Resting tension in arteries with PVT around half of the circumference (0.82 ± 0.03 N/m) was marginally higher (n = 112, P < 0.001, paired two‐tailed Student's t test) than in matched arteries without PVT (0.70 ± 0.02 N/m). Similarly, resting tension in arteries with PVT around the whole circumference (0.81 ± 0.07 N/m) was modestly higher (n = 13, P < 0.05, paired two‐tailed Student's t test) than in matched arteries without PVT (0.60 ± 0.04 N/m). In arteries wrapped in PVT, resting tension was 0.68 ± 0.04 N/m, which was not significantly different from 0.63 ± 0.03 N/m in matched arteries without PVT (n = 14, P = 0.31, paired two‐tailed Student's t test).
PVT inhibits agonist‐induced but not depolarization‐induced contractions
Concentration‐dependent constrictions of coronary septal arteries were observed in response to application of serotonin (Fig. 2 A), the stable thromboxane analogue U46619 (Fig. 2 B) and the selective α1‐adrenoceptor agonist phenylephrine (Fig. 2 C). Original traces showing constrictions to serotonin are provided in Fig. 2 D. For all three agonists, the contractile response was strongly attenuated when arteries were investigated with PVT (Fig. 2 A–C). The magnitude of the anti‐contractile effect depended on the amount of PVT present, as demonstrated by the observation that arteries with PVT around half of the circumference typically elicited greater contractile responses than arteries with PVT around the whole circumference (Fig. 2 A and C).
No difference in contractile response to elevated extracellular [K+] was observed between arteries with and without PVT (Fig. 2 E). This finding demonstrates that artery constriction to membrane depolarization is unaffected by PVT and suggests that the difference in agonist‐induced contractile responses between arteries with and without PVT (Fig. 2 A–C) is not a result of mechanical interference with artery constriction. When arteries were stimulated with 80 mm extracellular K+ in combination with 300 nm U46619 or 1 μm serotonin (Fig. 2 F), the elicited contractions in arteries without PVT were only modestly higher than contractions in response to 80 mm K+ alone (Fig. 2 E). Arteries with PVT around half of the circumference developed no additional tone when stimulated with serotonin or U46619 on top of 80 mM extracellular K+ (Fig. 2 E and F).
Anti‐contractile effects of PVT cannot be ascribed solely to diffusion hindrance
Anti‐contractile effects of PVT surrounding rat mesenteric arteries have been ascribed largely to PVT acting as a diffusion hindrance to agonists applied to the myograph chamber (Li et al. 2013). Cleaning one side of the coronary septal arteries of PVT, however, only modestly reduced the anti‐contractile effect (Fig. 2 A–C), even though it would be expected to dramatically reduce potential diffusion limitations imposed by the PVT. Furthermore, when PVT was wrapped loosely around the arteries (Fig. 2 G) or placed at the bottom of the myograph chamber without direct contact to the arteries (Fig. 2 H), the anti‐contractile influence of the PVT persisted even if the effect became gradually smaller. Taken together, these findings strongly support the idea that PVT releases one or more diffusible factors that inhibit agonist‐induced vasoconstriction.
Anti‐contractile effects of coronary artery PVT are distinct from adipocyte‐derived relaxing factors
Several putative anti‐contractile factors released from PVT (e.g. adiponectin, leptin, NO and H2S) have been proposed to act at least partly through activation of VSMC K+ channels (Li et al. 2013; Lohn et al. 2002; Verlohren et al. 2004; Lynch et al. 2013; Zavaritskaya et al. 2013; Kohn et al. 2012; Weston et al. 2013). Therefore, we measured VSMC membrane potentials (Fig. 3 A) in coronary septal arteries with and without PVT and tested pharmacologically the involvement of K+ channels, NO and H2S.
Figure 3. The VSMC membrane potential is unaffected by PVT, and the anti‐contractile effect of PVT persists after inhibition of K+ channels, H2S and NO synthesis and adenosine receptors .
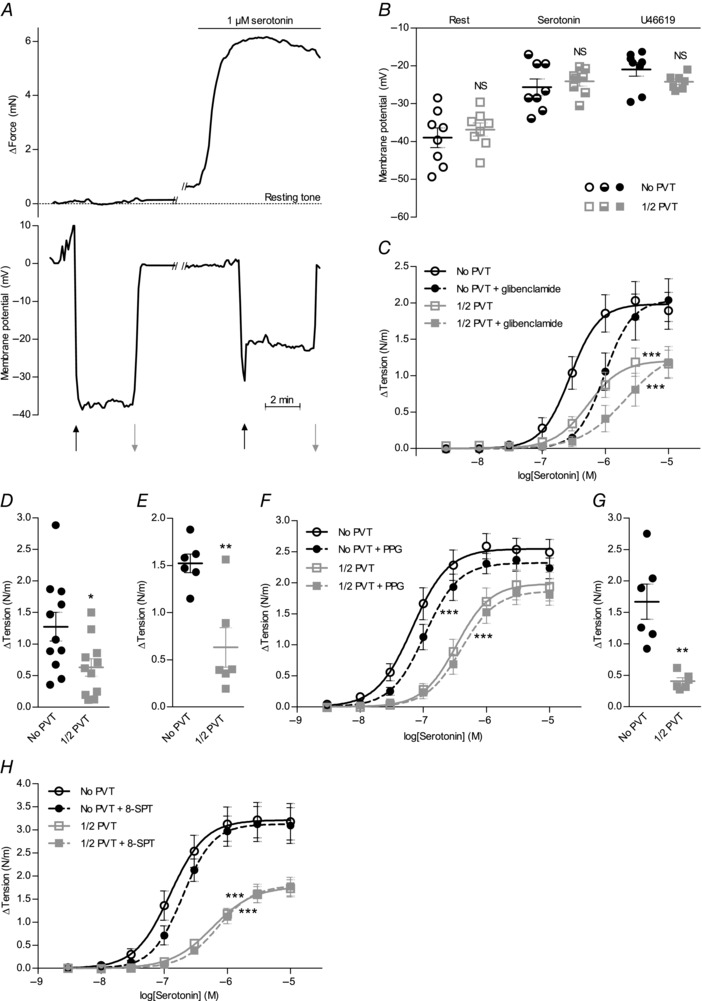
A,original traces of matched VSMC membrane potential and arterial force development during serotonin stimulation. B,VSMC membrane potentials at rest and during stimulation with 1 μm serotonin or 1 μm U46619 did not differ between coronary septal arteries with and without PVT (n = 7–8). C,the anti‐contractile effect of PVT around coronary septal arteries (n = 8) persists in the presence of 10 μm KATP channel inhibitor glibenclamide. Resting tension prior to the addition of serotonin was 0.80 ± 0.09 N/m (No PVT) and 0.72 ± 0.09 N/m (1/2 PVT) in the presence of glibenclamide compared to 0.81 ± 0.09 N/m (No PVT) and 0.84 ± 0.10 N/m (1/2 PVT) without glibenclamide. D and E,the increase in basal arterial tension following the addition of 10 μm Kv7 channel inhibitor XE991 (D) (n = 11) or 1 μm BK channel inhibitor paxillin (E) (n = 6) was reduced in arteries with PVT compared to arteries without PVT. No further basal tone development was seen if the concentration of paxillin was increased to 10 μm (data not shown). Resting tension was 0.60 ± 0.04 N/m (No PVT) and 0.61 ± 0.07 N/m (1/2 PVT) prior to the addition of XE991, and 0.57 ± 0.06 N/m (No PVT) and 0.74 ± 0.04 N/m (1/2 PVT) prior to addition of paxillin. F,the vasocontractile responses to serotonin were unaffected by 10 mm CSE inhibitor PPG in coronary septal arteries with and without PVT (n = 8). The slight rightward shifts of the concentration–response curves were similar to effects observed in time control experiments without addition of drug. Resting tension prior to the addition of serotonin was 0.81 ± 0.12 N/m (No PVT) and 0.61 ± 0.04 N/m (1/2 PVT) in the presence of PPG compared to 0.67 ± 0.04 N/m (No PVT) and 0.64 ± 0.04 N/m (1/2 PVT) without PPG. G,the increase in basal arterial tension following the addition of 100 μM NO‐synthase inhibitor l‐NAME was reduced in arteries with PVT compared to arteries without PVT (n = 6). Resting tension prior to addition of l‐NAME was 0.74 ± 0.11 N/m (No PVT) and 0.73 ± 0.08 N/m (1/2 PVT). H,vasocontractile responses to serotonin were unaffected by 100 μm adenosine receptor antagonist 8‐SPT in coronary septal arteries with and without PVT (n = 5). Resting tension prior to the addition of serotonin was 0.62 ± 0.02 N/m (No PVT) and 0.36 ± 0.03 N/m (1/2 PVT) in the presence of 8‐SPT compared to 0.65 ± 0.03 N/m (No PVT) and 0.53 ± 0.02 N/m (1/2 PVT) without 8‐SPT. The data were compared by a paired two‐tailed Student's t test (B, D, E and G) or fitted to sigmoidal curve fits and compared using extra sum‐of‐squares F tests (C, F and H). **P < 0.01, ***P < 0.001. NS, not significantly different vs. arteries without PVT.
Resting VSMC membrane potential and depolarization to 1 μm serotonin or U46619 did not differ between arteries with and without PVT (Fig. 3 B). These findings strongly suggest that changes in K+ channel activity do not contribute significantly to the anti‐contractile effects of PVT surrounding coronary arteries. The conclusion that cardiomyocyte‐rich PVT acts independently of K+ channel activation is supported by the maintained difference in contractile response between arteries with and without PVT upon stimulation with serotonin or U46619 in the presence of 80 mm extracellular K+ (Fig. 2 F) when the gradient for K+ is almost abolished. Further reinforcing this conclusion, the anti‐contractile effects of PVT persisted after inhibition of ATP‐sensitive KATP channels (Fig. 3 C), voltage‐gated Kv7 channels (Fig. 3 D) and large‐conductance Ca2+‐activated BK channels (Fig. 3 E), which have been suggested to mediate the effects of H2S (Zhao et al. 2001; Schleifenbaum et al. 2010), adiponectin (Lynch et al. 2013), leptin (Shanley et al. 2002) and NO (Feletou, 2009). A concentration of 10 μm glibenclamide is required for full inhibition of KATP channels in VSMCs (Quayle et al. 1995). Although this concentration of glibenclamide shifted the concentration–response curve for serotonin to the right (Fig. 3 C), which is consistent with additional unspecific effects (Wareing et al. 2006; Crosbie et al. 2000), the persistent difference between arteries with and without PVT suggests that KATP channels are not required for the anti‐contractile effect of PVT. The development of basal tone in response to the Kv7 channel blocker XE991 (Fig. 3 D) and the BK channel blocker paxillin (Fig. 3 E) prevented meaningful comparisons of agonist‐induced contractions, although the development of lower basal tone in coronary septal arteries with than without PVT suggests that Kv7 and BK channels are not prominently involved in mediating the anti‐contractile effects of cardiomyocyte‐rich PVT.
In time control experiments, resting tension was essentially stable in arteries with (Δtension = −0.08 ± 0.03 N/m) and without (Δtension = −0.04 ± 0.03 N/m) PVT over a 40 min observation period corresponding to the time between two consecutive stimulations. The anti‐contractile effect of PVT was also well‐maintained between a first (ΔAUC1/2 PVT – No PVT = −1.72 ± 0.70) and second (ΔAUC1/2 PVT – No PVT = −1.42 ± 0.73) cumulative serotonin stimulation (3 nm to 10 μm) performed 40 min apart (n = 6,P = 0.27, paired two‐tailed Student's t test). The addition of dl‐propargylglycine (PPG) to inhibit cystathionine γ‐lyase (CSE), which is the primary source of H2S synthesis in the vascular wall (Hosoki et al. 1997; Zhao et al. 2001), did not affect vasoconstriction to serotonin or the anti‐contractile effect of PVT (Fig. 3 F). The non‐selective NO‐synthase inhibitor N‐nitro‐l‐arginine methyl ester (l‐NAME) caused a significant increase in resting tension, which was of greater magnitude in coronary septal arteries without PVT than in matched arteries with PVT (Fig. 3 G). Taken together, these findings suggest that neither NO, nor H2S is prominently involved in the anti‐contractile effect of PVT in coronary septal arteries.
The effect of PVT was not mediated by adenosine because 100 μm of the adenosine receptor blocker 8‐(p‐sulphophenyl)theophylline (8‐SPT) did not affect vasoconstriction to serotonin in arteries with or without PVT (Fig. 3 H). By contrast, 100 μm 8‐SPT completely abolished the vasorelaxant response to exogenous adenosine: relative relaxation to 100 μm adenosine was 19 ± 3% under control conditions and −8 ± 1% in the presence of 8‐SPT (n = 4, P < 0.01, paired two‐tailed Student's t test).
PVT inhibits rho‐kinase‐dependent VSMC Ca2+ sensitivity
To determine the cellular signalling mechanisms by which PVT inhibits vasoconstriction, we next measured VSMC Ca2+ responses during serotonin stimulation. Original recordings based on confocal fluorescence microscopy of arteries dually loaded with Calcium Green‐1 and Fura Red are shown in Fig. 4 A. PVT did not significantly affect the resting VSMC [Ca2+] level (baseline fluorescence ratio was 0.52 ± 0.04 in arteries with PVT compared to 0.54 ± 0.04 in arteries without PVT; n = 7, P = 0.62, paired two‐tailed Student's t test) or the increase in intracellular [Ca2+] upon addition of serotonin (Fig. 4 B). These findings suggest that the effect of PVT on artery tone acts primarily via reduced VSMC Ca2+ sensitivity; accordingly, we found that constriction of rat coronary septal arteries was concentration‐dependently abrogated by the selective rho‐kinase inhibitor Y‐27632 (Fig. 4 C). Application of 1 μm Y‐27632 inhibited contractions in response to 10 μm serotonin to similar relative extents (n = 7, P = 0.50, paired two‐tailed Student's t test) in arteries without PVT (49.3 ± 6.9%) and in arteries with PVT around half the circumference (42.7 ± 10.0%), indicating that the sensitivity to Y‐27632 was unaffected by PVT. Application of 1 μm protein kinase C (PKC) inhibitor bisindolylmaleimide X (Bis‐10) inhibited contractions induced by serotonin more modestly (Fig. 4 D). Y‐27632 and Bis‐10 were used at concentrations previously shown to provide complete kinase inhibition (Davies et al. 2000). Our findings demonstrate that particularly the rho‐kinase pathway plays a key role in vasomotor control of rat coronary septal arteries. The almost complete inhibition of vasoconstriction with 10 μm Y‐27632, however, complicates pharmacological evaluation of whether the anti‐contractile effect of PVT is rho‐kinase‐dependent. Instead, we investigated the importance of altered rho‐kinase signalling by determining the level of MYPT phosphorylation at Thr850. MYPT is an immediate downstream target of the rho‐kinase and its phosphorylation was reduced by ∼40% in arteries wrapped in PVT compared to arteries without PVT when constricted by a concentration of serotonin leading to half‐maximal tension development in arteries without PVT (Fig. 4 E). Application of Y‐27632 attenuated MYPT phosphorylation (Fig. 4 E) at concentrations equivalent to those inhibiting vasoconstriction (Fig. 4 C).
Figure 4. Coronary septal arteries with PVT show reduced rho‐kinase‐dependent VSMC Ca2+ sensitivity .
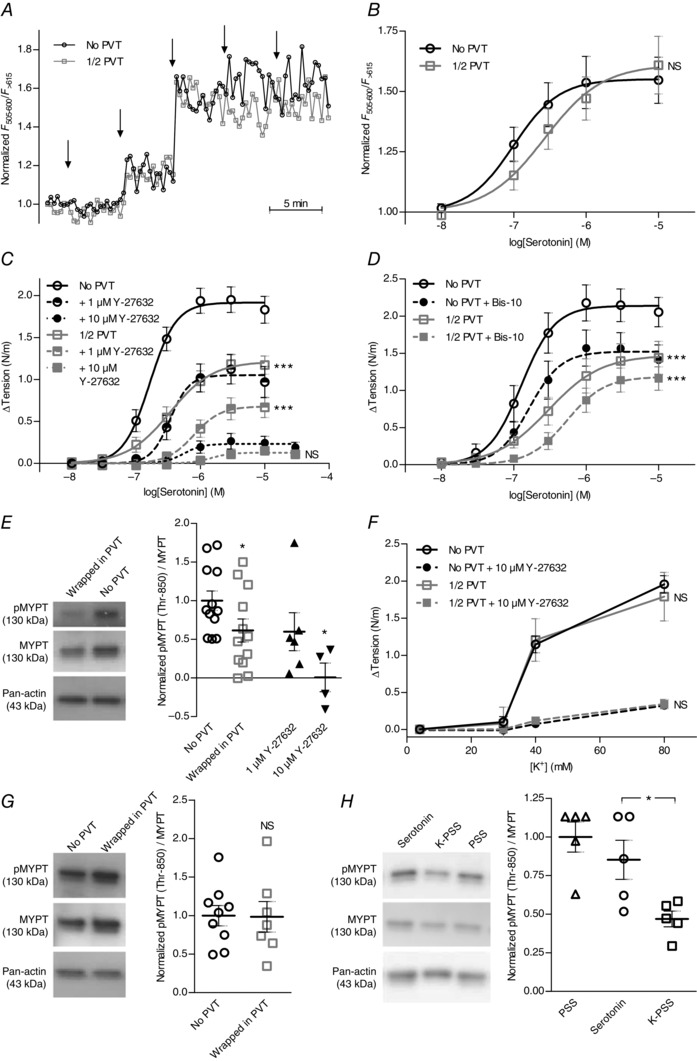
A,original traces of the relative changes in Ca2+‐dependent fluorescence in VSMCs of arteries with and without PVT during cumulative addition of serotonin. At the arrows, the concentration of serotonin was increased stepwise to 10 nm, 100 nm, 300 nm, 1 μm and 10 μm, respectively. B,average VSMC Ca2+ responses to serotonin were not significantly affected by PVT (n = 7). C,vasoconstriction to serotonin was concentration‐dependently abolished by rho‐kinase inhibitor Y‐27632 (n = 7–8) in arteries with and without PVT (P < 0.001). Resting tension prior to addition of serotonin was 0.64 ± 0.05 N/m (No PVT) and 0.68 ± 0.07 N/m (1/2 PVT) without Y‐27632, 0.60 ± 0.06 N/m (No PVT) and 0.47 ± 0.05 N/m (1/2 PVT) with 1 μm Y‐27632 and 0.58 ± 0.05 N/m (No PVT) and 0.49 ± 0.06 N/m (1/2 PVT) with 10 μm Y‐27632. D,vasoconstriction to serotonin was partly inhibited by 1 μm PKC‐inhibitor Bis‐10 (No PVT: P < 0.001, 1/2 PVT: P < 0.01) but the relative difference between arteries with and without PVT persisted following PKC inhibition (n = 8). Resting tension prior to addition of serotonin was 0.60 ± 0.03 N/m (No PVT) and 0.49 ± 0.03 N/m (1/2 PVT) in the presence of Bis‐10 compared to 0.65 ± 0.03 N/m (No PVT) and 0.66 ± 0.03 N/m (1/2 PVT) without Bis‐10. E,representative immunoblots and average relative levels of MYPT phosphorylation at Thr850 in arteries stimulated with a concentration of serotonin giving half‐maximal tension development in arteries without PVT (n = 12). The level of MYPT phosphorylation was reduced by ∼40% in arteries wrapped in PVT (n = 12) and concentration‐dependently abolished by Y‐27632 (n = 4–6). Levels of phosphorylation are expressed relative to serotonin‐stimulated arteries without PVT. F,vasocontractile responses to depolarization induced by elevated extracellular [K+] were unaffected by PVT but strongly attenuated by 10 μm rho‐kinase inhibitor Y‐27632 (n = 4). Resting tension at 4 mm extracellular K+ was 0.47 ± 0.06 N/m (No PVT) and 0.47 ± 0.05 N/m (1/2 PVT) in the presence of Y‐27632 compared to 0.54 ± 0.05 N/m (No PVT) and 0.77 ± 0.05 N/m (1/2 PVT) without Y‐27632. G,representative immunoblots and relative levels of MYPT phosphorylation at Thr850 in arteries stimulated with 40 mm extracellular K+ (n = 7–9). Phosphorylation was not significantly different between arteries without PVT and arteries wrapped in PVT. Levels of phosphorylation are expressed relative to arteries without PVT stimulated with K‐PSS. H,representative immunoblots and relative levels of MYPT phosphorylation at Thr850 in arteries without PVT (n = 5) under basal conditions (PSS), during stimulation with 1 μm serotonin and after activation with 40 mm extracellular K+ (K‐PSS). MYPT phosphorylation was high under resting conditions and serotonin stimulation but decreased markedly upon K+‐induced activation. Levels of phosphorylation are expressed relative to resting arteries without PVT. Data were compared by paired or unpaired two‐tailed Student's t tests (E and G), repeated measures one‐way ANOVA followed by Bonferroni post hoc tests (H) or fitted to sigmoidal curve fits and compared using extra sum‐of‐squares F tests (B, C, D and F). *P < 0.05, ***P < 0.001. NS, not significantly different vs. arteries without PVT under the same experimental conditions or as indicated.
Consistent with previous findings (Urban et al. 2003; Mita et al. 2002; Sakamoto et al. 2003), we found (Fig. 4 F) that 10 μm Y‐27632 strongly inhibited constriction in response to depolarization induced by elevated extracellular [K+]. Nonetheless, constriction to high extracellular [K+] was unaffected by PVT both in the presence and absence of Y‐27632 (Fig. 4 F). Consistent with these findings, we saw no difference in MYPT phosphorylation levels between arteries with and without PVT when constricted by 40 mm extracellular K+ (Fig. 4 G).
To explore the mechanisms of rho‐kinase activation in rat coronary septal arteries, we performed matched experiments where MYPT phosphorylation was determined in unstimulated arteries maintained in a normal physiological saline solution (PSS) and in arteries stimulated with either 1 μm serotonin or 40 mm extracellular K+ (K‐PSS). As shown in Fig. 4 H, MYPT phosphorylation was high in unstimulated coronary arteries. This is consistent with previous studies reporting that coronary septal arteries from Wistar rats develop substantial levels of myogenic tone, which is dependent on rho‐kinase activity (Kold‐Petersen et al. 2012), and matches the prominent basal tone development following K+ channel (Fig. 3 D and E) or NO‐synthase (Fig. 3 G) inhibition. The high basal level of MYPT phosphorylation was maintained in arteries stimulated with serotonin, whereas it decreased substantially when arteries were depolarized by elevated extracellular [K+] (Fig. 4 H). Although the mechanism for reduced rho‐kinase activity at elevated extracellular [K+] is currently undetermined, it is consistent with the modest additional tension development to U46619 and serotonin observed in the presence of 80 mm extracellular K+ (Fig. 2 F).
Taken together, our findings suggest that anti‐contractile factors released by PVT surrounding rat coronary septal arteries play a major role for regulation of vasomotor tone by inhibiting rho‐kinase signalling and agonist‐induced contractions.
PVT inhibits the basal vasorelaxant influence of the endothelium
To test the role of the endothelium for the anti‐contractile effect of the PVT, we denuded arteries of endothelium causing an almost complete inhibition of endothelium‐dependent vasorelaxation (Fig. 5 A). As shown in Fig. 5 B, the anti‐contractile effect of the PVT was accentuated after endothelial denudation: AUC increased by 0.15 ± 0.25 in arteries with PVT around half the circumference, whereas it increased by 1.07 ± 0.41 in arteries without PVT (P < 0.05, unpaired two‐tailed Student's t test). These findings show that the anti‐contractile effect of PVT surrounding coronary septal arteries is not endothelium‐dependent. Instead, PVT reduces the basal vasorelaxant influence of the endothelium.
Figure 5. Endothelial denudation accentuates the net anti‐contractile effect of PVT .
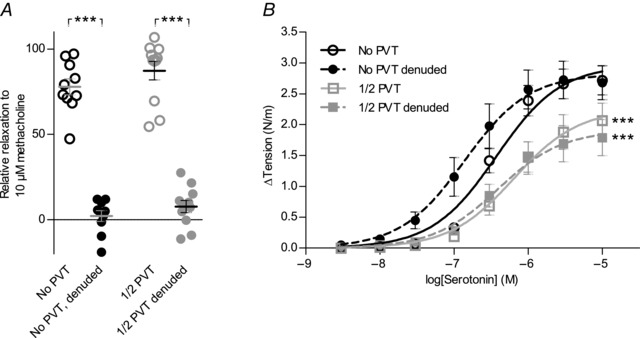
A,vasorelaxant responses to 10 μm methacholine before and after endothelial denudation (n = 10–11). The data were compared by a paired two‐tailed Student's t test. B,the net anti‐contractile effect of PVT was increased after endothelial denudation of coronary septal arteries (n = 10–11). Resting tension prior to addition of serotonin was 0.76 ± 0.09 N/m (No PVT) and 0.67 ± 0.08 N/m (1/2 PVT) in endothelium‐denuded arteries compared to 0.67 ± 0.06 N/m (No PVT) and 0.64 ± 0.06 N/m (1/2 PVT) in control arteries. The data were fitted to sigmoidal curve fits and compared using extra sum‐of‐squares F tests.
PVT has anti‐relaxant effects
We next tested whether the presence of PVT affected the relaxant responses of coronary septal arteries to endothelial agonists. ACh (Fig. 6 A) and the ACh‐esterase resistant analogue methacholine (Fig. 6 B) induced concentration‐dependent vasorelaxation, and the sensitivity to these cholinergic agonists was lower in arteries with than without PVT. Original traces are shown in Fig. 6 C.
Figure 6. Vasorelaxation to cholinergic stimulation is attenuated in arteries with PVT due to abolished H2S production .
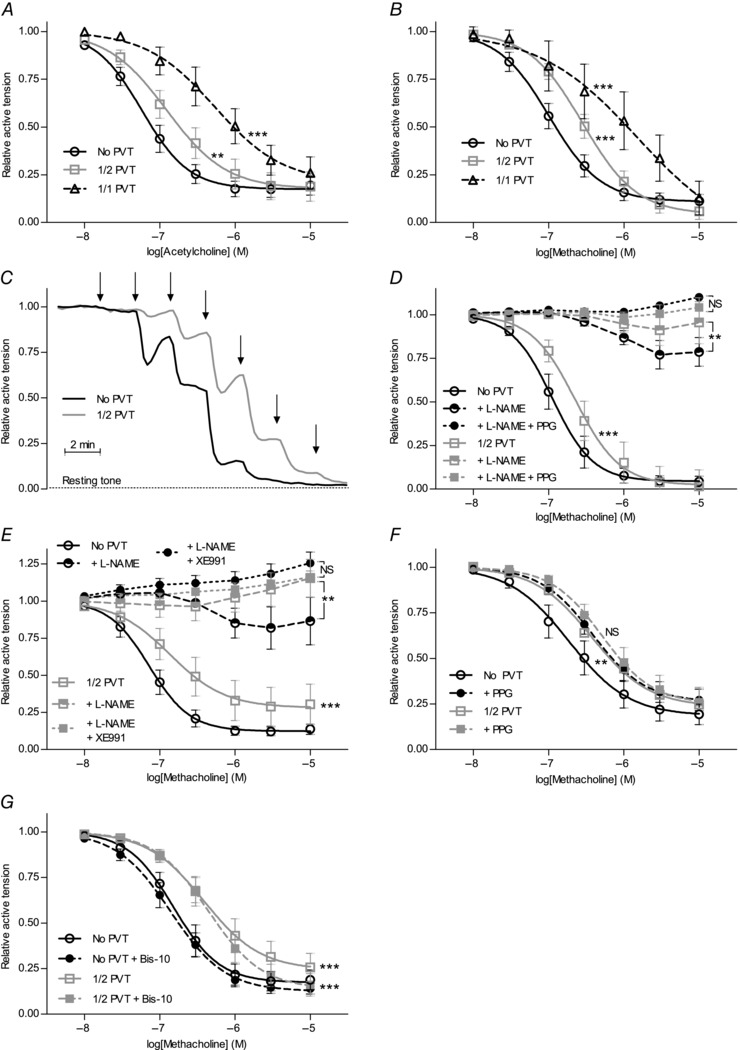
Vasorelaxant responses to ACh (A) (n = 6–13) and methacholine (B) (n = 5–17) were attenuated in U46619‐preconstricted arteries with PVT around the whole (1/1) or half (1/2) of the arterial circumference compared to arteries without PVT. C,original traces of vasorelaxant responses to cumulative application of methacholine in U46619‐preconstricted coronary septal arteries with and without PVT. At each arrow, the concentration of methacholine was increased by a half‐log step (between 10 nm and 10 μm). D and E,in the presence of 100 μm NO‐synthase inhibitor l‐NAME, the vasorelaxant response to methacholine was still significantly smaller in U46619‐preconstricted arteries with PVT compared to arteries without PVT. The l‐NAME‐insensitive vasorelaxant response to methacholine was sensitive to 10 mm CSE inhibitor PPG (D) (n = 6–8) and 10 μm Kv7 channel inhibitor XE991 (E) (n = 7–8). F,incubation with 10 mm CSE inhibitor PPG alone significantly attenuated the difference in vasorelaxation to methacholine stimulation between arteries with and without PVT (n = 14). G,vasorelaxation to methacholine in the presence of 1 μm PKC inhibitor Bis‐10 was persistently reduced in U46619‐preconstricted arteries with PVT compared to arteries without PVT (n = 8). Data were fitted to sigmoidal curve fits and compared using extra sum‐of‐squares F tests. Effects of pharmacological inhibitors on the methacholine responses were evaluated by comparing areas under the curves. **P < 0.01, ***P < 0.001. NS, not significantly different vs. arteries without PVT under the same conditions.
Relaxation to cholinergic stimulation was largely sensitive to 100 μm l‐NAME (Fig. 6 D and E). Particularly in arteries without PVT, an additional PPG‐sensitive (i.e. H2S‐dependent) component was observed (Fig. 6 D). Although the difference in relaxant response to cholinergic stimulation between arteries with and without PVT remained following NO‐synthase inhibition (Fig. 6 D and E), it was absent after inhibition of H2S production (Fig. 6 D and F). Incubation with PPG alone reduced the difference in relaxation (evaluated as ΔAUC) between arteries with and without PVT from −0.31 ± 0.14 to −0.08 ± 0.12 (Fig. 6 F). By contrast, the vasorelaxant response to methacholine was well‐maintained in time control experiments where the inhibitory effect of PVT persisted from the first (ΔAUC1/2 PVT – No PVT = −0.41 ± 0.11) to the second (ΔAUC1/2 PVT – No PVT = −0.60 ± 0.17) cumulative methacholine stimulation performed 40 min apart. Thus, incubation with PPG significantly abrogated methacholine‐induced relaxations compared to time control experiments (P < 0.05, unpaired two‐tailed Student's t test). The difference in vasorelaxant response after NO‐synthase inhibition between arteries with and without PVT was also abolished when Kv7 channels, which represent important targets for H2S (Martelli et al. 2013), were inhibited with 10 μm XE991 (Fig. 6 E).
Relaxant responses to the exogenous NO‐donors sodium nitroprusside (SNP, Fig. 7 A) and S‐nitroso‐N‐acetyl‐d,l‐penicillamine (SNAP) (Fig. 7 B) were not significantly different between arteries with and without PVT. The response of preconstricted arteries to the H2S‐donor NaHS was, as described by others (Ali et al. 2006), biphasic, with initial constriction followed by relaxation (Fig. 7 C). Consistent with this complex vasomotor response to NaHS, studies have shown the involvement of multiple cellular targets (Cheang et al. 2010; Tian et al. 2012; Ping et al. 2015). Still, investigators have proposed that Kv7 channels play a predominant role for vascular responses to endogenous H2S (Schleifenbaum et al. 2010). We found that Kv7‐dependent vasorelaxation to 1 mm NaHS, as calculated from the level of maximal contraction at 100 μm NaHS, was similar (P = 0.97, paired two‐tailed Student's t test) in arteries (n = 6) with (relative XE991‐sensitive relaxation = 0.31 ± 0.15) and without (relative XE991‐sensitive relaxation = 0.30 ± 0.10) adherent PVT. The abolished H2S‐mediated component of endothelium‐dependent vasorelaxation but relatively well‐maintained vasorelaxation to NaHS suggests that the production rather than the action of H2S is attenuated in arteries surrounded by PVT. Also, in contrast to the findings of a previous study in canine circumflex coronary arteries investigated with or without periadventitial adipose tissue (Payne et al. 2009), the anti‐relaxant effect of the PVT surrounding rat coronary septal arteries was not affected by PKC inhibition (Fig. 6 G).
Figure 7. Vasorelaxation to NO‐ and H2S‐donors are unaffected by PVT .
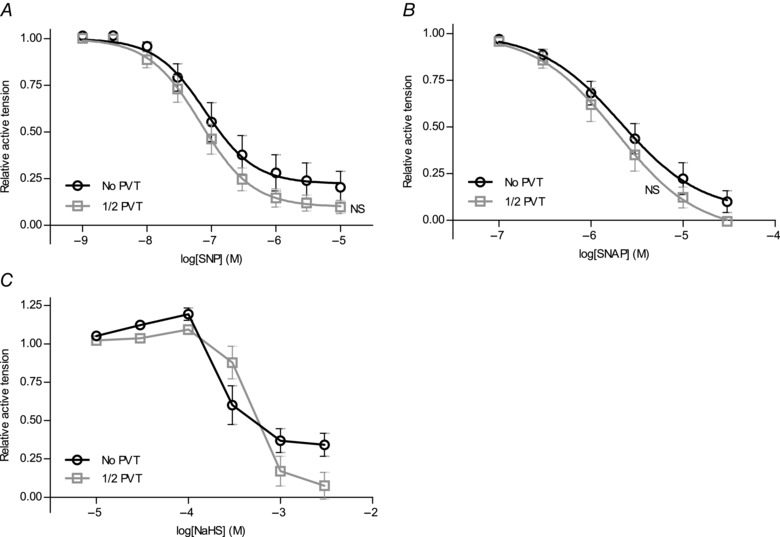
Vasomotor responses of preconstricted arteries with or without PVT to cumulative application of the NO‐donors SNP (A) (n = 6–13) or SNAP (B) (n = 7–11) or the H2S‐donor NaHS (C) (n = 12). Data were fitted to sigmoidal curve fits and compared using extra sum‐of‐squares F tests. NS, not significantly different vs. arteries without PVT.
The inability of cholinergic agonists (Fig. 6 A and B), SNP (Fig. 7 A) and SNAP (Fig. 7 B) to relax arteries below resting levels (i.e. to levels of relative tension below 0) both in arteries with and without PVT suggests that neither group of arteries in general developed active tone under resting conditions.
PVT inhibits endothelial function by lowering Ca2+ responses
To determine the mechanistic background for the reduced vasorelaxant response to methacholine in arteries with PVT, we measured EC Ca2+ responses by confocal microscopy using the Ca2+‐sensitive fluorophores Calcium Green‐1 and Fura Red loaded specifically into ECs of intact isolated coronary arteries (Fig. 8 A and B). Original traces of Ca2+ responses recorded in coronary arteries with and without PVT are shown in Fig. 8 C. The baseline Ca2+‐dependent fluorescence ratio did not differ (P = 0.58, unpaired two‐tailed Student's t test) between arteries with (0.54 ± 0.03, n = 17) and without (0.57 ± 0.03, n = 15) PVT. By contrast, the presence of PVT strongly attenuated the EC Ca2+ response to methacholine both under control conditions (Fig. 8 D) and after treatment with PPG (Fig. 8 E) or Y‐27632 (Fig. 8 F). The baseline Ca2+‐dependent fluorescence ratio in ECs also did not differ between arteries with and without PVT after treatment with PPG (No PVT: 0.78 ± 0.08, 1/2 PVT: 0.73 ± 0.08; n = 5; P = 0.63, unpaired two‐tailed Student's t test) or Y‐27632 (No PVT: 0.52 ± 0.05; 1/2 PVT: 0.49 ± 0.02; n = 6–8; P = 0.64, unpaired two‐tailed Student's t test). Notably, the EC Ca2+ response to NaHS was similar in arteries with and without PVT (Fig. 8 G).
Figure 8. Vasorelaxation to cholinergic stimulation is reduced in arteries with PVT because of lower endothelial Ca2+ responses .
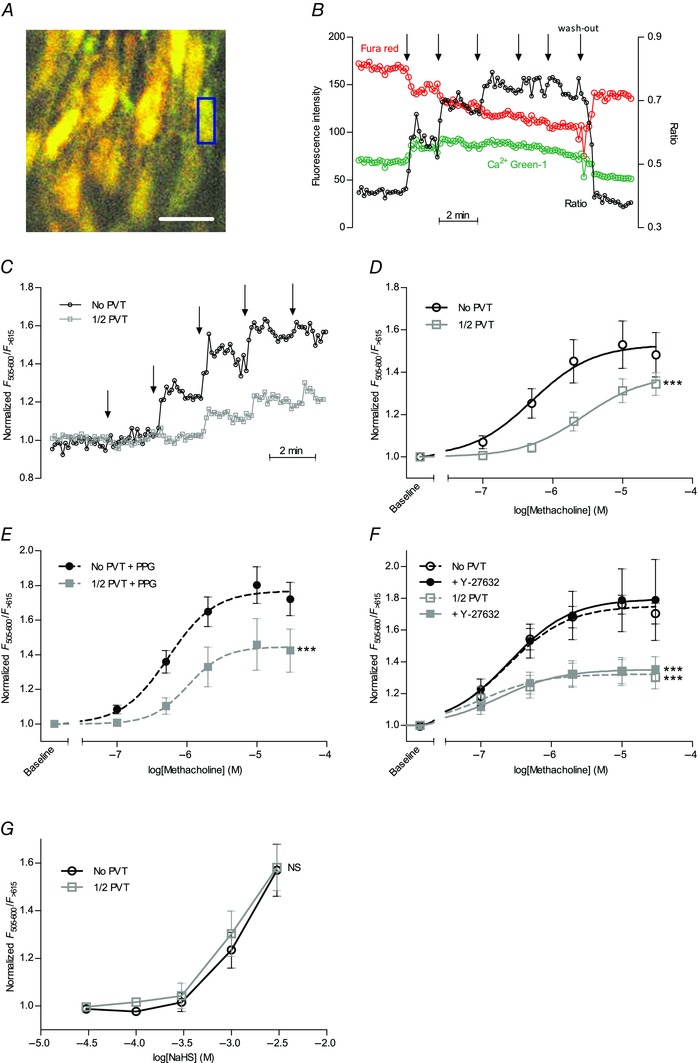
A,image of ECs in an isolated coronary septal artery loaded with Calcium Green‐1 and Fura Red. Individual ECs were marked up as regions of interest (exemplified by the blue rectangle), analysed for changes in Ca2+‐dependent fluorescence and averaged for each experiment. Scale bar represents 20 μm. B and C,original traces of relative changes in Ca2+‐dependent fluorescence ratio (between Calcium Green‐1 and Fura Red emissions) during cumulative addition of methacholine to arteries with and without PVT. At the arrows, the concentration of methacholine was increased stepwise to 100 nm, 500 nm, 2 μm, 10 μm and 30 μm, respectively. D–F,the increase in Ca2+‐dependent fluorescence upon methacholine stimulation under control conditions (D) (n = 6–7) in presence of 10 mm CSE inhibitor PPG (E) (n = 5) or with 10 μm rho‐kinase inhibitor Y‐27632 (F) (n = 6–8) was reduced in arteries with PVT compared to arteries without PVT. G,the increase in Ca2+‐dependent fluorescence upon application of the H2S‐donor NaHS was not affected in arteries with PVT (n = 5) compared to arteries without PVT (n = 6). Data were fitted to sigmoidal curve fits and compared using extra sum‐of‐squares F tests. **P < 0.01, ***P < 0.001. NS, not significantly different vs. arteries without PVT.
Together, these findings suggest that PVT releases one or more factors that interfere with EC Ca2+ handling and thereby modulate vasorelaxant responses to endothelial agonists.
Discussion
In the present study, we show that diffusible factors from PVT facilitate cross‐talk between coronary arteries and the surrounding cardiomyocyte‐rich tissue and also propose cellular signalling mechanisms that allow coronary arteries to respond to changes in the metabolic requirements of the surrounding cardiac tissue. We demonstrate that vasoconstriction of rat coronary arteries is reduced in the presence of cardiomyocyte‐rich PVT primarily as a result of inhibition of rho‐kinase‐dependent VSMC Ca2+ sensitivity (Figs 2, 3, 4). The PVT inhibits agonist‐induced contractions, whereas no effect is seen on contractions induced by depolarization with elevated extracellular [K+] (Fig. 2). The influence of PVT specifically on VSMC Ca2+ sensitivity is in contrast to previous studies from other vascular beds surrounded predominantly by adipose tissue: in mesenteric (Lynch et al. 2013; Li et al. 2013; Verlohren et al. 2004; Weston et al. 2013), skeletal (Zavaritskaya et al. 2013) and subcutaneous (Aghamohammadzadeh et al. 2013; Greenstein et al. 2009) arteries and in the aorta (Lohn et al. 2002; Kohn et al. 2012), perivascular adipose tissue activates VSMC K+ channels, and also causes VSMC hyperpolarization and vasorelaxation. In the present study, we see no effect of PVT on VSMC membrane potential at rest or during contraction (Fig. 3 B), which is consistent with PVT of coronary septal arteries acting independently of membrane excitability.
The few previous studies investigating the role of PVT around coronary arteries have focused on porcine and canine coronary circumflex arteries, which are surrounded primarily by adipose tissue (Payne et al. 2009; Bunker & Laughlin, 2010; Reifenberger et al. 2007). Adiponectin, H2S and leptin are prominent candidates for anti‐contractile factors released from adipocyte‐rich PVT (Momin et al. 2006; Dashwood et al. 2011; Greenstein et al. 2009). Perivascular synthesis of NO (Dashwood et al. 2007) or enhancement of endothelium‐derived NO in response to leptin (Sahin & Bariskaner, 2007) or adiponectin (Greenstein et al. 2009) has also been suggested. The rat coronary septal arteries used for the present study are surrounded by cardiac tissue separated from the vascular wall only by a thin layer of connective tissue containing few adipocytes (Fig. 1). Coronary septal arteries are influenced by anti‐contractile factors released from PVT (Fig. 2), although the pharmacological profile and lack of K+ channel dependency (Fig. 3) are not consistent with involvement of NO, adiponectin, leptin or H2S. Instead, the inhibition of contraction is caused by reduced rho‐kinase‐dependent VSMC Ca2+ sensitivity (Fig. 4). Considering the close approximation between the coronary septal arteries and the surrounding myocardial tissue, the findings of the present study are probably relevant for intramural coronary arteries, which contribute importantly to coronary vascular resistance.
Earlier studies have focused almost exclusively on anti‐contractile effects of PVT, although a study on canine coronary circumflex arteries suggested PVT‐dependent inhibition of vasorelaxation caused by a PKC‐mediated attenuation of NO‐synthase activity (Payne et al. 2009). The anti‐relaxant effect of PVT around rat coronary septal arteries, however, persists after inhibition of NO synthesis (Fig. 6 D and E) and is insensitive to PKC‐inhibition (Fig. 6 G). Instead, we find that the H2S‐dependent component of vasorelaxation induced by cholinergic stimulation is strongly inhibited by PVT (Fig. 6 D–F). Activation of the H2S‐generating enzyme CSE upon cholinergic stimulation is Ca2+‐dependent (Yang et al. 2008) and, in congruence with the reduced H2S signalling, we observe substantially blunted EC Ca2+ responses in coronary septal arteries with PVT (Fig. 8). Although our findings suggest that endothelial H2S production is very markedly attenuated by PVT, the endothelial NO production was proportionally much less affected. This suggests that CSE requires high levels of intracellular [Ca2+] for activation, whereas the substantial NO‐mediated vasorelaxation in arteries with PVT also suggests that, at reduced EC Ca2+ levels, the NO‐synthase is prominently activated to induce vasorelaxation. In congruence with this observation, we find development of basal tone upon treatment with l‐NAME (Fig. 3 G) consistent with basal production of NO at the low levels of EC [Ca2+] found in unstimulated arteries. By contrast, treatment with PPG did not cause an increase in basal tone, suggesting that H2S production mostly occurs when EC [Ca2+] is elevated above resting levels.
A previous study (Li et al. 2013) investigated the possibility that PVT inhibits vasomotor responses in vitro because it represents a diffusion barrier to agonists applied to the myograph bath. In the present study, we show that vasomotor effects of PVT could not be ascribed solely to diffusion hindrance because they were not dramatically reduced when PVT was removed from one side of the artery (Fig. 2 A–C) and were still present when PVT was wrapped around arteries (Fig. 2 G) or placed at the bottom of the myograph chamber (Fig. 2 H) without physical contact to the artery. Additionally, we found that endothelial denudation increased the net anti‐contractile effect of the PVT (Fig. 5), although it should not increase any potential diffusion hindrance.
The dual regulation of coronary artery tone by PVT, modifying both vasoconstriction and vasorelaxation, probably provides more dynamic control of vascular resistance. Other than its putative contribution to metabolic regulation of coronary blood flow, cross‐talk between cardiomyocytes and coronary arteries also purportedly contributes to ischaemic preconditioning (Bell & Yellon, 2012), although the signalling mechanisms involved have not been fully resolved.
In conclusion, we show that diffusible vasoactive factors released from cardiomyocyte‐rich PVT surrounding coronary septal arteries regulate arterial tone through distinct anti‐contractile and anti‐relaxant mechanisms. The exact nature of the diffusible factors is still unknown, although their inhibitory effect on artery constriction is caused by a lowering of rho‐kinase‐dependent VSMC Ca2+ sensitivity. The anti‐relaxant effects of the PVT result from inhibition of endothelium‐dependent vasorelaxation and are principally explained by attenuated EC Ca2+ responses and reduced H2S signalling. Our findings demonstrate that the modulation of vasomotor function previously described for perivascular adipose tissue surrounding arteries of different sources (including the aorta, mesenteric arteries, skeletal muscles, subcutaneous arteries and epicardial coronary arteries) also applies to other types of PVT, although the signalling pathways are different. We propose that the described signalling mechanisms permit cross‐talk between coronary arteries and cardiomyocyte‐rich PVT, and thus allow coronary arteries to respond to changes in the metabolic requirements of the surrounding cardiac tissue.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
Experiments were performed at the Department of Biomedicine, Aarhus University, Denmark. EB conceived the project. EB, FA and LB designed the experiments, as well as analysed and interpreted data. FA, LB and SK collected data. EB wrote the manuscript. All authors revised the manuscript for important intellectual content and approved the final version of the manuscript submitted for publication.
Funding
This work was supported by the Danish Council for Independent Research (grant numbers 10‐094816 and 12‐125922 to EB) and the Danish Heart Foundation (grant number 14‐R97‐A5321‐22809 to EB).
Acknowledgements
The authors would like to thank Jane Rønn and Jørgen Andresen for their expert technical assistance.
References
- Aghamohammadzadeh R, Greenstein AS, Yadav R, Jeziorska M, Hama S, Soltani F, Pemberton PW, Ammori B, Malik RA, Soran H & Heagerty AM (2013). Effects of bariatric surgery on human small artery function: evidence for reduction in perivascular adipocyte inflammation, and the restoration of normal anticontractile activity despite persistent obesity.J Am Coll Cardiol 62, 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali MY, Ping CY, Mok YY, Ling L, Whiteman M, Bhatia M & Moore PK (2006). Regulation of vascular nitric oxide in vitro and in vivo; a new role for endogenous hydrogen sulphide? Br J Pharmacol 149, 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RM & Yellon DM (2012). Conditioning the whole heart – not just the cardiomyocyte. J Mol Cell Cardiol 53, 24–32. [DOI] [PubMed] [Google Scholar]
- Bhattacharya I, Dragert K, Albert V, Contassot E, Damjanovic M, Hagiwara A, Zimmerli L, Humar R, Hall MN, Battegay EJ & Haas E (2013). Rictor in perivascular adipose tissue controls vascular function by regulating inflammatory molecule expression. Arterioscler Thromb Vasc Biol 33, 2105–2111. [DOI] [PubMed] [Google Scholar]
- Boedtkjer E, Kim S & Aalkjaer C (2013). Endothelial alkalinisation inhibits gap junction communication and endothelium‐derived hyperpolarisations in mouse mesenteric arteries. J Physiol 591, 1447–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boedtkjer E, Praetorius J, Matchkov VV, Stankevicius E, Mogensen S, Füchtbauer AC, Simonsen U, Fuchtbauer EM & Aalkjaer C (2011). Disruption of Na+,HCO3 −‐cotransporter NBCn1 (slc4a7) inhibits NO‐mediated vasorelaxation, smooth muscle Ca2+‐sensitivity and hypertension development in mice. Circulation 124, 1819–1829. [DOI] [PubMed] [Google Scholar]
- Bunker AK & Laughlin MH (2010). Influence of exercise and perivascular adipose tissue on coronary artery vasomotor function in a familial hypercholesterolemic porcine atherosclerosis model. J Appl Physiol 108, 490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheang WS, Wong WT, Shen B, Lau CW, Tian XY, Tsang SY, Yao X, Chen ZY & Huang Y (2010). 4‐aminopyridine‐sensitive K+ channels contributes to NaHS‐induced membrane hyperpolarization and relaxation in the rat coronary artery. Vascul Pharmacol 53, 94–98. [DOI] [PubMed] [Google Scholar]
- Crosbie AE, Vuylsteke A, Ritchie AJ, Latimer RD & Callingham BA (2000). Inhibitory effects of glibenclamide on the contraction of human arterial conduits used in coronary artery bypass surgery. J Pharm Pharmacol 52, 333–340. [DOI] [PubMed] [Google Scholar]
- Dashwood MR, Dooley A, Shi‐Wen X, Abraham DJ, Dreifaldt M & Souza DS (2011). Perivascular fat‐derived leptin: a potential role in improved vein graft performance in coronary artery bypass surgery. Interact Cardiovasc Thorac Surg 12, 170–173. [DOI] [PubMed] [Google Scholar]
- Dashwood MR, Dooley A, Shi‐Wen X, Abraham DJ & Souza DS (2007). Does periadventitial fat‐derived nitric oxide play a role in improved saphenous vein graft patency in patients undergoing coronary artery bypass surgery? J Vasc Res 44, 175–181. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M & Cohen P (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feletou M (2009). Calcium‐activated potassium channels and endothelial dysfunction: therapeutic options? Br J Pharmacol 156, 545–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fesus G, Dubrovska G, Gorzelniak K, Kluge R, Huang Y, Luft FC & Gollasch M (2007). Adiponectin is a novel humoral vasodilator. Cardiovasc Res 75, 719–727. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Takemori K, Su LY, An WS, Lu C, Sharma AM & Lee RM (2006). Perivascular adipose tissue promotes vasoconstriction: the role of superoxide anion. Cardiovasc Res 71, 363–373. [DOI] [PubMed] [Google Scholar]
- Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M, Laing I, Yates AP, Pemberton PW, Malik RA & Heagerty AM (2009). Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation 119, 1661–1670. [DOI] [PubMed] [Google Scholar]
- Hosoki R, Matsuki N & Kimura H (1997). The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun 237, 527–531. [DOI] [PubMed] [Google Scholar]
- Kohn C, Schleifenbaum J, Szijarto IA, Marko L, Dubrovska G, Huang Y & Gollasch M (2012). Differential effects of cystathionine‐gamma‐lyase‐dependent vasodilatory H2S in periadventitial vasoregulation of rat and mouse aortas. PLoS One 7, e41951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kold‐Petersen H, Brondum E, Nilsson H, Flyvbjerg A & Aalkjaer C (2012). Impaired myogenic tone in isolated cerebral and coronary resistance arteries from the goto‐kakizaki rat model of type 2 diabetes. J Vasc Res 49, 267–278. [DOI] [PubMed] [Google Scholar]
- Lee RM, Lu C, Su LY & Gao YJ (2009). Endothelium‐dependent relaxation factor released by perivascular adipose tissue. J Hypertens 27, 782–790. [DOI] [PubMed] [Google Scholar]
- Li R, Andersen I, Aleke J, Golubinskaya V, Gustafsson H & Nilsson H (2013). Reduced anti‐contractile effect of perivascular adipose tissue on mesenteric small arteries from spontaneously hypertensive rats: role of Kv7 channels. Eur J Pharmacol 698, 310–315. [DOI] [PubMed] [Google Scholar]
- Lohn M, Dubrovska G, Lauterbach B, Luft FC, Gollasch M & Sharma AM (2002). Periadventitial fat releases a vascular relaxing factor. FASEB J 16, 1057–1063. [DOI] [PubMed] [Google Scholar]
- Lynch FM, Withers SB, Yao Z, Werner ME, Edwards G, Weston AH & Heagerty AM (2013). Perivascular adipose tissue‐derived adiponectin activates BKCa channels to induce anticontractile responses. Am J Physiol Heart Circ Physiol 304, H786–H795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelli A, Testai L, Breschi MC, Lawson K, McKay NG, Miceli F, Taglialatela M & Calderone V (2013). Vasorelaxation by hydrogen sulphide involves activation of Kv7 potassium channels. Pharmacol Res 70, 27–34. [DOI] [PubMed] [Google Scholar]
- Mita M, Yanagihara H, Hishinuma S, Saito M & Walsh MP (2002). Membrane depolarization‐induced contraction of rat caudal arterial smooth muscle involves Rho‐associated kinase. Biochem J 364, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momin AU, Melikian N, Shah AM, Grieve DJ, Wheatcroft SB, John L, El GA, Desai JB, Nelson T, Driver C, Sherwood RA & Kearney MT (2006). Leptin is an endothelial‐independent vasodilator in humans with coronary artery disease: evidence for tissue specificity of leptin resistance. Eur Heart J 27, 2294–2299. [DOI] [PubMed] [Google Scholar]
- Mulvany MJ & Halpern W (1977). Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ Res 41, 19–26. [DOI] [PubMed] [Google Scholar]
- Payne GA, Bohlen HG, Dincer UD, Borbouse L & Tune JD (2009). Periadventitial adipose tissue impairs coronary endothelial function via PKC‐b‐dependent phosphorylation of nitric oxide synthase. Am J Physiol Heart Circ Physiol 297, H460–H465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping NN, Li S, Mi YN, Cao L & Cao YX (2015). Hydrogen sulphide induces vasoconstriction of rat coronary artery via activation of Ca2+ influx. Acta Physiol (Oxf) 214, 88–96. [DOI] [PubMed] [Google Scholar]
- Quayle JM, Bonev AD, Brayden JE & Nelson MT (1995). Pharmacology of ATP‐sensitive K+ currents in smooth muscle cells from rabbit mesenteric artery. Am J Physiol 269, C1112–C1118. [DOI] [PubMed] [Google Scholar]
- Reifenberger MS, Turk JR, Newcomer SC, Booth FW & Laughlin MH (2007). Perivascular fat alters reactivity of coronary artery: effects of diet and exercise. Med Sci Sports Exerc 39, 2125–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin AS & Bariskaner H (2007). The mechanisms of vasorelaxant effect of leptin on isolated rabbit aorta. Fundam Clin Pharmacol 21, 595–600. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Hori M, Izumi M, Oka T, Kohama K, Ozaki H & Karaki H (2003). Inhibition of high K+‐induced contraction by the ROCKs inhibitor Y‐27632 in vascular smooth muscle: possible involvement of ROCKs in a signal transduction pathway. J Pharmacol Sci 92, 56–69. [DOI] [PubMed] [Google Scholar]
- Schleifenbaum J, Kohn C, Voblova N, Dubrovska G, Zavarirskaya O, Gloe T, Crean CS, Luft FC, Huang Y, Schubert R & Gollasch M (2010). Systemic peripheral artery relaxation by KCNQ channel openers and hydrogen sulfide. J Hypertens 28, 1875–1882. [DOI] [PubMed] [Google Scholar]
- Shanley LJ, Irving AJ, Rae MG, Ashford ML & Harvey J (2002). Leptin inhibits rat hippocampal neurons via activation of large conductance calcium‐activated K+ channels. Nat Neurosci 5, 299–300. [DOI] [PubMed] [Google Scholar]
- Tian XY, Wong WT, Sayed N, Luo J, Tsang SY, Bian ZX, Lu Y, Cheang WS, Yao X, Chen ZY & Huang Y (2012). NaHS relaxes rat cerebral artery in vitro via inhibition of L‐type voltage‐sensitive Ca2+ channel. Pharmacol Res 65, 239–246. [DOI] [PubMed] [Google Scholar]
- Urban NH, Berg KM & Ratz PH (2003). K+ depolarization induces RhoA kinase translocation to caveolae and Ca2+ sensitization of arterial muscle. Am J Physiol Cell Physiol 285, C1377–C1385. [DOI] [PubMed] [Google Scholar]
- Verlohren S, Dubrovska G, Tsang SY, Essin K, Luft FC, Huang Y & Gollasch M (2004). Visceral periadventitial adipose tissue regulates arterial tone of mesenteric arteries. Hypertension 44, 271–276. [DOI] [PubMed] [Google Scholar]
- Wareing M, Greenwood SL, Fyfe GK, Baker PN & Taggart MJ (2006). Glibenclamide inhibits agonist‐induced vasoconstriction of placental chorionic plate arteries. Placenta 27, 660–668. [DOI] [PubMed] [Google Scholar]
- Weston AH, Egner I, Dong Y, Porter EL, Heagerty AM & Edwards G (2013). Stimulated release of a hyperpolarizing factor (ADHF) from mesenteric artery perivascular adipose tissue: involvement of myocyte BKCa channels and adiponectin. Br J Pharmacol 169, 1500–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH & Wang R (2008). H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma‐lyase. Science 322, 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudkin JS, Eringa E & Stehouwer CD (2005). "Vasocrine" signalling from perivascular fat: a mechanism linking insulin resistance to vascular disease. Lancet 365, 1817–1820. [DOI] [PubMed] [Google Scholar]
- Zavaritskaya O, Zhuravleva N, Schleifenbaum J, Gloe T, Devermann L, Kluge R, Mladenov M, Frey M, Gagov H, Fesus G, Gollasch M & Schubert R (2013). Role of KCNQ channels in skeletal muscle arteries and periadventitial vascular dysfunction. Hypertension 61, 151–159. [DOI] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y & Wang R (2001). The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J 20, 6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]