

Abstract
Visceral leishmaniasis (VL), caused by a protozoan parasite Leishmania donovani, is still a threat to mankind due to treatment failure, drug resistance and coinfection with HIV. The limitations of first-line drugs have led to the development of new strategies to combat this dreaded disease. Recently, we have shown the immunomodulatory property of Ara-LAM, a TLR2 ligand, against leishmanial pathogenesis. In this study, we have extended our study to the effect of Ara-LAM on regulatory T cells in a murine model of VL. We observed that Ara-LAM-treated infected BALB/c mice showed a strong host-protective Th1 immune response due to reduced IL-10 and TGF-β production, along with marked decrease in CD4+ CD25+ Foxp3+ GITR+ CTLA4+ regulatory T cell (Treg) generation and activation. The reduction in Foxp3 expression was due to effective modulation of TGF-β-induced SMAD signaling in Treg cells by Ara-LAM. Moreover, we demonstrated that Ara-LAM-induced IRF1 expression in the Treg cells, which negatively regulated foxp3 gene transcription, resulting in the reduced immunosuppressive activity of Treg cells. Interestingly, irf1 gene knockdown completely abrogated the effect of Ara-LAM on Treg cells. Thus, these findings provide detailed mechanistic insight into Ara-LAM-mediated modulation of Treg cells, which might be helpful in combating VL.
Keywords: Leishmania donovani, Ara-LAM, regulatory T cell, TGF-β, IRF1, Foxp3
This report depicts the role of an immunomodulator, Ara-LAM, in restricting regulatory T-cell generation and activation during murine leishmaniasis, which results in the generation of effector T cell and disease regression.
Graphical Abstract Figure.
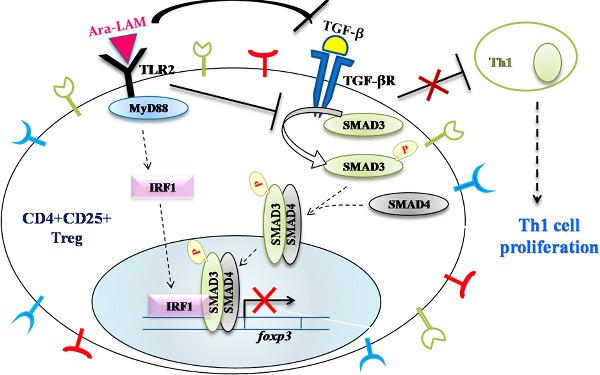
This report depicts the role of an immunomodulator, Ara-LAM, in restricting regulatory T-cell generation and activation during murine leishmaniasis, which results in the generation of effector T cell and disease regression.
INTRODUCTION
Visceral leishmaniasis (VL), a neglected tropical disease caused by the protozoan parasite Leishmania donovani, is characterized by an acute infection in the liver, whereas a lifelong chronic infection in the spleen (Stanley and Engwerda 2007; Sundar and Chakravarty 2013). Leishmania resides and replicates as amastigote in myeloid cells of the host, such as macrophages and dendritic cells (DCs), and survives by interfering or skewing the host-protective T-cell responses (Stanley and Engwerda 2007). Such interference with the T-cell response was attributed to impaired Ag processing, reduced MHC class II (MHC-II) expression and subverted costimulatory molecule expression (Stanley and Engwerda 2007; Sundar and Chakravarty 2013). There is currently no protective vaccine against VL, and first-line drug treatments are imperfect because of long courses of treatment, significant toxicity and emergence of drug resistant parasite (Okwor and Uzonna 2013; Vanaerschot et al. 2014). Therefore, new treatments against VL along with strategies to improve the safety and efficacy of the current drugs are urgently needed.
TLRs are recognized as important ancient receptors for the initiation of efficient immune responses against several pathogenic diseases (Faria, Reis and Lima 2012). Thus, they are often targeted for developing different anti-pathogenic immunotherapies. We characterized a novel TLR2 ligand, arabinosylated lipoarabinomannan (Ara-LAM)—a Mycobacterium smegmatis cell-wall glycolipid (Majumder et al. 2008), for effective anti-tubercular as well as antileishmanial immunoprophylaxis (Majumder et al. 2008; Bhattacharya et al. 2010). Ara-LAM induces TLR2 expression and reciprocally regulates mitogen-activated protein kinase activation and proinflammatory cytokine production in a TLR2-dependent manner (Bhattacharya et al. 2010, 2011). It also enhances the IFN-γ responsiveness of the parasitized macrophages through JAK1/JAK2-STAT1-mediated signaling by counterregulating IRF4 and IRF8 transcription factors and induces nitric oxide synthase-dependent killing of amastigotes, the intracellular form of the parasite (Chowdhury et al. 2015). IRFs comprise a very important group of transcription factors that modulate host defense through different interferon signaling (Constantinescu et al. 1998; Honda and Taniguchi 2006; Ozato, Tailor and Kubota 2007). Enhanced IFN-γ responsiveness and IRF8 generation by Ara-LAM pretreatment lead to better antigen presentation and robust T-cell proliferation in L. donovani-infected mice (Chowdhury et al. 2015).
Among different subpopulations of T cells, CD4+CD25+ regulatory T cells (Tregs) suppress immune responses of other cell types (Gupta et al. 2011). They constitutively express surface CD25 and the transcriptional regulator Foxp3. Other characteristic markers of Tregs are glucocorticoid-induced tumor necrosis factor receptor (GITR) and cytotoxic T lymphocyte antigen 4 (CTLA4) (Sakaguchi et al. 1995; Lee et al. 1997; Read, Malmstrom and Powrie 2000; Shimizu et al. 2002; Gupta et al. 2011; Majumder et al. 2014). Tregs have been reported to play an important role in the progression of leishmanial disease predominantly by suppressing Th1 immune responses (Belkaid et al. 2002; Gupta et al. 2011; Majumder et al. 2014) and inducing high levels of immunosuppressive Th2 cytokines secretion such as IL-10 and TGF-β (Majumder et al. 2014). IL-10, a classical Th2 cytokine is produced by many different cell types including CD4+CD25+ T cells during human VL (Nylén et al. 2007). But in active murine VL, TGF-β is the principle disease promoting cytokine secreted from CD4+CD25+ Tregs (Rodrigues et al. 2009). Like IL-10, TGF-β has several downregulatory effects on the host immune system; it inhibits TNF-α and IFN-γ production from activated T cells, decreases the nitric oxide production and also abrogates the antigen presenting function of host macrophages (Gorelik, Constant and Flavell 2002; Peng et al. 2004; Majumder et al. 2014). Treatment with TGF-β-neutralizing antibodies to susceptible mice results in resistance to Leishmania infection (Majumder et al. 2014). Besides, TGF-β is also important for the in vivo expansion of CD4+CD25+ Treg cells (Peng et al. 2004). Studies with TGF-β knockout mice have shown that Foxp3+ Treg cells are defective in their suppressor function (Majumder et al. 2014). Effective TGF-β signaling in Tregs leads to enhanced Foxp3 expression via activation of the SMAD proteins (Fantini et al. 2004; Gupta et al. 2011). As Ara-LAM is found to induce IFN-γ secreting T-cell generation via macrophage activation, this leads to significant parasite clearance and host protection. However, it remains to be examined whether Ara-LAM treatment can affect Treg generation. Although, several reports suggest that TLR2 controls regulatory T-cell generation and function both in vitro and in vivo (Sutmuller et al. 2006; Fragale et al. 2008; Lal et al. 2011).
Herein, we demonstrate that Ara-LAM pretreatment reduced the elevated frequency of immunosuppressive CD4+CD25+ Treg cells along with its specific phenotypes (Foxp3, GITR and CTLA4) in Leishmania-infected mice. It differentially regulated different immunoregulatory cytokines such as IFN-γ, IL-10 and TGF-β in a cell-specific manner. Among these, TGF-β production and its signaling in CD4+CD25+ Foxp3+Treg cells were prominently downregulated by Ara-LAM pretreatment in L. donovani-infected mice, which might be responsible for the subversion of the suppressive activity of the Treg cells. Moreover, Ara-LAM abrogated the Foxp3 gene transcription by reciprocally regulating SMAD3 and IRF1, the two important transcription factors involved in Foxp3 gene expression. Interestingly, IRF1 silencing resulted in the inhibition of Ara-LAM-mediated Treg suppression in L. donovani-infected mice. Therefore, these results suggest that Ara-LAM confers protection against experimental VL by suppressing the function of Treg cell via IRF1.
MATERIALS AND METHODS
Ethics statement
In the present study, female and male BALB/c mice (18–20 g) were used, which were acclimatized for 15 days in polypropylene cages in the Animal House facility, with standard food and water ad libitum. All the animal experiments were conducted in accordance with the OECD guidelines, accepted by the Committee for the Purpose of Control and Supervision on Experiments on Animals (CPCSEA), Thiruvanmiyur, Chennai, India, and as per the approval of the Institutional Animal Ethical Committee (Bose Institute, Kolkata, Registration Number: 1796/PO/ERe/S/14/CPCSEA). The surgical procedures were performed under Ketamine hydrochloride (100 mg kg−1 i.m.) anesthesia, and all efforts were made to minimize the suffering of the animals.
Animals and parasites
BALB/c mice (both sex, 6–8 weeks old) were purchased from the National Centre for Laboratory Animal Sciences, India. Leishmania donovani (strain MHOM/IN/1983/AG83) was maintained in Medium 199 (Sigma) with 10% fetal calf serum (FCS; Gibco, Grand Island, NY) and passage through BALB/c mice to maintain the virulence. Stationary-phase promastigotes, obtained by suitable transformation, were used for infecting BALB/c mice according to the animal-use protocols approved by the institutional animal ethics committee.
Isolation and purification of Ara-LAM
Ara-LAM was isolated as described elsewhere (Majumder et al. 2008). The noncytotoxic dose of Ara-LAM was found to be 3 μg ml−1 (Bhattacharya et al. 2010).
Preparation of TLR2-shRNA and IRF1-shRNA
For in vivo gene silencing, TLR2-specific and IRF1-specific short hairpin oligos (shRNA∼50 bases) were synthesized with a nine base loop sequences in the middle and a terminator sequence (five to six Ts) at the 3′ end and inserted in the multiple cloning site of pSilencer 1.0 U6 (mouse) plasmid vector having mouse U6 promoter (Ambion Inc., Grand Island, NY). Scrambled shRNA was used as control shRNA.
In vivo experiments
BALB/c mice were treated with Ara-LAM (30 μg intraperitonially) 2 days before L. donovani infection (injected with 1 × 107 parasites/mice, through tail vein). TLR2-shRNA, IRF1-ShRNA or control shRNA (100 μg mice−1) were administered through tail vein, prior to Ara-LAM treatment and infection. Mice were sacrificed 28 days after infection; the splenic and hepatic parasite loads (expressed in Leishman-Donovan units) were enumerated under a microscope. Isolated splenocytes were cultured in RPMI 1640 medium plus 10% (FCS) for cytokine profiling, gene expression study and T-cell proliferation assay.
CD4+ T-cell purification
Splenic CD4+ T cells (purity ∼95% as ascertained by FACS) from differently treated mice were isolated by positive selection using CD4+ IMag beads, according to the manufacturer's instructions (BD Biosciences). For further separation, total CD4+ T cells were isolated by negative selection using magnetic beads followed by positive selection using anti-CD25 magnetic beads on a magnetic separator column into CD4+CD25+ and CD4+CD25− populations as per manufacturer's suggested protocol (MagCellect Treg isolation kit, R&D Systems). The purities of both CD4+CD25+ and CD4+CD25− T cells were routinely >90%.
Flow cytometry
CD4+ T cells were stained with phycoerythrin (PE)-labeled anti-CD25 antibody and fluorescein isothiocyanate (FITC)-labeled anti-Foxp3 antibody (Gupta et al. 2011). For intracellular cytokine staining, purified CD4+CD25+ T cells from soluble leishmanial antigen (SLA) stimulated and Brefeldin A (10 mg ml−1) treated splenocytes were permeabilized (0.1% saponin) and stained with anti-mouse Foxp3-FITC and anti-mouse TGFβ-PE antibodies (Santa Cruz Biotech, Santa Cruz, CA). Cells were analyzed using a FACS Verse flow cytometer (Becton Dickinson, San Diego, CA).
Isolation of RNA and reverse transcriptase polymerase chain reaction
Total RNA extracted from both CD4+CD25+ and CD4+CD25− T cells (TRI reagent; Sigma) were reverse transcribed using Revert Aid M-MuLV Reverse Transcriptase (Fermentas) and semi-quantitative polymerase chain reaction (PCR) was performed using Perkin Elmer Gen Amp PCR system 2400. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as reference. Sequences of the PCR primers are listed in Table 1. The reaction conditions consisted of an initial activation step (5 min at 95°C) and cycling step (denaturation for 30 s at 94°C, annealing for 30 s at 58°C and extension for 1 min at 72°C for 35 cycles). PCR-amplified product was subsequently size fractioned on 2% agarose gel, stained with ethidium bromide and visualized under UV light.
Table 1.
Sequences of the PCR primers.
| Gene | Sequences of primers |
|---|---|
| Foxp3 | Forward 5′-CGTACACCCAGGAAAGACAG-3′ |
| Reverse 5′-ATCCAGGAGATGATCTGCTTG-3′ | |
| GITR | Forward 5′-GACGGTCACTGCAGACTTTG-3′ |
| Reverse 5′-GCCAT GACCAGGAAGATGAC-3′ | |
| CTLA4 | Forward 5′-GGACGCAGATTTATGTCATTGATC- 3′ |
| Reverse 5′-CCAAGCTAACTGCGACAAGGA-3′ | |
| IFN-γ | Forward 5′-AGCTCTTCCTCATGGCTGTTTC-3′ |
| Reverse 5′-TGTTGCTGATGGCCTGATTGT-3′ | |
| IL-10 | Forward 5′-ACTTGGGTTGCCAAGCCTTAT-3′ |
| Reverse 5′-ATCACTCTTCACCTGCTCCACT-3′ | |
| TGF-β | Forward 5′-GGATACCAACTATTGCTTCAGCTCC-3′ |
| Reverse 5′-AGGCTCCAAATATAGG GGCAGGGTC-3′ | |
| IRF-1 | Forward 5′- CAGAGGAAAGAGAGAAAGTCC-3′ |
| Reverse 5′- CACACGGTGACAGTGCTGG-3′ | |
| IRF-4 | Forward 5′- GTGACTGTGCCCTGGCTTAT-3' |
| Reverse 5′- TGGACATGATCTGGGCAACC-3' | |
| IRF-8 | Forward 5′-AACTGTGCTCTGGGCTCATC-3' |
| Reverse5′- CCTCCGGGAAGTGTCCCTTA-3' | |
| Foxp3 promoter | Forward 5'-GTGGTGAGGGGAAGAAATCA-3' |
| Reverse 5'-GATGAGTGTGTGCGCTGATAA-3' | |
| GAPDH | Forward 5′-GTTGTCTCCTGCGACTTCAACA-3′ |
| Reverse 5′-TCTCTTGCTCAGTGTCCTTGCT-3′ |
Proliferation assay and cytokine ELISA
In the absence or presence of increasing numbers of splenic CD4+CD25+ Treg cells, splenic responder CD4+CD25− T cells (5 × 105) and T-depleted, mitomycin C-treated syngeneic APCs (5 × 105) were cultured for 4 days in 96-well, round-bottom plates as described earlier (Majumder et al. 2014). SLA (10 μg ml−1) was added to the culture for stimulation. In in vitro experiment, WT or shRNA-treated Treg cells (isolated from naive BALB/c mice) were cocultured with naive CD4+CD25+ responder T cells (Th) for 3 days, in the presence of soluble anti-CD3 (1 μg ml−1) and T-depleted, mitomycin C-treated, syngeneic APCs. Ara-LAM (3 μg ml−1) was added at the start of the coculture. In both in vivo and in vitro experiments, one microcurie of [3H]thymidine was added 18 h before harvesting, and 3H-Thymidine uptake, as an index of proliferation, was measured using a liquid scintillation counter (Tri-Carb 2800TR; Perkin Elmer). Supernatants were collected from the coculture of responder CD4+CD25− and CD4+CD25+ Treg cells (1:1) at 24 h (for IL-2) or 72 h (for IFN-γ). In some cases, splenocytes (2 × 106 cells/ml per well), CD4+CD25− Tcells (1 × 106 cells/ml per well) or CD4+CD25+ Treg cells (1 × 106 cells/ml per well) from different sets of treatment were stimulated with SLA (10 μg ml−1) for 72 h. The levels of cytokines in supernatants were determined by specific ELISAs (BD Biosciences and R&D Systems).
Preparation of cell lysate and immunoblot analysis
Cell lysates were prepared as described in our previous reports (Bhattacharya et al. 2010). Equal amounts of protein (50 μg) were subjected to 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis, and immunoblotting for phosphorylated forms of different SMAD protein were performed (Bhattacharya et al. 2010).
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed using the ChIP assay kit following the manufacturers protocol (Millipore, Billerica, MA) as described elsewhere (Bhattacharya et al. 2011). With the extracted DNA, PCR was conducted using foxp3 promoter-specific primers, specifically IRF1 and SMAD3-binding sites. PCR-amplified product was resolved on 2% agarose gel, stained with ethidium bromide and visualized under UV light.
Statistical analysis
A minimum of three mice were used per group for in vivo experiments. Data, including densitometry analysis, represented as means ± SD, are from one of three representative experiments. One-way ANOVA was used to assess the significance of differences between the mean values for control and experimental groups. A difference with P < 0.05 was considered significant, and a difference with P < 0.001 was considered highly significant.
RESULTS
Effect of Ara-LAM on frequency and phenotype of Treg cells in Leishmania-infected BALB/c mice
We have already reported that pre-treatment with Ara-LAM confers significant protection in L. donovani-infected mice via a Th1-polarized anti-parasite response (Bhattacharya et al. 2010, 2011; Chowdhury et al. 2015). However, it is unclear whether Ara-LAM treatment can modulate the Treg cells that are involved in disease progression during leishmaniasis. To address these questions, the frequency and phenotype of splenic Treg cell population were first evaluated by FACS analysis. We observed that there was a marked decrease (*P < 0.001) from 21.43% to 11.71% in CD4+CD25+Foxp3+ Treg cells in splenocytes isolated from Ara-LAM-treated infected mice in comparison to infected sets (Fig. 1A). Similarly, other Treg cell-specific markers like Foxp3, CTLA4 and GITR showed a consistent decrease in splenic CD4+ CD25+ Treg cells isolated from Ara-LAM-treated infected sets as compared to infected sets (Fig. 1B). Semiquantitative RT-PCR analysis showed 4-fold decrease in Foxp3, 3-fold decrease in CTLA4 and 2.5-fold decrease in GITR gene expression in Ara-LAM-treated infected sets in comparison to infected sets (Fig. 1B). Surprisingly, TLR2 silencing by TLR2-specific shRNA (Fig. S1, Supporting Information) abrogated the effect of Ara-LAM on Treg frequency and phenotype in infected mice, confirming again that Ara-LAM is a potent TLR2 ligand (Fig. 1A and B).
Figure 1.

Effect of Ara-LAM pretreatment on the frequency and phenotype of T regulatory cells. (A) CD4+ T cells (1 × 106) were purified by MACS from spleen of infected and indicated treatment groups of mice 28 days after infection, plated aseptically followed by fixation and staining for PE-conjugated CD25 and FITC-conjugated Foxp3 as mentioned in the section ‘Materials and Methods’ and analyzed by flow cytometry. Data are from one of three representative experiments. (B) In a separate experiment, CD4+CD25+ Treg cells (2 × 105) were purified from spleen of differently treated mice by MACS as described in the section ‘Materials and Methods’ and collected in TRIZOL for mRNA extraction. The expression of Foxp3, GITR and CTLA4 mRNAs was analyzed by semiquantitative RT-PCR. The data are representative of three independent experiments, which yielded similar results.
Ara-LAM treatment abrogated the suppressive activity of CD4+CD25+ Treg cells in L. donovani-infected mice
Regulatory T cells are known to propagate the disease by suppressing the responder T cells proliferation. To examine whether the Ara-LAM treatment could modulate the suppressive function of CD4+CD25+ Foxp3+ Treg cells towards responder T cells, we performed coculture experiments using CD4+CD25+ and CD4+CD25− T cells purified from spleen of differently treated mice. In response to SLA stimulation, the proliferation of responder CD4+CD25− T cells was measured in the presence of T-depleted, mitomycin C-treated, syngeneic APCs by thymidine incorporation assay. Results suggested that in untreated infected sets, CD4+CD25+ Treg cells efficiently suppressed the proliferation of responder CD4+CD25− T cells in a dose-dependent manner. Whereas CD4+CD25+ Treg cells, from Ara-LAM-treated infected mice, could not suppress the proliferation of responder CD4+CD25− T cells as compared with untreated infected sets (Fig. 2A). This T-cell proliferation was supported by enhanced IL-2 and IFN-γ production. Herein we found that CD4+CD25+ Treg cells from Ara-LAM-treated mice, when cocultured with CD4+CD25− responder T cells (1:1), could not abrogate the release of IL-2, which was otherwise suppressed by the CD4+CD25+ Treg cells isolated from infected mice (Fig. 2B). Moreover, we found enhanced IFN-γ secretion in the coculture set of CD4+CD25+ Treg cells from Ara-LAM-treated mice, but CD4+CD25+ Treg cells from L. donovani-infected mice significantly abrogated the IFN-γ production from responder CD4+CD25− T cells supporting parasite-induced Treg activity (Fig. 2C). Our in vivo findings were further supported by in vitro suppression assay, where we observed that in vitro Ara-LAM treatment reduced the suppressive activity of Treg cells towards the responder CD4+CD25− T cells (Fig. S2, Supporting Information).
Figure 2.

Ara-LAM treatment inhibited the suppressive activity of T regulatory cells during VL. (A) CD4+CD25− T cells and CD4+CD25+ T cells were purified from spleen of differently treated mice (section ‘Materials and methods’) after 28 days post-infection. CD4+CD25− responder T cells (5 × 105 cells) and T-depleted, mitomycin C-treated, syngeneic APCs (5 × 105 cells) were stimulated with SLA in the absence or presence of increasing numbers of splenic CD4+CD25+ Treg cells for 4 days. Proliferation was measured by an 18 h [3H]thymidine incorporation assay. Data represent means ± SD for three animals per group. *P < 0.001 for the comparison with infected mice. (B and C) The supernatants were collected at 24 h for IL-2 (B) or 72 h for IFN-γ (C) following stimulation with SLA, and level of cytokines was determined by ELISA. Data represent means ± SD for triplicate sets. *P < 0.001 for the comparison with infected mice.
Effect of Ara-LAM on cytokine secretion from T regulatory cells in L. donovani-infected BALB/c mice
There are several pieces of evidence suggesting that activity of CD4+CD25+ Treg cells in vivo involves some immunosuppressive cytokines (Gupta et al. 2011; Majumder et al. 2014). So we compared the cytokine profile of Ara-LAM-treated infected CD4+CD25+ Treg cells with the profile of untreated infected counterparts. Data suggested that proinflammatory cytokine IFN-γ was downregulated, but anti-inflammatory cytokine IL-10 and TGF-β were upregulated in both CD4+CD25+ Treg cells and CD4+CD25− T responder cells from L. donovani-infected mice as compared to WT mice (Fig. 3A). Interestingly, CD4+CD25+ Treg cells were found to be a better source of TGF-β but not IL-10 production as compared to its CD4+CD25− counterpart. By contrast, Ara-LAM treatment enhanced the IFN-γ production and inhibited the infection induced IL-10 and TGF-β generation in both cell types. Notably, CD4+CD25− T cells from Ara-LAM-treated mice produced significantly higher amount of IFN-γ in comparison with CD4+CD25+ Treg cells. Here we also found that Ara-LAM mediated regulation of all the three cytokines (at both mRNA and protein levels) in both cell types was TLR2 dependent (Fig. 3B–D).
Figure 3.

Ara-LAM differentially regulated Th1 and Th2 cytokine generation in various cell types during VL. (A) CD4+CD25− T cells and CD4+CD25+ T cells were purified from spleen of differently treated mice by MACS (section ‘Materials and Methods’) and collected in TRIZOL for mRNA extraction. The expression of IFN-γ, IL-10 and TGF-β was analyzed by semiquantitative RT-PCR. The data are representative of three independent experiments yielding similar results. (B–D) In a separate experiment, CD4+CD25− T cells and CD4+CD25+ T cells were stimulated with SLA and supernatant was collected at 72 h for determining the level of IFN-γ, IL-10 and TGF-β secretion by ELISA. Data represent means ± SD for triplicate sets. *P < 0.001 and **P < 0.05 for the comparison with infected mice.
Ara-LAM mediated abrogation of the TGF-β signaling in Treg cells during L. donovani infection
Corroborating with previous reports (Nylén et al. 2007; Majumder et al. 2014), our observation indicated that in infected mice IL-10 was mainly produced by CD4+CD25− T cells. However, CD4+CD25+ Treg cells produced significant amount of TGF-β in L. donovani-infected mice, which was inhibited by Ara-LAM treatment (Fig. 3D). Now, it is well known that TGF-β is an important mediator of Treg functioning. As Ara-LAM downregulated both TGF-β and Foxp3 expression in Treg, we wanted to examine whether this Ara-LAM mediated suppression involves the regulation of TGF-β signaling during infection. Thus, we treated the mice with a specific inhibitor of TGF-β signaling, SB431542 (Gupta et al. 2011), followed by Leishmania infection and compared its effect with that of Ara-LAM treatment. In both cases, we found significantly low TGF-β secretion from Treg cells of infected mice. Interestingly, Ara-LAM mediated inhibition of Foxp3 expression was likely to be stronger as compared to SB431542 (Fig. 4A and B). We also analyzed CD4+CD25+ Treg cells, isolated from different groups of animals, for TGF-β secreting Foxp3+ Treg cells. We observed an elevated level (76.64%) of TGF-β secreting Foxp3+ Treg cell population in infected mice, which was significantly diminished (32.9%) in Ara-LAM-treated infected mice. Here also SB431542 was found to be less effective than Ara-LAM (43.09%) (Fig. 4C).
Figure 4.

Effect of Ara-LAM treatment on TGF-β signaling in T regulatory cells. (A and B) BALB/c mice were pretreated with SB-431542(SB) (10 mg/kg body weight, i.v.) for 1 day or with respective shRNAs (i.v.) for 2 days followed by Ara-LAM treatment and Leishmania challenge. CD4+CD25+ Treg cells (2 × 105), purified from spleen of differently treated mice (see section ‘Materials and Methods’) 4 weeks post-infection, were collected in TRIZOL for mRNA extraction and semiquantitative RT-PCR to study TGF-β and Foxp3 mRNA expression or were stimulated with SLA for 72 h, after which the cell supernatants were collected for estimation of TGF-β (B) by ELISA. Data represent means ± SD for triplicate sets. *P < 0.001 for the comparison with infected mice. (C) In a separate experiment, CD4+CD25+ Treg cells (1 × 106), isolated from spleen of infected and indicated treatment groups of mice 28 days after infection, plated aseptically followed by fixation and staining for PE-conjugated TGF-β and FITC-conjugated Foxp3 as mentioned in the section ‘Materials and Methods’ and analyzed by flow cytometry. Data are from one of three representative experiments. Bar diagram represents means ± SD for triplicate sets. *P < 0.001 and **P < 0.05 for the comparison with infected mice. (D) In a separate experiment, similarly isolated CD4+CD25+ Treg cells (2 × 106) were stimulated with SLA for 30 min, followed by cell lysis and western blotting with antibodies against p-SMAD3, p-SMAD4, SMAD3, SMAD4 and GAPDH as described in the section ‘Materials and Methods’. The data are representative of three independent experiments yielding similar results. Bar diagram represents densitometric fold change in arbitrary unit.
For further confirmation of this Ara-LAM mediated inhibition of TGF-β signaling, we studied the involvement of SMAD3, one of the key TGF-β signaling-activated SMADs, and SMAD4, the Co-SMAD in Treg cells (Fantini et al. 2004; Gupta et al. 2011). By western blot analysis it was observed that after 30 min of SLA stimulation, maximum phosphorylation of SMAD3 and SMAD4 was observed in CD4+CD25+ Treg cells from infected mice (Fig. 4D). However, Ara-LAM treatment efficiently abrogated their phosphorylation and activation. We also found that this Ara-LAM mediated deactivation of SMAD proteins were TLR2 dependent and was comparable with the effect of SB431542, a classical TGF-β signaling inhibitor (Fig. 4D), thereby suggesting that Ara-LAM regulates the TGF-β signaling by downregulating the activation of SMAD proteins.
Foxp3 was a direct target of IRF1 in CD4+CD25+ Treg cells in Ara-LAM-treated infected mice
It was previously reported that SMAD3 is an important transcription factor for Foxp3 gene expression regulated by TGF-β signaling (Fragale et al. 2008; Lal et al. 2011). In our study, we observed that Ara-LAM abrogated the TGF-β signaling mediated SMAD3 activation and Foxp3 expression in a TLR2-dependent manner. But, whether this is the only mechanism or some other factors are also involved in this Ara-LAM mediated regulation of foxp3 gene is still not clear. Previously Lal et al. (2011) suggested that TLR2-induced inhibition of Foxp3 is dependent on Myd88 and IRF1. IRF1 binds to the IRF-E present in the proximal promoter and intronic enhancer (near SMAD3-binding site) region at the foxp3 locus, and negatively regulates the transcription of foxp3 to suppress Treg cell function. Similarly, Fragale et al. (2008) reported that IRF-1 directly regulates transcriptional activity of the foxp3 gene promoter and CD4+CD25− T cells from IRF-1−/− mice are prone to be converted into CD4+CD25+Foxp3+ Treg with a higher suppressive activity than WT cells.
Earlier we have reported that Ara-LAM is a potent TLR2 ligand and it can reciprocally regulate IRF4 and IRF8 during leishmanial infection (Chowdhury et al. 2015). To examine the Ara-LAM mediated regulation of IRF1 in infected mice, we studied these three IRFs in both CD4+CD25+ Treg cells and CD4+CD25− responder T cells. We found that IRF4 was induced by infection in both cell types, which was downregulated by Ara-LAM. However, Ara-LAM mediated induction of IRF8 was only observed in CD4+CD25− T cells, but not in CD4+CD25+ Treg cells. In contrast, IRF1 was significantly induced by Ara-LAM treatment in CD4+CD25+ Treg cells as well as CD4+CD25− T cells, suggesting the possible involvement of IRF1 in Ara-LAM mediated Treg cell regulation (Fig. 5A). Moreover, a prominent binding of IRF1 to the Foxp3 promoter was seen in Treg cells from Ara-LAM-treated mice, which is comparable with the effect of PGN, a known TLR2 ligand (Fig. 5B). Not only that, Ara-LAM treatment also decreased their acetylated histone 3 (AcH3) at this promoter region (Fig. 5D). On the other hand, Leishmania infection induced SMAD3 binding to the foxp3 promoter, which was found to be significantly diminished by Ara-LAM treatment (Fig. 5C).
Figure 5.

Ara-LAM downregulated foxp3 gene expression by IRF1 mediated suppression in Tregs during L. donovani infection. (A) CD4+CD25− T cells and CD4+CD25+ T cells were purified from spleen of differently treated mice by MACS (section ‘Materials and Methods’) and collected in TRIZOL for mRNA extraction. The expression of IRF1, IRF4 and IRF8 was analyzed by semiquantitative RT-PCR. The data are representative of three independent experiments yielding similar results. (B–D) CD4+CD25+Tregs, from spleen of differently treated mice, were purified as described earlier. IRF1 binding at Foxp3 promoter (IP:IRF1), SMAD3 binding at Foxp3 promoter (IP:SMAD3) and histone 3 acetylation at the Foxp3 promoter were analyzed by ChIP assay. Data represent means ± SD for three sets of experiments.
IRF1 silencing resulted in the inhibition of Ara-LAM mediated Treg cell suppression during Leishmania infection
To further confirm the involvement of IRF1 in the Ara-LAM mediated Treg cell suppression during L. donovani infection, we knocked down the IRF1 gene in mice by IRF1-shRNA followed by Ara-LAM treatment and L. donovani infection. After 28 days, we analyzed Treg generation along with its activation. We observed that, in IRF1–shRNA-treated infected mice, Ara-LAM failed to suppress the CD4+CD25+ Treg generation (Fig. 6A) and its activation markers such as Foxp3, CTLA4 and GITR were induced in these set of Tregs (Fig. 6B). Moreover, IRF1 silencing reversed the Ara-LAM mediated abrogation of Treg suppressive activity and reduced the proliferation of responder T cells (Fig. 6C), which is critical for the resistance against leishmanial pathogenesis. Thus, we observed a higher parasite load in both liver and spleen of IRF1 silenced Ara-LAM-treated infected mice in comparison of Ara-LAM-treated infected mice (Fig. 6D and E).
Figure 6.

Effect of IRF1 silencing in the Ara-LAM mediated regulation of T regulatory cells during VL. (A) CD4+ T cells (1 × 106) were purified by MACS from spleen of differently treated mice 28 days after infection, plated aseptically followed by fixation and staining for PE-conjugated CD25 and FITC-conjugated Foxp3 and analyzed by flow cytometry. Data are from one of three representative experiments. (B) In a separate experiment, CD4+CD25+ Treg cells (2 × 106), purified from spleen of differently treated mice by MACS, were collected in TRIZOL for mRNA extraction. The expression of Foxp3, GITR and CTLA4 mRNAs was analyzed by semiquantitative RT-PCR. The data are representative of three independent experiments, which yielded similar results. (C) CD4+CD25− T cells and CD4+CD25+ T cells were purified from spleen of differently treated mice (section ‘Materials and methods’) after 28 days post-infection. CD4+CD25− responder T cells (5 × 105 cells) and T-depleted, mitomycin C-treated, syngeneic APCs (5 × 105 cells) were stimulated with SLA in the presence of splenic CD4+CD25+ T cells (1:1) for 4 days. Proliferation was measured by an 18 h [3H]thymidine incorporation assay. Data represent means ± SD for three animals per group. *P < 0.001, **P < 0.05 for the comparison with infected mice. (D and E) In a separate experiment, mice were sacrificed after 28 days of infection. Levels of parasite burden in liver and spleen were expressed in Leishman–Donovan units (LDUs). Data represent means ± SD for three animals per group. *P < 0.001, **P < 0.05 for the comparison with infected mice.
All the above observations collectively demonstrated that Ara-LAM, a TLR2 ligand, downregulated Treg cell generation and activation in a TLR2-dependent manner. Ara-LAM not only inhibited the TGF-β signaling in Treg cells, moreover IRF1 plays a critical role in Ara-LAM mediated Treg cell suppression.
DISCUSSION
Development of fatal condition in visceral leishmaniasis is mainly associated with lack of proper treatment and emergence of drug resistance (Stanley and Engwerda 2007; Sundar and Chakravarty 2013; Okwor and Uzonna 2013; Vanaerschot et al. 2014). It is ascribed by parasite-driven Th2 cytokine production which multiplies its impact by creating an immunosuppresive milleu (Stanley and Engwerda 2007; Gupta et al. 2011; Sundar and Chakravarty 2013; Okwor and Uzonna 2013; Majumder et al. 2014; Vanaerschot et al. 2014). Treg cells are major source of immunosuppression in Leishmania infection (Belkaid et al. 2002; Rodrigues et al. 2009; Gupta et al. 2011; Majumder et al. 2014). Hence, identification of the molecules that control Treg cell differentiation and function is important not only for understanding the host immune responses but also in developing immunomodulators. On the other hand, we have established Ara-LAM as a potent immunemodulator, which induces proinflammatory functions in a TLR2-dependent manner reversing the impaired antileishmanial and antitubercular cell-mediated immune responses (Majumder et al. 2008; Bhattacharya et al. 2010, 2011; Chowdhury et al. 2015). However Ara-LAM mediated regulation of the Treg cells during the active disease was yet to be discovered. In this study, we silenced the tlr2 and irf1 gene expression in BALB/c mice by their specific shRNAs and their knockdown efficacy was confirmed at mRNA level (Fig. S1, Supporting Information). However, for better understanding of the Ara-LAM mediated Treg cell regulation, further experiments using a Treg-specific deletion of TLR2 (FoxP3cre × TLR-2 fl/fl) would be the next step in addressing this aspect.
Literatures suggested that an absence of IFN-γ results in an increase in Treg cells along with enhanced expression of Foxp3 (Lee et al. 1997). Previously, we reported that Ara-LAM induces IFN-γ secreting CD4+ T cells in Leishmania-infected BALB/c mice (Bhattacharya et al. 2010, 2011; Chowdhury et al. 2015). This can be correlated with impaired Treg cell functioning during leishmanial pathogenesis. Indeed, we observed that Ara-LAM pretreatment reduced both the frequency and immunosuppressive nature of CD25+Foxp3+CD4+ Treg cells in the infected mice (Fig. 1). This in turn, accompanied with effector T-cell proliferation both in vivo (Fig. 2) and in vitro (Fig. S2, Supporting Information), suggesting that Ara-LAM might directly act on regulatory T cells resulting in reduced suppression.
Functioning of Treg cells is the major determinant in various diseases (Szczepanski et al. 2009; Boschetti et al. 2011; Gupta et al. 2011). Recent reports suggest that CD4+CD25− T cells generated IL-10 plays a significant role in the progression of murine VL (Levings et al. 2002; Nylén et al. 2007). Our results also indicated that Ara-LAM treatment significantly inhibited the secretion of IL-10 from CD4+CD25− T cells, which could be crucial for the parasite clearance together with induction of IFN-γ secretion (Fig. 3). On the other hand, CD4+CD25+ Treg cells from infected mice secrete TGF-β helping in the multiplication of the parasite inside the host cells (Fig. 3) and creating a positive feedback loop for Treg cell generation and induction of FoxP3, GITR and CTLA4 in those cells (Suffia et al. 2006; Zheng et al. 2006; Majumder et al. 2014). Our results suggest that Ara-LAM inhibited TGF-β production from CD4+CD25+Foxp3+ Treg cells and reduced their suppressive activity by inhibiting Foxp3 expression (Fig. 4), which is critical for the resistance against Leishmania (Suffia et al. 2006). Subsequently, SMAD proteins are important regulators of efficient TGF-β signaling (Fantini et al. 2004; Gupta et al. 2011; Majumder et al. 2014). Our results indicated that Ara-LAM pretreatment restricted the parasite-induced enhanced phosphorylation of SMAD3 and SMAD4, which was comparable with the effect of SB431542, a TGF-β signaling inhibitor. However, under TLR2 silenced condition, the effects of Ara-LAM on TGF-β signaling were significantly abrogated (Fig. 4).
In response to various signals, the signature molecule of Treg cells, Foxp3, is epigenetically regulated by different regulatory elements, including c-Rel, p65, NFAT, CREB, SMAD, STAT, etc. (Lal and Bromberg 2009; Ruan et al. 2009). Besides, both IFN-γ administration and TLR2 stimulation lead to Treg cell suppression through a PGN/TLR2/Myd88/IRF-1 pathway (Coccia et al. 2000; Sutmuller et al. 2006; Lal et al. 2011). IRF1, a key regulator of IFN-γ mediated Th1 response, can act as both transcriptional activator and repressor depending on stimulations (Kirchhoff et al. 2000; Nishikawa et al. 2005; Lal et al. 2011). A similar report suggests that IRF-1 can directly repress Foxp3 expression and Treg cell development (Fragale et al. 2008; Lal et al. 2011). Commensurate with this, our observations showed that Ara-LAM treatment induced IRF1expression in both CD4+CD25+ Treg cells and CD4+CD25− T responder cells. In Treg cells, activated IRF1 then downregulated foxp3 gene transcription by binding to its promoter region. We also observed that Ara-LAM suppressed foxp3 gene expression by inhibiting SMAD3 binding to foxp3 locus and reducing its histone acetylation (Fig. 5). Knockdown of the irf1 gene in BALB/c mice prior to Ara-LAM treatment and L. donovani infection reversed the effect of Ara-LAM on generation and functioning of Treg cell and resulted in the restoration of intracellular parasites in liver and spleen (Fig. 6).
Taken together, by inhibiting TGF-β signaling and induction of IRF1 in Treg cells, the immunomodulator, Ara-LAM modulates immunosuppressive nature and frequency of regulatory T cells and induces a pro-host Th1response, opening up a broader avenue for other diseases where Treg cells are involved in disease pathogenesis.
Supplementary Material
Acknowledgments
We are grateful to The Director, Bose Institute (Kolkata) for providing space and other infrastructural facilities. We cordially thank Mr. Prabal Gupta and Central instrument facility, Bose Institute, for their technical assistance.
SUPPLEMENTARY DATA
FUNDING
This work was supported by Department of Science and Technology, Govt. of India. We acknowledge the Council of Scientific and Industrial Research (CSIR), Government of India, New Delhi for providing fellowship to Bidisha Paul Chowdhury.
Conflict of interest. None declared.
REFERENCES
- Belkaid Y, Piccirillo CA, Mendez S, et al. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–7. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- Bhattacharya P, Bhattacharjee S, Gupta G, et al. Arabinosylated lipoarabinomannan–mediated protection in Visceral leishmaniasis through up regulation of toll-like receptor-2 signaling: an immunoprophylactic approach. J Inf Dis. 2010;202:145–55. doi: 10.1086/653210. [DOI] [PubMed] [Google Scholar]
- Bhattacharya P, Gupta G, Majumder S, et al. Arabinosylated lipoarabinomannan skews Th2 phenotype towards Th1 during Leishmania infection by chromatin modification: involvement of MAPK signaling. PloS One. 2011;6:e24141. doi: 10.1371/journal.pone.0024141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boschetti G, Nancey S, Sardi F, et al. Therapy with anti-TNFα antibody enhances number and function of Foxp3(+) regulatory T cells in inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17:160–70. doi: 10.1002/ibd.21308. [DOI] [PubMed] [Google Scholar]
- Chowdhury BP, Bandyopadhyay S, Das S, et al. The host-protective effect of arabinosylated lipoarabinomannan against Leishmania donovani infection is associated with restoration of IFN-γ responsiveness. PLoS One. 2015;10:e0117247. doi: 10.1371/journal.pone.0117247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coccia EM, Stellacci E, Marziali G, et al. IFNgamma and IL-4 differently regulate inducible NO synthase gene expression through IRF-1 modulation. Int Immunol. 2000;12:977–85. doi: 10.1093/intimm/12.7.977. [DOI] [PubMed] [Google Scholar]
- Constantinescu CS, Hondowicz BD, Elloso MM, et al. The role of IL-12 in the maintenance of an established Th1 immune response in experimental leishmaniasis. Eur J Immunol. 1998;28:2227–33. doi: 10.1002/(SICI)1521-4141(199807)28:07<2227::AID-IMMU2227>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Fantini MC, Becker C, Monteleone G, et al. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25-Tcells through Foxp3 induction and downregulation of Smad7. J Immunol. 2004;172:5149–53. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
- Faria MS, Reis FC, Lima AP. Toll-like receptors in Leishmania infections: guardians or promoters? J Parasitol Res. 2012;2012:1–12. doi: 10.1155/2012/930257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragale A, Gabriele L, Stellacci E, et al. IFN regulatory factor-1negatively regulates CD4+CD25+ regulatory T cell differentiation by repressing Foxp3 expression. J Immunol. 2008;181:1673–82. doi: 10.4049/jimmunol.181.3.1673. [DOI] [PubMed] [Google Scholar]
- Gorelik L, Constant S, Flavell RA. Mechanism of transforming growth factor b-induced inhibition of T helper type1 differentiation. J Exp Med. 2002;195:1499–505. doi: 10.1084/jem.20012076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta G, Majumdar S, Adhikari A, et al. Treatment with IP-10 induces host-protective immuneresponse by reg- ulating the Tregulatory cell functioning in Leishmania donovani-infected mice. Med Microbiol Immunol. 2011;200:241–53. doi: 10.1007/s00430-011-0197-y. [DOI] [PubMed] [Google Scholar]
- Honda K, Taniguchi T. IRFs: master regulators of signaling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6:644–58. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- Kirchhoff S, Oumard A, Nourbakhsh M, et al. Interplay between repressing and activating domains defines the transcriptional activity of IRF-1. Eur J Biochem / FEBS. 2000;267:6753–61. doi: 10.1046/j.1432-1033.2000.01750.x. [DOI] [PubMed] [Google Scholar]
- Lal G, Bromberg JS. Epigenetic mechanisms of regulation of Foxp3 expression. Blood. 2009;114:3727–35. doi: 10.1182/blood-2009-05-219584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal G, Yin N, Xu J, et al. Distinct inflammatory signals have physiologically divergent effects on epigenetic regulation of Foxp3 expression and Treg function. Am J Transplant. 2011;11:203–14. doi: 10.1111/j.1600-6143.2010.03389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Han Y, Lu HT, et al. TGF-b suppresses IFN- g induction of classII-MHC gene expression by inhibition classII transactivator messenger RNA expression. J Immunol. 1997;158:2065–75. [PubMed] [Google Scholar]
- Levings MK, Sangregorio R, Sartirana C, et al. Human CD25+CD4+ T suppressor cell clones produce transforming growth factor beta, but not interleukin10, and are distinct from type1 Tregulatory cells. J Exp Med. 2002;196:1335–46. doi: 10.1084/jem.20021139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder N, Bhattacharjee S, Dey R, et al. Arabinosylated lipoarabinomannan modulates the impaired cell mediated immune response in Mycobacterium tuberculosis H37Rv infected C57BL/6 mice. Microbes Infect. 2008;10:349–57. doi: 10.1016/j.micinf.2007.12.013. [DOI] [PubMed] [Google Scholar]
- Majumder S, Bhattacharjee A, Chowdhury BP, et al. Antigen-pulsed CpG-ODN-activated dendritic cells induce host-protective immune response by regulating the T regulatory cell functioning in Leishmania donovani-infected mice: critical role of CXCL10. Front Immunol. 2014;5:261. doi: 10.3389/fimmu.2014.00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa H, Kato T, Tawara I, et al. IFN-γ controls the generation/ activation of CD4+CD25+ regulatory T cells in antitumor immune response. J Immunol. 2005;175:4433–40. doi: 10.4049/jimmunol.175.7.4433. [DOI] [PubMed] [Google Scholar]
- Nylén S, Maurya R, Eidsmo L, et al. Splenic accumulation of IL-10mRNA in T cells distinct from CD4+CD25+(Foxp3) regulatory T cells in human visceral leishmaniasis. J Exp Med. 2007;204:805–17. doi: 10.1084/jem.20061141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okwor I, Uzonna JE. The immunology of Leishmania/HIV co-infection. Immunol Res. 2013;56:163–71. doi: 10.1007/s12026-013-8389-8. [DOI] [PubMed] [Google Scholar]
- Ozato K, Tailor P, Kubota T. The interferon regulatory factor family in host defense: mechanism of action. J Biol Chem. 2007;282:20065–9. doi: 10.1074/jbc.R700003200. [DOI] [PubMed] [Google Scholar]
- Peng Y, Laouar Y, Li MO, et al. TGF-beta regulates in vivo expansion of Foxp3-expressing CD4+CD25+ regulatory Tcells responsible for protection against diabetes. P Natl Acad Sci USA. 2004;101:4572–7. doi: 10.1073/pnas.0400810101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen4 plays an essential role in the function of CD25(+)CD4(+)regulatory cells that control intestinal inflammation. J Exp Med. 2000;192:295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues OR, Marques C, Soares-Clemente M, et al. Identification of regulatory T cells during experimental Leishmania infantum infection. Immunobiology. 2009;214:101–11. doi: 10.1016/j.imbio.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Ruan Q, Kameswaran V, Tone Y, et al. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity. 2009;31:932–40. doi: 10.1016/j.immuni.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Sakaguchi N, Asano M, et al. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–64. [PubMed] [Google Scholar]
- Shimizu J, Yamazaki S, Takahashi T, et al. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self tolerance. Nat Immunol. 2002;3:135–42. doi: 10.1038/ni759. [DOI] [PubMed] [Google Scholar]
- Stanley AC, Engwerda CR. Balancing immunity and pathology in visceral leishmaniasis. Immunol Cell Biol. 2007;85:138–47. doi: 10.1038/sj.icb7100011. [DOI] [PubMed] [Google Scholar]
- Suffia IJ, Reckling SK, Piccirillo CA, et al. Infected site-restricted Foxp3+ natural regulatory T cells are specific for microbial antigens. J Exp Med. 2006;203:777–88. doi: 10.1084/jem.20052056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar S, Chakravarty J. Leishmaniasis: an update of current pharmacotherapy. Expert Opin Pharmaco. 2013;14:53–63. doi: 10.1517/14656566.2013.755515. [DOI] [PubMed] [Google Scholar]
- Sutmuller RP, den Brok MH, Kramer M, et al. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116:485–94. doi: 10.1172/JCI25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepanski MJ, Szajnik M, Czystowska M, et al. Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leukemia. Clin Cancer Res. 2009;15:3325–32. doi: 10.1158/1078-0432.CCR-08-3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanaerschot M, Dumetz F, Roy S, et al. Treatment failure in leishmaniasis: drug-resistance or another (epi-) phenotype? Expert Rev Anti-Infe. 2014;12:937–46. doi: 10.1586/14787210.2014.916614. [DOI] [PubMed] [Google Scholar]
- Zheng SG, Wang JH, Stohl W, et al. TGF-beta requires CTLA-4 early after T cell activation to induce FoxP3 and generate adaptive CD4+CD25+ regulatory cells. J Immunol. 2006;176:3321–9. doi: 10.4049/jimmunol.176.6.3321. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.