

Abstract
Pertussis, or whooping cough, is a highly contagious respiratory disease that is caused by the Gram-negative bacterium Bordetella pertussis, which is transmitted exclusively from human to human. While vaccination against B. pertussis has been successful, replacement of the whole cell vaccine with an acellular component vaccine has correlated with reemergence of the disease, especially in adolescents and infants. Based on their presumed importance in mediating adherence to host tissues, filamentous hemagglutinin (FHA) and fimbria (FIM) were selected as components of most acellular pertussis vaccines. In this review, we describe the biogenesis of FHA and FIM, recent data that show that these factors do, in fact, play critical roles in adherence to respiratory epithelium, and evidence that they also contribute to persistence in the lower respiratory tract by modulating the host immune response. We also discuss shortcomings of whole cell and acellular pertussis vaccines and the possibility that FHA and FIM could serve as effective protective antigens in next-generation vaccines.
Keywords: Bordetella pertussis, filamentous hemagglutinin, fimbriae, immunity
Bordetella fimbriae and filamentous hemagglutinin are critical adhesins and are potential protective antigens for next-generation vaccines.
Graphical Abstract Figure.
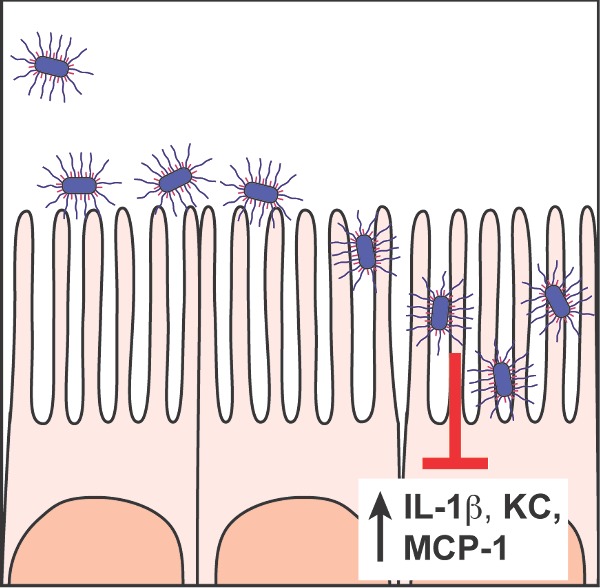
Bordetella fimbriae and filamentous hemagglutinin are critical adhesins and are potential protective antigens for next-generation vaccines.
INTRODUCTION
Pertussis (aka whooping cough) is caused by the fastidious Gram-negative bacterium Bordetella pertussis (Melvin et al. 2014). This highly contagious respiratory disease is most severe in infants and young children. Pertussis was widespread until the late 1940s, with annual case rates as high as 270 000 in the United States and fatality rates at about 10% (Clark 2014). Whole cell vaccines (wP), composed of killed B. pertussis organisms and introduced in the late 1940s, were hugely successful and morbidity and mortality rates plummeted throughout the 1950s and 1960s in areas with high vaccine coverage (Clark 2014). Adverse publicity in the 1970s, however, led to decreased compliance and prompted the development of acellular component vaccines (aP) that are composed of one to five purified pertussis proteins [chemically inactivated pertussis toxin (PT), filamentous hemagglutinin (FHA), pertactin (PRN), and/or fimbriae of serotype 2 or 3 (FIM2, FIM3)]. aP vaccines were introduced in the 1990s and are the only pertussis vaccines currently licensed in the USA and most other developed countries. Coincident with the transition to the aP vaccines, case rates and regional pertussis epidemics have increased steadily, with pertussis disease incidence in 2012 reaching a level last seen in the 1950s (Clark 2014). Recent disease rates have increased most dramatically in adolescents and epidemiological studies indicate that immunity from aP vaccines is not as durable as initially thought (Klein et al. 2012; Misegades et al. 2012; Witt, Katz and Witt 2012; Tartof et al. 2013; Koepke et al. 2014; McGirr and Fisman 2015). Consequently, there is renewed interest in understanding molecular mechanisms of pertussis pathogenesis and the immune response that is generated upon natural infection with the goal of developing a new generation of safe yet effective pertussis vaccines.
Two B. pertussis surface structures that have featured prominently in the development of pertussis vaccines are FHA and FIM. Both were identified initially as hemagglutinins or agglutinogens responsible for determining the serotype of B. pertussis strains. Agglutinogens 2 and 3 were subsequently determined to be pili or fimbriae (FIM2 and FIM3) and FHA was determined to be a large non-fimbrial filamentous hemagglutinating molecule (Ashworth, Irons and Dowsett 2006). Despite little supportive data at the time, both FHA and FIM were presumed to mediate adherence of B. pertussis to host tissues and therefore there was an effort to ensure that wP vaccines were made from B. pertussis strains producing both FIM2 and FIM3 (as well as FHA) and that aP vaccines included these antigens. In this review, we discuss current knowledge of the function of FHA and FIM in the ability of B. pertussis to establish respiratory infection and to influence the development of immunity and considerations for the development of new pertussis vaccines. Although most studies have been conducted using B. pertussis or proteins derived from B. pertussis, the human-restricted host range of this organism has hampered studies aimed at determining the roles of specific virulence factors in disease pathogenesis and the induction of immunity. Several groups have therefore included B. bronchiseptica in their analyses because this extremely closely related organism naturally infects nearly all mammals and therefore pathogenesis can be investigated in the context of a natural bacteria–host interaction. Our review will therefore also summarize data obtained from studies with B. bronchiseptica.
FHA AND FIM BIOGENESIS
Electron microscopy and structural analyses indicate that FHA, which is both cell associated and released into culture supernatants, is an ∼4 × 40 nm β-helix with a globular domain at one end and a molecular mass of ∼240 kDa (Makhov et al. 1994; Kajava et al. 2001). FHA is exported to the cell surface by the two-partner secretion (TPS) pathway. TPS is a broadly used mechanism by which Gram-negative bacteria export large, β-helical, exoproteins to the cell surface using outer membrane, channel-forming, β-barrel proteins and FHA and its cognate outer membrane transporter protein, FhaC, are its prototypical members (Mazar and Cotter 2007; Jacob-Dubuisson et al. 2013). FHA is first synthesized as an ∼375 kDa preproprotein called FhaB (Fig. 1 ). It is translocated across the cytoplasmic membrane by the general Sec system via a signal sequence that contains an extended signal peptide region (ESPR) at its N-terminus (Chevalier et al. 2004). ESPRs are present on many TPS exoproteins and autotransporter proteins and although they appear to be important for controlled secretion of these large proteins, their exact function is unclear (Desvaux et al. 2006, 2007). After removal of the ESPR-containing signal sequence, the N-terminal ∼250 aa ‘TPS domain’ of FhaB initiates export across the outer membrane by interacting with the periplasmically located polypeptide transport associated (POTRA) domains of FhaC (Hodak et al. 2006; Delattre et al. 2011; Baud et al. 2014). Export of FhaB proceeds in an N- to C-terminal direction, with the N-terminus remaining associated with FhaC such that a hairpin structure forms and elongates on the cell surface (Mazar and Cotter 2006). All TPS exoproteins are predicted to contain large β-helical regions, especially near their N-termini, and it has been postulated that folding of the stable β-helix facilitates export by contributing to a Brownian ratchet mechanism (Jacob-Dubuisson et al. 2013). Once approximately 2500 aa of FhaB have been exported to the surface, a region called the prodomain N-terminus (PNT) prevents further translocation and hence the C-terminal ∼1200 aa of FhaB (called the prodomain) remains in the periplasm (Noël et al. 2012). Specific subdomains within the prodomain are important for preventing degradation by an unknown protease, influencing the folding of the C-terminal domain of mature FHA (called the MCD) on the cell surface, and contributing somehow to resistance to phagocytic cell clearance (Noël et al. 2012; Melvin et al. 2015) (discussed below). Degradation of the prodomain in the periplasm and cleavage N-terminal to the prodomain by the surface-localized autotransporter SphB1 results in the formation of the mature ∼240 kDa FHA molecule that is oriented with its C-terminus (the MCD) located distally from the cell surface (Coutte et al. 2001; Mazar and Cotter 2006). A substantial amount of FHA is ultimately released from the cell. Neither the mechanism of FHA release nor whether FHA release is important for infection is known.
Figure 1.

FHA biogenesis. FhaB is an ∼370 kDa polypeptide. After removal of the 71 aa signal sequence, the TPS domain of FhaB (gold) interacts with the POTRA domains of FhaC (blue), initiating translocation across the outer membrane (om). The N-terminal ∼2500 aa of FhaB fold into a β-helix on the cell surface. The C-terminal proline-rich region (PRR) may interact with a periplasmic protein or protein domain, and the extreme C-terminal domain (ECT) inhibits an unknown protease. The N-terminus of the prodomain (PNT, dark green) prevents translocation of the prodomain across the om. In response to an unknown signal, the ECT releases inhibition of the unknown protease and the prodomain is degraded. FhaB is also processed by the serine protease SphB1 (purple) to form ‘mature’ ∼240 kDa FHA. FHA is released from the cell surface by an unknown mechanism.
Bordetella FIM are members of the type 1 pili family. These long, thin, hair-like structures are composed of thousands of pilin subunits. The structures are ∼7.5 nm in diameter and can be several hundred nanometers in length (Heck, Trus and Steven 1996). Type I pili are exported by the chaperone–usher mechanism, which has been extensively studied in uropathogenic Escherichia coli (UPEC) (see Busch, Phan and Waksman 2015 for a review). Based on aa sequence similarity, the Bordetella FimB, FimC and FimD proteins are hypothesized to function as the periplasmic chaperone, the outer membrane usher and the tip adhesin, respectively (Willems, van der Heide and Mooi 1992). The major pilins in Bordetella are Fim2 and Fim3. Other major pilins (FimX, FimN and FimA) are also encoded by most strains, but their production has not been detected. According to the current model based on UPEC pili (Busch, Phan and Waksman 2015), the pilin, chaperone and usher proteins are translocated across the cytoplasmic membrane by the Sec system. The pilin subunits bind to the chaperone, FimB in Bordetella, by a mechanism called donor strand complementation in which an N-terminal extension (Nte) region of the pilin fits within a groove in the chaperone to complete an immunoglobulin-like domain. The chaperone delivers the pilin subunits to the usher, where donor strand exchange (transfer of the Nte of the pilin from the chaperone to another pilin protein) and translocation of a growing chain of pilin subunits across the outer membrane occur (Fig. 2). The helical nature of the pilus rod is thought to contribute to the ability of the pili to withstand shear forces while maintaining adhesion because although it is rigid, it can uncoil under stress, acting like a spring or shock absorber (Thanassi, Bliska and Christie 2012). This feature would likely be important during adherence of Bordetella to beating cilia in the trachea and bronchi.
Figure 2.
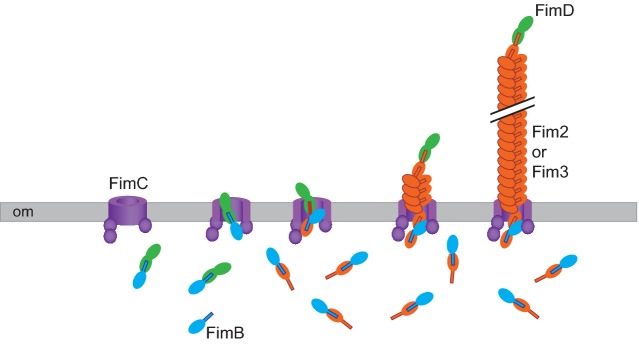
FIM biogenesis. The FimC protein is predicted to be an integral outer membrane (om) β-barrel usher protein. FimB is predicted to function as a chaperone that binds FimD, Fim2 and Fim3 proteins, preventing their degradation in the periplasm and directing them to FimC for translocation across the om. Within FimC, the FimB chaperone transfers the fimbrial subunit to the next subunit in the channel by donor strand exchange, resulting in elongation of the FIM structure.
Genes encoding the exoproteins and outer membrane transporters of most TPS systems are adjacent and cotranscribed (Jacob-Dubuisson et al. 2013). Similarly, genes required for type 1 pili biogenesis are typically clustered and coregulated (Busch, Phan and Waksman 2015). In Bordetella, however, the fimA, fimB, fimC and fimD genes are located between fhaB and fhaC (Fig. 3). Moreover, fimD and fhaC overlap such that they are both transcriptionally and translationally coupled. A single promoter 5′ to the fimB gene appears to be responsible for transcription of the fimBCDfhaC operon (Willems, van der Heide and Mooi 1992). While the fimA gene is intact in B. bronchiseptica (and B. parapertussis) strains, it is missing its 5′ end and promoter region in B. pertussis strains (Fig. 3 and Boschwitz et al. 1997). Although fimA is predicted to encode a major pilin subunit, FimA-containing FIM have not been described. All B. pertussis and B. bronchiseptica strains that have been characterized produce FIM composed of Fim2, Fim3 or both, the structural genes for which are located elsewhere on the chromosome. The fhaB-fimA-fimBCDfhaC gene cluster is adjacent to and divergent from the bvgAS operon, which encodes the BvgAS two-component regulatory system that activates transcription of all known protein virulence factor-encoding genes (Cotter and DiRita 2000; Cummings et al. 2006). When the BvgAS system is active (such as when the bacteria are within the mammalian respiratory tract; Cotter and Miller 1994; Merkel et al. 1998), BvgA is phosphorylated and activates transcription of the fhaB and fimB promoters, as well as the bvgAS P1 promoter, by binding high-affinity binding sites (Boucher et al. 1997, 2003; Cotter and Jones 2003). Transcription of the fim2 and fim3 genes is also activated by BvgAS (Chen et al. 2010). Hence, FHA and FIM are amongst the first virulence factors produced when the bacteria move from Bvg− phase conditions (ex vivo) to Bvg+ phase conditions (in vivo). The fact that the bvgAS, fhaB and fimBCDfhaC genes are clustered and coregulated suggests that they play similarly important and perhaps codependent or coordinated roles during infection.
Figure 3.

The bvgAS-fhaB-fimA-fimBCDfhaC gene cluster. The arrangement of the bvgAS-fhaB-fimA-fimBCDfhaC genes on the chromosome is shown. The pointed end of the box for each gene represents the 3′ end. Promoters are shown as arrows.
FHA AND FIM FUNCTION
The locus responsible for FHA production was first identified in 1983 as the site of a Tn5 transposon in a B. pertussis mutant that was unable to agglutinate sheep erythrocytes (Weiss et al. 1983). Comparison of this strain, or strains containing deletion mutations in fhaB, with wild-type B. pertussis for their ability to adhere to ciliated epithelium (Relman et al. 1989; Prasad et al. 1993), non-ciliated epithelium (van den Berg et al. 1999), Chinese hamster ovary cells (Relman et al. 1989; Aricò et al. 1993), monocytes (Relman et al. 1989; Prasad et al. 1993; Ishibashi, Claus and Relman 1994) and neutrophils (Mobberley-Schuman and Weiss 2005) suggested that FHA plays an important role in mediating adherence of the bacteria to host cells. Similarly, studies with strains containing either a large deletion mutation in fhaB or in which the fhaB and fhaC genes were expressed in a ΔbvgS mutant indicated that FHA is both necessary and sufficient for B. bronchiseptica to adhere to a variety of cells lines in vitro (Cotter et al. 1998; Mattoo, Miller and Cotter 2000). Although early preparations of anti-FHA antibodies often contained antibodies against other Bordetella factors, blocking studies also implicated FHA as an important adhesin in vitro (Sato et al. 1981; Redhead 1985; Prasad et al. 1993; van den Berg et al. 1999; Julio et al. 2009).
Analyses aimed at identifying domains within FHA that mediate interactions with host cells initially implicated a region near the N-terminus of FHA (named the heparin-binding domain, HBD) in binding sulfated carbohydrates such as heparin and haparan sulfate and in agglutinating sheep erythrocytes. This region contains consensus heparin-binding sites of fibronectin (Cardin et al. 1991; Busby et al. 1995) and FHA purified from culture supernatants was shown to interact with heparin-sepharose (Menozzi et al. 1994). Moreover, incubation with heparin decreased B. pertussis adherence to HeLa cells (Menozzi et al. 1994). However, the current model for FHA secretion places the HBD near the bacterial surface where it would likely be inaccessible to host cell molecules (Mazar and Cotter 2006; Noël et al. 2012), casting doubt about whether these interactions occur in vivo. Residues 1141–1279 were named the carbohydrate recognition domain because a monoclonal antibody that recognized an epitope in this region blocked the binding of FHA to glycolipids and reduced binding of B. pertussis to rabbit ciliated epithelial cells, which could also be blocked by glycolipids (Tuomanen et al. 1988; Prasad et al. 1993). A polyclonal antibody raised against a polypeptide containing this region did not, however, block binding of B. pertussis or B. bronchiseptica to either rat lung epithelial (L2) cells or J774A.1 macrophage-like cells (Julio et al. 2009). FHA also contains an RGD triplet centered at aa 1098 (numbering based on FHA from B. pertussis) which was implicated in adherence to monocytes (Relman et al. 1990; Aricò et al. 1993; Ishibashi, Claus and Relman 1994) by interacting with leukocyte response integrin-integrin associated protein (LRI-IAP) complexes and inducing surface presentation of CR3 (aka CD11b/CD18) (Ishibashi, Claus and Relman 1994). However, a B. bronchiseptica strain producing FHA with RAE in place of RGD was indistinguishable from wild-type bacteria in its ability to establish persistent infection in rats and mice (Julio et al. 2009). Focus on the N-terminal regions of FHA was due in part to the assumption that FHA was oriented with its N-terminus extended distally from the cell surface. Secretion and topology studies in 2006 (Mazar and Cotter 2006), however, showed that the N-terminus remains anchored at the cell surface and the C-terminus of mature FHA (the MCD) is located distally, placing it in an optimal location to interact with host cells. Antibody blocking studies and mutational analyses demonstrated the importance of the MCD in binding to epithelial cells and macrophages in vitro and for FHA function in vivo (Julio et al. 2009), and therefore it seems likely that FHA mediates interactions with host cells during infection primarily via the MCD, although roles for other regions of FHA have not been definitively ruled out. Identifying the molecule(s) with which FHA interacts in vivo remains an important and elusive goal.
Although fimbriae have been shown to function as important adhesins for many bacterial pathogens, evidence for FIM mediated-adherence in Bordetella has been lacking. Several studies investigating the role of FIM in B. pertussis were conducted in the 1990s (Hazenbos et al. 1994, 1995; Geuijen et al. 1997), but all of the strains used in these experiments were also defective for FHA production and hence conclusions about the role of FIM could not be drawn. Rodriguez et al. (2006) reported that anti-FIM antibodies could block attachment of B. pertussis to epithelial cells, but FIM-deficient bacteria were not included in the analysis as a control. A B. bronchiseptica strain lacking FIM due to an in-frame deletion mutation of the fimBCD genes but unaltered for FHA production was indistinguishable from wild-type bacteria in its ability to adhere to a broad range of cell lines in vitro, providing evidence against a role for FIM in adherence (Mattoo, Miller and Cotter 2000). However, this strain was defective in adherence to ciliated rabbit tracheal explants, suggesting that FIM may mediate adherence specifically to cilia (Edwards, Groathouse and Boitano 2005). Given the lack of evidence for FIM-mediated adherence in vitro, it is not surprising that a host cell receptor has not been identified. While studies using purified fimbrial subunits suggested that FIM may interact with sulfated sugars (Geuijen et al. 1998), confirmatory experiments with intact fimbriae have not been reported.
To avoid caveats associated with working with cultured cell lines in vitro and to investigate the role of FIM and FHA in adherence to respiratory epithelium in a biologically relevant context, we recently developed an in vivo adherence assay using B. bronchiseptica and mice (Scheller et al. 2015). We inoculated mice with 50 μl of PBS containing 7.5 × 104 cfu of wild-type or mutant bacteria, performed bronchoalveolar lavage (BAL) 30 minutes later, and determined the number of CFU recovered in the BAL fluid and retained in the lungs post-BAL. These experiments revealed equally critical roles for FHA and FIM in adherence to tissue within the lower respiratory tract; neither factor could mediate adherence without the other as the ΔfhaB and ΔfimBCD mutants were as defective as each other, as a ΔfhaBΔfimBCD double mutant, and as a ΔbvgS mutant in this assay. Determining where wild-type, ΔfhaB and ΔfimBCD, mutants localized within the lungs when not removed by BAL provided additional insight into FHA and FIM function. FHA-deficient bacteria, like wild-type bacteria, localized to major airways (bronchi and bronchioles), while FIM-deficient bacteria localized predominantly to alveoli. These data suggest that FIM mediates adherence specifically to ciliated epithelium (without FIM, the inoculated bacteria bypass the bronchi and bronchioles and reach the alveoli), but that this adherence alone is not sufficient to resist removal by BAL. Similarly, FHA-mediated adherence in the alveoli either does not occur or is insufficiently strong to resist BAL. Hence, we propose a model in which FIM adhere specifically, but with low or moderate affinity, to cilia, bringing the bacteria close enough for FHA to bind to these cells with high affinity that is sufficient for the bacteria to resist BAL in our experiments and mucociliary clearance during infection.
Roles for FIM and FHA in adherence to ciliated epithelium in the lower respiratory tract are consistent with the data obtained from inoculating rats and pigs intranasally with a small volume of PBS containing a relatively small number of bacteria. In these natural-host colonization models, FHA-deficient and FIM-deficient B. bronchiseptica are recovered only from the nasal cavity (the initial site of inoculation), while wild-type bacteria colonize the nasal cavity initially, then the trachea within about 5 days and they persist in both the nasal cavity and the lower respiratory tract (predominantly the trachea) indefinitely (Cotter et al. 1998; Mattoo, Miller and Cotter 2000; Nicholson, Brockmeier and Loving 2009). Experiments with high-dose, large-volume-inoculation mouse models suggest that FIM and FHA perform functions beyond simply mediating adherence to host cells. Both ΔfhaB and ΔfimBCD mutants induce a more robust inflammatory response, characterized by high levels of the proinflammatory cytokine interleukin (IL)-1β, and the neutrophil and macrophage chemokines KC and MCP-1, respectively, in the lungs at 1–3 days post-inoculation compared with wild-type bacteria (Henderson et al. 2012; Scheller et al. 2015). Examination of hematoxylin and eosin-stained lung sections reveal correspondingly increased inflammatory cell infiltrate in the lungs of mice inoculated with the mutants compared with those from mice inoculated with wild-type bacteria. In addition, the ΔfhaB and ΔfimBCD mutants are cleared from the lungs within 2 weeks post-inoculation while wild-type bacteria typically persist for a month or more in this model. These data suggest that FIM and FHA are required for wild-type B. bronchiseptica to suppress the initial inflammatory response to infection and that doing so facilitates their persistence. Coinoculation experiments suggest that in addition to influencing the intensity of the inflammatory response that develops, FHA and FIM also contribute to the ability of B. bronchiseptica to defend itself from clearance by the elicited innate immune response (Inatsuka, Julio and Cotter 2005; Henderson et al. 2012; Scheller et al. 2015). When wild-type and ΔfhaB bacteria were coinoculated into mice, the level of proinflammatory cytokines and chemokines present in the lungs 1–3 days post-inoculation was similar to that of mice inoculated with only wild-type bacteria, suggesting that the wild-type bacteria suppressed the overall inflammatory response. However, although the ΔfhaB mutant persisted longer in the coinoculation experiment than when inoculated alone, it was still cleared faster than the coinoculated wild-type bacteria, suggesting that it is defective at defending itself against clearance by the recruited phagocytic cells or other components of the early immune response. Coinoculation of wild-type and FIM-deficient bacteria also resulted in proinflammatory cytokine and chemokine levels similar to those induced by wild-type bacteria, but the persistence defect of the ΔfimBCD strain was as severe as when inoculated alone. Considering the results of the bacterial localization studies, these data suggest that complementation by wild-type bacteria requires close proximity; wild-type bacteria could ‘rescue’ the ΔFHA mutant because they also localize to major airways while wild-type bacteria were unable to rescue ΔFIM mutants because the ΔFIM mutants localize to alveoli.
Most recently, we found evidence that it is full-length FhaB, rather than mature ∼240 kDa FHA, that is important for mediating resistance to clearance by the elicited inflammatory response (Melvin et al. 2015). We constructed and characterized B. bronchiseptica strains that produce FhaB proteins lacking either the PRR, the ECT or both of these domains that are located at the end of the full-length FhaB protein (Fig. 1). The ΔPRR strain was indistinguishable from wild-type bacteria in its ability to produce full-length FhaB (aside from the FhaB protein missing its PRR domain in the mutant), to process FhaB to FHA and to release FHA into culture supernatants in vitro. The ΔPRR strain was also indistinguishable from wild-type bacteria in its ability to mediate adherence to cell lines in vitro and to respiratory epithelium in vivo and it suppressed the initial inflammatory response to infection similarly to wild-type bacteria. The ΔPRR mutant was defective, however, in its ability to persist in the lower respiratory tract; it was cleared from the lungs of mice as quickly as the ΔfhaB strain. Because this mutant produces mature FHA that is indistinguishable from that produced by wild-type bacteria in our assays, and because the FhaB prodomain is degraded rapidly and does not exist as a stand-alone polypeptide, these results implicate a role for full-length FhaB in persistence, specifically in resistance to clearance by the early immune response. We hypothesize that the PRR domain is involved in delivering a toxin (perhaps adenylate cyclase toxin) to phagocytic inflammatory cells in response to binding that is mediated by the MCD region of full-length FhaB.
Overall, data from comparing wild-type and mutant B. bronchiseptica strains in natural-host animal models support a model in which wild-type bacteria use FIM for initial adherence to ciliated epithelial cells in the lower respiratory tract and then FHA for tight adherence to these cells. Colonization of the respiratory epithelium induces inflammation, likely due to recognition of LPS and other MAMPs on, or released by, the bacteria by TLRs on host cells, resulting in the production of proinflammatory cytokines and chemokines. Neutrophils, macrophages, dendritic cells and lymphocytes are then recruited to the site of infection and as these cells attempt to clear the bacteria, they may also release cytokines and chemokines, amplifying the inflammatory response. Wild-type B. bronchiseptica (and likely also B. pertussis) suppress the inflammatory response in a manner dependent on FIM and FHA, but whether they do so through interactions with epithelial cells, inflammatory cells or both and the mechanisms involved are unknown. Wild-type bacteria, in a manner dependent on the PRR of FhaB, also appear to be capable, to some extent, of resisting clearance by recruited phagocytic cells, which contributes to bacterial persistence in the lower respiratory tract (Melvin et al. 2015). The bacteria are ultimately cleared, however, in a manner dependent on the adaptive immune response (Harvill et al. 1999).
FHA AND FIM: PROTECTIVE ANTIGENS?
Antibodies were initially thought to be of prime importance for protection against pertussis. Consequently, with the rationale that antibodies against adhesins would block colonization, there was an effort to use B. pertussis strains producing both FIM2 and FIM3 in the production of whole cell vaccines and when acellular vaccines were developed, FIM and FHA were considered to be important, if not essential, components. However, the aP and wP vaccines induce serum antibody responses to vaccine components that are similar in magnitude to those induced by infection, yet human case control and cohort studies indicate that immunity induced by aP vaccination is not as durable as immunity induced by wP vaccination, which is not as durable as immunity induced by infection with B. pertussis, (Plotkin 2013; Edwards and Berbers 2014; Acosta et al. 2015). Moreover, serological correlates of protection were never established in clinical vaccine trials. Attention has therefore turned to understanding the role of cell-mediated immunity in controlling pertussis. These studies have shown that immunization with aP vaccines induces primarily a Th2 or mixed Th1/Th2 response, while immunization with wP vaccines and infection with B. pertussis induce primarily a Th1 response (Zepp et al. 1996; Ausiello et al. 1997, 1999; He et al. 1998; Ryan et al. 1998; Edwards and Berbers 2014). The hypothesis that a Th1, and possibly a Th17, type cell-mediated immune response is required for protection from pertussis is also supported by data from mouse studies (Mills et al. 1993; Barbic et al. 1997; Leef et al. 2000; Ross et al. 2013). Because most of the human studies were conducted before the appreciation of Th17 cells, the induction of this T-cell subpopulation in response to wP vaccination or B. pertussis infection in humans is unknown. Recent studies, however, attempted but failed to detect B. pertussis antigen-specific Th17 cells in children immunized with aP vaccines, supporting the conclusion that aP vaccines induce exclusively Th2 or Th1/Th2 responses (Stenger et al. 2010; Schure et al. 2012, 2013) and strengthening the hypothesis that cell-mediated immunity, and not antibodies, is essential for protection.
Insight into the specific contributions of types of immune responses induced by vaccination versus infection has come from studies using the recently developed baboon model. This model, using Papio anubis, accurately recapitulates many features of human pertussis, including paroxysmal cough, lymphocytosis and aerosol transmission (Warfel, Beren and Merkel 2012a,b). Baboons infected with B. pertussis developed strong, long-lasting Th1 and Th17 responses (Warfel et al. 2012b; Warfel, Zimmerman and Merkel 2014). After recovery from the initial infection, the baboons were completely protected from both disease (cough and lymphocytosis) and colonization upon challenge with B. pertussis. Baboons immunized with wP (in the form of DTwP) also developed both Th1 and Th17 responses, although they were not as strong as those induced in response to infection (Warfel, Zimmerman and Merkel 2014). DTwP-vaccinated baboons were protected from disease, but not from colonization, although they cleared infection in about half the time as naïve animals (∼2 weeks versus 1 month). Baboons immunized with DTaP developed a mixed Th1/Th2 response. They were also protected from disease, but not colonization; in fact, they cleared infection as slowly as naïve animals. Most importantly and perhaps revealing with regard to the current reemergence of pertussis, baboons immunized with DTaP and then challenged were capable of transmitting infection to naïve animals. All baboons that were either infected with B. pertussis or immunized with wP or aP vaccines generated high titers of serum anti-Bordetella antibodies, demonstrating that, consistent with the studies on humans, differences in serum antibody titer do not account for differences in protection. Warfel and colleagues concluded from the baboon (and human) studies that antibodies may mediate protection against disease while Th1 and Th17 type cell-mediated immune responses mediate protection against colonization (Warfel, Zimmerman and Merkel 2014; Warfel and Edwards 2015). While this explanation is plausible, it does not fully account for the fact that baboons that had been infected previously were completely protected from colonization while baboons vaccinated with wP vaccines were not, yet both had generated Th1 and Th17 responses. It is possible that the difference in protection from colonization in convalescent and wP-vaccinated animals stems from differences in the magnitude of the Th1 and Th17 responses induced, or differences in the specific antigens to which these responses were directed. However, another possibility that has not been investigated is that protection from colonization is mediated by mucosal IgA antibodies and that parenteral immunization with either aP or wP vaccines fails to generate a sufficiently strong mucosal antibody response to afford this protection. Our recent data demonstrating the essentiality of FHA and FIM for adherence to respiratory epithelium suggest that mucosal antibodies against either or both of these factors would block adherence and therefore protect against colonization and, consequently, disease. In support of this hypothesis, two studies using an intranasal inoculation protocol have shown that FHA could induce a protective immune response in mice (Alonso et al. 2002; Knight et al. 2006). We therefore believe that efforts towards developing safe and efficacious pertussis vaccines should include obtaining a complete understanding of the immune responses that are induced by natural infection as well as those induced by wP and aP vaccines for comparison and that these studies should include measurements of mucosal IgA levels. A finding that mucosal antibodies to FHA and /or FIM are sufficient for protection against colonization would have a dramatic impact on vaccine development as effort could be focused on inducing this specific type of response, perhaps even focused on specific domains of FHA and FIM.
Acknowledgments
We thank members of our laboratory for helpful discussions. Work on Bordetella in the Cotter lab is supported by the National Institutes of Health (AI094991).
Conflict of interest. None declared.
REFERENCES
- Acosta AM, DeBolt C, Tasslimi A, et al. Tdap vaccine effectiveness in adolescents during the 2012 Washington State pertussis epidemic. Pediatrics. 2015;135:981–9. doi: 10.1542/peds.2014-3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso S, Reveneau N, Pethe K, et al. Eighty-kilodalton N-terminal moiety of Bordetella pertussis filamentous hemagglutinin: adherence, immunogenicity, and protective role. Infect Immun. 2002;70:4142–7. doi: 10.1128/IAI.70.8.4142-4147.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aricò B, Nuti S, Scarlato V, et al. Adhesion of Bordetella pertussis to eukaryotic cells requires a time-dependent export and maturation of filamentous hemagglutinin. P Natl Acad Sci USA. 1993;90:9204–8. doi: 10.1073/pnas.90.19.9204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth LAE, Irons LI, Dowsett AB. Antigenic relationship between serotype-specific agglutinogen and fimbriae of Bordetella pertussis. Infect Immun. 2006;37:1278–81. doi: 10.1128/iai.37.3.1278-1281.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausiello CM, Lande R, Urbani F, et al. Cell-mediated immune responses in four-year-old children after primary immunization with acellular pertussis vaccines. Infect Immun. 1999;67:4064–71. doi: 10.1128/iai.67.8.4064-4071.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausiello CM, Urbani F, la Sala A, et al. Vaccine- and antigen-dependent type 1 and type 2 cytokine induction after primary vaccination of infants with whole-cell or acellular pertussis vaccines. Infect Immun. 1997;65:2168–74. doi: 10.1128/iai.65.6.2168-2174.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbic J, Leef MF, Burns DL, et al. Role of gamma interferon in natural clearance of Bordetella pertussis infection. Infect Immun. 1997;65:4904–8. doi: 10.1128/iai.65.12.4904-4908.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud C, Guérin J, Petit E, et al. Translocation path of a substrate protein through its Omp85 transporter. Nat Commun. 2014;5:1–9. doi: 10.1038/ncomms6271. [DOI] [PubMed] [Google Scholar]
- Boschwitz JS, van der Heide HG, Mooi FR, et al. Bordetella bronchiseptica expresses the fimbrial structural subunit gene fimA. J Bacteriol. 1997;179:7882–5. doi: 10.1128/jb.179.24.7882-7885.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher PE, Maris AE, Yang M-S, et al. The response regulator BvgA and RNA polymerase alpha subunit C-terminal domain bind simultaneously to different faces of the same segment of promoter DNA. Mol Cell. 2003;11:163–73. doi: 10.1016/s1097-2765(03)00007-8. [DOI] [PubMed] [Google Scholar]
- Boucher PE, Murakami K, Ishihama A, et al. Nature of DNA binding and RNA polymerase interaction of the Bordetella pertussis BvgA transcriptional activator at the fha promoter. J Bacteriol. 1997;179:1755–63. doi: 10.1128/jb.179.5.1755-1763.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busby TF, Argraves WS, Brew SA, et al. Heparin binding by fibronectin module III-13 involves six discontinuous basic residues brought together to form a cationic cradle. J Biol Chem. 1995;270:18558–62. doi: 10.1074/jbc.270.31.18558. [DOI] [PubMed] [Google Scholar]
- Busch A, Phan G, Waksman G. Molecular mechanism of bacterial type 1 and P pili assembly. Philos T R Soc A. 2015;373:20130153. doi: 10.1098/rsta.2013.0153. [DOI] [PubMed] [Google Scholar]
- Cardin AD, Demeter DA, Weintraub HJ, et al. Molecular design and modeling of protein-heparin interactions. Methods Enzymol. 1991;203:556–83. doi: 10.1016/0076-6879(91)03030-k. [DOI] [PubMed] [Google Scholar]
- Chen Q, Decker KB, Boucher PE, et al. Novel architectural features of Bordetella pertussis fimbrial subunit promoters and their activation by the global virulence regulator BvgA. Mol Microbiol. 2010;77:1326–40. doi: 10.1111/j.1365-2958.2010.07293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier N, Moser M, Koch H-G, et al. Membrane targeting of a bacterial virulence factor harbouring an extended signal peptide. J Mol Microb Biot. 2004;8:7–18. doi: 10.1159/000082076. [DOI] [PubMed] [Google Scholar]
- Clark TA. Changing pertussis epidemiology: everything old is new again. J Infect Dis. 2014;209:978–81. doi: 10.1093/infdis/jiu001. [DOI] [PubMed] [Google Scholar]
- Cotter PA, DiRita VJ. Bacterial virulence gene regulation: an evolutionary perspective. Annu Rev Microbiol. 2000;54:519–65. doi: 10.1146/annurev.micro.54.1.519. [DOI] [PubMed] [Google Scholar]
- Cotter PA, Jones AM. Phosphorelay control of virulence gene expression in Bordetella. Trends Microbiol. 2003;11:367–73. doi: 10.1016/s0966-842x(03)00156-2. [DOI] [PubMed] [Google Scholar]
- Cotter PA, Miller JF. BvgAS-mediated signal transduction: analysis of phase-locked regulatory mutants of Bordetella bronchiseptica in a rabbit model. Infect Immun. 1994;62:3381–90. doi: 10.1128/iai.62.8.3381-3390.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter PA, Yuk MH, Mattoo S, et al. Filamentous hemagglutinin of Bordetella bronchiseptica is required for efficient establishment of tracheal colonization. Infect Immun. 1998;66:5921–9. doi: 10.1128/iai.66.12.5921-5929.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutte L, Antoine R, Drobecq H, et al. Subtilisin-like autotransporter serves as maturation protease in a bacterial secretion pathway. EMBO J. 2001;20:5040–8. doi: 10.1093/emboj/20.18.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings CA, Bootsma HJ, Relman DA, et al. Species- and strain-specific control of a complex, flexible regulon by Bordetella BvgAS. J Bacteriol. 2006;188:1775–85. doi: 10.1128/JB.188.5.1775-1785.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delattre A-S, Saint N, Clantin B, et al. Substrate recognition by the POTRA domains of TpsB transporter FhaC. Mole Microbiol. 2011;81:99–112. doi: 10.1111/j.1365-2958.2011.07680.x. [DOI] [PubMed] [Google Scholar]
- Desvaux M, Scott-Tucker A, Turner SM, et al. A conserved extended signal peptide region directs posttranslational protein translocation via a novel mechanism. Microbiology. 2007;153:59–70. doi: 10.1099/mic.0.29091-0. [DOI] [PubMed] [Google Scholar]
- Desvaux ML, Cooper LM, Filenko NA, et al. The unusual extended signal peptide region of the type V secretion system is phylogenetically restricted. FEMS Microbiol Lett. 2006;264:22–30. doi: 10.1111/j.1574-6968.2006.00425.x. [DOI] [PubMed] [Google Scholar]
- Edwards JA, Groathouse NA, Boitano S. Bordetella bronchiseptica adherence to cilia is mediated by multiple adhesin factors and blocked by surfactant protein A. Infect Immun. 2005;73:3618–26. doi: 10.1128/IAI.73.6.3618-3626.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards KM, Berbers GAM. Immune responses to pertussis vaccines and disease. J Infect Dis. 2014;209(Suppl 1):S10–5. doi: 10.1093/infdis/jit560. [DOI] [PubMed] [Google Scholar]
- Geuijen CA, Willems RJ, Bongaerts M, et al. Role of the Bordetella pertussis minor fimbrial subunit, FimD, in colonization of the mouse respiratory tract. Infect Immun. 1997;65:4222–8. doi: 10.1128/iai.65.10.4222-4228.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geuijen CA, Willems RJ, Hoogerhout P, et al. Identification and characterization of heparin binding regions of the Fim2 subunit of Bordetella pertussis. Infect Immun. 1998;66:2256–63. doi: 10.1128/iai.66.5.2256-2263.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvill ET, Cotter PA, Yuk MH, et al. Probing the function of Bordetella bronchiseptica adenylate cyclase toxin by manipulating host immunity. Infect Immun. 1999;67:1493–500. doi: 10.1128/iai.67.3.1493-1500.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazenbos WL, van den Berg BM, Geuijen CW, et al. Binding of FimD on Bordetella pertussis to very late antigen-5 on monocytes activates complement receptor type 3 via protein tyrosine kinases. J Immunol. 1995;155:3972–8. [PubMed] [Google Scholar]
- Hazenbos WL, van den Berg BM, van't Wout JW, et al. Virulence factors determine attachment and ingestion of nonopsonized and opsonized Bordetella pertussis by human monocytes. Infect Immun. 1994;62:4818–24. doi: 10.1128/iai.62.11.4818-4824.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Tran Minh NN, Edelman K, et al. Cytokine mRNA expression and proliferative responses induced by pertussis toxin, filamentous hemagglutinin, and pertactin of Bordetella pertussis in the peripheral blood mononuclear cells of infected and immunized schoolchildren and adults. Infect Immun. 1998;66:3796–801. doi: 10.1128/iai.66.8.3796-3801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck DV, Trus BL, Steven AC. Three-dimensional structure of Bordetella pertussis fimbriae. J Struct Biol. 1996;116:264–9. doi: 10.1006/jsbi.1996.0041. [DOI] [PubMed] [Google Scholar]
- Henderson MW, Inatsuka CS, Sheets AJ, et al. Contribution of Bordetella filamentous hemagglutinin and adenylate cyclase toxin to suppression and evasion of interleukin-17-mediated inflammation. Infect Immun. 2012;80:2061–75. doi: 10.1128/IAI.00148-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodak H, Clantin B, Willery E, et al. Secretion signal of the filamentous haemagglutinin, a model two-partner secretion substrate. Mol Microbiol. 2006;61:368–82. doi: 10.1111/j.1365-2958.2006.05242.x. [DOI] [PubMed] [Google Scholar]
- Inatsuka CS, Julio SM, Cotter PA. Bordetella filamentous hemagglutinin plays a critical role in immunomodulation, suggesting a mechanism for host specificity. P Natl Acad Sci USA. 2005;102:18578–83. doi: 10.1073/pnas.0507910102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi Y, Claus S, Relman DA. Bordetella pertussis filamentous hemagglutinin interacts with a leukocyte signal transduction complex and stimulates bacterial adherence to monocyte CR3 (CD11b/CD18) J Exp Med. 1994;180:1225–33. doi: 10.1084/jem.180.4.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob-Dubuisson F, Guérin J, Baelen S, et al. Two-partner secretion: as simple as it sounds? Res Microbiol. 2013;164:583–95. doi: 10.1016/j.resmic.2013.03.009. [DOI] [PubMed] [Google Scholar]
- Julio SM, Inatsuka CS, Mazar J, et al. Natural-host animal models indicate functional interchangeability between the filamentous haemagglutinins of Bordetella pertussis and Bordetella bronchiseptica and reveal a role for the mature C-terminal domain, but not the RGD motif, during infection. Mol Microbiol. 2009;71:1574–90. doi: 10.1111/j.1365-2958.2009.06623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajava AV, Cheng N, Cleaver R, et al. Beta-helix model for the filamentous haemagglutinin adhesin of Bordetella pertussis and related bacterial secretory proteins. Mol Microbiol. 2001;42:279–92. doi: 10.1046/j.1365-2958.2001.02598.x. [DOI] [PubMed] [Google Scholar]
- Klein NP, Bartlett J, Rowhani-Rahbar A, et al. Waning protection after fifth dose of acellular pertussis vaccine in children. N Eng J Med. 2012;367:1012–9. doi: 10.1056/NEJMoa1200850. [DOI] [PubMed] [Google Scholar]
- Knight JB, Huang YY, Halperin SA, et al. Immunogenicity and protective efficacy of a recombinant filamentous haemagglutinin from Bordetella pertussis. Clin Exp Immunol. 2006;144:543–51. doi: 10.1111/j.1365-2249.2006.03097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepke R, Eickhoff JC, Ayele RA, et al. Estimating the effectiveness of tetanus-diphtheria-acellular pertussis vaccine (Tdap) for preventing pertussis: evidence of rapidly waning immunity and difference in effectiveness by Tdap brand. J Infect Dis. 2014;210:942–53. doi: 10.1093/infdis/jiu322. [DOI] [PubMed] [Google Scholar]
- Leef M, Elkins KL, Barbic J, et al. Protective immunity to Bordetella pertussis requires both B cells and CD4(+) T cells for key functions other than specific antibody production. J Exp Med. 2000;191:1841–52. doi: 10.1084/jem.191.11.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGirr A, Fisman DN. Duration of pertussis immunity after DTaP immunization: a meta-analysis. Pediatrics. 2015;135:331–43. doi: 10.1542/peds.2014-1729. [DOI] [PubMed] [Google Scholar]
- Makhov AM, Hannah JH, Brennan MJ, et al. Filamentous hemagglutinin of Bordetella pertussis. A bacterial adhesin formed as a 50-nm monomeric rigid rod based on a 19-residue repeat motif rich in beta strands and turns. J Mol Biol. 1994;241:110–24. doi: 10.1006/jmbi.1994.1478. [DOI] [PubMed] [Google Scholar]
- Mattoo S, Miller JF, Cotter PA. Role of Bordetella bronchiseptica fimbriae in tracheal colonization and development of a humoral immune response. Infect Immun. 2000;68:2024–33. doi: 10.1128/iai.68.4.2024-2033.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazar J, Cotter PA. Topology and maturation of filamentous haemagglutinin suggest a new model for two-partner secretion. Mol Microbiol. 2006;62:641–54. doi: 10.1111/j.1365-2958.2006.05392.x. [DOI] [PubMed] [Google Scholar]
- Mazar J, Cotter PA. New insight into the molecular mechanisms of two-partner secretion. Trends Microbiol. 2007;15:508–15. doi: 10.1016/j.tim.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Melvin JA, Scheller EV, Miller JF, et al. Bordetella pertussis pathogenesis: current and future challenges. Nat Publishing Group. 2014;12:274–88. doi: 10.1038/nrmicro3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melvin JA, Scheller EV, Noël CR, et al. New insight into filamentous hemagglutinin secretion reveals a role for full-length FhaB in Bordetella virulence. mBio. 2015;6:e01189–15. doi: 10.1128/mBio.01189-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menozzi FD, Boucher PE, Riveau G, et al. Surface-associated filamentous hemagglutinin induces autoagglutination of Bordetella pertussis. Infect Immun. 1994;62:4261–9. doi: 10.1128/iai.62.10.4261-4269.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkel TJ, Stibitz S, Keith JM, et al. Contribution of regulation by the bvg locus to respiratory infection of mice by Bordetella pertussis. Infect Immun. 1998;66:4367–73. doi: 10.1128/iai.66.9.4367-4373.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills KH, Barnard A, Watkins J, et al. Cell-mediated immunity to Bordetella pertussis: role of Th1 cells in bacterial clearance in a murine respiratory infection model. Infect Immun. 1993;61:399–410. doi: 10.1128/iai.61.2.399-410.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misegades LK, Winter K, Harriman K, et al. Association of childhood pertussis with receipt of 5 doses of pertussis vaccine by time since last vaccine dose, California, 2010. JAMA. 2012;308:2126–32. doi: 10.1001/jama.2012.14939. [DOI] [PubMed] [Google Scholar]
- Mobberley-Schuman PS, Weiss AA. Influence of CR3 (CD11b/CD18) expression on phagocytosis of Bordetella pertussis by human neutrophils. Infect Immun. 2005;73:7317–23. doi: 10.1128/IAI.73.11.7317-7323.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson TL, Brockmeier SL, Loving CL. Contribution of Bordetella bronchiseptica filamentous hemagglutinin and pertactin to respiratory disease in Swine. Infect Immun. 2009;77:2136. doi: 10.1128/IAI.01379-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noël CR, Mazar J, Melvin JA, et al. The prodomain of the Bordetella two-partner secretion pathway protein FhaB remains intracellular yet affects the conformation of the mature C-terminal domain. Mol Microbiol. 2012;86:988–1006. doi: 10.1111/mmi.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin SA. Complex correlates of protection after vaccination. Clin Infect Dis. 2013;56:1458–65. doi: 10.1093/cid/cit048. [DOI] [PubMed] [Google Scholar]
- Prasad SM, Yin Y, Rodzinski E, et al. Identification of a carbohydrate recognition domain in filamentous hemagglutinin from Bordetella pertussis. Infect Immun. 1993;61:2780–5. doi: 10.1128/iai.61.7.2780-2785.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redhead K. An assay of Bordetella pertussis adhesion to tissue-culture cells. J Med Microbiol. 1985;19:99–108. doi: 10.1099/00222615-19-1-99. [DOI] [PubMed] [Google Scholar]
- Relman D, Tuomanen E, Falkow S, et al. Recognition of a bacterial adhesion by an integrin: macrophage CR3 (alpha M beta 2, CD11b/CD18) binds filamentous hemagglutinin of Bordetella pertussis. Cell. 1990;61:1375–82. doi: 10.1016/0092-8674(90)90701-f. [DOI] [PubMed] [Google Scholar]
- Relman DA, Domenighini M, Tuomanen E, et al. Filamentous hemagglutinin of Bordetella pertussis: nucleotide sequence and crucial role in adherence. P Natl Acad Sci USA. 1989;86:2637–41. doi: 10.1073/pnas.86.8.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez ME, Hellwig SMM, Perez Vidakovics MLA, et al. Bordetella pertussis attachment to respiratory epithelial cells can be impaired by fimbriae-specific antibodies. FEMS Immunol Med Mic. 2006;46:39–47. doi: 10.1111/j.1574-695X.2005.00001.x. [DOI] [PubMed] [Google Scholar]
- Ross PJ, Sutton CE, Higgins S, et al. Relative contribution of Th1 and Th17 cells in adaptive immunity to Bordetella pertussis: towards the rational design of an improved acellular pertussis vaccine. PLoS Pathog. 2013;9:e1003264–14. doi: 10.1371/journal.ppat.1003264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan M, Murphy G, Ryan E, et al. Distinct T-cell subtypes induced with whole cell and acellular pertussis vaccines in children. Immunology. 1998;93:1–10. doi: 10.1046/j.1365-2567.1998.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Izumiya K, Sato H, et al. Role of antibody to leukocytosis-promoting factor hemagglutinin and to filamentous hemagglutinin in immunity to pertussis. Infect Immun. 1981;31:1223–31. doi: 10.1128/iai.31.3.1223-1231.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller EV, Melvin JA, Sheets AJ, et al. Cooperative roles for fimbria and filamentous hemagglutinin in Bordetella adherence and immune modulation. mBio. 2015;6:e00500–15. doi: 10.1128/mBio.00500-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schure RM, Hendrikx LH, de Rond LGH, et al. T-cell responses before and after the fifth consecutive acellular pertussis vaccination in 4-year-old Dutch children. Clin Vaccine Immunol. 2012;19:1879–86. doi: 10.1128/CVI.00277-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schure RM, Hendrikx LH, de Rond LGH, et al. Differential T- and B-cell responses to pertussis in acellular vaccine-primed versus whole-cell vaccine-primed children 2 years after preschool acellular booster vaccination. Clin Vaccine Immunol. 2013;20:1388–95. doi: 10.1128/CVI.00270-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenger RM, Smits M, Kuipers B, et al. Impaired long-term maintenance and function of Bordetella pertussis specific B cell memory. Vaccine. 2010;28:6637–46. doi: 10.1016/j.vaccine.2010.06.118. [DOI] [PubMed] [Google Scholar]
- Tartof SY, Lewis M, Kenyon C, et al. Waning immunity to pertussis following 5 doses of DTaP. Pediatrics. 2013;131:e1047–52. doi: 10.1542/peds.2012-1928. [DOI] [PubMed] [Google Scholar]
- Thanassi DG, Bliska JB, Christie PJ. Surface organelles assembled by secretion systems of Gram-negative bacteria: diversity in structure and function. FEMS Microbiol Rev. 2012;36:1046–82. doi: 10.1111/j.1574-6976.2012.00342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuomanen E, Towbin H, Rosenfelder G, et al. Receptor analogs and monoclonal antibodies that inhibit adherence of Bordetella pertussis to human ciliated respiratory epithelial cells. J Exp Med. 1988;168:267–77. doi: 10.1084/jem.168.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg BM, Beekhuizen H, Willems RJ, et al. Role of Bordetella pertussis virulence factors in adherence to epithelial cell lines derived from the human respiratory tract. Infect Immun. 1999;67:1056–64. doi: 10.1128/iai.67.3.1056-1062.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfel JM, Beren J, Kelly VK, et al. Nonhuman primate model of pertussis. Infect Immun. 2012b;80:1530–6. doi: 10.1128/IAI.06310-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfel JM, Beren J, Merkel TJ. Airborne transmission of Bordetella pertussis. J Infect Dis. 2012a;206:902–6. doi: 10.1093/infdis/jis443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfel JM, Edwards KM. ScienceDirectPertussis vaccines and the challenge of inducing durable immunity. Curr Opin Immunol. 2015;35:48–54. doi: 10.1016/j.coi.2015.05.008. [DOI] [PubMed] [Google Scholar]
- Warfel JM, Zimmerman LI, Merkel TJ. Acellular pertussis vaccines protect against disease but fail to prevent infection and transmission in a nonhuman primate model. P Natl Acad Sci USA. 2014;111:787–92. doi: 10.1073/pnas.1314688110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss AA, Hewlett EL, Myers GA, et al. Tn5-induced mutations affecting virulence factors of Bordetella pertussis. Infect Immun. 1983;42:33–41. doi: 10.1128/iai.42.1.33-41.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems RJ, van der Heide HG, Mooi FR. Characterization of a Bordetella pertussis fimbrial gene cluster which is located directly downstream of the filamentous haemagglutinin gene. Mol Microbiol. 1992;6:2661–71. doi: 10.1111/j.1365-2958.1992.tb01443.x. [DOI] [PubMed] [Google Scholar]
- Witt MA, Katz PH, Witt DJ. Unexpectedly limited durability of immunity following acellular pertussis vaccination in preadolescents in a North American outbreak. Clin Infect Dis. 2012;54:1730–5. doi: 10.1093/cid/cis287. [DOI] [PubMed] [Google Scholar]
- Zepp F, Knuf M, Habermehl P, et al. Pertussis-specific cell-mediated immunity in infants after vaccination with a tricomponent acellular pertussis vaccine. Infect Immun. 1996;64:4078–84. doi: 10.1128/iai.64.10.4078-4084.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]