

Abstract
Ziconotide (Prialt®), a synthetic version of the peptide ω-conotoxin MVIIA found in the venom of a fish-hunting marine cone snail Conus magnus, is one of very few drugs effective in the treatment of intractable chronic pain. However, its intrathecal mode of delivery and narrow therapeutic window cause complications for patients. This review will summarize progress in the development of small molecule, non-peptidic mimics of Conotoxins and a small number of other venom peptides. This will include a description of how some of the initially designed mimics have been modified to improve their drug-like properties.
Keywords: venom peptides, toxins, conotoxins, peptidomimetics, N-type calcium channel, Cav2.2
1. Introduction
A wide range of species from the animal kingdom produce venom for use in capturing prey or for self-defense. Animal venoms typically consist of a complex mixture of proteins, peptides and small molecules [1]. Venom peptides in particular have been intensively studied following the discovery of fascinating pharmacological properties of peptides isolated from marine mollusks and terrestrial snakes and arthropods. The sheer number of bioactive venom peptides produced by species from the animal kingdom is astronomical [1] and these represent a vast and largely untapped source of new research tools and therapeutic leads. Peptides found in the venom of the fish-hunting cone snails of the genus Conus have been the focus of considerable attention, largely due to their often powerful and selective modulating effects on human ion channels. While a synthetic version of one of these Conus peptides (ω-conotoxin MVIIA, Ziconotide (Prialt®)) is now in limited clinical use, the biophysical properties of peptides in general makes them unsuitable for use as drugs. Conus peptides have, however, served as the inspiration for a number of bioactive discovery programs where the objective is to develop drug-like small molecules that mimic the biological activity of the natural peptide. Presented in this review is a discussion of the approaches used by researchers in this area and progress to date. Compared to conotoxin mimics, the mimicry of venom peptides from other sources has been more limited. Publically disclosed efforts with snake peptides and a peptide from the sun anemone will be described. The most successful of all of these programs has been the development of Tirofiban (Aggrastat®), an anti-platelet drug used to reduce the rate of thrombotic cardiovascular events and inspired by a tripeptide fragment of an anti-coagulant found in the venom of the African saw-scaled viper Echis carinatus.
2. Peptidomimetics
Peptide mimics or peptidomimetics are compounds that possess similar biological activity and/or 3D topography as a native peptide or protein fragment. Peptidomimetics typically interact with a biological target in a similar manner to the native peptide and as a result they exert a similar biological effect [2]. Peptidomimetics are commonly designed to overcome the bioavailability and instability issues associated with peptides and proteins. Three distinct types of peptidomimetics have been classified [3,4,5]. Type-I peptidomimetics, also known as structural mimetics, are peptide backbone mimetics, where the local topography is analogous to the native peptide and functionalities required to interact with the enzyme or receptor are present. Type-II peptidomimetics, also known as functional mimetics, interact with the enzyme or receptor of interest in a related way to the active peptide but are not necessarily structurally related to the peptide. Type-III peptidomimetics, also known as functional-structural mimetics, have a core scaffold structure that is different from the active peptide, and from which projects amino acid side chain mimics in a well-defined spatial orientation, simulating the way the native peptide interacts with the biological target.
One approach to the development of type-III mimetics involves the mimicry of the α,β-bond vectors of the amino acid residues responsible for a peptide’s biological activity. This method was promulgated by Bartlett and Lauri [6] and is the fundamental tenet of the Computer-Assisted Vector Evaluation and Target Design (CAVEAT) program [7]. Much of the biological activity elicited by an active peptide results from the interaction of the side chain termini with the receptor, and when detailed structural information about this interaction is absent, it is very difficult to predict the exact spatial orientation of these termini when bound. The position and direction of the first side chain bonds that project from the peptide backbone, the α,β-bond vectors, are however, usually much better defined. If a reliable solution state structure or computational model of the peptide is available, the α,β-bond vector approach can be used to design type-III mimetics. At their core, these compounds have a semi-rigid molecular scaffold from which bonds that are positioned to simulate the location and direction of the α,β-bond vectors of the key amino acids side chains on the active peptide are projected. Amino acid side mimics are attached to α,β-bond replicas with the expectation that, as with the active peptide, the termini of these functionalities will find the desired binding site within the receptor.
3. Pain Blocking Conotoxins and Their Mimetics
3.1. Conotoxins
Conotoxins are neurotoxic peptides isolated from the venom of marine cone snails from the genus Conus. To date over 700 species from this genus have been described, with the venom of each containing a unique and diverse mixture of pharmacologically active compounds [8,9,10,11]. Conotoxins are small, structurally defined peptides that range in size from eight to thirty amino acid residues and typically have one or more disulfide bonds. The fish-hunting cone snails have the most poisonous venom and there have been over 30 deaths as a result of cone snails envenomation [10]. The majority of these deaths are due to the cone snail Conus geographus, whose sting can cause paralysis within 30 min and coma or death within five hours [12]. Interestingly, in these unfortunate cases the victim has been reported to die a “painless death” [10]. The potency and analgesic activity of the Conus venoms has motivated a number of research groups to isolate and identify the pharmacologically active components in the venom, with Olivera and coworkers being pioneers in this field. There are a number of excellent reviews that summarize this research [13,14,15,16,17]. The comprehensive pharmacology of conotoxin peptides [9,10,16,17,18,19,20,21,22] has also been comprehensively reviewed, and hence will not be described here in detail.
The nomenclature for conotoxins was first proposed in 1985 [23] and further refined in 1988 [15]: When the conotoxin has a known mode of action, then a Greek letter prefix is used to designate its pharmacological action. This is then followed by “-conotoxin”, a one or two letter code to assign the Conus species, a Roman numeral to indicate the Cys arrangement and finally an upper case letter that allows conotoxins of the same class, isolated from the same species, to be differentiated [13,15,23]. For example, ω-conotoxin GVIA targets voltage gated calcium channels (ω), is a peptide from the Conus species Conus geographus (G) and has a class VI Cys pattern (C–C–CC–C–C). Conotoxins of particular relevance to this article are the ω-conotoxins and μ-conotoxins. More recently conotoxins have been classed according to their superfamily. Peptides within a superfamily have a highly conserved amino acid sequence and share the same disulfide connectivity [24,25]. The ω-conotoxins belong to the O1-superfamily of which κ-, δ-, and µO-conotoxins are also members, and the μ-conotoxins are members of the M-superfamily. For a recent review on conotoxin gene superfamilies, see Robinson and Norton [26].
ω-Conotoxins contain six Cys residues and three disulfide bridges. To date 21 ω-conotoxins peptides have been identified with net charges ranging from +5 to +7 [21,27,28]. The most well studied ω-conotoxins are the N-type calcium channel (Cav2.2) blockers MVIIA and GVIA, and the P/Q-type calcium channel (Cav2.1) blocker CVID. The amino acid sequences of these peptides are shown in Figure 1 as well as the sequence of MVIIC, a Cav2.1 blocker. The backbone structure of ω-conotoxins MVIIA, GVIA and CVID, determined from Nuclear Magnetic Resonance (NMR) spectroscopy experiments, are shown in Figure 2.
Figure 1.

Single letter amino acid sequences of ω-conotoxin MVIIA, MVIIC, CVID and GVIA; disulfide bridges are indicated by solid lines, O = hydroxyproline.
Figure 2.

Three-dimensional structures of ω-conotoxins that are relevant to this review. The peptide backbone structure is shown as a yellow tube, the side chain residues are shown as thin tubes and are colored according to the atom type, and hydrogen atoms are not shown. The amino acid side chains thought to be important for biological activity are highlighted. (a) ω-conotoxin GVIA (PDB code: 2cco); (b) ω-conotoxin MVIIA (PDB code: 1ttk); and (c) ω-conotoxin CVID (coordinates obtained from reference [29]).
Voltage-gated calcium ion channels (VGCC) control a number of physiological functions including chronic and neuropathic pain. For recent reviews on the topic see references [30,31,32,33,34,35,36,37]. VGCC can be broadly divided into two classes; the low-voltage activated and high-voltage activated channels. The N-type (Cav2.2) and P/Q-type calcium channels (Cav2.1) are members of the high-voltage activated channels with the ω-conotoxins selectively targeting these channels. The ability of conotoxins such as MVIIA, GVIA and CVID to selectively block Cav2.2 channels is of significant interest as these channels are implicated in refractory pain states such as neuropathic pain. By contrast, Cav2.1 channels occur at the neuromuscular junction and the inhibition of these channels can produce a number of nasty pathological effects. ω-Conotoxin MVIIC is known to inhibit Cav2.1 channels.
Μ-Conotoxins contain six Cys residues, three disulfide bridges, and have a class III Cys pattern (CC–C–C–CC). To date, 20 μ-conotoxins peptides have been identified [38]. μ-Conotoxins selectively block voltage-gated sodium channels (VGSCs).
3.2. Commonly Used Assays
Before discussing the development of conotoxin mimetics as potential pain blockers, a summary of typical bioassays used to assess these mimetics is presented here.
3.2.1. Rat Vas Deferens
It has been shown that the twitch contractions of the rat vas deferens, in response to electrical field simulation, are controlled by release of neurotransmitters governed by the Cav2.2 channel [14,18,39]. These electrically induced muscle contractions are inhibited by ω-conotoxin GVIA and it is proposed that this is due to the peptide binding to the presynaptic sites on the Cav2.2 channels [14,18,39]. Consequently, this assay was used in early studies to assess the structure–function of ω-conotoxin GVIA [40,41] and CVID [42], and the potency of peptidomimetics of ω-conotoxin GVIA [43].
3.2.2. Radioligand Displacement Assay
The radioligand binding assay was first used by Snyder and coworkers in 1988 to characterize the binding of ω-conotoxin GVIA in the brain, using membranes from rat brains and 125I-labeled ω-conotoxin GVIA [44]. This technique has been extended to the development of a radioligand displacement assay, which can be used to gauge the ability of compounds to bind to rat brain Cav2.2 channels [45]. After the preparation of crude rat brain membrane, the labeled peptide and a range of ligand concentrations are added [45], and a dose-response is then determined by counting the residual 125I-labeled ω-conotoxin GVIA that remains associated with the membrane. The binding affinity of small molecule mimetics of ω-conotoxin GVIA to the Cav2.2 channel has been determined by this method [46,47,48,49,50,51]. It is assumed that a compound that is able to displace 125I-labeled ω-conotoxin GVIA from rat brain membrane does so by binding to the same part of the Cav2.2 ion channel as GVIA and hence should have the same pain-blocking effect. This may not always be true as other factors such as allosteric effects could cause the labeled GVIA to be released from the brain membrane. Nonetheless, this assay has served as a useful initial screen for Cav2.2 channel affinity and has led to the discovery of compounds with functional activity in, for example, electrophysiological assays.
3.2.3. FLIPR assay with SH-SY5Y Neuroblastoma Cells
In recent years a functional FLIPR (Fluorescence Imaging Plate Reader) assay has replaced the radioligand displacement assay as the high-throughput assay of choice for the screening of Cav2.2 blockers. Neuroblastoma cells are a good cellular model for monitoring VGCC [52]. The magnitude of intracellular calcium flux elicited by the addition of KCl and CaCl2 is determined by measuring the change in fluorescence of a calcium-sensitive dye and can be used to gauge the level of inhibition of calcium channels by added compounds. For Cav2.2 inhibition studies, SH-SY5Y neuroblastoma cells that endogenously express human Cav2.2 channels in a physiologically relevant context, are briefly incubated with varying concentrations of the compounds of interest, in the presence of saturating concentrations of the L-type calcium channel blocker nifedipine. The Ca2+ responses produced by KCl-depolarization are measured and dose-response curves enable calculation of IC50 values [51,53,54]. The major drawbacks of the radioligand displacement assay—The requirement for radioisotopes and the fact that it is not a functional assay—are negated with the FLIPR assay. Furthermore, the FLIPR assay results are reported to be a better predictor of activity in electrophysiological assays (for information on electrophysiological methods see below). Estimated IC50 values for the functional inhibition of calcium channels obtained from the FLIPR assay and electrophysiology experiments have, in some cases, been observed to be in close agreement, whereas the IC50 values obtained from the radioligand displacement assay can over estimate functional activity by up to an order of magnitude [51].
3.2.4. IMR-32 Human Neuroblastoma Cell Assay
IMR-32 human neuroblastoma cells were first used to investigate the binding of ω-conotoxins to VGCC in 1988 (see reference [55] and later references [52,56]). The IMR-32 human neuroblastoma cell assay has some similarities to the SH-SY5Y neuroblastoma cell assay described above, both are fluorescent based assays, intracellular calcium flux elicited by the addition of K+, and both assays include the L-type calcium channel blocker nifedipine. To date, there is only one report of this assay being used to determine the binding affinity of small molecule mimics of an ω-conotoxin and in this assay Indo-1 is used as the indicator [57].
3.2.5. Electrophysiological Methods—Patch Clamp Technique
Electrophysiological methods have long been considered the “gold-standard” technique for studying ion channels. In the patch clamp technique, whole cell electrophysiological recordings are made using a glass micropipette electrode that is “clamped” to the surface of a cell expressing the ion channel of interest. For voltage gated ion channels like Cav2.2, the channel can be cycled from the resting to the activated state by varying the electrical potential from a holding potential of ~−80 mV to more depolarized test potentials, and the current generated under the voltage clamp measured. The inhibition of such channel currents by potential channel blocking compounds can be observed by adding the compounds to the extra-cellular solution. Dose responses can be used to assess the relative effectiveness of channel blockers. Since the patch clamp technique can be used to cycle voltage-gated ion channels through resting, depolarized and inactivated forms, a compound’s effect on each of these states can be measured. This information can be used to optimize compounds for particular channel states relevant to certain disease pathologies like, for example, neuropathic pain.
3.3. ω-Conotoxin Mimetics
3.3.1. ω-Conotoxin MVIIA
The first report of a low molecular weight type-III peptidomimic of ω-conotoxin MVIIA was by the group of Horwell from Parke-Davis (now Pfizer) [58]. The 1H NMR solution structure of the peptide revealed the backbone structure of the peptide [59] and it was known that the residues required for binding were Lys2, Arg10, Leu11, Tyr13 and Arg21 (Neurex Corporation) [60]. Utilizing this information, Horwell et al. designed a dendritic scaffold that would enable the mimicry of three key amino acid residues [Arg10, Leu11 and Tyr13]. The first generation compound, the synthesized alkylphenyl ether (compound 1 in Figure 3) based on a phloroglucinol core [58], gave poor inhibition (19% at 10 μM) in an N-type IMR-32 human neuroblastoma cell assay [57]. The second generation compounds (2 and 3), whose design was also supported by molecular modeling, gave improved activity (IC50 values of 3.3 and 2.7 μM) [57]. This report was one of the first to use in silico design to assist with the construction of active compounds, thereby enabling the pharmacophore of the native peptide to be better replicated. The flexibility of the projected residues in these mimics was integral to their design as it was expected that their fluxional nature would allow them to “sample” a wider region of conformational space and hence would be more likely to able to adopt an optimal ion channel binding conformation.
Figure 3.

Non-peptide mimetics containing a dendritic scaffold. The dendritic scaffold is highlighted in blue. IC50 values are for Cav2.2 activity in the IMR-32 assay [57,58].
In a follow-up report, three dendroid molecules were synthesized using the core of 8-hydroxy-2-(1H)-quinolinone attached to a 5-hydroxymethyl resorcinol (4–6, Figure 4) [61]. While these compounds were designed to be mimics of ω-conotoxin MVIIA, their ability to inhibit Cav2.2 channels has not been reported.
Figure 4.

Non-peptide mimetic of ω-conotoxin MVIIA, using the scaffold core of 8-hydroxy-2-(1H)-quinolinone and 5-hydroxymethyl resorcinol. The scaffold core is highlighted in green [61].
3.3.2. ω-Conotoxin CVID
It has been shown that Leconotide, a synthetic version of CVID (also known as AM336 and CNSB004), is the most selective ω-conotoxin for the N-type (Cav2.2) over the P/Q-type (Cav2.1) calcium channel with 700,000-fold selectivity observed [42]. Furthermore, the native peptide has been shown to have a wider therapeutic window than Ziconotide (Prialt®) and has fewer cardiovascular side effects [62,63]. Lewis and coworkers developed small molecule cyclic pentapeptides as type-I mimics of ω-conotoxin CVID [64]. The 1H NMR solution structure of ω-conotoxin CVID showed that this conotoxin has two hydrogen bonds between loop 2 and 4 that stabilize loop 2 and provide extra rigidity [42]. Lewis and coworkers determined the Cα–Cβ vectors of the residues believed to be required for binding to the Cav2.2 channel, concentrating on residues 9–14 that are found in loop 2 [64]. Simplification of this pharmacophore to the Cα–Cβ of residues 9–14 enabled virtual screening of a library of compounds and identification of the cyclic pentapeptide framework containing one or more D-amino acids (Figure 5). In total, twenty cyclic pentapeptides of this first series were synthesized and tested for their binding affinity to the Cav2.2 channel using the radioligand displacement assay with 125I-GVIA. Two analogues [c-AKlMy] and [c-aKlMY] (L-amino acids are shown as uppercase letters and D-amino acids are represented by lowercase letters), gave IC50 values of 40 and 60 μM respectively. Based on these results a second series was designed and tested. This series, [c-KlPyK], [c-lKPyK] and [c-lPKyK], had improved rigidity due to the introduction of a proline residue and also possessed a second positive charge through the inclusion of a second lysine residue. The IC50 values for the second series of peptides ranged from 16 to 480 μM. The most potent analogue was [c-KlPyK] (compound 7 in Figure 5, IC50 = 16 μM). These compounds were also tested for their selectivity for N-type (Cav2.2) over the P/Q-type (Cav2.1) voltage gated calcium channel by testing their binding affinity in the radioligand displacement assay with 125I-MVIIC. ω-Conotoxin MVIIC is known to inhibit Cav2.1. None of the peptides were found to displace 125I-MVIIC at concentrations below 300 μM and it was thus concluded that these cyclic peptides were selective for N-type voltage gated calcium channels.
Figure 5.

(a) Diagram of the three-dimensional structure of ω-conotoxin CVID with sidechains of residues 9–14 shown, coordinates obtained from reference [29]. (b) Backbone and sidechains of residue 9–14 alone; (c) Structure of [c-KlPyK] 7, the most potent analogue discovered (L-amino acids are shown as uppercase letters and D-amino acids are represented by lowercase letters). The IC50 value was determined using the radioligand displacement assay with 125I-GVIA [64].
As the cyclic pentapeptides discussed above mimic the binding of ω-conotoxin CVID and are selective against for N-type calcium channels, there was great hope that small molecules could be designed and developed to mimic ω-conotoxin CVID. However, since this original report by Lewis and coworkers in 2004 [64] there have been no further reports of CVID peptidomimetics.
3.3.3. ω-Conotoxin GVIA
Of all the ω-conotoxins, ω-conotoxin GVIA has been the subject of the most reports of type-III mimetics, firstly by the group of Baell and more recently from the group of Duggan and Tuck. The initial report of a type-III GVIA peptidomimetic was by Baell et al. in 2001 [43]. Three non-planar, rigid scaffolds were designed using the data from the NMR solution structure of ω-conotoxin GVIA, utilizing the Cα–Cβ bond vector approach combined with interactive de novo design. It was known that the residues Lys2 and Tyr13 were crucial for potency and to a lesser extent the residues Arg17, Tyr22 and Lys24 [40]. With this in mind, Baell et al. concluded that a three-point mimic must include Lys2 and Tyr13, but it was unclear which of the remaining three residues should be included. Guided by molecular modeling and scaffold design, it was decided that the third residue would be Arg17. Mimetics based on two out of the three proposed scaffolds, the benzothiazole derivative 8 and the anthranilamide derivative 9, were synthesized and were found to have activity in the rat vas deferens assay, with IC50 values of 68 μM and 98 μM, respectively, being obtained (see Figure 6 for relevant compound structures). Subsequent testing of these compounds for Cav2.2 activity using the radioligand displacement assay with 125I-GVIA gave the IC50 values of 1.9 μM [46] and 3.5 μM [47], respectively. An overlay of the benzothiazole and anthranilamide core scaffolds respectively with the NMR solution structure of ω-conotoxin GVIA is shown in Figure 7. This figure demonstrates that the benzothiazole mimetics and the anthranilamide mimics possess the correct geometry to ensure that the side chain mimics are positioned to replicate the location and direction of the α,β-bond vectors of the key amino acids side chains.
Figure 6.

Initial type-III peptidomimetics of ω-conotoxin GVIA. Cav2.2 activity was determined using the rat vas deferens assay [43] and later using the radioligand displacement assay with 125I-GVIA [46,47]. The core scaffold is colored blue.
Figure 7.

Backbone structure of ω-conotoxin GVIA (yellow) with the amino acid side chains Lys2, Tyr13, Arg17 shown in teal; (a) overlaid with a model of the benzothiazole mimetic 8 [28]—Reproduced with permission; and (b) overlaid with X-ray crystal structure of the anthranilamide scaffold core without side chain mimics. The α,β-bond vectors are shown in purple and the bonds mimicking the α,β-bond vectors are shown in yellow [47]—Reproduced with permission.
Encouraged by the activity of benzothiazole derivative 8 and the anthranilamide derivative 9, Baell, Duggan and coworkers synthesized analogues of both in order to start to build up structure-activity relationships [46,47]. Three benzothiazole analogues were tested for their binding affinity to the Cav2.2 channel using the radioligand displacement assay with 125I-GVIA. The structures of two analogues are shown in Figure 8 [46]. Conversion of the original Tyr side chain to a Phe mimic, by removal of the phenolic OH group, gave compound 10, which resulted in a negligible loss of activity (Figure 8). Limited activity loss was also observed when the guanidinium moiety was truncated to a primary amine (compound 11). Interestingly, these benzothiazole analogues (8, 10 and 11) showed a 20–25-fold selectivity for the N-type (Cav2.2) over the P/Q-type (Cav2.1) voltage gated calcium channel. In the anthranilamide series, truncation of the guanidinium to the primary amine was also investigated (Figure 8) [47]. Compound 9 was 30-fold more selective for Cav2.2 over Cav2.1. A negligible loss of activity was observed for 12 and this analogue was observed to be 13-fold selective for Cav2.2 over Cav2.1.
Figure 8.

Exploration of the benzothiazole and anthranilamide scaffolds, Cav2.2 activity was determined using the radioligand displacement assay with 125I-GVIA [46,47]. The scaffold core is colored blue.
Further investigation of the benzothiazole scaffold by Duggan and Tuck resulted in the synthesis of the truncated analogues 13–16 [50]. Previous work had shown that rotation around the N-benzyl bond resulted in two main conformations; the Arg17 side chain mimic could point up or down with respect to the benzothiazole ring and hence this side chain had the potential to mimic the Lys2 side chain of GVIA. Truncation of compound 10 resulted in analogues that have a significantly lower molecular weight, and could be synthesized in 6–7 fewer steps (Figure 9). However, the truncated Tyr13-Lys2 mimic 13 was found to be inactive. The corresponding guanidylated compound 14 showed improved activity, however this was still ten-fold lower than that of the three-point mimic 10. Conversely, the truncated Tyr13-Arg17 mimic 15 showed comparable activity to the parent analogue 10, though taking the guanidinium functional group back to a primary amine (compound 16) resulted in a significant loss of activity. This finding confirmed the important contribution that the guanidinium group makes to the observed activity of these benzothiazole compounds and showed that the truncated mimic, a two-point mimic, could have comparable activity to a more complex, higher molecular weight compound.
Figure 9.

Truncation of the original benzothiazole scaffold 10 resulting in two-point mimetics 13–16. Cav2.2 activity was determined using the radioligand displacement assay with 125I-GVIA [50].
Additional work by Duggan and Tuck led to the synthesis of two amino-anthranilamide analogues that incorporate an amide linkage in the Arg17 mimic rather than the ether linkage (compounds 17 and 18, Figure 10). However, while Cav2.2 activity was retained, the potency was relatively weak and as such no further analogues of this type were investigated [48]. It was noted that long-chain aliphatic amines like dodecylamine had strong calcium channel affinity [65], and in light of this Duggan and Tuck undertook a comprehensive study into the structure activity relationship (SAR) relevant to the alkyl chain length of the anthranilamide derivative 12. By varying the alkyl length of the ether and amide it was discovered that the optimum linker length was n = 3 and n = 7, respectively, although other linker lengths were well tolerated. They also explored removal of the p-hydroxyl substituent in 12 and replacement of the p-hydroxyl substituent with a p-fluoro substituent [49]. Fluorine, a hydroxyl surrogate, is advantageous as phenoxy groups are very quickly metabolized in the liver and replacement of the phenoxy group with fluorine prevents this metabolism. This study also investigated if a single guanidinium group was required for good activity or if diguanidinylated compounds were appropriate. It was found that the diguanidinylated compounds were the most active, deletion of the hydroxyl substituent had little influence on activity and the analogue with the p-fluoro substituent was the most active compound prepared.
Figure 10.

Exploration of the anthranilamide scaffold, only selected compounds are shown. Cav2.2 activity was determined using the radioligand displacement assay with 125I-GVIA [48,49].
Recognizing that effective voltage-gated calcium channel blockers Z160, a reformulated form of NP118809 [66], and Abbott’s A-1048400 [67] contain a diphenylmethylpiperazine moiety, Duggan and Tuck investigated substitution of the phenoxyaniline substituent with diphenylmethylpiperazine in the anthranilamide class of compounds [51]. Such a replacement was expected to limit the toxicity of these compounds. A number of analogues were synthesized and tested for their ability to block neuronal calcium channels in non-functional and functional assays (Figure 11). This was the first report that compared the Cav2.2 activity determined using the radioligand displacement assay, the FLIPR assay (fluorescence-based calcium response assay with SH-SY5Y neuroblastoma cells) and a whole-cell patch clamp electrophysiology assay. Pleasingly, the mimetics identified to be active in the radioligand displacement assay were also found to elicit functional inhibition of intracellular calcium responses in SH-SY5Y neuroblastoma cells and calcium currents in HEK293 cells stably expressing human Cav2.2 channels. These results justify the overall approach employed here to identify selective Cav2.2 channel blockers—Which used the structure of ω-conotoxin GVIA to design inhibitors and the 125I-GVIA radioligand displacement assay as an initial screening tool for Cav2.2 activity.
Figure 11.
Further exploration of the anthranilamide class of compounds where the phenoxyaniline substituent has been replaced with a diphenylmethylpiperazine moiety [51].
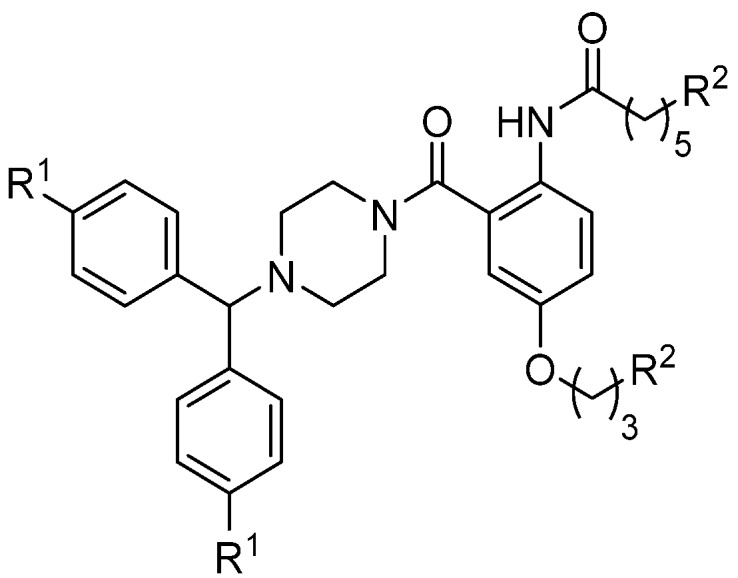
| Compd. No. | R1 | R2 | Radioligand Displacement Assay IC50 (μM) | SH-SY5Y Neuroblastoma Cells IC50 (μM) | HEK293 hCav2.2 + β3 + α2δ1 IC50 (μM) |
|---|---|---|---|---|---|
| 25 | H | NH2 | 214 | 160 | 232 |
| 26 | H | N=C(NH2)2 | 12 | 206 | 299 |
| 27 | F | NH2 | 65 | 286 | 288 |
| 28 | F | N=C(NH2)2 | 16 | 156 | 156 |
In a recent study, Duggan and Tuck investigated molecular modifications of the anthranilamides, in the hope of improving physiochemical properties while maintaining Cav2.2 activity. The anthranilamide-based mimetics were simplified by deletion of one of the amino side chain mimics and the influence of the substitution pattern of the central aromatic ring was explored [54]. Compounds 29–34 (Figure 12) were evaluated for their ability to inhibit Cav2.2 calcium responses in SH-SY5Y cells. Compound 30 has been tested for its ability to block calcium currents in HEK293 cells stably expressing human Cav2.2 channels, giving an IC50 of 36 µM [54]. The mode of action of this set of compounds is being further investigated in patch clamp electrophysiology experiments, and these results will be reported in the near future.
Figure 12.

Truncation and SAR exploration of the anthranilamide scaffold. Cav2.2 activity was determined using the FLIPR SH-SY5Y neuroblastoma cells assay [54].
3.4. μ-Conotoxin Mimetics
μ-Conotoxin KIIIA, from Conus kinoshitai, is a 16 residue, three-disulfide bridged, peptide that blocks several voltage-gated sodium channel isoforms and has been shown to have potent analgesic activity in the mouse model for pain [68,69]. Structure–function activity studies determined that the key residues required for KIIIA’s activity against voltage-gated sodium channels were Lys7, Trp8, Arg10, Asp11, His12, and Arg14 [69]. Additionally, the solution NMR structure of μ-conotoxin KIIIA revealed that 5 of the 6 key residues (the exception was Arg14) were located on an α-helix. It was thus concluded that the α-helix plays an important role in the interaction of μ-conotoxin KIIIA with the voltage-gated sodium channels [70,71]. Recently type-III peptidomimetics of μ-conotoxin KIIIA have been designed with the aid of an in silico de novo strategy [72]. Using the Cα–Cβ bond vector approach Baell and coworkers devised a diketopiperazine carboxamide scaffold that would mimic the Cα–Cβ bond vectors of the residues Lys7, Trp8 and His12. A key part of the design is the use of the amide link to the His12 mimic, thus resulting in an internal H-bond that causes the molecule to adopt the conformation required to correctly mimic the Cα–Cβ bond vectors of the His12 sidechain. In this study two such mimetics were synthesized (compounds 35 and 36, Figure 13) and tested for their ability to block the Nav1.7 channel (patch clamp assay). While both compounds only weakly blocked the Nav1.7 channel they are the first type-III peptidomimetics developed based on the native structure of μ-conotoxin KIIIA. It was reported that peptidomimetic 35 will serve as a template for future studies. A second scaffold, which has a relatively simple design, was developed to mimic the Arg10 and Arg14 residues in μ-conotoxin KIIIA [73]. Compound 37 blocked the Nav1.7 channel relatively weakly.
Figure 13.

Non-peptide mimetics of μ-conotoxin KIIIA by Baell and coworkers. The diketopiperazine carboxamide scaffold is highlighted in blue in 35 and 36. Activity is block of the Nav1.7 channel (patch clamp assay) [72,73].
4. Snake Venom Peptides and Their Mimetics
4.1. Echistatin
Tirofiban (Aggrastat®, MK-0383; L-700,462, 38 Figure 14) a small molecule peptidomimetic antiplatelet drug, was discovered by researchers from Merck in the early 1990s [74,75,76,77,78,79] and granted FDA approval in 1998. The development of Tirofiban was based on the 3D-structure of Echistatin (disintegrin), which is a derivative of an anticoagulant found in the venom of the African saw-scaled viper Echis carinatus. Researchers from Merck designed small molecule mimetics of the cell adhesion motif RGD (Arg-Gly-Asp), which was identified as the recognition element required for the antiplatelet activity possessed by Echistatin. Merck’s in-house sample collection was searched for compounds that had carboxylate and amino functionalities separated by distances of 10–20 Å. The latter corresponds to the approximate distance between the termini of the Arg and Asp side chains in RGD. Tirofiban was the end result of optimization of the initial hit from this screen [74,75].
Figure 14.
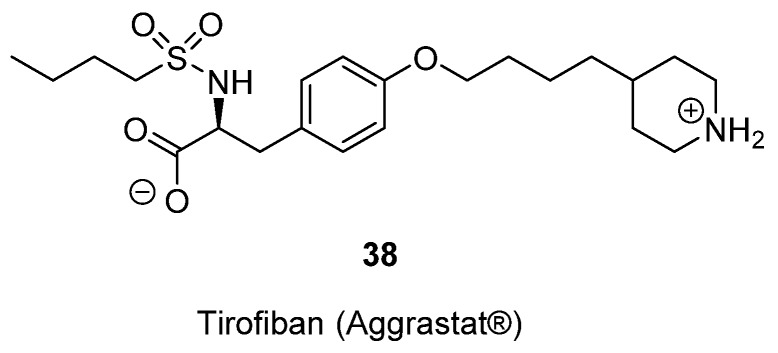
The chemical structure of Tirofiban 38.
4.2. Erabutoxin B
Erabutoxin B, found in the venom of the broad-banded blue sea snake, is a 62 amino acid protein bearing four disulfide bridges. It has high affinity for the post-synaptic nicotinic acetylcholine receptor (nAChR) [80,81]. Compounds that act on the nAChR have the potential to be treatments for nicotine addiction, to relieve the effects of Alzheimer’s disease or be muscle relaxants. Kahn and coworkers utilized the X-ray structure of erabutoxin B [82,83], and the knowledge that residues 31–34 (Asp, Phe, Arg, Gly) that form a β-turn are essential for binding to the nAChR, to design a type-III erabutoxin mimetic [84], guided by molecular modeling. This led to the hexahydro-indolizinone scaffold being chosen as the mimetic core. The side chain residues of Asp31 and Arg33 in erabutoxin B were appropriately mimicked as either a carboxylate or an ammonium salt respectively (compound 39, Figure 15). This seminal work is the first report of molecular modeling guided design of a small molecule mimic of a peptide or protein found in the venom of snakes or cone snails. Unfortunately, no biological evaluation of this mimetic has been reported and hence it is unclear if this is a true type-III mimetic of erabutoxin.
Figure 15.
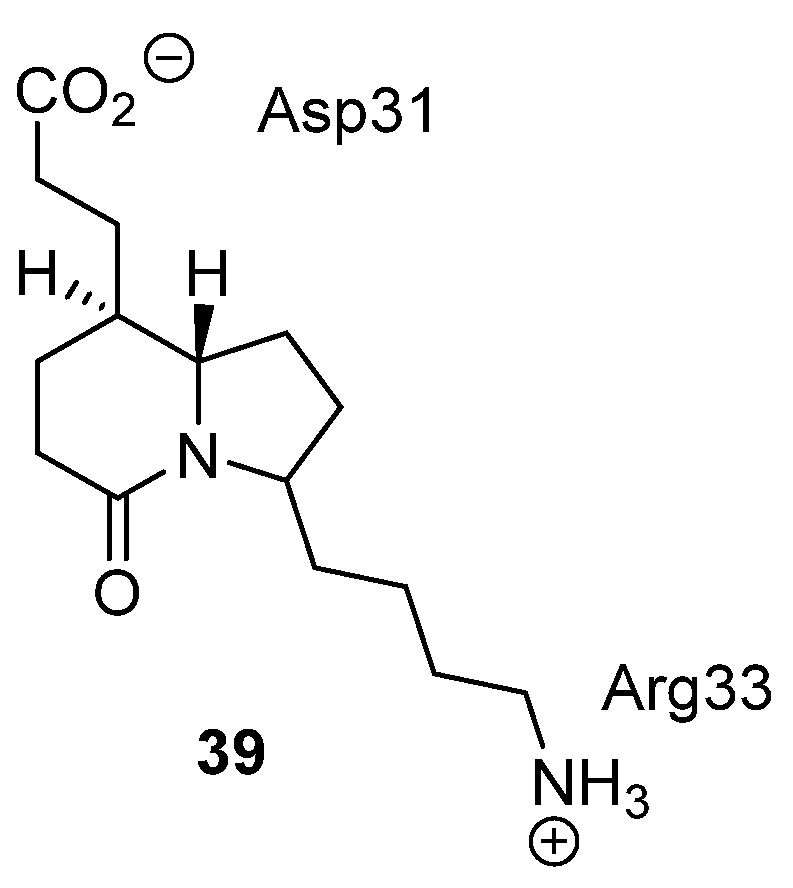
The chemical structure of an erabutoxin mimic 39.
5. Sun Anemone Toxin
Stichodactyla toxin (ShK), from the sea anemone Stichodactyla helianthus, is a 35 residue peptide that binds strongly to a number of voltage gated potassium channels [85]. The peptide has three disulfide linkages that produce a structurally well-defined molecule. ShK is known to have three separate binding sites [86,87]. The determination of the NMR solution structure allowed mapping of the potassium channel binding surface and thus identified the residues responsible for binding [88]. The toxin was found to contain a discontinuous (Arg11 and Phe27) and contiguous (Lys22, Tyr23 and Arg24) binding surface but its rigid nature makes it an attractive target for the rational design of type-III mimetics. Baell et al. utilized the previously mentioned the α,β-bond vector approach to devise suitable mimetics. Initially, the contiguous residues (Lys22, Tyr23 and Arg24) were investigated with a core N-benzylhydantoin scaffold. For synthetic simplicity, the Tyr sidechain mimic was replaced with a Phe mimic, as it was known that the OH group of Tyr23 is not required for binding [86]. This initial compound was inactive (structure not shown), presumably because the hydantoin ring orientates the phenyl ring (Tyr23 mimic) in an orientation that it is perpendicular to the way Tyr23 is displayed in the ShK toxin [89]. Subsequently the Lys22-Tyr23-Arg11 pharmacophore was investigated using a quinoxaline-dione ring, however this compound was also inactive [89]. Use of the 1,7-disubstituted 2-aminobenzimidazole core with appropriate functionalization allowed the development of hydroxylated and unhydroxylated compounds 40 and 41 (Figure 16). These compounds showed the same activity in voltage-gated Kv1.3 potassium channels patch clamp assay, thus confirming that the OH group of Tyr23 is not required for binding. The biological results from the first generation of compounds enabled pharmacophore-based database mining. Three nonpeptide hits were subsequently tested for activity, but were found to be inactive. While these first generation molecules were relatively weak binders, Baell and coworkers “built on” these results with their second-generation peptidomimetics [90]. For this study, they focused on the contiguous residues (Lys22, Tyr23 and Arg24), thus allowing for a smaller mimetic. Again, using the α,β-bond vectors of the Lys22 and Tyr23 side chains from the NMR solution structure of ShK, an N-alkylated indole-7-carboxamido scaffold was designed (compound 42). Further derivatization at the 4-position allowed for additional mimicry of the Arg24 sidechain residue. The activity of this compound was marginally better than that observed for the first-generation mimetics. It was noted that the production of a library of mimetics based around this the indole might lead to compounds with improved activity.
Figure 16.

Peptidomimetics of ShK that contain the (a) 2-aminobenzimidazole scaffold, compounds 40 and 41 [89]; and (b) the N-alkylated indole-7-carboxamido scaffold, compound 42 [90]. The scaffold structure is highlighted in blue. Activity was determined using a voltage-gated Kv1.3 potassium channels patch clamp assay.
6. Summary and Conclusions
The stupendous number of pharmacologically active substances present in animal venoms is a largely untapped source of new bioactives. Peptides form a significant component of these actives but due to problems associated with their bioavailability, these compounds have rarely reached the clinic. The development of lower molecular weight organic compounds that mimic the action of some of these peptides—Peptidomimetics—has been one approach aimed at overcoming bioavailablity problems. Peptides found in the venom from the Conus marine snails have received by far the most attention in this regard. This has largely focused on the mimicry of the neuronal calcium ion channel blocking ability of the ω-conotoxins. The ω-conotoxins are exquisitely selective and potent modulators of voltage gated calcium ion channels and the design and construction of small molecules that approach the activity of these peptides has been a challenging task. Nonetheless, progress has been made with ω-conotoxin GVIA mimics, with the most recently developed compounds showing considerable promise. Mimicry of active peptides from snake and anemone venom has also been undertaken. The initial hit that led to the antiplatelet drug Tirofiban was inspired by an active peptide found in the venom of the broad-banded blue sea snake.
There are numerous avenues to the discovery of hits with the potential to be developed into potent bioactives, including the screening of natural and synthetic compound collections, analoging of known drugs and fragment-based approaches. Where little is known about the structure of the biological target, which is the case with many neuronal ion channels, the mimicry of venom peptides remains an attractive option that is expected to be fruitful for many years to come.
Acknowledgments
The Monash-CSIRO Collaborative Research Support Scheme, Monash University and CSIRO are acknowledged for providing funding to support K.L.T. and P.J.D.’s continued research in pain-blocker discovery. Jonathan Baell is thanked for first attracting P.J.D. to the field of conotoxin mimetics. The ongoing support of K.L.T. and P.J.D.’s research by Richard Lewis by providing access to his ion channel assays and through many valuable discussions is greatly appreciated.
Author Contributions
P.J.D and K.L.T. contributed equally to this review.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Kaas Q., Craik D. Bioinformatics-aided venomics. Toxins. 2015;7:2159–2187. doi: 10.3390/toxins7062159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vagner J., Qu H., Hruby V.J. Peptidomimetics, a synthetic tool of drug discovery. Curr. Opin. Chem. Biol. 2008;12:292–296. doi: 10.1016/j.cbpa.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silverman R.B., Holladay M.W. Lead Discovery and Lead Modification. In: Holladay R.B., Silverman M.W., editors. The Organic Chemistry of Drug Design and Drug Action. 3rd ed. Academic Press; Boston, MA, USA: 2014. pp. 19–122. [Google Scholar]
- 4.Trabocchi A., Guarna A. Peptidomimetics in Organic and Medicinal Chemistry. John Wiley & Sons, Ltd.; New York, NY, USA: 2014. The basics of peptidomimetics; pp. 1–17. [Google Scholar]
- 5.Ripka A.S., Rich D.H. Peptidomimetic design. Curr. Opin. Chem. Biol. 1998;2:441–452. doi: 10.1016/S1367-5931(98)80119-1. [DOI] [PubMed] [Google Scholar]
- 6.Lauri G., Bartlett P. CAVEAT: A program to facilitate the design of organic molecules. J. Comput.-Aided Mol. Des. 1994;8:51–66. doi: 10.1007/BF00124349. [DOI] [PubMed] [Google Scholar]
- 7.CAVEAT. [(accessed on 1 September 2015)]. Available online: http://www.cchem.berkeley.edu/pabgrp/Data/caveat.html.
- 8.Olivera B.M. E.E. Just Lecture, 1996: Conus venom peptides, receptor and ion channel targets, and drug design: 50 million years of neuropharmacology. Mol. Biol. Cell. 1997;8:2101–2109. doi: 10.1091/mbc.8.11.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewis R.J., Dutertre S., Vetter I., Christie M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012;64:259–298. doi: 10.1124/pr.111.005322. [DOI] [PubMed] [Google Scholar]
- 10.Livett B.G., Gayler K.R., Khalil Z. Drugs from the sea: Conopeptides as potential therapeutics. Curr. Med. Chem. 2004;11:1715–1723. doi: 10.2174/0929867043364928. [DOI] [PubMed] [Google Scholar]
- 11.Olivera B.M. Peptide neurotoxins from fish-hunting cone snails. Science. 1985;230:1338–1343. doi: 10.1126/science.4071055. [DOI] [PubMed] [Google Scholar]
- 12.Cruz L.J., White J. Clinical toxicology of Conus snail stings. In: Julian W., Jurg M., editors. Handbook of Clinical Toxicology of Animal Venoms and Poisons. CRC Press; Boca Raton, FL, USA: 1995. pp. 117–128. [Google Scholar]
- 13.Olivera B.M., Cruz L.J. Conotoxins, in retrospect. Toxicon. 2001;39:7–14. doi: 10.1016/S0041-0101(00)00157-4. [DOI] [PubMed] [Google Scholar]
- 14.Olivera B.M., Miljanich G.P., Ramachandran J., Adams M.E. Calcium channel diversity and neurotransmitter release: The ω-Conotoxins and ω-Agatoxins. Annu. Rev. Biochem. 1994;63:823–867. doi: 10.1146/annurev.bi.63.070194.004135. [DOI] [PubMed] [Google Scholar]
- 15.Gray W.R., Olivera B.M., Cruz L.J. Peptide toxins from venomous Conus snails. Annu. Rev. Biochem. 1988;57:665–700. doi: 10.1146/annurev.bi.57.070188.003313. [DOI] [PubMed] [Google Scholar]
- 16.Norton R.S., Olivera B.M. Conotoxins down under. Toxicon. 2006;48:780–798. doi: 10.1016/j.toxicon.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 17.Han T.S., Teichert R.W., Olivera B.M., Bulaj G. Conus venoms—A rich source of peptide-based therapeutics. Curr. Pharm. Des. 2008;14:2462–2479. doi: 10.2174/138161208785777469. [DOI] [PubMed] [Google Scholar]
- 18.Hirata H., Albillos A., Fernández F., Medrano J., Jurkiewicz A., García A.G. ω-Conotoxins block neurotransmission in the rat vas deferens by binding to different presynaptic sites on the N-type Ca2+ channel. Eur. J. Pharmacol. 1997;321:217–223. doi: 10.1016/S0014-2999(96)00951-X. [DOI] [PubMed] [Google Scholar]
- 19.Lewis R.J. Conotoxins as selective inhibitors of neuronal ion channels, receptors and transporters. IUBMB Life. 2004;56:89–93. doi: 10.1080/15216540410001668055. [DOI] [PubMed] [Google Scholar]
- 20.Halai R., Craik D.J. Conotoxins: Natural product drug leads. Nat. Prod. Rep. 2009;26:526–536. doi: 10.1039/b819311h. [DOI] [PubMed] [Google Scholar]
- 21.Schroeder C., Lewis R. ω-Conotoxins GVIA, MVIIA and CVID: SAR and clinical potential. Mar. Drugs. 2006;4:193–214. doi: 10.3390/md403193. [DOI] [Google Scholar]
- 22.Akondi K.B., Muttenthaler M., Dutertre S., Kaas Q., Craik D.J., Lewis R.J., Alewood P.F. Discovery, Synthesis, and Structure–Activity Relationships of Conotoxins. Chem. Rev. 2014;114:5815–5847. doi: 10.1021/cr400401e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cruz L.J., Gray W.R., Yoshikami D., Olivera B.M. Conus venoms: A rich source of neuroactive peptides. Toxin Rev. 1985;4:107–132. doi: 10.3109/15569548509014416. [DOI] [Google Scholar]
- 24.Lin H., Li Q.-Z. Predicting conotoxin superfamily and family by using pseudo amino acid composition and modified Mahalanobis discriminant. Biochem. Biophys. Res. Commun. 2007;354:548–551. doi: 10.1016/j.bbrc.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 25.Kaas Q., Westermann J.-C., Craik D.J. Conopeptide characterization and classifications: An analysis using ConoServer. Toxicon. 2010;55:1491–1509. doi: 10.1016/j.toxicon.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 26.Robinson S.D., Norton R.S. Conotoxin gene superfamilies. Mar. Drugs. 2014;12:6058–6101. doi: 10.3390/md12126058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nielsen K.J., Schroeder T., Lewis R. Structure-activity relationships of ω-conotoxins at N-type voltage-sensitive calcium channels. J. Mol. Recognit. 2000;13:55–70. doi: 10.1002/(SICI)1099-1352(200003/04)13:2<55::AID-JMR488>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 28.Baell J.B., Duggan P.J., Lok Y.P. ω-Conotoxins and Approaches to Their Non-Peptide Mimetics. Aust. J. Chem. 2004;57:179–185. doi: 10.1071/CH03242. [DOI] [Google Scholar]
- 29.ConoServer. [(accessed on 1 September 2015)]. Available online: http://www.conoserver.org/?page=card&table=structure&id=138.
- 30.Winquist R.J., Pan J.Q., Gribkoff V.K. Use-dependent blockade of Cav2.2 voltage-gated calcium channels for neuropathic pain. Biochem. Pharmacol. 2005;70:489–499. doi: 10.1016/j.bcp.2005.04.035. [DOI] [PubMed] [Google Scholar]
- 31.Vink S., Alewood P.F. Targeting voltage-gated calcium channels: Developments in peptide and small-molecule inhibitors for the treatment of neuropathic pain. Br. J. Pharmacol. 2012;167:970–989. doi: 10.1111/j.1476-5381.2012.02082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pexton T., Moeller-Bertram T., Schilling J.M., Wallace M.S. Targeting voltage-gated calcium channels for the treatment of neuropathic pain: A review of drug development. Expert Opin. Investig. Drugs. 2011;20:1277–1284. doi: 10.1517/13543784.2011.600686. [DOI] [PubMed] [Google Scholar]
- 33.Belardetti F., Zamponi G.W. Calcium channels as therapeutic targets. WIREs Membr. Transp. Signal. 2012;1:433–451. doi: 10.1002/wmts.38. [DOI] [Google Scholar]
- 34.Perret D., Luo Z.D. Targeting voltage-gated calcium channels for neuropathic pain management. Neurotherapeutics. 2009;6:679–692. doi: 10.1016/j.nurt.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McGivern J.G. Targeting N-type and T-type calcium channels for the treatment of pain. Drug Discov. Today. 2006;11:245–253. doi: 10.1016/S1359-6446(05)03662-7. [DOI] [PubMed] [Google Scholar]
- 36.Rahman W., Dickenson A.H. Voltage gated sodium and calcium channel blockers for the treatment of chronic inflammatory pain. Neurosci. Lett. 2013;557(Pt A):19–26. doi: 10.1016/j.neulet.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 37.Lee M.S. Recent progress in the discovery and development of N-type calcium channel modulators for the treatment of pain. Prog. Med. Chem. 2014;53:147–186. doi: 10.1016/B978-0-444-63380-4.00004-4. [DOI] [PubMed] [Google Scholar]
- 38.Green B.R., Bulaj G., Norton R.S. Structure and function of μ-conotoxins, peptide-based sodium channel blockers with analgesic activity. Future Med. Chem. 2014;6:1677–1698. doi: 10.4155/fmc.14.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maggi C., Patacchini R., Santicioli P., Lippe I., Giuliani S., Geppetti P., del Bianco E., Selleri S., Meli A. The effect of omega conotoxin GVIA, a peptide modulator of the N-type voltage sensitive calcium channels, on motor responses produced by activation of efferent and sensory nerves in mammalian smooth muscle. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1988;338:107–113. doi: 10.1007/BF00174856. [DOI] [PubMed] [Google Scholar]
- 40.Lew M.J., Flinn J.P., Pallaghy P.K., Murphy R., Whorlow S.L., Wright C.E., Norton R.S., Angus J.A. Structure-function relationships of ω-conotoxin GVIA: Synthesis, structure, calcium channel binding, and functional assay of alanine substituted analogues. J. Biol. Chem. 1997;272:12014–12023. doi: 10.1074/jbc.272.18.12014. [DOI] [PubMed] [Google Scholar]
- 41.Flinn J.P., Pallaghy P.K., Lew M.J., Murphy R., Angus J.A., Norton R.S. Roles of key functional groups in ω-conotoxin GVIA. Eur. J. Biochem. 1999;262:447–455. doi: 10.1046/j.1432-1327.1999.00383.x. [DOI] [PubMed] [Google Scholar]
- 42.Lewis R.J., Nielsen K.J., Craik D.J., Loughnan M.L., Adams D.A., Sharpe I.A., Luchian T., Adams D.J., Bond T., Thomas L., et al. Novel ω-conotoxins from Conus catus discriminate among neuronal calcium channel subtypes. J. Biol. Chem. 2000;275:35335–35344. doi: 10.1074/jbc.M002252200. [DOI] [PubMed] [Google Scholar]
- 43.Baell J., Forsyth S., Gable R., Norton R., Mulder R. Design and synthesis of type-III mimetics of ω-conotoxin GVIA. J. Comput.-Aided Mol. Des. 2001;15:1119–1136. doi: 10.1023/A:1015930031890. [DOI] [PubMed] [Google Scholar]
- 44.Wagner J., Snowman A., Biswas A., Olivera B., Snyder S. Omega-conotoxin GVIA binding to a high-affinity receptor in brain: Characterization, calcium sensitivity, and solubilization. J. Neurosci. 1988;8:3354–3359. doi: 10.1523/JNEUROSCI.08-09-03354.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nielsen K.J., Adams D.A., Alewood P.F., Lewis R.J., Thomas L., Schroeder T., Craik D.J. Effects of chirality at Tyr13 on the structure-activity relationships of ω-conotoxins from Conus magus. Biochemistry. 1999;38:6741–6751. doi: 10.1021/bi982980u. [DOI] [PubMed] [Google Scholar]
- 46.Baell J.B., Duggan P.J., Forsyth S.A., Lewis R.J., Lok Y.P., Schroeder C.I. Synthesis and biological evaluation of nonpeptide mimetics of ω-conotoxin GVIA. Bioorg. Med. Chem. 2004;12:4025–4037. doi: 10.1016/j.bmc.2004.05.040. [DOI] [PubMed] [Google Scholar]
- 47.Baell J.B., Duggan P.J., Forsyth S.A., Lewis R.J., Lok Y.P., Schroeder C.I., Shepherd N.E. Synthesis and biological evaluation of anthranilamide-based non-peptide mimetics of ω-conotoxin GVIA. Tetrahedron. 2006;62:7284–7292. doi: 10.1016/j.tet.2006.05.041. [DOI] [Google Scholar]
- 48.Duggan P.J., Faber J.M., Graham J.E., Lewis R.J., Lumsden N.G., Tuck K.L. Synthesis and Cav2.2 binding data for non-peptide mimetics of ω-conotoxin GVIA based on a 5-amino-anthranilamide core. Aust. J. Chem. 2008;61:11–15. doi: 10.1071/CH07327. [DOI] [Google Scholar]
- 49.Andersson A., Baell J.B., Duggan P.J., Graham J.E., Lewis R.J., Lumsden N.G., Tranberg C.E., Tuck K.L., Yang A. ω-Conotoxin GVIA mimetics based on an anthranilamide core: Effect of variation in ammonium side chain lengths and incorporation of fluorine. Bioorg. Med. Chem. 2009;17:6659–6670. doi: 10.1016/j.bmc.2009.07.063. [DOI] [PubMed] [Google Scholar]
- 50.Duggan P.J., Lewis R.J., Lok Y.P., Lumsden N.G., Tuck K.L., Yang A. Low molecular weight non-peptide mimics of ω-conotoxin GVIA. Bioorg. Med. Chem. Lett. 2009;19:2763–2765. doi: 10.1016/j.bmcl.2009.03.130. [DOI] [PubMed] [Google Scholar]
- 51.Tranberg C.E., Yang A., Vetter I., McArthur J.R., Baell J.B., Lewis R.J., Tuck K.L., Duggan P.J. ω-Conotoxin GVIA mimetics that bind and inhibit neuronal Cav2.2 ion channels. Mar. Drugs. 2012;10:2349–2368. doi: 10.3390/md10102349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Passafaro M., Clementi F., Sher E. Metabolism of omega-conotoxin-sensitive voltage-operated calcium channels in human neuroblastoma cells: Modulation by cell differentiation and anti-channel antibodies. J. Neurosci. 1992;12:3372–3379. doi: 10.1523/JNEUROSCI.12-09-03372.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sousa S.R., Vetter I., Ragnarsson L., Lewis R.J. Expression and pharmacology of endogenous Cav channels in SH-SY5Y human neuroblastoma cells. PLoS ONE. 2013;8:e59293. doi: 10.1371/journal.pone.0059293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gleeson E., Graham J., Spiller S., Vetter I., Lewis R., Duggan P., Tuck K. Inhibition of N-Type Calcium Channels by Fluorophenoxyanilide Derivatives. Mar. Drugs. 2015;13:2030–2045. doi: 10.3390/md13042030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sher E., Pandiella A., Clementi F. ω-Conotoxin binding and effects on calcium channel function in human neuroblastoma and rat pheochromocytoma cell lines. FEBS Lett. 1988;235:178–182. doi: 10.1016/0014-5793(88)81258-4. [DOI] [PubMed] [Google Scholar]
- 56.Carbone E., Sher E., Clementi F. Ca currents in human neuroblastoma IMR32 cells: Kinetics, permeability and pharmacology. Pflügers Arch. 1990;416:170–179. doi: 10.1007/BF00370239. [DOI] [PubMed] [Google Scholar]
- 57.Menzler S., Bikker J.A., Suman-Chauhan N., Horwell D.C. Design and biological evaluation of non-peptide analogues of omega-conotoxin MVIIA. Bioorg. Med. Chem. Lett. 2000;10:345–347. doi: 10.1016/S0960-894X(99)00699-X. [DOI] [PubMed] [Google Scholar]
- 58.Menzler S., Bikker J.A., Horwell D.C. Synthesis of a non-peptide analogue of omega-conotoxin MVIIA. Tetrahedron Lett. 1998;39:7619–7622. doi: 10.1016/S0040-4039(98)01657-8. [DOI] [Google Scholar]
- 59.Kohno T., Kim J.I., Kobayashi K., Kodera Y., Maeda T., Sato K. Three-dimensional structure in solution of the calcium channel blocker omega-conotoxin MVIIA. Biochemistry. 1995;34:10256–10265. doi: 10.1021/bi00032a020. [DOI] [PubMed] [Google Scholar]
- 60.Nadasdi L., Yamashiro D., Chung D., Tarczy-Hornoch K., Adriaenssens P., Ramachandran J. Structure-activity analysis of a Conus peptide blocker of N-type neuronal calcium channels. Biochemistry. 1995;34:8076–8081. doi: 10.1021/bi00025a013. [DOI] [PubMed] [Google Scholar]
- 61.Guo Z.-X., Cammidge A.N., Horwell D.C. Dendroid peptide structural mimetics of ω-conotoxin MVIIA based on a 2(1H)-quinolinone core. Tetrahedron. 2000;56:5169–5175. doi: 10.1016/S0040-4020(00)00179-4. [DOI] [Google Scholar]
- 62.Smith M.T., Cabot P.J., Ross F.B., Robertson A.D., Lewis R.J. The novel N-type calcium channel blocker, AM336, produces potent dose-dependent antinociception after intrathecal dosing in rats and inhibits substance P release in rat spinal cord slices. Pain. 2002;96:119–127. doi: 10.1016/S0304-3959(01)00436-5. [DOI] [PubMed] [Google Scholar]
- 63.Wright C.E., Robertson A.D., Whorlow S.L., Angus J.A. Cardiovascular and autonomic effects of ω-conotoxins MVIIA and CVID in conscious rabbits and isolated tissue assays. Br. J. Pharmacol. 2000;131:1325–1336. doi: 10.1038/sj.bjp.0703701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schroeder C., Smythe M., Lewis R. Development of small molecules that mimic the binding of ω-conotoxins at the N-type voltage-gated calcium channel. Mol. Divers. 2004;8:127–134. doi: 10.1023/B:MODI.0000025656.79632.86. [DOI] [PubMed] [Google Scholar]
- 65.Beedle A.M., Zamponi G.W. Block of voltage-dependent calcium channels by aliphatic monoamines. Biophys. J. 2000;79:260–270. doi: 10.1016/S0006-3495(00)76288-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zamponi G.W., Feng Z.-P., Zhang L., Pajouhesh H., Ding Y., Belardetti F., Pajouhesh H., Dolphin D., Mitscher L.A., Snutch T.P. Scaffold-based design and synthesis of potent N-type calcium channel blockers. Bioorg. Med. Chem. Lett. 2009;19:6467–6472. doi: 10.1016/j.bmcl.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 67.Scott V.E., Vortherms T.A., Niforatos W., Swensen A.M., Neelands T., Milicic I., Banfor P.N., King A., Zhong C., Simler G., et al. A-1048400 is a novel, orally active, state-dependent neuronal calcium channel blocker that produces dose-dependent antinociception without altering hemodynamic function in rats. Biochem. Pharmacol. 2012;83:406–418. doi: 10.1016/j.bcp.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 68.Bulaj G., West P.J., Garrett J.E., Watkins M., Zhang M.-M., Norton R.S., Smith B.J., Yoshikami D., Olivera B.M. Novel conotoxins from Conus striatus and Conus kinoshitai selectively block TTX-resistant sodium channels. Biochemistry. 2005;44:7259–7265. doi: 10.1021/bi0473408. [DOI] [PubMed] [Google Scholar]
- 69.Zhang M.-M., Green B.R., Catlin P., Fiedler B., Azam L., Chadwick A., Terlau H., McArthur J.R., French R.J., Gulyas J., et al. Structure/function characterization of μ-conotoxin KIIIA, an analgesic, nearly irreversible blocker of mammalian neuronal sodium channels. J. Biol. Chem. 2007;282:30699–30706. doi: 10.1074/jbc.M704616200. [DOI] [PubMed] [Google Scholar]
- 70.Khoo K.K., Feng Z.-P., Smith B.J., Zhang M.-M., Yoshikami D., Olivera B.M., Bulaj G., Norton R.S. Structure of the analgesic μ-conotoxin KIIIA and effects on the structure and function of disulfide deletion. Biochemistry. 2009;48:1210–1219. doi: 10.1021/bi801998a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Khoo K.K., Gupta K., Green B.R., Zhang M.-M., Watkins M., Olivera B.M., Balaram P., Yoshikami D., Bulaj G., Norton R.S. Distinct disulfide isomers of μ-conotoxins KIIIA and KIIIB block voltage-gated sodium channels. Biochemistry. 2012;51:9826–9835. doi: 10.1021/bi301256s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brady R.M., Zhang M., Gable R., Norton R.S., Baell J.B. De novo design and synthesis of a μ-conotoxin KIIIA peptidomimetic. Bioorg. Med. Chem. Lett. 2013;23:4892–4895. doi: 10.1016/j.bmcl.2013.06.086. [DOI] [PubMed] [Google Scholar]
- 73.Brady R., Baell J., Norton R. Strategies for the development of conotoxins as new therapeutic leads. Mar. Drugs. 2013;11:2293–2313. doi: 10.3390/md11072293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hartman G.D., Egbertson M.S., Halczenko W., Laswell W.L., Duggan M.E., Smith R.L., Naylor A.M., Manno P.D., Lynch R.J. Non-peptide fibrinogen receptor antagonists. 1. Discovery and design of exosite inhibitors. J. Med. Chem. 1992;35:4640–4642. doi: 10.1021/jm00102a020. [DOI] [PubMed] [Google Scholar]
- 75.Egbertson M.S., Chang C.T.C., Duggan M.E., Gould R.J., Halczenko W., Hartman G.D., Laswell W.L., Lynch J.J., Lynch R.J. Non-peptide fibrinogen receptor antagonists. 2. Optimization of a tyrosine template as a mimic for Arg-Gly-Asp. J. Med. Chem. 1994;37:2537–2551. doi: 10.1021/jm00042a007. [DOI] [PubMed] [Google Scholar]
- 76.Lynch J.J., Cook J.J., Sitko G.R., Holahan M.A., Ramjit D.R., Mellott M.J., Stranieri M.T., Stabilito I.I., Zhang G., Lynch R.J. Nonpeptide glycoprotein IIb/IIIa inhibitors. 5. Antithrombotic effects of MK-0383. J. Pharmacol. Exp. Ther. 1995;272:20–32. [PubMed] [Google Scholar]
- 77.Peerlinck K., de Lepeleire I., Goldberg M., Farrell D., Barrett J., Hand E., Panebianco D., Deckmyn H., Vermylen J., Arnout J. MK-383 (L-700,462), a selective nonpeptide platelet glycoprotein IIb/IIIa antagonist, is active in man. Circulation. 1993;88:1512–1517. doi: 10.1161/01.CIR.88.4.1512. [DOI] [PubMed] [Google Scholar]
- 78.Barrett J.S., Murphy G., Peerlinck K., Lepeleire I.D., Gould R.J., Panebianco D., Hand E., Deckmyn H., Vermylen J., Arnout J. Pharmacokinetics and pharmacodynamics of MK-383, a selective non-peptide platelet glycoprotein-IIb/IIIa receptor antagonist, in healthy men. Clin. Pharmacol. Ther. 1994;56:377–388. doi: 10.1038/clpt.1994.152. [DOI] [PubMed] [Google Scholar]
- 79.Cook J.J., Bednar B., Lynch J.J., Gould R.J., Egbertson M.S., Halczenko W., Duggan M.E., Hartman G.D., Lo M.-W., Murphy G.M., et al. Tirofiban (Aggrastat®) Cardiovasc. Drug Rev. 1999;17:199–224. doi: 10.1111/j.1527-3466.1999.tb00015.x. [DOI] [Google Scholar]
- 80.Lee C.Y. Chemistry and pharmacology of polypeptide toxins in snake venoms. Annu. Rev. Pharmacol. 1972;12:265–286. doi: 10.1146/annurev.pa.12.040172.001405. [DOI] [PubMed] [Google Scholar]
- 81.Tu A.T. Neurotoxins of animal venoms: Snakes. Annu. Rev. Biochem. 1973;42:235–258. doi: 10.1146/annurev.bi.42.070173.001315. [DOI] [PubMed] [Google Scholar]
- 82.Tsernoglou D., Petsko G.A. The crystal structure of a post-synaptic neurotoxin from sea snake at 2.2 Å resolution. FEBS Lett. 1976;68:1–4. doi: 10.1016/0014-5793(76)80390-0. [DOI] [PubMed] [Google Scholar]
- 83.Low B.W., Potter R., Jackson R.B., Tamiya N., Sato S. X-ray crystallographic study of the erabutoxins and of a diiodo derivative. J. Biol. Chem. 1971;246:4366–4368. [PubMed] [Google Scholar]
- 84.Kahn M., Chen B., Zieske P. The design and synthesis of a nonpeptide mimic of erabutoxin. Heterocycles. 1987;25:29–31. doi: 10.3987/S-1987-01-0029. [DOI] [Google Scholar]
- 85.Castañeda O., Sotolongo V., Amor A.M., Stöcklin R., Anderson A.J., Harvey A.L., Engström Å., Wernstedt C., Karlsson E. Characterization of a potassium channel toxin from the Caribbean sea anemone Stichodactyla helianthus. Toxicon. 1995;33:603–613. doi: 10.1016/0041-0101(95)00013-c. [DOI] [PubMed] [Google Scholar]
- 86.Pennington M.W., Mahnir V.M., Krafte D.S., Zaydenberg I., Byrnes M.E., Khaytin I., Crowley K., Kem W.R. Identification of three separate binding sites on SHK toxin, a potent inhibitor of voltage-dependent potassium channels in human T-lymphocytes and rat brain. Biochem. Biophys. Res. Commun. 1996;219:696–701. doi: 10.1006/bbrc.1996.0297. [DOI] [PubMed] [Google Scholar]
- 87.Rauer H., Pennington M., Cahalan M., Chandy K.G. Structural conservation of the pores of calcium-activated and voltage-gated potassium channels determined by a sea anemone toxin. J. Biol. Chem. 1999;274:21885–21892. doi: 10.1074/jbc.274.31.21885. [DOI] [PubMed] [Google Scholar]
- 88.Tudor J.E., Pallaghy P.K., Pennington M.W., Norton R.S. Solution structure of ShK toxin, a novel potassium channel inhibitor from a sea anemone. Nat. Struct. Mol. Biol. 1996;3:317–320. doi: 10.1038/nsb0496-317. [DOI] [PubMed] [Google Scholar]
- 89.Baell J., Harvey A., Norton R. Design and synthesis of type-III mimetics of ShK toxin. J. Comput.-Aided Mol. Des. 2002;16:245–262. doi: 10.1023/A:1016310602813. [DOI] [PubMed] [Google Scholar]
- 90.Harvey A.J., Gable R.W., Baell J.B. A three-residue, continuous binding epitope peptidomimetic of ShK toxin as a Kv1.3 inhibitor. Bioorg. Med. Chem. Lett. 2005;15:3193–3196. doi: 10.1016/j.bmcl.2005.05.014. [DOI] [PubMed] [Google Scholar]