

Abstract
BACKGROUND
Prostaglandin E2 (PGE2) is an essential intrafollicular regulator of ovulation. In contrast with the one-gene, one-protein concept for synthesis of peptide signaling molecules, production and metabolism of bioactive PGE2 requires controlled expression of many proteins, correct subcellular localization of enzymes, coordinated PGE2 synthesis and metabolism, and prostaglandin transport in and out of cells to facilitate PGE2 action and degradation. Elevated intrafollicular PGE2 is required for successful ovulation, so disruption of PGE2 synthesis, metabolism or transport may yield effective contraceptive strategies.
METHODS
This review summarizes case reports and studies on ovulation inhibition in women and macaques treated with cyclooxygenase inhibitors published from 1987 to 2014. These findings are discussed in the context of studies describing levels of mRNA, protein, and activity of prostaglandin synthesis and metabolic enzymes as well as prostaglandin transporters in ovarian cells.
RESULTS
The ovulatory surge of LH regulates the expression of each component of the PGE2 synthesis-metabolism-transport pathway within the ovulatory follicle. Data from primary ovarian cells and cancer cell lines suggest that enzymes and transporters can cooperate to optimize bioactive PGE2 levels. Elevated intrafollicular PGE2 mediates key ovulatory events including cumulus expansion, follicle rupture and oocyte release. Inhibitors of the prostaglandin-endoperoxide synthase 2 (PTGS2) enzyme (also known as cyclooxygenase-2 or COX2) reduce ovulation rates in women. Studies in macaques show that PTGS2 inhibitors can reduce the rates of cumulus expansion, oocyte release, follicle rupture, oocyte nuclear maturation and fertilization. A PTGS2 inhibitor reduced pregnancy rates in breeding macaques when administered to simulate emergency contraception. However, PTGS2 inhibition did not prevent pregnancy in monkeys when administered to simulate monthly contraceptive use.
CONCLUSION
PTGS2 inhibitors alone may be suitable for use as emergency contraceptives. However, drugs of this class are unlikely to be effective as monthly contraceptives. Inhibitors of additional PGE2 synthesis enzymes or modulation of PGE2 metabolism or transport also hold potential for reducing follicular PGE2 and preventing ovulation. Approaches which target multiple components of the PGE2 synthesis-metabolism-transport pathway may be required to effectively block ovulation and lead to the development of novel contraceptive options for women. Therapies which target PGE2 may also impact disorders of the uterus and could also have benefits for women's health in addition to contraception.
Keywords: contraception; ovary; prostaglandin E2, cyclooxygenase-2; follicle
Introduction
There are many contraceptive options available for women today. However, none block ovulation as the primary method of pregnancy prevention. Selective inhibition of ovulation is an important target for the development of new contraceptives. Ovulation inhibition without alteration of ovarian steroid hormone synthesis would be particularly valuable. This approach would minimize or eliminate the side effects experienced by many women who use current hormonal contraceptives but still provide protection against undesired pregnancy.
Ovulation results from many functional and structural changes which occur within the dominant or Graafian follicle (reviewed in Espey and Lipner (1994) and Wassarman and Albertini (1994), Fig. 1). Following the ovulatory surge of LH, follicle cells synthesize progesterone in place of estrogen as the primary steroid hormone. The oocyte resumes meiosis and completes the first meiotic cell division, then arrests to await fertilization. Cumulus granulosa cells loosen their intercellular junctions and produce a novel extracellular matrix, leading to cumulus expansion and detachment of the cumulus-oocyte complex from the follicle wall. Mural granulosa cells and theca cells begin to differentiate into luteal cells; theca and other stromal cells invade the granulosa cells as the follicle undergoes luteinization. Well-controlled proteolysis promotes tissue remodeling throughout the luteinizing follicle, with extensive degradation of connective tissue selectively at the follicle apex. Removal of the ovarian surface epithelium and granulosa cells at the follicle apex contributes to formation of the ovulatory canal, follicle rupture and release of the cumulus-oocyte complex.
Figure 1.
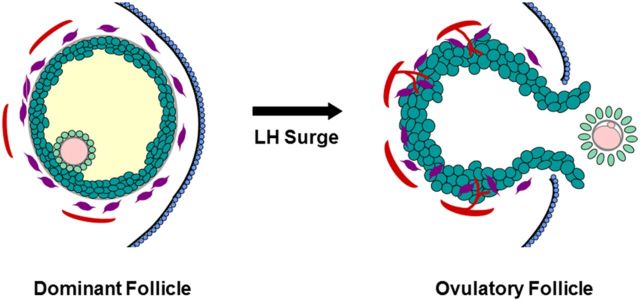
Structural changes in the dominant follicle during ovulation. Before the LH surge, the oocyte (pink) is surrounded by the zona pellucida (gray) and tight cumulus granulosa cells (light green); mural granulosa cells (dark green) surround the follicular fluid (yellow). Outside the granulosa cell basement membrane (gray), stromal components include theca cells (purple) and small vessels (red). The follicle apex is covered by the ovarian surface epithelium (blue). In response to the LH surge, the oocyte resumes meiosis and produces the first polar body. The cumulus expands and detaches from the mural granulosa cells. Luteinization includes hypertrophy of granulosa cells and invasion of theca cells and new vessels into the granulosa cell layer. A breach in the ovarian surface epithelium, underlying stroma, and mural granulosa cells results in rupture at the follicle apex. Mural granulosa cells protrude through the rupture site to form an ovulatory stigmata. The ovulatory canal permits release of the cumulus-oocyte complex (COC).
The ovulatory LH surge initiates these changes within the follicle. However, only theca and the outermost mural granulosa cells express large numbers of LH receptors (Peng et al., 1991; Yung et al., 2014). For ovulation to be successful, the LH signal must be conveyed within the remainder of the follicle by paracrine factors.
Prostaglandin E2 (PGE2) is an essential paracrine mediator of the LH surge. The LH surge stimulates an elevation in intrafollicular PGE2, which controls the timing of key ovulatory events (Richards, 1997). Rodents either treated with prostaglandin synthesis inhibitors or lacking expression of key prostaglandin synthesis enzymes experienced reduced rates of cumulus expansion, follicle rupture, oocyte maturation and oocyte release (Armstrong and Grinwich, 1972; Downs and Longo, 1982, 1983; Sogn et al., 1987; Lim et al., 1997; Mikuni et al., 1998; Takahashi et al., 2006). Subsequent studies identified PGE2 as the key ovulatory prostaglandin since mice lacking expression of the PGE2 receptor PTGER2 experienced ovulatory failure (Hizaki et al., 1999; Tilley et al., 1999). Administration of prostaglandin synthesis inhibitors also blocked ovulatory events in rabbits, cows and primates (Wallach et al., 1975a; De Silva and Reeves, 1985; Espey et al., 1986). The specific role of PGE2 in primate ovulation was demonstrated using an ablate-and-replace approach. Injection of a prostaglandin synthesis inhibitor directly into a dominant macaque follicle prevented cumulus expansion, follicle rupture and oocyte release in response to the LH surge; injection of the inhibitor concomitant with PGE2 restored these ovulatory events (Duffy and Stouffer, 2002).
The role of PGE2 to regulate progesterone production by the luteinizing follicle is somewhat controversial. PGE2 does promote progesterone production by mouse and rat follicles (Sogn et al., 1987; Elvin et al., 2000). In contrast, inhibition of PGE2 synthesis in macaque and bovine ovulatory follicles does not compromise progesterone production in response to the LH surge (Duffy and Stouffer, 2002; Peters et al., 2004; Kim et al., 2014). Clearly PGE2 mediates some, but not all, ovulatory actions of the LH surge in these larger mammals.
The essential role of prostaglandins in ovulation has been appreciated for decades (Tsafriri et al., 1972). A second cyclooxygenase enzyme (COX2, now called prostaglandin-endoperoxide synthase 2 (PTGS2)) was discovered in the early 1990s (Vane et al., 1998). Soon thereafter, this enzyme was determined to be the LH-stimulated cyclooxygenase responsible for PGE2 production by rat ovulatory follicles (Wong and Richards, 1991). Subsequent advances in molecular biology techniques have facilitated detailed investigation of cyclooxygenases, additional enzymes, and other key molecules involved in PGE2 synthesis, metabolism, and transport (Sirois et al., 2004).
PGE2 synthesis, metabolism and transport in the ovarian follicle
Prostaglandins are categorized as belonging to the 1, 2 or 3 series, based on the number of double bonds in the molecular structure (Fig. 2). Arachidonic acid is the most abundant prostaglandin precursor and results in formation of prostaglandins of the 2 series. Thus, prostaglandins of the 2 series (e.g. PGE2, PGF2α, PGD2, PGI2) predominate in mammalian cells. Prostaglandin synthesis can be divided into three steps: liberation of arachidonic acid from membrane phospholipids via a phospholipase, conversion of arachidonic acid to the short-lived intermediate PGH2 by a dual function enzyme with peroxidase and cyclooxygenase activities, and synthesis of bioactive prostaglandin from PGH2 by a specific prostaglandin synthase.
Figure 2.

Enzymes involved in PGE2 synthesis and metabolism. Arachidonic acid is cleaved from membrane phospholipids by a phospholipase A2 (PLA2). A cyclooxygenase (PTGS1 or PTGS2) converts arachidonic acid to prostaglandin H2 (PGH2). PGH2 can be converted to PGE2 by a PGE2 synthase (PTGES) or to PGF2α by aldo-keto reductase 1C3 (AKR1C3). PGE2 is metabolized to PGF2α by an enzyme with 9-keto reductase activity (AKR1C1 or AKR1C2). PGE2 is metabolized to 15-keto-PGE2 by prostaglandin dehydrogenase (HPGD). For each conversion, enzyme listed in boldface is the predominant form in the ovulatory follicle. Molecular structures are provided to highlight the conversion made by each enzyme. PGE1, PGE2 and PGE3 are distinguished by the number of double bonds in the molecular structure.
Phospholipase A2
Phospholipases cleave fatty acids from membrane phospholipids (Fig. 2). There are five major groups of phospholipases capable of cleaving fatty acids from the second position of membrane phospholipids (PLA2s): cytosolic PLA2, secreted PLA2, calcium-independent PLA2, lipoprotein-associated PLA2 and lysosomal PLA2. These five groups are distinguished from each other by their structural properties, function, and requirement for calcium as a co-factor for enzymatic activity (Murakami et al., 2011). Three forms of PLA2 can cleave arachidonic acid from the second position of membrane phospholipids: the cytosolic PLA2 (PLA2G4A) and two forms of secreted PLA2 (PLA2G2A and PLA2G5) (Table I) (Balsinde et al., 2002).
Table I.
Enzymes and transporters in PGE2 synthesis, metabolism and transport.
| Category | Gene name | Common name | Function | Other comments |
|---|---|---|---|---|
| Phospholipase A2 | PLA2G4A | cPLA2 | Cleavage of arachidonic acid from membrane phospholipids | Calcium promotes translocation from cytosol to intracellular membranes; predominates in granulosa cells |
| PLA2G2A | sPLA2IIA | Cleavage of arachidonic acid from membrane phospholipids | Can be activated by cPLA2 | |
| PLA2G5 | sPLA2V | Cleavage of arachidonic acid from membrane phospholipids | Can be activated by cPLA2 | |
| Cyclooxygenase; prostaglandin G/H synthase | PTGS1 | COX1 | Converts arachidonic acid to PGH2 | Constitutive expression |
| PTGS2 | COX2 | Converts arachidonic acid to PGH2 | Inducible expression; predominates in granulosa cells | |
| PTGS1 | COX3, COX1b | Converts arachidonic acid to PGH2 | Splice variant of COX1; may lack enzymatic activity | |
| Prostaglandin E synthase | PTGES | mPGES1 | Converts PGH2 to PGE2 | Associated with intracellular membranes; predominates in granulosa cells |
| PTGES2 | mPGES2 | Converts PGH2 to PGE2 | Associated with intracellular membranes | |
| PTGES3 | cPGES | Converts PGH2 to PGE2 | Cytosolic enzyme | |
| Aldo Keto reductases | AKR1C1 | 20α-HSD | Converts PGE2 to PGF2α | Minimal PGE-F isomerase activity in granulosa cells; significant 20α-HSD activity |
| AKR1C2 | PGE-F isomerase, 9-keto reductase | Converts PGE2 to PGF2α | Primary PGE-F isomerase activity in granulosa cells | |
| AKR1C3 | PGFS | Converts PGH2 to PGF2α | Primary PGF2α synthase in granulosa cells | |
| Prostaglandin dehydrogenase | HPGD | PGDH | Converts many prostaglandins to 15-keto metabolites | Prostaglandin inactivation |
| Prostaglandin transporters | SLCO2A1 | PGT, OATP | Facilitates prostaglandin movement across plasma membrane | Direction of prostaglandin movement is cell dependent |
| ABCC4 | MRP4 | Facilitates prostaglandin movement across plasma membrane | Prostaglandin movement out of cells predominates |
cPLA2, cytosolic phospholipase A2; sPLA2IIA, secreted phospholipase A2 group 2A; sPLA2V, secreted phospholipase A2 group 5; COX1, cyclooxygenase 1; PGH2, prostaglandin H2; COX2, cyclooxygenase 2; COX2, cyclooxygenase 3; COX1b, cyclooxygenase 1b; mPGES1, microsomal prostaglandin E synthase 1; PGE2, prostaglandin E2; mPGES2, microsomal prostaglandin E synthase 2; cPGES, cytosolic prostaglandin E synthase; 20α-HSD, 20α-hydroxysteroid dehydrogenase; PGF2α, prostaglandin F2α; PGE-F, conversion of prostaglandins of the E series to prostaglandins of the F series; PGFS, prostaglandin F synthase; PGDH, prostaglandin dehydrogenase; PGT, prostaglandin transporter; OATP, organic anion transporting polypeptide; MRP4, multiple drug resistance protein 4.
PLA2G4A has been identified as the key PLA2 for PGE2 synthesis within the ovulatory follicle. While expression of PLA2G2A and PLA2G5 in the ovulatory follicle is low and unchanging, PLA2G4A expression in granulosa cells increases in response to the ovulatory gonadotrophin surge in mice, rats, cows, monkeys and humans ((Kurusu et al., 1998a, b, 2012; Duffy et al., 2005a; Diouf et al., 2006; Wissing et al., 2014) and Fig. 3A, E and I). PLA2G4A and PLA2G5 expression is also reported in theca cells of mouse ovulatory follicles (Kurusu et al., 2012). Elevated follicular PGE2 synthesis correlates with increased PLA2G4A mRNA and protein in these species (Kurusu et al., 1998b, 2012; Duffy et al., 2005a; Diouf et al., 2006). Peak PLA2G4A activity in rat ovarian tissue (Bonney and Wilson, 1993; Ben-Shlomo et al., 1997) and monkey granulosa cells (Duffy et al., 2005a) occurs in the hours just prior to ovulation. PGE2 synthesis, ovulation rate, fertilization rate and overall reproductive success are compromised in rats given a PLA2G4A inhibitor (Kurusu et al., 1998a) and in mice lacking PLA2G4A expression (Bonventre et al., 1997; Song et al., 2002; Kurusu et al., 2012), supporting the conclusion that PLA2G4A contributes significantly to ovulatory events.
Figure 3.

PGE2 synthesis and metabolism enzyme mRNA, protein, and activity in primate ovulatory follicles. All cells and tissues were obtained after ovarian stimulation before (0 h) and 12, 24, or 36 h after administration of an ovulatory dose of hCG. Ovulation is anticipated 37–42 h after the ovulatory gonadotrophin stimulus, so cell/tissue collections at times 0–36 h after hCG span the ovulatory period. Levels of mRNA (A–D), protein (E–H) and activity (I and J) for PLA2G4A (A, E and I), PTGS2 (B and F), PTGES (E and G), and 15-hydroxyprostaglandin dehydrogenase (HPGD) (D, H and J) in monkey granulosa cells are shown. PGE2 (K) and 15-keto PGE2 metabolites (PGEM; L) levels in follicular fluid from monkey ovulatory follicles are shown. (M) Relative levels of PGE2 synthesis and PGE2 metabolism enzyme activities indicate that metabolism predominates early in the ovulatory period while synthesis predominates late in the ovulatory period. In (A)–(D), (G), and (I–L), groups with no common letters (a,b,c) are different by analysis of variance and appropriate post hoc analysis. In (E) and (F), an = antrum, gc = granulosa cell, st = stroma, asterisk indicates luteinizing granulosa cell layer, arrowhead indicates stromal (possibly theca) cell immunopositive for PTGS2. In (I), nd = not determined. See text for details; republished with permissions from Duffy and Stouffer (2001), Duffy et al. (2005a, b, c) and Duffy (2011).
Intracellular location influences PLA2 activity. In response to elevated intracellular calcium, PLA2G4A translocates from the cytosol to intracellular membranes, such as the nuclear envelope and endoplasmic reticulum, where this enzyme has access to arachidonic acid and is, therefore, catalytically active (Clark et al., 1991; Schievella et al., 1995; Stahelin et al., 2003). Crosstalk between PLA2G4A and secreted forms of PLA2 suggests that the activity of these enzymes may be interrelated. For example, secreted forms of PLA2 can play a permissive role in activation of PLA2G4A (Han et al., 2003). In other studies, PLA2G4A enhanced the activity of secreted PLA2s, perhaps by causing secreted forms of PLA2 to relocate to the plasma membrane, where these enzymes show maximal catalytic activity (Murakami et al., 1998). PLA2G4A expression and activity are clearly regulated by the LH surge in ovulatory follicles and directly involved in ovulatory events. In addition, PLA2G4A may promote the activity of secreted forms of PLA2 present in follicular cells to enhance overall PGE2 synthesis.
Cyclooxygenase
The cyclooxygenase enzyme possesses two distinct enzymatic activities. In the presence of a cyclooxygenase enzyme, arachidonic acid undergoes two structural conversions (Vane et al., 1998) (Fig. 2). The cyclooxygenase activity creates the ring at the midpoint of the arachidonic acid molecule that gives the prostaglandin its characteristic hairpin structure and results in the formation of a very unstable prostaglandin intermediary, PGG2. Very rapidly the peroxidase activity reduces a hydroperoxide group to a hydroxyl, forming PGH2. PGH2 is a transient molecule that is rapidly converted to more stable, bioactive prostaglandins by specific prostaglandin synthases.
Two forms of cyclooxygenase have been identified (Table I). PTGS1 (commonly referred to as COX1) and PTGS2 (commonly referred to as COX2) are products of different genes but share significant sequence homology and catalytic activities (reviewed in Vane et al. (1998)). PTGS1 is a constitutively-expressed enzyme found in many tissues. This enzyme is often referred to as the ‘housekeeping’ form of the cyclooxygenase enzyme and modulates aspects of routine functions such as platelet aggregation and gastric acid secretion. In contrast, PTGS2 expression increases rapidly in selected cells in response to specific stimuli, so PTGS2 is often referred to as the ‘inducible’ form of cyclooxygenase. PTGS2 is involved in mediating inflammation as well as essential events in reproduction, including ovulation and implantation. A cyclooxygenase enzyme originally identified as COX3 or COX1b has been reclassified as a splice variant of PTGS1 (Chandrasekharan et al., 2002). PTGS1 and PTGS2 proteins are located in the perinuclear area and are associated with the membranes of the nuclear envelope and endoplasmic reticulum (Morita et al., 1995), as are other key enzymes involved in prostaglandin synthesis.
The essential role of cyclooxygenase activity in ovulatory events was appreciated as early as the 1970s, when cyclooxygenase inhibitors such as indomethacin were shown to prevent follicle rupture and oocyte release in rats, cows and monkeys (Armstrong and Grinwich, 1972; Wallach et al., 1975a; De Silva and Reeves, 1985). The single form of cyclooxygenase known at that time (PTGS1) was present in the ovary, but enzyme protein levels did not correlate well with the rapid rise in follicular prostaglandin synthesis (Bauminger and Lindner, 1975; Hedin et al., 1987). After the identification of the second form of cyclooxygenase (PTGS2) in the 1980s, it was rapidly determined that PTGS2 was responsible for periovulatory prostaglandin production by the ovarian follicle (reviewed in Sirois et al. (2004)).
Details of PTGS2 expression in ovarian follicles are now well understood. PTGS2 expression by granulosa cells of pre-ovulatory follicles is low to nondetectable before the LH surge; PTGS2 mRNA and protein levels increase after the LH surge to reach peak levels near the expected time of ovulation in rodents, domestic animals, and monkeys ((Wong and Richards, 1991; Sirois, 1994; Sirois and Dore, 1997; Duffy and Stouffer, 2001) and Fig. 3B and F). PTGS2 has been detected in human granulosa cells (Narko et al., 1997), and gene array studies of human granulosa cells show a significant increase in PTGS2 levels after administration of an ovulatory dose of gonadotrophin (Wissing et al., 2014). Increased granulosa cell PTGS2 expression occurs before ovulation and precedes rising follicular PGE2 concentrations, leading some to conclude that PTGS2 catalyzes the rate-limiting step in follicular PGE2 production and determines the time of ovulation (Richards, 1997). PTGS2 is also expressed by theca cells of monkey ovulatory follicles (Duffy and Stouffer, 2001; Duffy et al., 2005b), though comparison of prostaglandin production by monkey granulosa cells and theca cells suggests that theca make a minimal contribution to prostaglandin concentrations in primate follicles (Duffy and Stouffer, 2003; Duffy et al., 2005b).
The development of drugs which preferentially inhibit PTGS2 over PTGS1 allowed selective inhibition of PTGS2-mediated events in vitro and in vivo (Fitzgerald and Patrono, 2001). Studies using PTGS2-selective inhibitors (Mikuni et al., 1998) as well as in mice lacking PTGS2 expression (Lim et al., 1997) confirmed that this form of cyclooxygenase is responsible for production of ovulatory PGE2 in mammalian follicles. Mice lacking PTGS2 expression or activity have reduced rates of cumulus expansion, ovulation and fertilization. These findings, along with studies of PTGS2 inhibitor administration in cows and monkeys, confirm that PTGS2 activity is required for key ovulatory events, including cumulus expansion, follicle rupture and oocyte maturation in preparation for fertilization (Peters et al., 2004; Hester et al., 2010; Duffy and VandeVoort, 2011).
In contrast, follicular PTGS1 is not required for ovulation. Early studies demonstrated the expression of the single known form of cyclooxygenase (PTGS1) in ovarian tissues (Hedin et al., 1987; Curry et al., 1990). After discovery of the second form of cyclooxygenase (PTGS2) and development of highly-selective antibodies, researchers confirmed that follicular PTGS1 expression was low and did not increase rapidly in response to the ovulatory LH surge (Sirois, 1994; Sirois and Dore, 1997; Duffy and Stouffer, 2001). Furthermore, mice lacking PTGS1 did not display ovulatory defects (Langenbach et al., 1995). Taken together, these findings support the importance of LH-induced PTGS2 expression for successful ovulation.
Prostaglandin E synthesis
Specific prostaglandin synthases convert PGH2 into bioactive prostaglandins (Table I). Production of bioactive PGE2 from PGH2 can occur via two pathways. A family of three prostaglandin E synthases converts PGH2 into PGE2. PGF2α (which is formed via several routes) can also be converted to PGE2 via an enzyme with PGE-F isomerase (9-keto reductase) activity.
The primary prostaglandin E synthase, PTGES, is expressed in the granulosa cells of ovulatory follicles of mice, cows, monkeys and humans ((Filion et al., 2001; Guan et al., 2001; Duffy et al., 2005b; Sun et al., 2006) and Fig. 3C and G). PTGES mRNA and protein levels in granulosa cells increase in response to the ovulatory gonadotrophin surge in cows and monkeys (Filion et al., 2001; Duffy et al., 2005b). hCG-stimulated increase in PTGES was also reported in human granulosa cells (Wissing et al., 2014). PTGES was detected in theca of monkeys and possibly cows (Filion et al., 2001; Duffy et al., 2005b) but not in theca of mouse ovulatory follicles (Sun et al., 2006). Other enzymes with prostaglandin E synthase activity (PTGES2 and PTGES3) are also expressed by granulosa cells of ovulatory follicles from several species, but levels do not change in response to the ovulatory gonadotrophin surge (Duffy et al., 2005b; Sun et al., 2006).
Co-transfection studies confirm that PTGES and PTGS2 are associated with intracellular membranes and preferentially co-operate for synthesis of PGE2 (Murakami et al., 2000). This contrasts with the cytosolic location of PTGES3, which typically co-operates with PTGS1 to synthesize prostaglandins (Murakami et al., 2002). PTGES2 is associated with intracellular membranes and can functionally couple with either PTGS1 or PTGS2 (Murakami et al., 2003a). However, PTGS1 and PTGES2 are typically described as ‘housekeeping’ enzymes, and expression levels do not change in response to provocative stimuli. In the monkey ovarian follicle, expression of PTGES2 does not increase after the ovulatory gonadotrophin surge, suggesting that PTGES is responsible for the rise in periovulatory PGE2 within the follicle ((Duffy and Stouffer, 2001; Duffy et al., 2005b) and Fig. 2).
Prostaglandin dehydrogenase
PGE2 is converted to biologically-inactive prostaglandin metabolites, with 15-hydroxyprostaglandin dehydrogenase (HPGD) catalyzing the rate-limiting step in prostaglandin inactivation (Murakami et al., 2011) (Table I; Fig. 2). PGE2 in the bloodstream likely has limited biological relevance (Legler et al., 2010). Prostaglandins, including PGE2, typically act in an autocrine or paracrine manner, near the site of synthesis. For this reason, local conversion of PGE2 to inactive prostaglandin metabolites is an important mechanism to control bioactive PGE2 levels at the cellular and tissue level.
Prostaglandin dehydrogenase activity was described in rabbit ovaries in 1987 (Schlegel et al., 1987). Male mice lacking HPGD expression are fertile (Coggins et al., 2002). However, female mice lacking HPGD expression are infertile (Coggins et al., 2002), suggesting a key role for this enzyme in female reproduction. More recently, HPGD has been suggested as a regulator of follicular PGE2 and ovulatory events. HPGD is expressed by bovine, monkey and human ovulatory follicles ((Duffy et al., 2005c; Sayasith et al., 2007; Thill et al., 2009; Peluffo et al., 2014) and Fig. 3D and H). The ovulatory gonadotrophin surge increased granulosa cell levels of HPGD mRNA and protein in monkey and bovine follicles, with increased HPGD expression also reported in bovine theca cells (Duffy et al., 2005c; Sayasith et al., 2007; Peluffo et al., 2014). In monkey granulosa cells, HPGD protein and activity levels peaked midway through the periovulatory interval, with low HPGD protein and activity present in follicles just before ovulation (Duffy et al., 2005c; Duffy, 2011). Follicular levels of prostaglandin E metabolites rise before levels of PGE2 (Duffy et al., 2005c), suggesting that PGE2 metabolism outpaces PGE2 synthesis immediately after the ovulatory gonadotrophin surge (Fig. 3K and L). Falling HPGD protein and activity correlate with peak follicular PGE2 levels just before ovulation ((Sirois, 1994; Duffy et al., 2005c) and Fig. 3M). Taken together, these data support the concept that HPGD activity is a key component regulating PGE2 levels in the ovulatory follicle.
The aldo-keto reductase family of enzymes
The aldo-keto reductase enzymes are structurally-similar enzymes which are products of different genes (Table I). High sequence homologies between AKR1C enzymes and the ability of these enzymes to act on multiple substrates have complicated the study of these enzymes. An analysis of the activities of human recombinant AKR1C enzymes assessed members of this family for prostaglandin synthesis and steroidogenesis activities (Nishizawa et al., 2000). AKR1C2 has primarily 9-keto prostaglandin reductase/isomerase activity, which interconverts PGE2 and PGF2α (Fig. 2). AKR1C3 is the classical PGF synthase (Suzuki-Yamamoto et al., 1999). This enzyme converts PGH2 and PGD2 to PGF2α and 9α,11β-PGF2α, respectively; both forms of PGF2α can activate the PGF2α receptor (Sugimoto et al., 1994). AKR1C enzymes also possess steroidogenic activities. For example, AKR1C1 has limited 9-keto prostaglandin reductase activity and is thought to be primarily a 20α-hydroxysteroid dehydrogenase, which converts progesterone to a less active metabolite.
Activity of AKR1C family members (either 9-keto prostaglandin reductase or 20α-hydroxysteroid dehydrogenase activities) has been reported in ovaries of sheep, rats, horses and monkeys (Murdoch and Farris, 1988; Iwata et al., 1990; Brown et al., 2006; Nanjidsuren et al., 2011). However, significant differences exist in the expression pattern and role of these enzymes in different species. For example, AKR1C1 is highly expressed in rat ovaries, and the 20α-hydroxysteroid dehydrogenase activity of this enzyme is involved in regulation of luteolysis (Stocco et al., 2000). Luteolysis is controlled by fundamentally different mechanisms in rodents and primates; AKR1C1 does not play this role in human ovaries. Monkey ovaries do contain AKR1C1 mRNA and protein (Nanjidsuren et al., 2011). Granulosa cells have been identified as the primary site of AKR1C1/AKR1C2 expression in monkey ovaries (Dozier et al., 2008). In horse ovulatory follicles, both granulosa and theca cells express a closely-related equine enzyme, AKR1C23 (Brown et al., 2006). Use of selective inhibitors confirmed that both monkey and human granulosa cells contain primarily AKR1C2 activity, and both mRNA and activity increased in response to the ovulatory LH surge (Dozier et al., 2008). Further studies using radiolabeled prostaglandin precursors and selective inhibitors demonstrated that granulosa cell AKR1C2 has unidirectional activity to convert PGE2 to PGF2α ((Dozier et al., 2008) and Fig. 2). However, the significance of this conversion in ovulatory events is unknown.
Studies performed in the 1970s and 1980s suggested that both PGE2 and PGF2α mediated ovulatory events within the ovarian follicle. Follicular levels of both prostaglandins rise in response to the LH surge (Sirois, 1994; Sirois and Dore, 1997; Duffy and Stouffer, 2001; Duffy et al., 2005c), and co-administration of either prostaglandin during cyclooxygenase inhibition restored ovulation in species as diverse as rats, sheep, and monkeys (Wallach et al., 1975a, b; Murdoch et al., 1986; Sogn et al., 1987; Janson et al., 1988). Pure preparations of prostaglandins were difficult to obtain during this era, and it is likely that more than one prostaglandin was replaced during cyclooxygenase inhibition in these early studies. PGF2α receptors are expressed by follicular cells in many species (Ristimaki et al., 1997; Sayasith et al., 2006; Bridges and Fortune, 2007; Kim et al., 2015), but PGF2α receptors capable of signal transduction have not been reported in granulosa cells of any species. Furthermore, normal ovulatory events were observed in mice lacking expression of the PGF2α receptor (Sugimoto et al., 1997). Taken together, these data support the conclusion that PGF2α does not regulate essential ovulatory events in mammals, including monkeys and women. PGF2α accumulation in the follicle may be the result of PGE2 metabolism via the AKR1C2 enzyme. Since monkey granulosa cell 9-keto prostaglandin reductase activity increases in response to the ovulatory LH surge (Dozier et al., 2008), AKR1C2 may serve a metabolic role, converting ovulatory PGE2 into other prostaglandins.
PGE2 transport
In addition to enzymes involved in prostaglandin synthesis and metabolism, prostaglandin transporters have been implicated in the control of prostaglandin bioactivity. As described above, prostaglandins are synthesized intracellularly. Prostaglandin receptors can be located on the plasma membrane (Narumiya et al., 1999), in which case prostaglandins in the extracellular fluid have access to the ligand binding domain of the receptor. However, prostaglandin receptors can also be located on intracellular membranes (Bhattacharya et al., 1999), in which case cytoplasmic prostaglandins have access to the ligand binding domain. Prostaglandins are converted to inactive metabolites by the cytoplasmic enzyme HPGD as detailed above. For these reasons, prostaglandin movement across the plasma membrane has important implications for both bioactivity and inactivation.
At physiological pH, bioactive prostaglandins are negatively charged. Prostaglandins are somewhat lipid-soluble and can diffuse out of the cell of synthesis in response to both a concentration and an electrochemical gradient, although with limited efficiency (Schuster, 2002; Chi et al., 2006). Very important to the movement of prostaglandins are two families of plasma membrane transporters, the multiple drug resistance proteins (MRPs) and the organic anion transporting polypeptides (OATPs), which have been shown to facilitate prostaglandin movement across plasma membranes.
MRPs likely assist with prostaglandin efflux, or exit, from the cell of synthesis (Reid et al., 2003) (Table I). MRP4 (gene name ABCC4) is a transmembrane, ATP-dependent pump which moves many small molecules, including PGE2, from the intracellular to the extracellular side of the plasma membrane. Information on follicular expression of MRPs and, specifically, the ABCC4 transporter is very limited. ABCC4 mRNA increased after the ovulatory gonadotrophin surge in human granulosa cells (Wissing et al., 2014), so this transporter may be available to assist with PGE2 efflux from granulosa cells during the ovulatory interval.
Prostaglandins can also move across the plasma membrane via a prostaglandin transporter (PTG, a member of the OATP family), now referred to by the gene name SLCO2A1 (Lu et al., 1996) (Table I). This protein is predicted to have 12-membrane spanning domains and functions as a lactate/prostaglandin exchanger (Chan et al., 2002). The direction of prostaglandin movement is likely sensitive to lactate levels, such that an elevated rate of glycolysis enhances prostaglandin uptake by cells. Prostaglandin movement into and out of cells via this transporter have both been reported (Schuster, 2002). However, most recent studies indicate that SLCO2A1 preferentially moves prostaglandins from the extracellular fluid into the cytosol. Cells transfected with SLCO2A1 showed an increased rate of prostaglandin influx (Nomura et al., 2004; Holla et al., 2008). Importantly, SLCO2A1 has been shown to enhance PGE2 influx into cells which express HPGD. Absence of SLCO2A1 expression or SLCO2A1 blockade led to accumulation of PGE2 extracellularly and decreased conversion of PGE2 into 15-keto metabolites (Nomura et al., 2004; Chi et al., 2006; Holla et al., 2008; Kochel and Fulton, 2015). These and similar studies support the concept that SLCO2A1 functions primarily in prostaglandin influx and facilitates prostaglandin metabolism.
Expression of SLCO2A1 has been examined in cells of the ovulatory follicle. The ovulatory gonadotrophin surge increased SLCO2A1 in granulosa cells but decreased SLCO2A1 in theca cells obtained from bovine ovulatory follicles (Bridges and Fortune, 2007). In a recent study comparing gene expression in human granulosa cells before and 36 h after an ovulatory dose of hCG, mRNA levels of SLCO2A1 were elevated 27-fold in response to hCG compared with pre-hCG levels (Wissing et al., 2014), but SLCO2A1 levels were not different in cumulus granulosa cells collected before and after hCG administration (Yerushalmi et al., 2014). Importantly, protein expression for this prostaglandin transporter by follicular granulosa cells has not been confirmed in any mammalian species, nor has the direction of prostaglandin transport via SLCO2A1 in granulosa cells been directly examined.
Coordinating PGE2 synthesis, metabolism and transport
Control of expression and activity of individual enzymes and transporters is essential for effective regulation of cellular responses to PGE2. Additional features, including co-localization of enzymes within the cell, coordinated expression of enzymes and transporters, and coordinated regulation of enzyme activity, can further enhance changes in PGE2 activity within target tissues. Rapid increases in PGE2 synthesis and responses are essential for successful ovulation and in some cases have been examined in the ovarian follicle. However, these relationships are best understood in the context of cancer models.
Enzymes involved in PGE2 synthesis are located in the same region of the cell, which leads to functional coupling of these enzymes to preferentially produce PGE2 over other eicosanoids (Fig. 4). PLA2G4A, PTGS2 and PTGES are most often described as located in the endoplasmic reticulum or nuclear envelope. PLA2G4A translocates from the cytosol to these membranes in response to rising intracellular calcium (Clark et al., 1991; Schievella et al., 1995; Stahelin et al., 2003). In contrast, PTGS2 is an integral membrane protein found in the nuclear envelope and endoplasmic reticulum (Spencer et al., 1998). Immunofluorescence and other techniques consistently show PTGES located in or near the endoplasmic reticulum/nuclear envelope, but the mechanism by which PTGES associates specifically with intracellular membranes is unknown (Murakami et al., 2003b).
Figure 4.
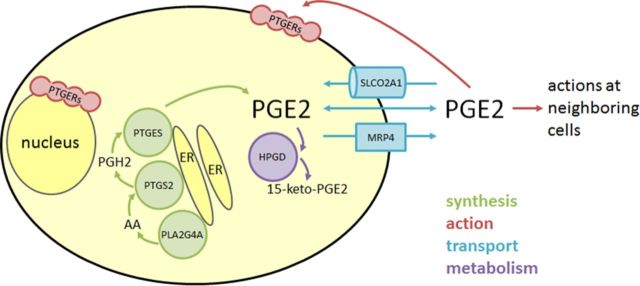
PGE2 synthesis, action, transport and metabolism in granulosa cells. PGE2 is synthesized by the enzymes PLA2G4A, PTGS2, and PTGES which are associated with intracellular membranes including the endoplasmic reticulum (ER). Intermediates in PGE2 synthesis include arachidonic acid (AA) and PGH2. Functional PGE2 receptors (PTGERs) can be located in the plasma membrane or nuclear envelope, so both intracellular and extracellular PGE2 can bind to receptors and initiate signal transduction. PGE2 can exit and enter a cell via passive diffusion through the plasma membrane or facilitated diffusion via transporters such as SLCO2A1 and MRP4. Bioactive PGE2 is converted to the inactive PGE metabolite 15-keto-PGE2 through the activity of the cytoplasmic enzyme HPGD.
The membranes of the endoplasmic reticulum and nuclear envelope are the source of the phospholipid substrate for PLA2G4A activity. PLA2G4A cleaves arachidonic acid from phospholipids in these intracellular membranes and generates a locally-high concentration of arachidonic acid in the perinuclear region to serve as a substrate for PTGS2 activity. Similarly, PGH2 produced by PTGS2 is at high concentration near the endoplasmic reticulum and nuclear envelope, so PTGES can rapidly and efficiently convert this common prostaglandin precursor into PGE2. In this way, locally-high concentrations of precursors are available to these specific enzymes and may contribute to preferential production of PGE2 over other eicosanoids.
PGE2 transport and metabolism may also co-operate synergistically to efficiently remove PGE2 from the extracellular fluid. For example, Nomura and colleagues demonstrated that conversion of PGE2 to inactive 15-keto metabolites was most efficient when intact cells expressed both the prostaglandin transporter SLCO2A1 and metabolic enzyme HPGD (Nomura et al., 2004). These and similar studies highlight the potential for functional coupling between transport and metabolism to rapidly decrease PGE2 levels (Kochel and Fulton, 2015).
Simultaneously increasing or decreasing expression or activity of multiple enzymes and transporters can rapidly regulate levels of PGE2. This is best understood in cancers, where high levels of PGE2 promote tumor growth and metastasis (reviewed in Greenhough et al. (2009)). Coordinated increased expression of PLA2G4 and PTGS2 has been observed in many cancers and cancer cell lines; similarly, PTGS2 expression has been shown to be decreased when PLA2G4 expression is ablated (reviewed in Murakami et al. (2011)). In mice lacking PLA2G4A, brain levels of PTGS2, PTGS2 protein, and PGE2 were significantly reduced, providing evidence that PLA2G4A activity enhances PTGS2 activity (Bosetti and Weerasinghe, 2003). Coordinated regulation of PTGS2 and HPGD may also maintain elevated levels of bioactive prostaglandins (Tai et al., 2006). HPGD expression and activity often decrease in parallel with increased expression of PTGS2 and other PGE2 synthesis enzymes in cancer cells (Backlund et al., 2005; Tai et al., 2007; Liu et al., 2008). When this occurs, elevated PGE2 synthesis, coupled with decreased PGE2 metabolism, further elevates PGE2 levels, a situation which parallels the changing expression of key enzymes which regulate PGE2 levels within the ovulatory follicle.
Coordinated regulation of enzyme mRNA, protein, and activity may delay the rise in follicular PGE2 levels, leading to a long ovulatory interval, as seen in large mammalian species. The PGE2 concentration in follicular fluid increases rapidly late in the ovulatory interval, reaching 0.1–1 µM just before ovulation in species with longer ovulatory intervals, including primates, horses, and cows ((Liu et al., 1997; Sirois and Dore, 1997; Duffy et al., 2005c) and Fig. 3K). In monkeys, follicle levels of PLA2G4A, PTGS2, and PTGES rise after the LH surge and remain at high levels throughout the ovulatory interval (Duffy et al., 2005a, b; Peluffo et al., 2014). While granulosa cell HPGD mRNA, protein, and activity are high immediately after the LH surge, HPGD expression and activity decrease to low/nondetectable levels at the time ovulation (Duffy et al., 2005c; Duffy, 2011). Reciprocal expression of PGE2 synthesis enzymes and HPGD has also been observed in granulosa cells of horses (Sirois and Dore, 1997; Filion et al., 2001; Diouf et al., 2006; Sayasith et al., 2007) and is similar to observations made in cultured cell lines and cancers (Tai et al., 2006).
Several mechanisms of cooperation of enzyme expression and activity may contribute to control of PGE2 in ovulatory follicles. We have proposed that elevated HPGD activity early in the ovulatory interval delays the rise in follicular PGE2 levels, thereby lengthening the interval between the LH surge and ovulation ((Duffy et al., 2005c) and Fig. 3M). An alternative was suggested by Sirois and colleagues, who demonstrated that transcriptional regulators present prior to the LH surge can delay PTGS2 expression in granulosa cells of horses (Liu et al., 2000). Kurusu and colleagues demonstrated that PLA2G4A activity was required for induction of granulosa cell PTGS2 expression in rodents (Kurusu et al., 2012). However, PLA2G4A activity was measured only very late in the primate ovulatory interval (Fig. 3I), so a role for PLA2G4A activity to increase PTGS2 activity may be a feature of the ovulatory process in species with relatively short (12–16 h) ovulatory intervals.
Cyclooxygenase inhibitors as potential contraceptives
Luteinized unruptured follicles (LUFs) have long been associated with ovulatory dysfunction and infertility (reviewed in Smith et al. (1996)). By the 1980s, clinicians linked the use of non-steroidal anti-inflammatory drugs (NSAIDs, which inhibit cyclooxygenase activity) to decreased follicular prostaglandin production and anovulation (Killick and Elstein, 1987; Priddy et al., 1989, 1990; Smith et al., 1996). Case reports described women who failed to conceive while taking NSAIDs for rheumatological and other conditions (Akil et al., 1996; Mendonca et al., 2000). These women were counseled to discontinue NSAID use, and the majority conceived without additional medical intervention (Akil et al., 1996; Mendonca et al., 2000). These instances of reversible infertility were attributed to ovulation failure due to cyclooxygenase inhibition.
These observations have provided a foundation for studies oriented towards contraceptive development, focusing on pharmacologic inhibition of prostaglandin production in women (recently reviewed in Weiss and Gandhi (2014)) and macaques. Humans and macaques share many features of the ovulatory cascade, such as maturation of a single dominant follicle and release of a single oocyte during a true menstrual cycle. Importantly, women and macaques experience similar timing of ovulatory events, with ovulation expected 37–42 h after the endogenous ovulatory surge of LH or administration of hCG as an ovulatory gonadotrophin stimulus.
As noted earlier, prostaglandin-mediated ovulatory changes include cumulus expansion, oocyte maturation in preparation for fertilization, follicle rupture and oocyte release into the oviduct. Importantly, disruption of prostaglandin synthesis does not alter the timing or magnitude of the ovulatory surge of LH (Pall et al., 2001; Hester et al., 2010; Jesam et al., 2010, 2014). While elevated intrafollicular progesterone is required for successful ovulation (Hibbert et al., 1996), prostaglandins do not appear to regulate ovarian steroidogenesis, including progesterone synthesis, in women and monkeys (Pall et al., 2001; Duffy and Stouffer, 2002; Bata et al., 2006; Hester et al., 2010; Jesam et al., 2010; Edelman et al., 2013; Kim et al., 2014). Inhibition of prostaglandin synthesis can reduce or prevent structural luteinization of the ovarian follicle (Duffy and Stouffer, 2002; Hester et al., 2010; Kim et al., 2014), but this does not significantly impact steroid hormone synthesis by the follicle/young corpus luteum. Importantly, disruption of prostaglandin synthesis can result in normal serum LH and steroid hormone levels, despite ovulation failure (Duffy and Stouffer, 2002; Hester et al., 2010; Kim et al., 2014).
Studies to evaluate ovulatory success or failure in macaques can include use of invasive techniques. For example, the ovary bearing the ovulatory follicle can be removed, and histological sections can be directly examined for the presence of an oocyte to confirm ovulation or anovulation. Studies in macaques have demonstrated that prostaglandin synthesis inhibitor administration can result in failure of cumulus expansion, failure of follicle rupture, or dysfunctional release of the oocyte into the stroma surrounding the ovulatory follicle (Hester et al., 2010).
Studies evaluating ovulatory success or failure in women must rely on indirect assessments, such as ovarian ultrasonography. Ultrasound can be used to monitor the growth of the dominant follicle. A significant decrease in the diameter of the leading follicle reflects the loss of follicular fluid when the follicle ruptures or collapses. It is important to note that the condition of the cumulus and location of the oocyte cannot currently be determined by ultrasound. However, the decrease in follicular size based on ultrasound has been used as presumptive indication of ovulation.
Prostaglandin synthesis inhibitors have been considered for use as both emergency contraceptives and monthly contraceptives. During typical contraceptive use, the day of the LH surge is unknown relative to the day of intercourse. Prostaglandin synthesis must be inhibited during the days immediately following the LH surge in order to prevent ovulation. The period during which intercourse can result in pregnancy is the 5–6 days leading up to and including the day of ovulation for both women and macaques (Dukelow and Bruggemann, 1979; Behboodi et al., 1991; Wilcox et al., 1995). This is due, in part, to the period of sperm viability within the female reproductive tract, which is estimated at 5–6 days (Wilcox et al., 1995). Therefore, a delay of ovulation for 5–6 days should prevent interaction between the oocyte and viable sperm and, therefore, embryo formation after a single instance of intercourse. For this reason, a 5–6 day course of treatment is often used as a model for emergency contraception. Models of monthly contraception have included either daily drug administration or treatment during menstrual cycle days 5–22. Treatment during this window will include the 5–6 critical days for ovulation inhibition for women with a cycle length within the normal range of 21–35 days (Office on Women's Health, 2009).
Human studies
In an early study, Killick and Elstein assessed the importance of cyclooxygenase activity for successful ovulation in women (Killick and Elstein, 1987). The non-selective cyclooxygenase inhibitors indomethacin and azapropazone were given orally to female volunteers for a total of 5 days during natural menstrual cycles, beginning when the follicular diameter reached 16 mm; hCG was given to initiate ovulatory events when the leading follicle reach 18 mm in diameter (Table II). A follicle which displayed a rapid decrease in diameter within 48 h of hCG was considered ovulatory. A follicle with an echo-free appearance, which continued to increase in diameter at subsequent measurements, was considered to be a LUF. Indomethacin and, to a lesser extent, azapropazone increased the percentage of LUF cycles experienced by women. A paired analysis of serum progesterone levels did reveal that individual women experienced slightly lower progesterone in the days immediately following hCG administration during LUF cycles when compared with cycles where timely follicle collapse was consistent with ovulation. However, overall serum progesterone was not different between placebo and treatment cycles at any time after hCG administration (Killick and Elstein, 1987). This study provided early evidence that inhibition of cyclooxygenase activity may reduce rates of ovulation in women.
Table II.
Ovulation inhibition with cyclooxygenase inhibitors: studies in women.
| COX inhibitor | Target enzyme | Major outcome measure (s) (success rate) | Additional outcome measures | Contraceptive model (monthly, emergency) | Lead author and year of publication |
|---|---|---|---|---|---|
| Indomethacin | PTGS1 and PTGS2 | Formation of LUF with indomethacin (88%) and placebo (11%) | Serum progesterone | N/A | Killick, 1987 |
| Azapropazone | PTGS1 and PTGS2 | Formation of LUF with azapropazone (50%) and placebo (11%) | Serum progesterone | N/A | Killick, 1987 |
| Indomethacin | PTGS1 and PTGS2 | Delay of follicle collapse (83%) | Serum FSH, LH, estrogen, progesterone | N/A | Athanasiou, 1996 |
| Rofecoxib | PTGS2 | Delay of follicle collapse at least 48 h (67%) | Serum FSH, LH, estrogen, progesterone | N/A | Pall, 2001 |
| Ibuprofen | PTGS1 and PTGS2 | Interval between LH surge and follicle collapse was longer with ibuprofen (27%) compared with placebo | Serum LH, progesterone | N/A | Uhler, 2001 |
| Meloxicam | PTGS2 | Follicle collapse was delayed an average of 5 days | Serum progesterone | N/A | Bata, 2006 |
| Meloxicam | PTGS2 | Dysfunctional ovulation at low (50%) and high (91%) doses of meloxicam | N/A | Emergency contraception | Jesam, 2010 |
| Celecoxib | PTGS2 | Dysfunction ovulation when celecoxib administration began before (25%) and after (20%) LH surge | Serum LH, estrogen, progesterone | Emergency contraception | Edelman, 2013 |
| Meloxicam | PTGS2 | Dysfunctional ovulation with meloxicam+levonorgesterol (88%) and levonorgesterol only (66%) | Serum progesterone | Emergency contraception | Massai, 2007 |
| Meloxicam | PTGS2 | Dysfunctional ovulation at low (55%) and high (78%) doses of meloxicam when administered on cycle days 5–22 | Serum LH, estrogen, progesterone | Monthly contraception | Jesam, 2014 |
LUF, luteinized unruptured follicle; N/A, not applicable.
The discovery of PTGS2 and identification of this enzyme as the key cyclooxygenase in the ovulatory follicle spurred testing of cyclooxygenase inhibitors with high selectivity for PTGS2 over PTGS1. The PTGS2 selective inhibitors rofecoxib (Pall et al., 2001), meloxicam (Bata et al., 2006; Jesam et al., 2010) and celecoxib (Edelman et al., 2013) have been tested for their ability to delay follicle collapse in women experiencing natural menstrual cycles. Rofecoxib administration began when the dominant follicle reached 14–16 mm diameter and continued for a total of 9 days (Pall et al., 2001). The mean day of follicle collapse was Day 1 after peak LH in placebo cycles. Of women receiving rofecoxib, 67% experienced delayed follicle collapse on average at Day 4 after peak LH, with 33% of women experiencing follicle collapse within 48 h after peak LH, similar to women receiving placebo (Fig. 5). In a similar study, meloxicam treatment began about 2 days before peak LH and continued for a total of 5 days of treatment (Bata et al., 2006). Follicular diameter remained elevated for at least 5 days in women receiving meloxicam, but follicle collapse occurred within 2 days of the estimated day of peak LH in women receiving placebo. A subsequent study by this group confirmed that the maximal therapeutic dose of meloxicam (30 mg/day for 5 days) was more likely to result in delayed or absent follicle collapse than a lower dose (15 mg/day for 5 days) (Jesam et al., 2010). Celecoxib treatment resulted in high rates of apparently normal ovulatory events, regardless of whether celecoxib administration was initiated before (70%) or after (75%) the LH surge (Edelman et al., 2013). Serum levels of FSH, LH, estrogen, and progesterone were not different between placebo and PTGS2 inhibitor-treatment cycles (Pall et al., 2001; Bata et al., 2006; Jesam et al., 2010; Edelman et al., 2013). These studies indicate that inhibition of ovulation was most successful when PTGS2 inhibitor administration was initiated before the LH surge and when using a maximal therapeutic dose of PTGS2 inhibitor. PTGS2 inhibitors consistently did not alter serum steroids or gonadotrophins. While a few studies of PTGS2 inhibitors achieved reduction in ovulation rates comparable to available steroidal methods of emergency contraception (Glasier, 2013), PTGS2 inhibitors alone did not consistently block ovulation with contraceptive efficacy.
Figure 5.

Ultrasound evaluation of ovulatory failure in women. An ovarian follicle in a woman receiving placebo was at 19 mm diameter on the day of the LH peak (A) and showed decreased follicular diameter (follicle collapse; consistent with follicle rupture) on the day after peak LH (B). An ovarian follicle in a woman receiving the PTGS2 inhibitor rofecoxib was at 22 mm diameter on the day of peak LH (C) and was at 50 mm diameter on Day 10 after peak LH (D). With PTGS2 inhibitor treatment, the follicle continued to increase in diameter after the LH surge (no follicle collapse; failure of follicle rupture). In these ultrasound images, ovarian follicles are black circles, with dotted lines to indicate measurement of follicular diameters. All panels are at approximately the same magnification. Republished with permissions from Pall et al. (2001).
In contrast to the PTGS2 inhibitors discussed above, steroidal contraceptives utilize different cellular mechanisms to block ovulatory events (ESHRE Capri Workshop Group, 2015). Massai and colleagues investigated whether a PTGS2 inhibitor could enhance the ability of the progestin levonorgestrel to reduce rates of ovulation in a model of emergency contraception (Massai et al., 2007). As an emergency contraceptive, levonorgestrel most effectively prevents pregnancy when administered before the onset of the LH surge (Gemzell-Danielsson et al., 2013). PTGS2 inhibitors block prostaglandin production, which occurs after the LH surge. Therefore, it is reasonable to suggest that the combination of levonorgestrel and a PTGS2 inhibitor may block ovulation more reliably than either drug alone. While 50% of women receiving levonorgestrel experienced follicle collapse within 2 days of the LH surge, consistent with normal ovulation, only 19% of women receiving levonorgestrel plus meloxicam experienced normal ovulatory events (Massai et al., 2007). Although preliminary, these data provide support for the utility of PTGS2 inhibitors as a component of a multi-drug emergency contraceptive.
Finally, Jesam and colleagues (Jesam et al., 2014) administered meloxicam on Days 5–22 of the menstrual cycle to simulate use as a monthly contraceptive. Meloxicam at low and high therapeutic doses resulted in either delayed ovulation or formation of a LUF at rates of 55 and 78%, respectively. While serum levels of LH were within the normal range, progesterone levels were lower in women experiencing delayed ovulation or LUFs when compared with progesterone levels in women experiencing normal ovulation. This study did not include a placebo group to obtain normal values for ovulatory success or serum hormone levels. However, the longer time of PTGS2 inhibitor administration did not appear to improve ovulation inhibition when compared with the shorter treatment intervals described in previous reports. The authors of this study concluded that ovulation in 22% of cycles is unlikely to prevent pregnancies with an acceptable efficacy for contraceptive use.
Monkey studies
While ultrasound examinations of women can provide indirect evidence of ovulation, studies conducted in macaques are able to directly confirm ovulatory success and failure. The ability to remove the ovary bearing the dominant follicle at controlled times after the ovulatory gonadotrophin surge, serially section the entire ovary, and examine each tissue section for an oocyte and evidence of follicle rupture to the exterior of the ovary provides a direct measure of ovulatory success or failure. Macaque studies can also provide information regarding the mechanism by which a cyclooxygenase inhibitor delays or prevents ovulation. Finally, macaques in breeding situations provide an outstanding preclinical model for pregnancy studies.
Studies conducted in the 1970s by Wallach and colleagues demonstrated that systemic administration of the general cyclooxygenase inhibitor indomethacin could decrease ovulation in macaques ((Wallach et al., 1975a) and Table III). More recent studies examining the ability of cyclooxygenase inhibitors to block ovulatory events in primates used direct ovarian administration to examine only local actions of prostaglandins on ovulatory events. Intrafollicular injection of vehicle or indomethacin was performed the day before or the day of the LH surge; ovaries removed 3 days later were evaluated for histological evidence of follicle rupture and presence/absence of an oocyte within the ovulatory follicle (Duffy and Stouffer, 2002). In this study, all vehicle-injected follicles were devoid of oocytes and showed evidence of successful follicle rupture (Duffy and Stouffer, 2002). In contrast, 50% of indomethacin-injected follicles contained oocytes, even though many of these ovaries showed evidence of small breeches in the ovarian surface consistent with follicle rupture (Duffy and Stouffer, 2002). In a subsequent study, intrafollicular administration of vehicle or indomethacin was coupled with controlled administration of gonadotrophins to permit removal of ovaries precisely 48 h after the ovulatory gonadotrophin stimulus, or 8 h after the expected time of ovulation (Kim et al., 2014). In this study, all vehicle-injected follicles lacked oocytes and showed clear evidence of follicle rupture. In contrast, all indomethacin-injected follicles contained oocytes surrounded by unexpanded cumulus (Kim et al., 2014). In both studies, injection of indomethacin plus PGE2 restored follicle rupture, oocyte release, and presumably cumulus expansion, confirming a key role for locally-synthesized, cyclooxygenase-derived PGE2 in these ovulatory events (Duffy and Stouffer, 2002; Kim et al., 2014).
Table III.
Ovulation inhibition and pregnancy prevention with cyclooxygenase inhibitors: studies in monkeys.
| COX inhibitor | Target enzyme | Major outcome measure (s) (success rate) | Additional outcome measures | Contraceptive model (monthly, emergency) | Lead author and year of publication |
|---|---|---|---|---|---|
| Indomethacin | PTGS1 and PTGS2 | Prevention of ovulation sites with indomethacin | Serum estrogen | N/A | Wallach, 1975 |
| Indomethacin | PTGS1 and PTGS2 | Prevention of follicle rupture (50%) and oocyte release (50%) with indomethacin | Serum LH, estrogen, progesterone | N/A | Duffy, 2001 |
| Meloxicam | PTGS2 | Prevention of oocyte release (73%) and cumulus expansion (36%) with meloxicam | Serum LH, estrogen, progesterone | Emergency contraception | Hester, 2010 |
| Indomethacin | PTGS1 and PTGS2 | Prevention of follicle rupture (100%), cumulus expansion (100%), and oocyte release (100%) with indomethacin | Ovarian steroidogenic enzymes; serum estrogen, progesterone | N/A | Kim, 2014 |
| Meloxicam | PTGS2 | Meloxicam prevented 80% of anticipated pregnancies | N/A | Emergency contraception | McMann, 2013 |
| Meloxicam | PTGS2 | No prevention of pregnancy with meloxicam every day administration | N/A | Monthly contraception | McMann, 2013 |
| Meloxicam | PTGS2 | No prevention of pregnancy with meloxicam administration on cycle days 5–22 | N/A | Monthly contraception | McMann, 2013 |
| Meloxicam | PTGS2 | No prevention of pregnancy with meloxicam administration for 5 days periovulatory | N/A | Monthly contraception | McMann, 2013 |
N/A, not applicable.
The potential utility of the PTGS2 inhibitor meloxicam as an emergency contraceptive was also investigated in the monkey model. Oral meloxicam administration for 5 days around the time of ovulation resulted in 73% of oocytes remaining trapped within a luteinizing follicle 2 days after the predicted day of ovulation (Hester et al., 2010), evidence of ovulatory failure. Evaluation of histological sections indicated that retention of the oocyte was due to a failure of cumulus expansion, follicle rupture, or both. Importantly, there was no difference between placebo and meloxicam treatment cycles in terms of serum levels of LH, estradiol or progesterone (Hester et al., 2010).
The paradigm of 5 day oral meloxicam administration was used to evaluate contraceptive efficacy in monkeys after a single instance of breeding to simulate emergency contraceptive use (McCann et al., 2013). Pregnancies were observed in 6.5% of meloxicam treatment menstrual cycles, compared with pregnancies in 33.3% of placebo menstrual cycles, resulting in a contraceptive efficacy of 80% in this preclinical model.
Timing of meloxicam administration relative to the LH surge impacted ovulatory events. For example, oocyte release failed in 86% of monkey ovaries when oral meloxicam administration was initiated before the LH surge; oocyte release failed in just 50% of ovaries when meloxicam administration began on or after the anticipated day of the LH surge (Hester et al., 2010). These findings suggest that PTGS2 inhibitors can sufficiently delay or prevent ovulation only when administered within a relatively narrow and specific window of time.
Finally, oral meloxicam was investigated as a potential monthly contraceptive in breeding monkeys (McCann et al., 2013). Three periods of meloxicam administration were utilized: 5 days around the time of ovulation, cycle days 5–22, and every day administration. The rationale for these periods of treatment was the same as described above for studies in women. None were effective at preventing pregnancy as pregnancy rates in all groups (75–100%) were comparable to, or exceeded, expected pregnancy rates for macaques with unrestricted breeding opportunities (Tardif et al., 2012).
Overall, PTGS2 inhibitors delayed or prevented follicle collapse in women and delayed or prevented oocyte release in monkeys in about 75% of menstrual cycles when administered at the most effective doses. Furthermore, the PTGS2 inhibitor meloxicam prevented 80% of anticipated pregnancies in breeding monkeys when administered to simulate use of emergency contraception. Drugs of this class do not significantly alter serum levels of ovarian steroid hormones, suggesting that a PTGS2 inhibitor may have minimal hormonal side effects. For these reasons, PTGS2 inhibitors may hold promise as potential emergency contraceptives. However, high pregnancy rates in a preclinical study in monkeys suggest that PTGS2 inhibitors alone are not suitable for monthly or routine contraceptive use.
Conclusions, perspectives and potential novel contraceptive targets
Current research has focused on cyclooxygenase as a target for potential contraceptives. Drugs which inhibit PTGS2 activity are generally safe and effective. Inhibitors with either high or modest selectivity for PTGS2 are widely available, and some are sold over the counter (Fitzgerald and Patrono, 2001). Safety concerns have been raised regarding the use of PTGS2 inhibitors as contraceptives. Drugs of this class can cause adverse events, most notably deep vein thrombosis and myocardial infarction (reviewed in Fitzgerald and Patrono (2001)). All NSAIDs, whether sold over the counter or by prescription, have a similar, slightly elevated risk of these adverse cardiovascular events (Helin-Salmivaara et al., 2006). Safety data for use of NSAIDs are generally obtained from populations of individuals who routinely use drugs of this class. These individuals, and especially users of PTGS2 selective inhibitors, are typically older and, therefore, outside the age range of contraceptive users (Layton et al., 2003a, b). The limited data available for use in young women (15–40 years) suggest that the incidence of these adverse events is extremely low in individuals in the demographic which includes contraceptive users (Layton et al., 2003a, b). It is important to note that commonly-used oral and transdermal hormonal contraceptives also have similar low but significant rates of these adverse cardiovascular events (Petitti et al., 1997; Tanis et al., 2001; Vandenbroucke et al., 2001; Baillargeon et al., 2005). Pregnancy itself poses risks of adverse cardiovascular events and other health issues (Centers for Disease Control, 2014), so the risk associated with contraceptive use should be balanced by the risk posed by an unintended pregnancy. Additional safety data obtained from women 15–40 years of age would be needed to fully assess the safety of PTGS2 inhibitors compared with the adverse events associated with current hormonal contraceptives as well as the health risks of pregnancy.
Multiple enzymatic targets may exist for development of novel contraceptives to block the ovulatory rise in follicular PGE2. To date, work has focused on inhibition of PTGS2, since it is generally assumed that PTGS2 catalyzes the rate-limiting step in follicular PGE2 production. However, late increases in PLA2G4A activity and PTGES protein in monkey follicles (Fig. 3I and G) suggest that factors in addition to PTGS2 activity may determine the timing of the PGE2 rise within follicular fluid. Finally, maintenance of elevated HPGD activity or alteration of PGE2 transport may dysregulate PGE2 accumulation within the follicle and present additional options to disrupt ovulatory processes.
As discussed above, PLA2G4A is necessary for ovulatory PGE2 production. PLA2G4A activity in monkey granulosa cells is detected only just before ovulation, correlating with peak PGE2 levels and leading to the suggestion that PLA2G4A activity may be rate-limiting in follicular PGE2 synthesis (Duffy et al., 2005a). In some tissues, inhibitors of PLA2G4A reduce PGE2 synthesis as effectively as PTGS2 inhibitors (Ono et al., 2002). Blockade of PLA2G4A activity may effectively reduce PGE2 production in the ovulatory follicle as well and has potential as a component of a contraceptive. While older PLA2G4A inhibitors were appropriate only for in vitro mechanistic experiments and animal studies, there has been a recent resurgence of interest in the identification of PLA2G4A inhibitors for treatment of human disease (Dennis et al., 2011; Murakami et al., 2011). An antisense strategy has also been used to reduce PLA2G4A expression in mice (Raichel et al., 2008). PLA2G4A has been implicated in numerous inflammatory conditions as well as conditions with characteristics of inflammatory reactions, including diabetes and atherosclerosis (Raichel et al., 2008; Wang et al., 2010; Kanter and Bornfeldt, 2013). Ovulation has also been characterized as an inflammatory process (Espey, 1994). The observation that PLA2G4A activity may be needed to optimally stimulate PTGS2 expression in the ovulatory follicle (Kurusu et al., 2012) suggests that coupling a PLA2G4A inhibitor with an inhibitor of PTGS2 may provide a highly effective method of lowering PGE2 production by the follicle and reducing ovulatory success as a potential method of contraception.
PGE2 is well established as the key prostaglandin which stimulates ovulatory events. For this reason, PTGES is another promising target for contraceptive development. Several naturally-derived products are selective for inhibition of PTGES without affecting PTGS1 or PTGS2, including compounds derived from green tea, turmeric and St. John's wort (Chang and Meuilette, 2011). Synthetic PTGES inhibitors have also been developed, with IC50 values in the micromolar to nanomolar range (Korotkova and Jakobsson, 2014). However, few have been tested in vivo (reviewed in Norberg et al. (2013)), and there are no reports of PTGES inhibitors in human clinical trials. Some inhibitors of PTGES also inhibit cyclooxygenase or lipoxygenase activity, and this lack of specificity decreased interest in further development of these compounds (Chang and Meuilette, 2011). Given the role of PTGS2 (discussed above) and perhaps lipoxygenases (Mikuni et al., 1988; Kurusu et al., 2009) in ovulation, the ability of these compounds to inhibit multiple enzymes implicated in ovulatory events suggests that they may have unique potential for the development of new contraceptives which limit the synthesis of multiple classes of eicosanoids.
Modulation of HPGD activity may be an additional target for contraceptive development. Inhibition of HPGD activity would likely lead to an increase in follicular PGE2 levels early in the ovulatory interval (see Fig. 3J and K) and dysregulate ovulatory events. While untested, an early rise in follicular PGE2 could lead to premature oocyte activation, incomplete cumulus expansion, and release of a cumulus-oocyte-complex unsuitable for fertilization. For example, PGE2 is necessary for cumulus expansion in primate follicles (Kim et al., 2014; Peluffo et al., 2014), and altered intrafollicular PGE2 concentrations correlate with delayed nuclear maturation and reduced fertilization rates (Duffy et al., 2010; Duffy and VandeVoort, 2011). Similarly, imbalances of proteolytic enzymes and their endogenous regulators could disrupt the proper selection of a rupture site or the timing of follicle rupture relative to other ovulatory events. Perhaps more promising is the observation that the growth of some cancers is stimulated by PGE2, and elevated expression of PTGS2 with reciprocal lowering of HPGD expression has been described in cancer cells and tissues (Backlund et al., 2005; Tai et al., 2007; Liu et al., 2008). Similar reciprocal expression of PTGS2 and HPGD has been reported in the ovulatory follicle (discussed above). Certain NSAIDs inhibit PTGS2 activity while also increasing the activity of HPGD (Tai, 2011). In these cases, drug administration would decrease PGE2 synthesis while increasing PGE2 metabolism, utilizing two complementary mechanisms to lower local PGE2 levels. Finally, some NSAIDs reduce the activity of ABCC4 efflux transporter (Reid et al., 2003), decreasing prostaglandin levels in the extracellular fluid. Regulation of multiple steps in the prostaglandin synthesis/metabolism/transport pathway has been suggested as a possible chemotherapeutic option (Na et al., 2011; Tai, 2011). Whether accomplished with a single drug or multi-drug combination of a PTGS2 inhibitor, HPGD activity enhancer, and/or transport blocker, this approach may limit PGE2 accumulation in the follicle and, therefore, prevent ovulation more effectively than a PTGS2 inhibitor alone.
Work is underway to identify potential pharmaceuticals which selectively inhibit AKR enzymatic activity. AKR1C family members are structurally-similar, so development of inhibitors which are highly selective for a single AKR1C family member has been challenging. Recent reviews describe the process of identifying suitable compounds (Bauman et al., 2005; Brozic et al., 2011). In the ovarian follicle, AKR1C2 or AKR1C3 inhibition would likely raise intrafollicular PGE2 levels by decreasing conversion of PGE2 or PGH2, respectively, to PGF2α. The overall effect of AKR1C2 or AKR1C3 inhibition on ovulation would likely be to prematurely activate PGE2-dependent changes in the follicle, similar to those described above for HPGD inhibition. While AKR1C inhibitors are in the early stages of development, the AKR family of enzymes remains a possible target for modulating ovulatory PGE2 levels within the ovarian follicle.
It is important to note that inhibition of any enzyme activity may increase precursor levels and alter the eicosanoid output of follicular cells. For example, inhibition of PTGS2 activity may increase production of other arachidonic acid metabolites, such as the leukotrienes and hydroxy-eicosatetraenoic acids. Shunting of precursors to alternative pathways may lead to production of unanticipated eicosanoid products and directly alter ovulatory events.
Alteration in cellular PGE2 transport could lead to mistimed or absent ovulatory events. SLCO2A1 expression has been confirmed for bovine and human granulosa cells (Bridges and Fortune, 2007; Wissing et al., 2014), but a role for this prostaglandin transporter in regulation of follicular PGE2 levels has not been demonstrated. A key role for SLCO2A1 has been demonstrated in prostaglandin-dependent cancer progression, where loss-of-function mutations or decreased expression of the prostaglandin transporter SLCO2A1 increased cancer risk (Holla et al., 2008; Guda et al., 2014; Pereira et al., 2014). Small molecule inhibitors of prostaglandin transporters are in development (Norberg et al., 2013). Inhibition of granulosa cell SLCO2A1 function would potentially increase extracellular PGE2 and dysregulate ovulatory events. Classes of drugs which enhance expression of both SLCO2A1 and HPGD are being considered for treatment of prostaglandin-stimulated cancers (Greenhough et al., 2009). Increased expression of SLCO2A1 and HPGD would promote cellular PGE2 uptake and conversion to inactive PGE2 metabolites, decreasing PGE2 levels in the extracellular compartment. Therapeutics using this approach may also have potential as contraceptives.
Prostaglandin E2 receptors are also potential contraceptive targets. PGE2 acts via four distinct G-protein coupled receptors (PTGER1, PTGER2, PTGER3, PTGER4). These receptors are the products of different genes, have different affinities for PGE2, and utilize different signal transduction pathways (Narumiya et al., 1999). Ovulation failure in mice lacking PTGER2 expression focused attention on this receptor as the key mediator of PGE2 action within the ovulatory follicle (Hizaki et al., 1999; Tilley et al., 1999). Recent studies have demonstrated that all four PGE2 receptors are expressed within the primate ovulatory follicle, and each PTGER is unique in terms of its regulation by the ovulatory gonadotrophin surge and spacial distribution within the ovulatory follicle (Markosyan et al., 2006; Harris et al., 2011). Importantly, inhibition of follicular PGE2 synthesis and coadministration of an agonist selective for a single PTGER indicates that PTGER1, PTGER2, and PTGER4 can each mediate aspects of the ovulatory cascade, such as cumulus expansion, follicle rupture, and follicular angiogenesis (Kim et al., 2014; Trau et al., 2015). It is not known if PGE2 receptors are located in the plasma membrane or intracellular membranes of target cells within the follicle. The cellular location of PTGERs will determine if intracellular PGE2 or extracellular PGE2 is required to activate signal transduction and whether modulation of intracellular or extracellular PGE2 is important for the action of a potential contraceptive. A PTGER2 antagonist was recently shown to prevent a significant percentage of anticipated pregnancies in breeding monkeys (Peluffo et al., 2014), providing initial proof of concept that modulation of PTGER responses holds promise as a component of an effective contraceptive.
While this review focuses on manipulation of ovarian PGE2 to prevent pregnancy, manipulation of uterine PGE2 also has potential to improve women's health. Altered levels of PTGS1, PTGS2 and PGE2 have been implicated in endometriosis, dysmenorrhea, menorrhagia and dysfunctional uterine bleeding (Jabbour et al., 2006; Smith et al., 2007; Wu et al., 2010). NSAIDs have been used to treat these disorders, with mixed success (Crosignani et al., 2006; Marjoribanks et al., 2010; Lethaby et al., 2013). Targeted therapies to reduce or dysregulate PGE2 synthesis, metabolism, transport, or receptors may have benefits for women's health in addition to novel methods of contraception.
Overall, the studies discussed in this review suggest that PTGS2 inhibitors can delay, prevent, or cause dysfunctional ovulation, which may contribute to an effective contraceptive strategy. Use of a PTGS2 inhibitor alone holds the greatest potential for the development of an emergency contraceptive. A critical feature is the timing of PTGS2 inhibitor administration relative to the day of the LH surge since PTGS2 inhibitors impact ovulatory events most effectively when administered before the LH surge. PTGS2 inhibitors may impact cumulus expansion, oocyte maturation, follicle rupture, and ultimately oocyte release. While in vitro studies suggest that oocyte maturation and fertilization are compromised by prostaglandin depletion, monkey oocytes released in vivo are capable of fertilization and implantation despite continued PTGS2 inhibitor administration, as evidenced by the high rates of pregnancies noted in studies of breeding monkeys receiving meloxicam into the luteal phase (McCann et al., 2013). However, it is not known if pregnancies which occur during PTGS2 inhibitor administration would result in healthy fetuses if carried to term and delivered normally. A key feature of the utility of PTGS2 inhibitors as potential contraceptives appears to be the ability to delay ovulation beyond the 5–6 day period of sperm viability resulting from a single act of intercourse. Reintroduction of viable sperm with repeated intercourse likely results in fertilization and pregnancy in cases where ovulation does ultimately occur. PTGS2 inhibitors alone are unlikely to be suited for use as a monthly non-hormonal contraceptive. Drugs which inhibit other enzymes involved in prostaglandin synthesis, pharmaceuticals which alter PGE2 transport or metabolism, or PGE2 receptor antagonists may hold promise for the inhibition of ovulation. Finally, modulators of PGE2 synthesis, metabolism, transport, or response could be paired with steroidal approaches to improve overall contraceptive efficacy. Many steps within the pathway including PGE2 synthesis, transport, metabolism, and receptors are amenable to pharmacological manipulation. A strategy targeting multiple components of this pathway may be necessary to inhibit ovulatory events with sufficient efficacy to create a novel, pre-fertilization contraceptive option for women.
Author's role
D.M.D. is solely responsible for all aspects of this manuscript.
Funding
Studies in Dr. Duffy's lab were supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD; HD39872, HD054691 and HD071875), CONRAD (CIG-07-115 and CIG-10-129), and Eastern Virginia Medical School (EVMS).
Conflict of interest
None declared.
Acknowledgements
The author would like to thank Drs David F. Archer, Thomas E. Curry, Jr., and Joanne E. Fortune for reviewing this manuscript. Studies in Dr Duffy's lab were supported by generous donations of recombinant human gonadotrophins and GnRH antagonists by Merck & Co. (Whitehouse Station, NJ, USA) and Serono Reproductive Biology Institute (Rockland, MA, USA).
References
- Akil M, Amos RS, Stewart P. Infertility may sometimes be associated with NSAID consumption. Br J Rheumatol 1996;35:76–78. [DOI] [PubMed] [Google Scholar]
- Armstrong DT, Grinwich DL. Blockade of spontaneous and LH-induced ovulation in rats by indomethacin, an inhibitor of prostaglandin biosynthesis. Prostaglandins 1972;1:21–28. [DOI] [PubMed] [Google Scholar]
- Athanasiou S, Bourne TH, Khalid A, Okokon EV, Crayford TJ, Hagstrom HG, Campbell S, Collins WP. Effects of indomethacin on follicular structure, vascularity, and function over the periovulatory period in women. Fertil Steril 1996;65:556–560. [PubMed] [Google Scholar]
- Backlund MG, Mann JR, Holla VR, Buchanan FG, Tai HH, Musiek ES, Milne GL, Katkuri S, DuBois RN. 15-Hydroxyprostaglandin dehydrogenase is down-regulated in colorectal cancer. J Biol Chem 2005;280:3217–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillargeon JP, McClish DK, Essah PA, Nestler JE. Association between the current use of low-dose oral contraceptives and cardiovascular arterial disease: a meta-analysis. J Clin Endocrinol Metab 2005;90:3863–3870. [DOI] [PubMed] [Google Scholar]
- Balsinde J, Winstead MV, Dennis EA. Phospholipase A2 regulation of arachidonic acid mobilization. FEBS Lett 2002;531:2–6. [DOI] [PubMed] [Google Scholar]
- Bata MS, Al-Ramahi M, Salhab AS, Gharaibeh MN, Schwartz J. Delay of ovulation by meloxicam in healthy cycling volunteers: a placebo-controlled, double-blind, crossover study. J Clin Pharmacol 2006;46:925–932. [DOI] [PubMed] [Google Scholar]
- Bauman DR, Rudnick SI, Szewczuk LM, Gophishetty S, Penning TM. Development of nonsteroidal anti-inflammatory drug analogs and steroid carboxylates selective for human aldo-keto reductase isoforms: potential antineoplastic agents that work independently of cyclooxygenase isozymes. Mol Pharmacol 2005;67:60–68. [DOI] [PubMed] [Google Scholar]
- Bauminger A, Lindner HR. Periovulatory changes in ovarian prostaglandin formation and their hormonal control in the rat. Prostaglandins 1975;9:737–751. [DOI] [PubMed] [Google Scholar]
- Behboodi E, Kaz DF, Samuels SJ, Hendrickx AG, Lasley BL. The use of a urinary estrone conjugates assay for detection of optimal mating time in the cynomolgus macaque (Macaca fascicularis). J Med Primatol 1991;20:229–234. [PubMed] [Google Scholar]
- Ben-Shlomo I, Kol S, Ando M, Altman KR, Putowski LT, Rohan RM, Adashi EY. Ovarian expression, cellular localization, and hormonal regulation of rat secretory phospholipase A2: Increased expression by interleukin-1 and by gonadotropins. Biol Reprod 1997;57:217–225. [DOI] [PubMed] [Google Scholar]
- Bhattacharya M, Varma DR, Chemtob S. Nuclear prostaglandin receptors. Gene Ther Mol Biol 1999;4:323–338. [Google Scholar]
- Bonney RC, Wilson CA. Hormonal control of ovarian phospholipase A2 activity in rats. J Reprod Fertil 1993;99:201–208. [DOI] [PubMed] [Google Scholar]
- Bonventre JV, Huang Z, Teheri MR, O'Leary E, Li E, Moskowitz MA, Sapirstein A. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature 1997;390:622–625. [DOI] [PubMed] [Google Scholar]
- Bosetti F, Weerasinghe GR. The expression of brain cyclooxygenase-2 is down-regulated in the cytosolic phospholipase A2 knockout mouse. J Neurochem 2003;87:1471–1477. [DOI] [PubMed] [Google Scholar]
- Bridges PJ, Fortune JE. Regulation, action and transport of prostaglandins during the periovulatory period in cattle. Mol Cell Endocrinol 2007;263:1–9. [DOI] [PubMed] [Google Scholar]
- Brown KA, Boerboom D, Bouchard N, Dore M, Lussier JG, Sirois J. Human chorionic gonadotropin-dependent induction of an equine aldo-keto reductase (AKR1C23) with 20α-hydroxysteroid dehydrogenase activity during follicular luteinization in vivo. J Mol Endocrinol 2006;36:449–461. [DOI] [PubMed] [Google Scholar]
- Brozic P, Turk S, Rizner TL, Gobec S. Inhibitors of aldo-keto reductases AKR1C1-AKR1C4. Curr Med Chem 2011;18:2554–2565. [DOI] [PubMed] [Google Scholar]
- Centers_for_Disease_Control. Severe maternal morbidity in the United States 2014. http://wwwcdcgov/reproductivehealth/MaternalInfantHealth/SevereMaternalMorbidityhtml (14 September 2014, date last accessed).
- Chan BS, Endo S, Kanai N, Schuster VL. Identification of lactate as a driving force for prostanoid transport by prostaglandin transporter PGT. Am J Physiol Renal Physiol 2002;282:F1097–F1102. [DOI] [PubMed] [Google Scholar]
- Chandrasekharan NV, Dai H, Roos KLT, Evanson NK, Tomsik J, Elton TS, Simmons DL. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci USA 2002;99:13926–13931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HH, Meuilette EJ. Identification and development of mPGES-1 inhibitors: where we are at? Future Med Chem 2011;3:1909–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi Y, Khersonsky SM, Chang Y-T, Schuster VL. Identification of a new class of prostaglandin transporter inhibitors and characterization of their biological effects on prostaglandin E2 transport. J Pharmacol Exp Ther 2006;316:1346–1350. [DOI] [PubMed] [Google Scholar]
- Clark JD, Lin LL, Kriz RW, Ramesha CS, Sultzman LA, Lin AY, Milona N, Knopf JL. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca(2+)-dependent translocation domain with homology to PKC and GAP. Cell 1991;65:1043–1051. [DOI] [PubMed] [Google Scholar]
- Coggins KG, Latour A, Nguyen MS, Audoly L. Metabolism of PGE2 by prostaglandin dehydrogenase is essential for remodeling the ductus arteriosus. Nat Med 2002;8:91–92. [DOI] [PubMed] [Google Scholar]
- Crosignani P, Olive D, Bergqvist A, Luciano A. Advances in the management of endometriosis: an update for clinicians. Hum Reprod Update 2006;12:179–189. [DOI] [PubMed] [Google Scholar]
- Curry TEJ, Bryant C, Haddix AC, Clark MR. Ovarian prostaglandin endoperoxide synthase: cellular localization during the rat estrous cycle. Biol Reprod 1990;42:307–316. [DOI] [PubMed] [Google Scholar]
- Dennis EA, Cao J, Hsu Y-H, Magrioti V, Kokotos G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem Rev 2011;111:6130–6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva M, Reeves JJ. Indomethacin inhibition of ovulation in the cow. J Reprod Fertil 1985;75:547–549. [DOI] [PubMed] [Google Scholar]
- Diouf MN, Sayasith K, Lefebvre R, Silversides DW, Sirois J, Lussier JG. Expression of phospholipase A2 group IVA (PLA2G4A) is upregulated by human chorionic gonadotropin in bovine granulosa cells of ovulatory follicles. Biol Reprod 2006;74:1096–1103. [DOI] [PubMed] [Google Scholar]
- Downs SM, Longo FJ. Effects of indomethacin on preovulatory follicles in immature, superovulated mice. Am J Anat 1982;164:265–274. [DOI] [PubMed] [Google Scholar]
- Downs SM, Longo FJ. Prostaglandins and preovulatory follicular maturation in mice. J Exp Zool 1983;228:99–108. [DOI] [PubMed] [Google Scholar]
- Dozier BL, Watanabe K, Duffy DM. Two pathways for prostaglandin F2α synthesis by the primate periovulatory follicle. Reproduction 2008;136:53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy DM. Prostaglandin dehydrogenase (PGDH) in granulosa cells of primate periovulatory follicles is regulated by the ovulatory gonadotropin surge via multiple G proteins. Mol Cell Endocrinol 2011;20:119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy DM, Stouffer RL. The ovulatory gonadotrophin surge stimulates cyclooxygenase expression and prostaglandin production by the monkey follicle. Mol Hum Reprod 2001;7:731–739. [DOI] [PubMed] [Google Scholar]
- Duffy DM, Stouffer RL. Follicular administration of a cyclooxygenase inhibitor can prevent oocyte release without alteration of normal luteal function in rhesus monkeys. Hum Reprod 2002;17:2825–2831. [DOI] [PubMed] [Google Scholar]
- Duffy DM, Stouffer RL. Luteinizing hormone acts directly at granulosa cells to stimulate periovulatory processes: modulation of luteinizing hormone effects by prostaglandins. Endocrine 2003;22:249–256. [DOI] [PubMed] [Google Scholar]
- Duffy DM, VandeVoort CA. Maturation and fertilization of non-human primate oocytes are compromised by oral administration of a COX2 inhibitor. Fertil Steril 2011;95:1256–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy DM, Seachord CL, Dozier BL. An ovulatory gonadotropin stimulus increases cytosolic phospholipase A2 (cPLA2) expression and activity in granulosa cells of primate periovulatory follicles. J Clin Endocrinol Metab 2005a;90:5858–5865. [DOI] [PubMed] [Google Scholar]
- Duffy DM, Seachord CL, Dozier BL. Microsomal prostaglandin E synthase-1 (mPGES-1) is the primary form of PGES expressed by the primate periovulatory follicle. Hum Reprod 2005b;20:1485–1492. [DOI] [PubMed] [Google Scholar]
- Duffy DM, Dozier BL, Seachord CL. Prostaglandin dehydrogenase and prostaglandin levels in periovulatory follicles: implications for control of primate ovulation by PGE2. J Clin Endocrinol Metab 2005c;90:1021–1027. [DOI] [PubMed] [Google Scholar]
- Duffy DM, McGinnis LK, VandeVoort CA, Christenson LK. Mammalian oocytes are targets for prostaglandin E2 (PGE2) action. Reprod Biol Endocrinol 2010;8:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukelow WR, Bruggemann S. Characteristics of the menstrual cycle in nonhuman primates II. Ovulation and optimal mating times in macaques. J Med Primatol 1979;8:79–87. [DOI] [PubMed] [Google Scholar]
- Edelman AB, Jensen JT, Doom C, Hennebold JD. Impact of the prostaglandin synthase-2 inhibitor celecoxib on ovulation and luteal events in women. Contraception 2013;87:352–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvin JA, Yan C, Matzuk MM. Growth differentiation factor-9 stimulates progesterone synthesis in granulosa cells via a prostaglandin E2/EP2 receptor pathway. Proc Natl Acad Sci USA 2000;97:10288–10293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESHRE_Capri_Workshop_Group. Emergency contraception. Widely available and effective but disappointing as a public health intervention: a review. Hum Reprod 2015;30:751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espey LL. Current status of the hypothesis that mammalian ovulation is comparable to an inflammatory reaction. Biol Reprod 1994;50:233–238. [DOI] [PubMed] [Google Scholar]
- Espey LL, Lipner H. Ovulation. In: Knobil E, Neill JD. (eds) The Physiology of Reproduction. New York, NY: Raven Press, 1994, 725–780. [Google Scholar]
- Espey LL, Norris C, Saphire D. Effect of time and dose of indomethacin on follicular prostaglandins and ovulation in the rabbit. Endocrinology 1986;119:746–754. [DOI] [PubMed] [Google Scholar]
- Filion F, Bouchard N, Goff AK, Lussier JG, Sirois J. Molecular cloning and induction of bovine prostaglandin E synthase by gonadotropins in ovarian follicles prior to ovulation in vivo. J Biol Chem 2001;276:34323–34330. [DOI] [PubMed] [Google Scholar]
- Fitzgerald GA, Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med 2001;345:433–442. [DOI] [PubMed] [Google Scholar]
- Gemzell-Danielsson K, Rabe T, Cheng L. Emergency contraception. Gynecol Endocrinol 2013;29:1–14. [DOI] [PubMed] [Google Scholar]
- Glasier A. Emergency contraception: clinical outcomes. Contraception 2013;87:309–313. [DOI] [PubMed] [Google Scholar]
- Greenhough A, Smartt HJM, Moore AE, Roberts HR, Williams AC, Paraskeva C, Kaidi A. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009;30:377–386. [DOI] [PubMed] [Google Scholar]
- Guan Y, Zhang Y, Schneider A, Riendeau D, Mancini JA, Davis L, Komhoff M, Breyer RM, Breyer MD. Urogenital distribution of a mouse membrane-associated prostaglandin E2 synthase. Am J Physiol Renal Physiol 2001;281:F1173–F1177. [DOI] [PubMed] [Google Scholar]
- Guda K, Fink SP, Milne GL, Molyneaux N, Ravi L, Lewis SM, Dannenberg AJ, Montogmery CG, Zhang S, Willis J, et al. Inactivating mutation in the prostaglandin transporter gene, SLCO2A1, associated with familial digital clubbing, colon neoplasia, and NSAID resistance. Cancer Prev Res 2014;7:805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han WK, Sapirstein A, Hung CC, Alessandrini A, Bonventre JV. Cross-talk between cytosolic phospholipase A2 alpha (cPLA2 alpha) and secretory phospholipase A2 (sPLA2) in hydrogen peroxide-induced arachidonic acid release in murine mesangial cells: sPLA2 regulates cPLA2 alpha activity that is responsible for arachidonic acid release. J Biol Chem 2003;278:24153–24163. [DOI] [PubMed] [Google Scholar]
- Harris SM, Aschenbach LC, Skinner SM, Dozier BL, Duffy DM. Prostaglandin E2 receptors are differentially expressed in subpopulations of granulosa cells from primate periovulatory follicles. Biol Reprod 2011;85:916–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedin L, Gaddy-Kurten D, Kurten R, DeWitt DL, Smith WL, Richards JS. Prostaglandin endoperoxide synthase in rat ovarian follicles: content, cellular distribution, and evidence for hormonal induction preceding ovulation. Endocrinology 1987;121:722–731. [DOI] [PubMed] [Google Scholar]
- Helin-Salmivaara A, Virtanen A, Vesalainen R, Gronroos JM, Klaukka T, Idanpaan-Heikkila JE, Huupponen R. NSAID use and the risk of hospitalization for first myocardial infarction in the general population: a nationwide case-control study from Finland. Eur Heart J 2006;27:1657–1663. [DOI] [PubMed] [Google Scholar]
- Hester KE, Harper MJK, Duffy DM. Oral administration of the cyclooxygenase-2 (COX-2) inhibitor meloxicam blocks ovulation in non-human primates when administered to simulate emergency contraception. Hum Reprod 2010;25:360–367. [DOI] [PubMed] [Google Scholar]
- Hibbert ML, Stouffer RL, Wolf DP, Zelinski-Wooten MB. Midcycle administration of a progesterone synthesis inhibitor prevents ovulation in primates. Proc Natl Acad Sci USA 1996;93:1897–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hizaki H, Segi E, Sugimoto Y, Hirose M, Saji T, Ushikubi F, Matsuoka T, Noda Y, Tanaka T, Yoshida N, et al. Abortive expansion of the cumulus and impaired fertility in mice lacking the prostaglandin E receptor subtype EP 2. Proc Natl Acad Sci USA 1999;96:10501–10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holla VR, Backlund MG, Yang P, Newman RA, DuBois RN. Regulation of prostaglandin transporters in colorectal neoplasia. Cancer Prev Res 2008;1:93–99. [DOI] [PubMed] [Google Scholar]
- Iwata N, Inazu N, Satoh T. Changes in rat ovarian carbonyl reductase activity and content during the estrous cycle, and localization. Biol Reprod 1990;42:161–166. [DOI] [PubMed] [Google Scholar]
- Jabbour HN, Sales KJ, Milling Smith OP, Battersby S, Boddy SC. Prostaglandin receptors are mediators of vascular function in endometrial pathologies. Mol Cell Endocrinol 2006;252:191–200. [DOI] [PubMed] [Google Scholar]
- Janson PO, Brannstrom M, Holmes PV, Sogn J. Studies on the mechanism of ovulation using the model of the isolated ovary. Ann N Y Acad Sci 1988;541:22–29. [DOI] [PubMed] [Google Scholar]
- Jesam C, Salvatierra AM, Schwartz J, Croxatto HB. Suppression of follicular rupture with meloxicam, a cyclooxygenase inhibitor: potential for emergency contraception. Hum Reprod 2010;25:368–373. [DOI] [PubMed] [Google Scholar]
- Jesam C, Salvatierra AM, Schwartz JL, Fuentes A, Croxatto HB. Effect of oral administration of a continuous 18 day regimen of meloxicam on ovulation: experience of a randomized controlled trial. Contraception 2014;90:168–173. [DOI] [PubMed] [Google Scholar]
- Kanter JE, Bornfeldt KE. Inflammation and diabetes-accelerated atherosclerosis: myeloid cell mediators. Trends Endocrinol Metab 2013;24:137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killick S, Elstein M. Pharmacologic production of luteinized unruptured follicles by prostaglandin synthetase inhibitors. Fertil Steril 1987;47:773–777. [DOI] [PubMed] [Google Scholar]
- Kim SO, Harris SM, Duffy DM. Prostaglandin E2 (EP) receptors mediate PGE2-specific events in ovulation and luteinization within primate ovarian follicles. Endocrinology 2014;155:1466–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SO, Markosyan N, Pepe GJ, Duffy DM. Estrogen promotes luteolysis by redistributing prostaglandin F2α receptors within primate luteal cells. Reproduction 2015;149:453–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochel TJ, Fulton AM. Multiple drug resistance-associated protein 4 (MRP4), prostaglandin transporter (PTG), and 15-hydroxyprostaglandin dehydrogenase (15-PGDH) as determinants of PGE2 levels in cancer. Prostaglandins Other Lipid Mediat 2015;116–117C:99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkova M, Jakobsson P-J. Characterization of microsomal prostaglandin E synthase 1 inhibitors. Basic Clin Pharmacol Toxicol 2014;11:64–69. [DOI] [PubMed] [Google Scholar]
- Kurusu S, Iwao M, Kawaminami M, Hashimoto I. Involvement of cytosolic phospholipase A2 in the ovulatory process in gonadotropin-primed immature rats. Prostaglandins Leukot Essent Fatty Acids 1998a;58:405–411. [DOI] [PubMed] [Google Scholar]
- Kurusu S, Motegi S, Kawaminami M, Hashimoto I. Expression and cellular distribution of cytosolic phospholipase A2 in the rat ovary. Prostaglandins Leukot Essent Fatty Acids 1998b;58:399–404. [DOI] [PubMed] [Google Scholar]
- Kurusu S, Jinno M, Ehara H, Yonezawa T, Kawaminami M. Inhibition of ovulation by a lipoxygenase inhibitor involves reduced cyclooxygenase-2 expression and prostaglandin E2 production in gonadotropin-primed rats. Reproduction 2009;137:59–66. [DOI] [PubMed] [Google Scholar]
- Kurusu S, Sapirstein A, Bonventre JV. Group IVA phospholipase A2 optimizes ovulation and fertilization in rodents through induction of and metabolic coupling with prostaglandin endoperoxide synthase 2. FASEB J 2012;26:3800–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD, et al. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell 1995;83:483–492. [DOI] [PubMed] [Google Scholar]
- Layton D, Heeley E, Hughes K, Shakir SAW. Comparison of the incidence rates of thromboembolic events reported for patients prescribed rofecoxib and meloxicam in general practice in England using prescription-event monitoring (PEM) data. Rheumatology 2003a;42:1342–1353. [DOI] [PubMed] [Google Scholar]
- Layton D, Hughes K, Harris S, Shakir SAW. Comparison of the incidence rates of thromboembolic events reported for patients prescribed celecoxib and meloxicam in general practice in England using prescription-event monitoring (PEM) data. Rheumatology 2003b;42:1354–1364. [DOI] [PubMed] [Google Scholar]
- Legler DF, Bruckner M, Uetz-von Allmen E, Krause P. Prostaglandin E2 at new glance: novel insights into functional diversity offer therapeutic chances. Int J Biochem Cell Biol 2010;42:198–201. [DOI] [PubMed] [Google Scholar]
- Lethaby A, Duckitt K, Farquhar C. Non-steroidal anti-inflammatory drugs for heavy menstrual bleeding. Cochrane Database Syst Rev 2013;1:CD0004000. [DOI] [PubMed] [Google Scholar]
- Lim H, Paria BC, Das SK, Dinchuk JE, Langenbach R, Trzaskos JM, Dey SK. Multiple female reproductive failures in cyclooxygenase 2-deficient mice. Cell 1997;91:197–208. [DOI] [PubMed] [Google Scholar]
- Liu J, Carriere PD, Dore M, Sirois J. Prostaglandin G/H synthase-2 is expressed in bovine preovulatory follicles after the endogenous surge of luteinizing hormone. Biol Reprod 1997;57:1524–1531. [DOI] [PubMed] [Google Scholar]
- Liu J, Antaya M, Boerboom D, Lussier G, Silversides DW, Sirois J. The delayed activation of the prostaglandin G/H synthase-2 promoter in bovine granulosa cells is associated with down-regulation of truncated upstream stimulatory factor-2. J Biol Chem 2000;274:35037–35045. [DOI] [PubMed] [Google Scholar]
- Liu Z, Wang X, Lu Y, Han S, Zhang F, Zhai H, Lei T, Liang J, Wang J, Wu K, et al. Expression of 15-PGDH is downregulated by COX-2 in gastric cancer. Carcinogenesis 2008;29:1219–1227. [DOI] [PubMed] [Google Scholar]
- Lu R, Kanai N, Bao Y, Schuster VL. Cloning, in vitro expression, and tissue distribution of a human prostaglandin transporter cDNA (hPGT). J Clin Invest 1996;98:1142–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjoribanks J, Proctor M, Farquhar C, Derks RS. Nonsteroidal anti-inflammatory drugs for dysmenorrhoea. Cochrane Database Syst Rev 2010;1:CD001751. [DOI] [PubMed] [Google Scholar]
- Markosyan N, Dozier BL, Lattanzio FA, Duffy DM. Primate granulosa cell response via prostaglandin E2 receptors increases late in the periovulatory interval. Biol Reprod 2006;75:868–876. [DOI] [PubMed] [Google Scholar]
- Massai MR, Forcelledo ML, Brache V, Tejada AS, Salvatierra AM, Reyes MV. Does meloxicam increase the incidence of anovulation induced by single administration of levonorgestrel in emergency contraception? A pilot study. Hum Reprod 2007;22:434–439. [DOI] [PubMed] [Google Scholar]
- McCann NC, Lynch TJ, Kim SO, Duffy DM. The COX-2 inhibitor meloxicam prevents pregnancy when administered as an emergency contraceptive to nonhuman primates. Contraception 2013;88:744–748. [DOI] [PubMed] [Google Scholar]
- Mendonca LL, Khamashta MA, Nelson-Piercy C, Hunt BJ, Hughes GR. Non-steroidal anti-inflammatory drugs as a possible cause for reversible infertility. Rheumatology (Oxford) 2000;39:880–882. [DOI] [PubMed] [Google Scholar]
- Mikuni M, Yoshida M, Hellberg P, Peterson CA, Edwin SS, Brannstrom M, Peterson CM. The lipoxygenase inhibitor, nordihydroguaiaretic acid, inhibits ovulation and reduces leukotriene and prostaglandin levels in the rat ovary. Biol Reprod 1988;58:1211–1216. [DOI] [PubMed] [Google Scholar]
- Mikuni M, Pall M, Peterson CM, Hellberg P, Brannstrom M, Richards JS, Hedin L. The selective prostaglandin endoperoxide synthase-2 inhibitor, NS-398, reduces prostaglandin production and ovulation in vivo and in vitro in the rat. Biol Reprod 1998;59:1077–1083. [DOI] [PubMed] [Google Scholar]
- Morita I, Schindler M, Regier MK, Otto JC, Hori T, DeWitt DL, Smith WL. Different intracellular locations for prostaglandin endoperoxide H synthase-1 and -2. J Biol Chem 1995;270:10902–10908. [DOI] [PubMed] [Google Scholar]
- Murakami M, Shimbara S, Kambe T, Kuwata H, Winstead MV, Tischfield JA, Kudo I. The functions of five distinct mammalian phosphodiesterase A2s in regulating arachidonic acid release. Type IIa and type V secretory phospholipase A2s are functionally redundant and act in concern with cytosolic phospholipase A2. J Biol Chem 1998;273:14411–14423. [DOI] [PubMed] [Google Scholar]
- Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, Ikeda T, Fueki M, Ueno A, Oh-ishi S, et al. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem 2000;275:32783–32792. [DOI] [PubMed] [Google Scholar]
- Murakami M, Nakatani Y, Tanioka T, Kudo I. Prostaglandin E synthase. Prostaglandins Other Lipid Mediat 2002;68–69:383–399. [DOI] [PubMed] [Google Scholar]
- Murakami M, Nakashima K, Kamei D, Masuda S, Ishikawa Y, Ishii T, Ohmiya Y, Watanabe K, Kudo I. Cellular prostaglandin E2 production by membrane-bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J Biol Chem 2003a;278:37937–37947. [DOI] [PubMed] [Google Scholar]
- Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, Ikeda T, Fueki M, Ueno A, Oh-ishi S, et al. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem 2003b;275:32783–32792. [DOI] [PubMed] [Google Scholar]
- Murakami M, Taketomi Y, Miki Y, Sato H, Hirabayashi T, Yamamoto K. Recent progress in phospholipase A2 research: from cells to animals to humans. Prog Lipid Res 2011;50:152–192. [DOI] [PubMed] [Google Scholar]
- Murdoch WJ, Farris ML. Prostaglandin E2-9-ketoreductase activity of preovulatory ovine follicles. J Anim Sci 1988;66:2924–2929. [DOI] [PubMed] [Google Scholar]
- Murdoch WJ, Peterson TA, Van Kirk EA, Vincent DL, Inskeep EK. Interactive roles of progesterone, prostaglandins, and collagenase in the ovulatory mechanism of the ewe. Biol Reprod 1986;35:1187–1194. [DOI] [PubMed] [Google Scholar]
- Na H-K, Park J-M, Lee HG, Lee H-N, Myung S-J, Surh Y-J. 15-hydroxyprostaglandin dehydrogenase as a novel molecular target for cancer chemoprevention and therapy. Biochem Pharmacol 2011;82:1352–1360. [DOI] [PubMed] [Google Scholar]
- Nanjidsuren T, Naidansuren P, Park C-W, Park J-J, Yun S-J, Sim B-W, Kang M-H, Lee S-R, Chang K-T, Min K-S. Expression and localization of the 20α-hydroxysteroid dehydrogenase (HSD) enzyme in the reproductive tissues of the cynomolgus macaque Macaca fascicularis. J Steroid Biochem Mol Biol 2011;127:337–344. [DOI] [PubMed] [Google Scholar]
- Narko K, Ritvos O, Ristimaki A. Induction of cyclooxygenase-2 and prostaglandin F2α receptor expression by interleukin-1β in cultured human granulosa-luteal cells. Endocrinology 1997;138:3638–3644. [DOI] [PubMed] [Google Scholar]
- Narumiya S, Sugimoto T, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev 1999;79:1193–1226. [DOI] [PubMed] [Google Scholar]
- Nishizawa M, Nakajima T, Yasuda K, Kanzaki H, Sasaguri Y, Watanabe K, Ito S. Close kinship of human 20α-hydroxysteroid dehydrogenase gene with three aldo-keto reductase genes. Genes Cells 2000;5:111–125. [DOI] [PubMed] [Google Scholar]
- Nomura T, Lu R, Pucci ML, Schuster VL. The two-step model of prostaglandin signal termination: in vitro reconstitution with prostaglandin transporter and prostaglandin 15 dehydrogenase. Mol Pharmacol 2004;65:973–978. [DOI] [PubMed] [Google Scholar]
- Norberg JK, Sells E, Chang H-H, Alla SR, Zhang S, Meuillet EJ. Targeting inflammation: multiple innovative ways to reduce prostaglandin E2. Pharm Pat Anal 2013;2:265–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Office_on_Women's_Health. Menstruation and the Menstrual Cycle 2009. U.S._Department_of_Health_and_Human_Services, www.womenshealth.gov (24 March 2015, date last accessed).
- Ono T, Yamada K, Chikazawa Y, Ueno M, Nakamoto S, Okuno T, Seno K. Characterization of a novel inhibitor of cytosolic phospholipase A2alpha, pyrrophenone. Biochem J 2002;363:727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pall M, Friden BE, Brannstrom M. Induction of delayed follicular rupture in the human by the selective COX-2 inhibitor rofecoxib: A randomized double-blind study. Hum Reprod 2001;16:1323–1328. [DOI] [PubMed] [Google Scholar]
- Peluffo MC, Stanley J, Braeuer N, Rotgeri A, Fritzemeier K-H, Fuhrmann U, Buchmann B, Adevai T, Murphy MJ, Zelinski MB, et al. A prostaglandin E2 receptor antagonist prevents pregnancies during a preclinical contraceptive trail with female macaques. Hum Reprod 2014;29:1400–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng XR, Hsueh AJ, LaPolt PS, Bjersing L, Ny T. Localization of luteinizing hormone receptor messenger ribonucleic acid expression in ovarian cell types during follicle development and ovulation. Endocrinology 1991;129:3200–3207. [DOI] [PubMed] [Google Scholar]
- Pereira C, Queiros S, Galaghar A, Sousa H, Pimentel-Nunes P, Brandao C, Moreira-Dias L, Medeiros R, Dinis-Riberiro M. Genetic variability in key genes in prostaglandin E2 pathway (COX-2, HPGD, ABCC4 and SLCO2A1) and their involvement in colorectal cancer development. PLoS ONE 2014;9:e92000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters MW, Pursley JR, Smith GW. Inhibition of intrafollicular PGE2 synthesis and ovulation following ultrasound-mediated intrafollicular injection of the selective cyclooxygenase-2 inhibitor NS-398 in cattle. J Anim Sci 2004;82:1656–1662. [DOI] [PubMed] [Google Scholar]
- Petitti DB, Sidney S, Quesenberry CP, Jr, Bernstein A. Incidence of stroke and myocardial infarction in women of reproductive age. Stroke 1997;28:280–283. [DOI] [PubMed] [Google Scholar]
- Priddy AR, Killick SR, Elstein M, Morris J, Sullivan M, Patel L, Elder MG. Ovarian follicular fluid eicosanoid concentrations during the pre-ovulatory period in humans. Prostaglandins 1989;38:197–202. [DOI] [PubMed] [Google Scholar]
- Priddy AR, Killick SR, Elstein M, Morris J, Sullivan M, Patel L, Elder M. The effect of prostaglandin synthetase inhibitors on human preovulatory follicular fluid prostaglandin, thromboxane, and leukotriene concentrations. J Clin Endocrinol Metab 1990;71:235–242. [DOI] [PubMed] [Google Scholar]
- Raichel L, Berger S, Hadad N, Kachko L, Karter M, Szaingurten-Solodkin I, Williams RO, Feldmann M, Levy R. Reduction of cPLA2alpha overexpression: an efficient anti-inflammatory therapy for collagen-induced arthritis. Eur J Immunol 2008;38:2905–2915. [DOI] [PubMed] [Google Scholar]
- Reid G, Wielinga P, Zelcer N, van der Heijden I, Kuil A, de Haas M, Wijnholds J, Borst P. The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci USA 2003;100:9244–9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards JS. Editorial: sounding the alarm-does induction of prostaglandin endoperoxide synthase-2 control the mammalian ovulatory clock? Endocrinology 1997;138:4047–4048. [DOI] [PubMed] [Google Scholar]
- Ristimaki A, Jaatinen R, Ritvos O. Regulation of prostaglandin F2α receptor expression in cultured human granulosa-luteal cells. Endocrinology 1997;138:191–195. [DOI] [PubMed] [Google Scholar]
- Sayasith K, Bouchard N, Dore M, Sirois J. Molecular cloning and gonadotropin-dependent regulation of equine prostaglandin F2α receptor in ovarian follicles during the ovulatory process in vivo. Prostaglandins Other Lipid Mediat 2006;80:81–92. [DOI] [PubMed] [Google Scholar]
- Sayasith K, Bouchard N, Dore M, Sirois J. Cloning of equine prostaglandin dehydrogenase and its gonadotropin-dependent regulation in theca and mural granulosa cells of equine periovulatory follicles during the ovulatory process. Reproduction 2007;133:455–466. [DOI] [PubMed] [Google Scholar]
- Schievella AR, Regier MK, Smith WL, Lin LL. Calcium-mediated translocation of cytosolic phospholipase A2 to the nuclear envelope and endoplasmic reticulum. J Biol Chem 1995;270:30749–30754. [DOI] [PubMed] [Google Scholar]
- Schlegel W, Daniels D, Kruger S. Partial purification of prostaglandin E2–9-ketoreductase and prostaglandin-15-hydroxydehydrogenase from ovarian tissues of rabbits. Clin Physiol Biochem 1987;5:336–342. [PubMed] [Google Scholar]
- Schuster VL. Prostaglandin transport. Prostaglandins Other Lipid Mediat 2002;68–69:633–647. [DOI] [PubMed] [Google Scholar]
- Sirois J. Induction of prostaglandin endoperoxide synthase-2 by human chorionic gonadotropin in bovine preovulatory follicles in vivo. Endocrinology 1994;135:841–848. [DOI] [PubMed] [Google Scholar]
- Sirois J, Dore M. The late induction of prostaglandin G/H synthase-2 in equine preovulatory follicles supports its role as a determinant of the ovulatory process. Endocrinology 1997;138:4427–4434. [DOI] [PubMed] [Google Scholar]
- Sirois J, Sayasith K, Brown KA, Stock AE, Bouchard N, Dore M. Cyclooxygenase-2 and its role in ovulation: a 2004 account. Hum Reprod Update 2004;10:373–385. [DOI] [PubMed] [Google Scholar]
- Smith G, Roberts R, Hall C, Nuki G. Reversible ovulatory failure associated with the development of luteinized unruptured follicles in women with inflammatory arthritis taking non-steroidal anti-inflammatory drugs. Br J Rheumatol 1996;35:458–462. [DOI] [PubMed] [Google Scholar]
- Smith OP, Jabbour HN, Critchley HOD. Cyclooxygenase enzyme expression and E series prostaglandin receptor signalling are enhanced in heavy menstruation. Hum Reprod 2007;22:1450–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogn JH, Curry TE, Jr, Brannstrom M, Lemaire WJ, Koos RD, Papkoff H, Janson PO. Inhibition of follicle-stimulating hormone-induced ovulation by indomethacin in the perfused rat ovary. Biol Reprod 1987;36:536–542. [DOI] [PubMed] [Google Scholar]
- Song SH, Lim H, Paria BC, Matsumoto H, Swift L, Morrow J, Bonventre JV, Dey SK. Cytosolic phospholipase A2α is crucial for ‘on-time’ embryo implantation that directs subsequent development. Development 2002;129:2879–2889. [DOI] [PubMed] [Google Scholar]
- Spencer AG, Woods JW, Arakawa T, Singer II, Smith WL. Subcellular localization of prostaglandin endoperoxide H synthases-1 and -2 by immunoelectron microscopy. J Biol Chem 1998;273:9886–9893. [DOI] [PubMed] [Google Scholar]
- Stahelin RV, Rafter JD, Das S, Cho W. The molecular basis of differential subcellular localization of C2 domains of protein kinase C-alpha and group IVa cytosolic phospholipase A2. J Biol Chem 2003;278:12452–12460. [DOI] [PubMed] [Google Scholar]
- Stocco CO, Zhong L, Sugimoto Y, Ichikawa A, Lau LF, Gibori G. Prostaglandin F2alpha-induced expression of 20alpha-hydroxysteroid dehydrogenase involves the transcription factor NUR77. J Biol Chem 2000;275:37202–37211. [DOI] [PubMed] [Google Scholar]
- Sugimoto Y, Hasumoto K, Namba T, Irie A, Katsuyama M, Negishi M, Kakizuka A, Narumiya S, Ichikawa A. Cloning and expression of a cDNA for mouse prostaglandin F receptor. J Biol Chem 1994;269:1356–1360. [PubMed] [Google Scholar]
- Sugimoto Y, Yamasaki A, Segi E, Tsuboi K, Aze Y, Nishimura T, Oida H, Yoshida N, Tanaka T, Katsuyama M, et al. Failure of parturition in mice lacking the prostaglandin F receptor. Science 1997;277:681–683. [DOI] [PubMed] [Google Scholar]
- Sun T, Deng W-B, Diao H-L, Ni H, Bai Y-Y, Ma X-H, Xu L-B, Yang Z-M. Differential expression and regulation of prostaglandin E synthases in the mouse ovary during sexual maturation and luteal development. J Endocrinol 2006;189:89–101. [DOI] [PubMed] [Google Scholar]
- Suzuki-Yamamoto T, Nishizawa M, Fukui M, Okuda-Ashitaka E, Nakajima T, Ito S, Watanabe K. cDNA cloning, expression and characterization of human prostaglandin F synthase. FEBS Lett 1999;462:335–340. [DOI] [PubMed] [Google Scholar]
- Tai H-H. Prostaglandin catabolic enzymes as tumor suppressors. Cancer Metastasis Rev 2011;30:409–417. [DOI] [PubMed] [Google Scholar]
- Tai HH, Cho H, Tong M, Ding Y. NAD+-linked 15-hydroxyprostaglandin dehydrogenase: Structure and biological functions. Curr Pharm Des 2006;12:955–962. [DOI] [PubMed] [Google Scholar]
- Tai HH, Tong M, Ding Y. 15-Hydroxyprostglandin dehydrogenase (15-PGDH) and lung cancer. Prostaglandins Other Lipid Mediat 2007;83:203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Morrow JD, Wang H, Dey SK. Cyclooxygenase-2 derived prostaglandin E2 directs oocyte maturation by differentially influencing multiple signaling pathways. J Biol Chem 2006;281:37117–37129. [DOI] [PubMed] [Google Scholar]
- Tanis BC, Van Den Bosch MA, Kemmeren JM, Cats VM, Helmerhorst FM, Algra A, Van Der Graaf Y, Rosendaal FR. Oral contraceptives and the risk of myocardial infarction. N Engl J Med 2001;345:1787–1793. [DOI] [PubMed] [Google Scholar]
- Tardif S, Carville A, Elmore D, Williams LE, Rice K. Reproduction and breeding of nonhuman primates. In: Abee CR, Mansfield K, Tardif S, Morris T. (eds) Nonhuman Primates in Biomedical Research. Waltham, MA, USA: Academic Press (Elsevier), 2012, 197–249. [Google Scholar]
- Thill M, Becker S, Fischer D, Cordes T, Hornemann A, Diedrich K, Salehin D, Friedrich M. Expression of prostaglandin metabolising enzymes COX-2 and 15-PGDH and VDR in human granulosa cells. Anticancer Res 2009;29:3611–3618. [PubMed] [Google Scholar]
- Tilley SL, Audoly LP, Hicks EH, Kim HS, Flannery PJ, Coffman TM, Koller BH. Reproductive failure and reduced blood pressure in mice lacking the EP2 prostaglandin E2 receptor. J Clin Invest 1999;103:1539–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trau HA, Davis JS, Duffy DM. Angiogenesis in the primate ovulatory follicle is stimulated by luteinizing hormone via prostaglandin E2. Biol Reprod 2015;92:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsafriri A, Lindner HR, Lamprecht SA. Physiological role of prostaglandins in the induction of ovulation. Prostaglandins 1972;2:1–10. [DOI] [PubMed] [Google Scholar]
- Uhler ML, Hsu JW, Fisher SG, Zinaman MJ. The effect of nonsteroidal anti-inflammatory drugs on ovulation: a prospective, randomized clinical trial. Fertil Steril 2001;76:957–961. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke JP, Rosing J, Bloemenkamp KW, Middeldorp S, Helmerhorst FM, Bouma BN, Rosendaal FR. Oral contraceptives and the risk of venous thrombosis. N Engl J Med 2001;344:1527–1535. [DOI] [PubMed] [Google Scholar]
- Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol 1998;38:97–120. [DOI] [PubMed] [Google Scholar]
- Wallach EE, de la Cruz A, Hunt J, Wright KH, Stevens VC. The effect of indomethacin on hMG-hCG induced ovulation in the rhesus monkey. Prostaglandins 1975a;9:645–658. [DOI] [PubMed] [Google Scholar]
- Wallach EE, Bronson R, Hamada Y, Wright KH, Stevens VC. Effectiveness of prostaglandin F2α in restoration of hMG-hCG induced ovulation in indomethacin-treated rhesus monkeys. Prostaglandins 1975b;10:129–138. [DOI] [PubMed] [Google Scholar]
- Wang YX, Ulu A, Zhang LN, Hammock B. Soluble epoxide hydrolase in atherosclerosis. Curr Atheroscler Rep 2010;12:174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassarman PM, Albertini DF. The mammalian ovum. In: Knobil E, Neill JD. (eds) The Physiology of Reproduction. New York, NY: Raven Press, 1994, 79–122. [Google Scholar]
- Weiss EA, Gandhi M. Preferential cyclooxygenase 2 inhibitors as a nonhormonal method of emergency contraception: a look at the evidence. J Pharm Pract 2014;1:5. [DOI] [PubMed] [Google Scholar]
- Wilcox AJ, Weinberg CR, Baird DD. Timing of sexual intercourse in relation to ovulation. N Engl J Med 1995;333:1517–1521. [DOI] [PubMed] [Google Scholar]
- Wissing ML, Kristensen SG, Anderson CY, Mikkelsen AL, Host T, Borup R, Grondahl ML. Identification of new ovulation-related genes in humans by comparing the transcriptome of granulosa cells before and after ovulation triggering in the same controlled ovarian stimulation cycle. Hum Reprod 2014;29:997–1010. [DOI] [PubMed] [Google Scholar]
- Wong WYL, Richards JS. Evidence for two antigenically distinct molecular weight variants of prostaglandin H synthase in the rat ovary. Mol Endocrinol 1991;5:1269–1279. [DOI] [PubMed] [Google Scholar]
- Wu M-H, Lu C-W, Chuang P-C, Tsai S-J. Prostaglandin E2: the master of endometriosis? Exp Biol Med 2010;235:668–677. [DOI] [PubMed] [Google Scholar]
- Yerushalmi GM, Salmon-Divon M, Yung Y, Maman E, Kedem A, Ophir L, Elemento O, Coticchio G, Dal Canto M, Mignini Renzinu M, et al. Characterization of the human cumulus cell transcriptome during final follicular maturation and ovulation. Mol Hum Reprod 2014;20:719–735. [DOI] [PubMed] [Google Scholar]
- Yung Y, Aviel-Ronen S, Maman E, Rubinstein N, Avivi C, Orvieto R, Hourvitz A. Localization of luteinizing hormone receptor protein in the human ovary. Mol Hum Reprod 2014;20:844–849. [DOI] [PubMed] [Google Scholar]