

Abstract
Retinal neurodegenerative diseases are especially attractive targets for gene replacement therapy, which appears to be clinically effective for several monogenic diseases. X-linked forms of retinitis pigmentosa (XLRP) are relatively severe blinding disorders, resulting from progressive photoreceptor dysfunction primarily caused by mutations in RPGR or RP2 gene. With a goal to develop gene therapy for the XLRP-RP2 disease, we first performed detailed characterization of the Rp2-knockout (Rp2-KO) mice and observed early-onset cone dysfunction, which was followed by progressive cone degeneration, mimicking cone vision impairment in XLRP patients. The mice also exhibited distinct and significantly delayed falling phase of photopic b-wave of electroretinogram (ERG). Concurrently, we generated a self-complementary adeno-associated viral (AAV) vector carrying human RP2-coding sequence and demonstrated its ability to mediate stable RP2 protein expression in mouse photoreceptors. A long-term efficacy study was then conducted in Rp2-KO mice following AAV-RP2 vector administration. Preservation of cone function was achieved with a wide dose range over 18-month duration, as evidenced by photopic ERG and optomotor tests. The slower b-wave kinetics was also completely restored. Morphologically, the treatment preserved cone viability, corrected mis-trafficking of M-cone opsin and restored cone PDE6 expression. The therapeutic effect was achieved even in mice that received treatment at an advanced disease stage. The highest AAV-RP2 dose group demonstrated retinal toxicity, highlighting the importance of careful vector dosing in designing future human trials. The wide range of effective dose, a broad treatment window and long-lasting therapeutic effects should make the RP2 gene therapy attractive for clinical development.
Introduction
The last decade has witnessed impressive advances in taking fundamental discoveries from bench to bedside, with a major impact on public health (1,2). Stem cell-based therapies and chemical screening approaches for drug discovery are evolving at a rapid pace. Gene therapy is finally beginning to reveal consistent effectiveness clinically (3); e.g. impressive results have been obtained for several monogenic inherited diseases, including lipoprotein lipase deficiency and hemophilia B (4,5). Retinal diseases offer a distinct advantage for developing gene therapy approaches to neurodegeneration because of relatively easy access to target cells for delivery and noninvasive methods for diagnosis and assessment of treatment outcome. Improvement in visual function by gene replacement in patients with Leber congenital amaurosis (LCA) caused by RPE65 mutations has created enthusiasm for expanding the approach to other inherited retinal degeneration (6).
Retinitis pigmentosa (RP) is the most common inherited retinal neurodegenerative disease with a prevalence of roughly 1 in 4000 in humans, afflicting over one million individuals worldwide (7–9). Although phenotypically and genetically heterogeneous, RP often involves common pathological changes that begin with rod photoreceptor dysfunction and/or degeneration followed by or concurrent death of cone cells. As a result, RP patients usually experience early onset of night blindness, with progressive loss of peripheral and then central vision, eventually leading to blindness. Over 60 genes have been associated with RP (9,10), which can exhibit autosomal dominant, autosomal recessive or X-linked pattern of inheritance. X-linked forms of RP (XLRP) are relatively severe, accounting for 10–20% of total RP (11,12). Though six genetic loci have been associated (10,13–15), mutations in the Retinitis Pigmentosa GTPase Regulator (RPGR) and Retinitis Pigmentosa 2 (RP2) genes account for almost 80% of all XLRP (16,17). In addition to rod disease that is present in both RPGR and RP2 patients, many RP2 patients show early-onset macular atrophy and poor visual acuity (18), suggesting the impairment of cone function independent of rod death. Thus, both rods and cones are considered as the target cells for treatment. We recently demonstrated successful development of gene replacement therapy for RPGR-associated retinal degeneration (19,20); however, such an approach has not been reported for RP2 disease.
The human RP2 gene consists of five exons and encodes a protein of 350 amino acid residues (21). In the retina, RP2 protein is targeted predominantly to the plasma membrane (22), likely mediated by post-translational acylation at its N-terminal end (23,24). RP2 is reported to act as GTPase activating protein (GAP) for adenosine-5′-diphosphoribosylation (ADP-ribosylation) factor-like 3 (ARL3) (25), a microtubule-associated small GTPase that localizes to the connecting cilia of photoreceptors (22,26). RP2 is suggested to control protein trafficking to the cilia, as loss of RP2 function in Rp2-knockout (RP2-KO) mouse models resulted in mis-trafficking of M-cone opsin (27) and diminished G protein-coupled receptor kinase 1 (GRK1) and cone phosphodiesterase 6 (PDE6) in cone outer segments (OS) (28). How RP2 regulates M-opsin trafficking is unclear; however, the absence of RP2-GAP activity is supposed to increase the level of ARL3-GTP, thereby forcing PDE6D, a prenyl-binding protein, to assume a predominantly ‘closed’ conformation (28) and impede its interaction with prenylated cargo such as GRK1 and PDE6. Both published RP2-KO mouse models exhibited severe and progressive cone degeneration in addition to relatively mild rod degeneration (27,28), likely caused by impediment of protein trafficking in both cone and rod photoreceptors. A recently developed rp2-KO zebrafish model (29) further supports the role of RP2 in directing protein transport in photoreceptors.
A vast majority of RP2 mutations in humans are predicted to result in loss of function (30); therefore, RP2 gene replacement is a viable option for therapy. The small size of the human RP2-coding sequence (1050 base-pairs) and the availability of Rp2-KO mice (27,28) permitted us to evaluate the gene replacement strategy using adeno-associated viral (AAV) vector as a delivery vehicle. In the present study, we report the natural history of photoreceptor dysfunction/degeneration in the Rp2-KO mice and the design, generation and characterization of AAV type 8 (AAV8) vector carrying a photoreceptor-specific human RP2 gene expression cassette. We have conducted a long-term dose efficacy study in mice and established the proof of concept for AAV-mediated RP2 gene delivery to efficiently rescue cone degeneration. Our study provides the foundation for the design of future clinical trials in patients with RP2 disease.
Results
Progressive cone-dominant photoreceptor dysfunction and degeneration in Rp2-KO retina
Rp2-KO mice were generated by crossing Rp2 flox/flox mice with either ZP3-Cre (31) or CAG-Cre line (27,32). In ZP3-Cre line, Cre is expressed specifically in oocytes. Although Cre is ubiquitously expressed in CAG-Cre line, Cre expression on its own does not affect retinal function, as shown in our previous work (27). We have replicated these findings (Supplementary Material, Fig. S1). Addition of CAG-Cre transgene even in the Rp2-KO line has no further impact of the retina. RP2 exon 2 was deleted in the resulting Rp2-KO mouse line, and no RP2 protein was detectable in the retina and other tissues (27) (data not shown). We first monitored a large cohort of Rp2-KO mice and their wild-type (WT) littermates by electroretinogram (ERG) analysis during 18-month period. Amplitude of dark-adapted a-wave is mainly contributed by rods. Though cone-derived a-wave is relatively small under light-adapted conditions, b-wave is produced by the inner retina neurons and reflects the activity of cone system. We, therefore, used dark-adapted a-wave and light-adapted b-wave to represent rod and cone functions, respectively. Consistent with previous observations (27,28), the Rp2-KO mice exhibited significantly reduced amplitudes of dark-adapted a-wave and light-adapted b-wave through the entire duration of the experiments (Fig. 1A and Supplementary Material, Fig. S2). This ERG amplitude reduction happened even as early as 1 month of age in a small group of mice we monitored (data not shown), indicating functional impairment of both rods and cones at an early age. However, measurement of the ratio of KO to WT for ERG amplitudes revealed distinct dynamics between rod and cone functions in the KO mice over the 18-month period. The dark-adapted a-wave amplitude of KO relative to that of WT remained stable after 4 months of age without additional reduction, whereas the KO to WT ratio of light-adapted b-wave amplitude continuously declined at a nearly constant rate between 4 and 18 months (Fig. 1B). As a result, about 78% of rod ERG amplitude was preserved at 18 months compared with only 33% of cone ERG amplitude, demonstrating a more severe impairment of cone function in the KO mice. Additionally, the relatively mild impairment in rod function did not significantly impact the inner retina function since no difference was observed between KO and WT mice for dark-adapted b-wave with dim flash intensity (Supplementary Material, Fig. S2). Figure 1C shows representative ERG waveforms from a KO mouse and its WT littermate. The progressive worsening of cone function in the KO retina is also reflected by the pronounced reduction in the flicker response (Fig. 1C).
Figure 1.
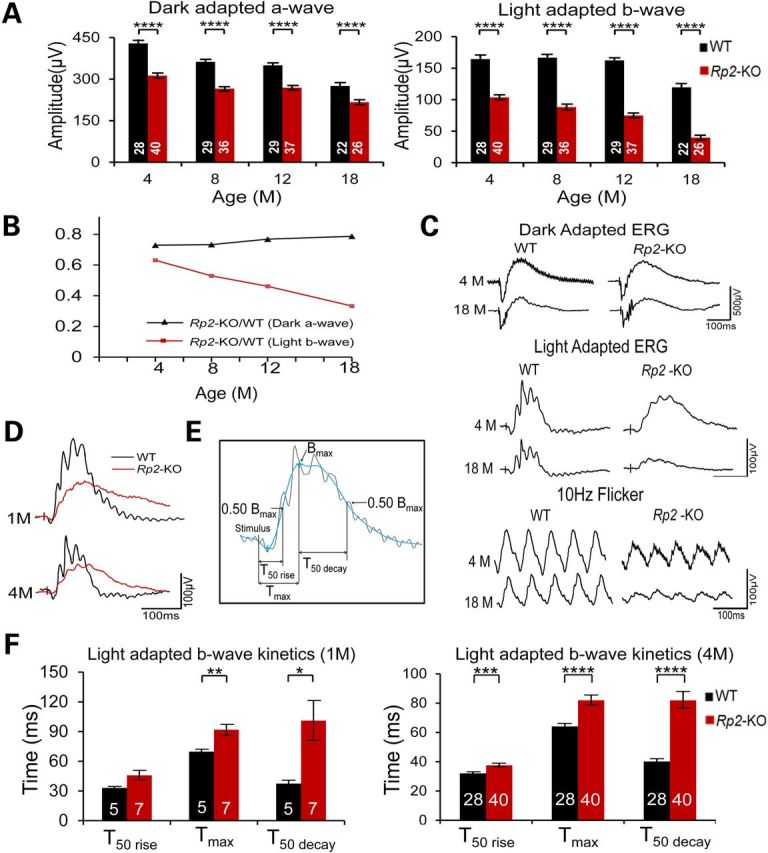
Progression of photoreceptor dysfunction in Rp2-KO mice. (A) ERG of Rp2-KO mice and their WT littermates over an 18-month period. The stimulus intensities for dark- and light-adapted ERGs were −4.0 to 3.0 and −1.0 to 2.0 log cd s/m2, respectively. Only the amplitudes with the highest flash stimuli are shown. The ERG response with a full range of flash stimuli is shown in the Supplementary Material, Figure S2. Significantly reduced amplitudes of dark-adapted a- (left penal) and light-adapted b-wave (right panel) were observed in Rp2-KO mice, starting from 4 months of age and through the entire duration. (B) The ratio between KO and WT of dark-adapted a-wave and light-adapted b-wave over the 18-month period. The ratio for light-adapted b-wave decreased at a nearly constant rate, whereas that for dark-adapted a-wave remained stable during 4–18 months of age. (C) Representative ERG traces including 10 Hz flicker at 4 and 18 months from one KO mouse and one WT littermate. (D) Overlay of light-adapted b-waves from an Rp2-KO and a WT mouse. (E) Illustration of the parameters used to characterize the kinetics of the light-adapted b-wave. T50 rise refers to the time from the stimulus onset to the 50% of the peak amplitude in the rising phase. Tmax refers to the time from the stimulus onset to the peak amplitude, same as b wave implicit time. T50 decay refers to the time from the peak amplitude to 50% of the peak amplitude in the falling phase. Calculations were based on filtered ERG trace shown in blue. The original ERG trace is shown in black. (F) Comparison between Rp2-KO and WT mice for light-adapted b-wave kinetics at 1 and 4 months. In addition to a significantly prolonged implicit time, the KO mice exhibit a much slower kinetics of the b-wave falling phase. In (A) and (F), the number of mice tested in each group is indicated by white lettering inside each bar. Data from WT and KO mice were compared by two-tailed unpaired t-test and represented as mean ± SEM. *P < 0.05, **P < 0.01, ****P < 0.0001.
A significant alteration in light-adapted b-wave kinetics was observed in Rp2-KO mice when compared with their WT littermates (Fig. 1D and F), consistent with a previous report (28). To assess the response kinetics, we measured the time it takes the b-wave to rise to 50% of its peak amplitude (T50 rise), the time to reach the peak amplitude (Tmax, same as implicit time) and the time to fall from the peak to 50% of the peak amplitude (T50 decay) (Fig. 1E). The 4-month-old KO mice displayed significantly longer time course in all three measurements than their WT littermates (Fig. 1F, right panel). In particular, the kinetics of the b-wave falling phase was distinctly slower in KO mice compared with WT, as reflected by much longer T50 decay time (40.1 ± 1.6 ms in WT versus 81.8 ± 5.5 ms in KO, mean ± SEM). This alteration in kinetics began early, as longer Tmax and T50 decay were already observed in 1-month-old KO mice (Fig. 1F, left panel). The kinetics difference between KO and WT mice appeared to be specific to cone system, as no such change was observed in dark-adapted b-wave under low flash stimulus intensity, which reflects the function of pure rod system (data not shown).
In concordance with the cone-mediated ERG changes, much fewer M- and S-cone opsin expressing cells were detected in the KO retina at 18 months of age compared with the WT retina (Fig. 2A). In contrast, rod cells did not show obvious defects. Similar levels of rhodopsin expression were observed in both KO and WT retinas with normal localization in OS, and the thickness of rod-dominant photoreceptor layer was not significantly altered even in the 18-month-old KO retina (Fig. 2B). The relatively mild rod dysfunction in the KO mice is likely caused by somewhat disorganized OS as revealed by ultrastructural analysis (27). Rod disorganization was not captured by our light microscopy analyses.
Figure 2.
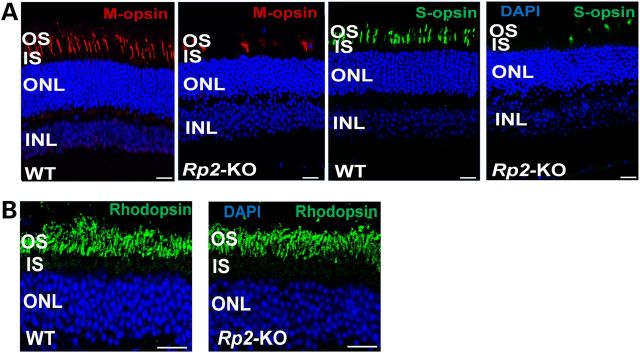
Cone degeneration in 18-month-old Rp2-KO mice. Age-matched WT C57/Bl6 mice were used as controls. (A) M- and S-opsin staining of retinal sections. Markedly reduced numbers of M- and S-opsin expressing cells in the KO retina were observed compared with those in the WT retina. (B) Rhodopsin staining of retinal sections. No obvious differences were observed between KO and WT mice in rhodopsin expression and its intracellular localization, and thickness of rod-dominant photoreceptor layer. Scale bar 20 μm. OS, outer segments; IS, inner segments; ONL, outer nuclear layer; INL, inner nuclear layer.
Stable expression of human RP2 protein in mouse photoreceptors by AAV8-RP2 vector
We designed and constructed an AAV vector carrying a human RP2 expression cassette that was composed of a photoreceptor-specific human rhodopsin kinase (RK) promoter (33), a Cytomegalovirus (CMV) and human β-globin hybrid intron, human RP2-coding sequence and the human β-globin polyadenylation site (20), flanked by two inverted terminal repeats (ITRs) from AAV serotype 2 (AAV2) (Fig. 3A). The RK promoter works efficiently in both rods and cones in mice, as previously reported (33). The RP2 expression cassette is <2 kb; therefore, we generated a self-complementary (sc) AAV vector capable of mediating an early and more efficient transgene expression compared with the conventional single-stranded (ss) vector (34–38). The sc vector was constructed by replacing one WT ITR by a mutant ITR in which the terminal resolution site and D sequence of AAV2 were deleted (34,35). The construct was packaged into AAV8, one of the naturally occurring AAV serotypes that transduce photoreceptors of mouse and non-human primate most efficiently (37,39,40), and was designated as AAV8-scRK-hRP2 vector.
Figure 3.
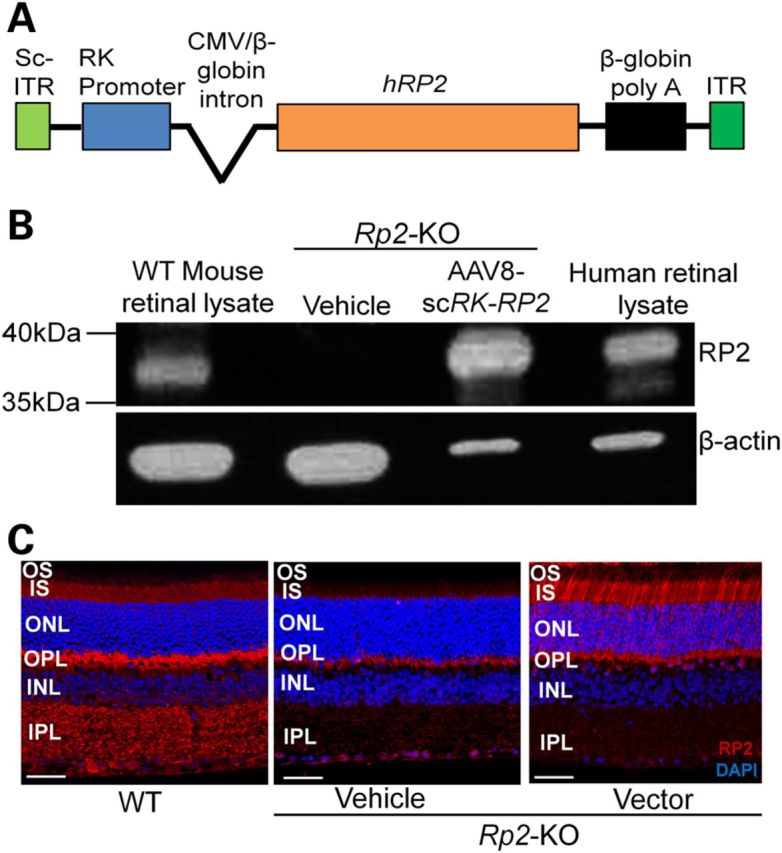
Human RP2 AAV vector and its expression in Rp2-KO retina. (A) Schematic representation of the vector. (B) Immunoblot analysis using an RP2 antibody that recognizes both mouse and human RP2 proteins. The retinal lysate from an Rp2-KO mouse receiving subretinal administration of the AAV8-scRK-hRP2 vector for 1 month revealed a ∼40 kDa protein band corresponding to the human RP2 protein, which is similar to that detectable in human retinal lysate. The retinal lysate from a WT mouse revealed a band that migrates slightly faster than that from the human. β-Actin was used as loading controls. (C) Immunostaining of retinal sections from Rp2-KO mice 1 month after they received subretinal injection of vector or vehicle, using an antibody against both mouse and human RP2 proteins. A WT C57/Bl6 retinal section was used as positive control. Endogenous mouse RP2 protein was detected in multiple layers in WT retina, including the IS, OPL and IPL, which was not seen in the Rp2-KO retina. The vector-expressed human RP2 protein was primarily observed at the IS and nuclei of photoreceptors. Scale bar 50 μm. OS, outer segments; IS, inner segments; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer.
We performed subretinal injections of the vector into RP2-KO mouse retina and examined the expression of RP2 protein 4 weeks later by immunoblot analyses of retinal extracts using a polyclonal antibody that recognizes both mouse and human RP2 proteins (Fig. 3B). While the vehicle-treated retina did not reveal any RP2-specific band, the vector-treated retina showed a protein of expected molecular weight (∼40 kDa), which was similar to that observed in the human retinal lysate. The endogenous mouse RP2 protein in the WT retina migrated somewhat faster than the human ortholog. As mouse and human RP2 proteins contain 347 and 350 residues, respectively, the electrophoretic mobility difference might reflect distinct amino acid composition and/or post-translational modification of the two proteins.
Endogenous mouse RP2 protein is detected in the photoreceptor inner segments (IS), as well as in other cell and plexiform layers (outer and inner plexiform layers, OPL and IPL, respectively) but was not present in the Rp2-KO retina. The vector-expressed human RP2 protein was primarily localized in the IS and nuclei of photoreceptors (Fig. 3C). Lack of RP2 immunostaining in other layers of the retina likely reflects the specificity of RK promoter and/or the inaccessibility of the vector to inner retina following subretinal administration. The vector-mediated RP2 expression was sustained throughout the entire 18-month study period (Supplementary Material, Fig. S3).
Preservation of cone function in Rp2-KO mice by AAV8-RP2 vector delivery over a wide dose range
The patients with RP2 mutations usually exhibit early-onset symptoms during the first or second decade of life. We, therefore, chose young adult (4- to 6-week-old) Rp2-KO mice to evaluate whether the vector treatment would rescue the disease. These mice received the vector injection subretinally in one eye at a dose of 1 × 108, 3 × 108 or 1 × 109 vector genomes (vg), with the contralateral eye receiving vehicle injection as control. A longitudinal ERG monitoring was performed until the mice reached 18 months of age. Given the large variation in ERG amplitudes among individual mice, two-tailed paired t-test was employed to compare the vector- and the vehicle-treated eyes. Rescue of cone function was achieved in both the 1 × 108 and the 3 × 108 vg/eye dose groups, as revealed by significantly higher light-adapted b-wave amplitudes in vector-treated eyes compared with fellow control eyes (Fig. 4A and Supplementary Material, Figs. S4 and S5). This therapeutic effect was observed as early as 4 months of age, the earliest time point of ERG examination, and lasted throughout the entire duration of the study period. Almost 75% (71–78%) of photopic b-wave amplitude was preserved in vector-treated eyes at 18 months in contrast to only ∼28% remaining in the control eyes. The waveforms of light-adapted ERG from a treated KO mouse and an age-matched WT mouse are shown in Figure 4B. In addition to preservation of light-adapted b-wave amplitude, the treatment completely corrected the alteration of b-wave kinetics in the KO retina, as revealed by nearly normal T50 rise, Tmax and T50 decay measured in the vector-treated eyes of 4-month-old mice (Fig. 4C). In contrast to the pronounced rescue of cone function, 1 × 108 vg/eye vector treatment appeared to be neither therapeutic nor toxic to rods, as no significant difference was observed between vector- and vehicle-treated eyes in rod ERG response (dark-adapted a-wave) during the 18-month study period (Supplementary Material, Fig. S4). Similarly, 3 × 108 vg/eye vector treatment had no obvious effect on rods in general, although slightly lower dark-adapted response was observed at certain time points (Supplementary Material, Fig. S5). The lack of therapeutic effect on rods may be explained by early onset (within 1 month of age and before vector administration, data not shown), though slower progression, of functional impairment in rods of Rp2-KO mice. Mice receiving the highest 1 × 109 vg/eye dose displayed retinal toxicity, which is discussed later.
Figure 4.
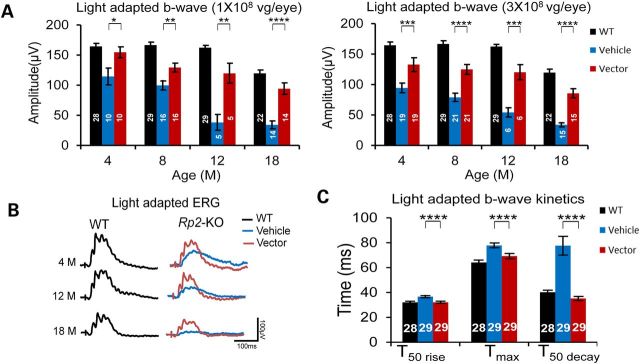
Preservation of ERG response in Rp2-KO mice that received vector treatment. Mice received unilateral injections of the AAV8-scRK-hRP2 vector and contralateral injections of vehicle at 4–6 weeks of age. (A) Light-adapted b-wave amplitude of Rp2-KO mice injected with 1 × 108 (left panel) or 3 × 108 vg/eye (right panel) of the vector. The stimulus intensities were from −1.0 to 2.0 log cd s/m2. Only the amplitude with the highest flash stimulus is shown. The ERG response with a full range of flash stimuli is shown in the Supplementary Material, Figure S4 and S5. Sustained higher amplitude was observed in vector-treated eyes than control eyes from 4 to 18 months of age. The number of mice tested in each group is indicated by white lettering inside each bar. The ages of the tested mice are indicated in the X-axis. ERG amplitudes from vector- and vehicle-injected eyes were compared by two-tailed paired t-test and represented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. ERG of age-matched WT mice shown in Figure 1 was used as reference. (B) Overlay of light-adapted ERG traces from vehicle- and 3 × 108 vg vector-injected eyes of an Rp2-KO mouse at different time points. ERG trace from an age-matched WT littermate is shown as control. While slower-b wave kinetics was observed in the vehicle-treated eye, the vector-treated eye displays a similar kinetics to that of WT. (C) Comparison between vehicle- and vector-treated eyes of Rp2-KO for their photopic b wave kinetics at 4 months. Age-matched WT littermates were used as references. While the vehicle-injected eyes exhibit a much slower kinetics of the b wave especially at the falling phase as indicated by the T50 decay, the vector-injected eyes display normal kinetics as the WT. Data from vector- and vehicle-injected eyes were compared by two-tailed paired t-test and represented as mean ± SEM. ***P < 0.001. In (A) and (C), the number of mice tested in each group is indicated by white lettering inside each bar.
The remarkable rescue of cone function at the dose of 1 × 108 or 3 × 108 vg/eye prompted us to explore whether a lower dose could still be efficacious. We, therefore, administered the vector to one group of mice with a dose of 5 × 107 vg/eye and examined them by ERG at 6.5 and 18 months of age. The vector-treated eyes still exhibited significantly higher light-adapted b-wave amplitude compared with control eyes, indicating the vector's potency even at this low dose (Supplementary Material, Fig. S6). The rescue of cone function was not biased toward M- or S-cones, since vector-treated eyes displayed comparable preservation of M- and S-cone-driven ERG responses (Supplementary Material, Fig. S7).
Rp2-KO mice treated with 1 × 108 vg or 3 × 108 vg vector dose were also subjected to optomotor test under photopic condition at 19 months of age. Although lower than WT controls, the optomotor response of the vector-treated eyes was significantly higher than that of the vehicle-treated eyes (Fig. 5), indicating a better-preserved cone-mediated visual behavior by gene replacement.
Figure 5.

Preservation of optomotor response in Rp2-KO mice following vector treatment. Optomotor test was performed on 19-month-old Rp2-KO mice that received subretinal injections of AAV8-scRK-hRP2 vector at 4–6 weeks of age. Mice were injected unilaterally with a dose of either 1 × 108 or 3 × 108 vg/eye, with vehicle injected to the contralateral eyes as controls. Age-matched WT littermates were used as references. The number of mice used in each group is indicated by white lettering inside each bar. Spatial resolution (expressed as cycles/degree) from vector- and vehicle- treated eyes were compared by paired two-tailed t-test. All the values in the bar graph were represented as mean ± SEM. *P < 0.05, **P < 0.01.
Maintenance of normal cone protein expression in Rp2-KO retina by RP2 gene delivery
M-opsin localizes to OS in WT cones but is mis-trafficked to IS, peri-nuclei and synaptic terminals in the control Rp2-KO cones (Fig. 6A), consistent with our previous findings (27). Normal subcellular localization of M-opsin was observed in vector-treated retina, suggesting that the treatment either prevented or reversed M-cone mis-trafficking (Fig. 6A). No obvious S-opsin mis-localization was noted in either vehicle- or vector-treated KO retina (Fig. 6B). Consistent with a recent report (28), cone PDE6 expression was almost undetectable in the OS of vehicle-treated KO retina, whereas vector-treated retina retained nearly normal expression of the protein (Fig. 6C). Two rod-specific proteins, rhodopsin and PDE6β, primarily localize to the OS in WT retina; their expression or localization in KO retina was not affected by vector treatment (Supplementary Material, Figs. S8 and S9).
Figure 6.
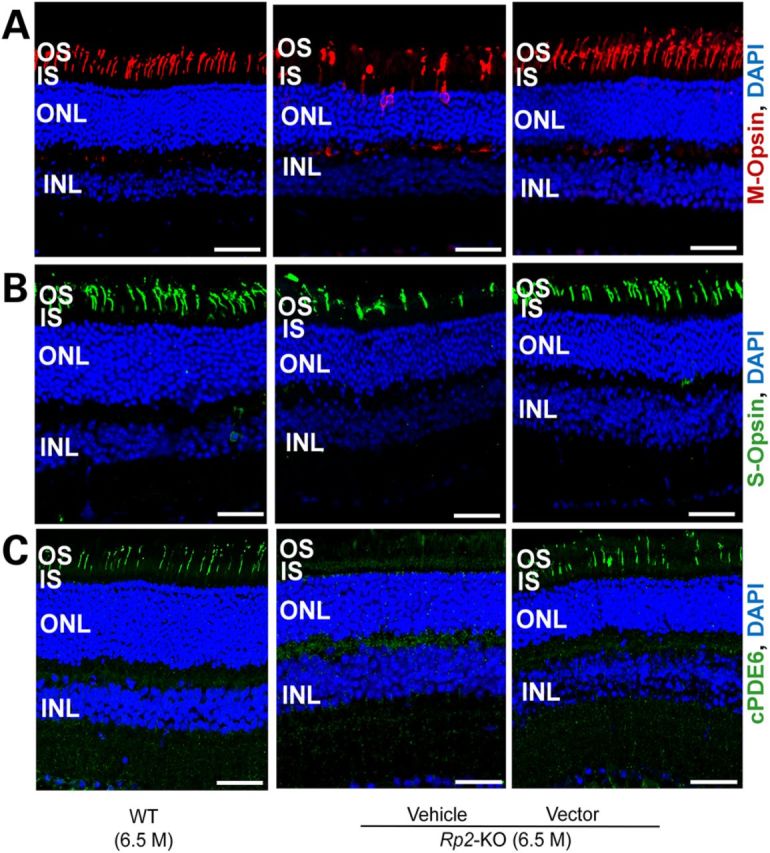
Correction of cone opsin mis-localization and restoration of cone PDE6 expression in Rp2-KO retina following vector treatment. Immunostaining of M-opsin (A), S-opsin (B) and cone PDE6 (C) of retinal sections from a 6.5-month-old Rp2-KO mouse that received vehicle or 5 × 107 vg AAV8-scRK-hRP2 vector administration at 4 weeks of age. Retinal sections from an age-matched WT C57/Bl6 mouse were used as controls. While M-opsin is mis-trafficked to OS, IS, nuclei and synaptic terminals in vehicle-treated retina, it is only localized to OS in vector-treated retina, similar to the WT mouse. Diminished cone PDE6 was observed in the vehicle-treated retina, whereas in the vector-treated retina, cone PDE6 restored and is localized to OS. Scale bar 50 μm. OS, outer segments; IS, inner segments; ONL, outer nuclear layer; INL, inner nuclear layer.
Pronounced cone preservation in vector-treated eyes was observed at 18 months, as revealed by a significantly higher number of peanut agglutinin (PNA)-stained cells in both superior and inferior retina (Fig. 7A and B) compared with those of the control eyes. Additionally, higher numbers of M- and S-cones were noticed in both the retinal whole mounts (Fig. 7A) and sections (Fig. 7C) in vector-treated eyes than in those of the control eyes.
Figure 7.

Long-term preservation of cone photoreceptors in Rp2-KO mice following vector treatment. (A) Immunofluorescence analysis of retinal whole mounts of an 18-month-old Rp2-KO mouse that received subretinal administration of vehicle or 1 × 108 vg AAV8-scRK-hRP2 vector at 4 weeks of age. Quantification of cone cells in retinal whole mounts (B) revealed a significantly higher number of cone cells in vector-treated eyes compared with vehicle-treated control eyes. The number of mice tested in each group is indicated by white lettering inside each bar. Data from vector- and vehicle-injected eyes were compared by two-tailed paired t-test and represented as mean ± SEM. **P < 0.01. (C) Retinal sections showing a higher number of S- and M-opsin expressing cells in vector-treated eye than in vehicle-treated eye. Scale bar 50 μm. OS, outer segments; IS, inner segments; ONL, outer nuclear layer; INL, inner nuclear layer.
Dose escalation studies of the AAV8-RP2 vector in Rp2-KO mice
Vector doses ranging from 5 × 107 to 3 × 108 vg/eye were efficacious in rescuing cone function and viability in Rp2-KO mice. These doses, though could not restore the mildly impaired rod function, were in general non-toxic to rods based on our ERG analyses (Supplementary Material, Figs. S4–S6). However, the dose of 1 × 109 vg/eye was apparently toxic to rods, as reflected by markedly reduced amplitudes of dark-adapted a- and b-waves (Fig. 8A and Supplementary Material, Fig. S11). Although this dose preserved cone function at 4 and 8 months, this beneficial effect diminished at 18 months, probably due to secondary cone cell death caused by eventual loss of rods. Immunofluorescence analyses at 18 months of age revealed much thinner or no outer nuclear layer (ONL) in multiple regions of the 1 × 109 vg-treated eye, in contrast to no obvious change in the 1 × 108 vg-treated eye (Fig. 8B). Therefore, the dose of 1 × 109 vg/eye was toxic to the retina.
Figure 8.

Retinal toxicity at high vector dose. (A) Dark- and light-adapted ERG responses at different time points in Rp2-KO mice treated with AAV8-scRK-hRP2 vector. Mice were injected unilaterally with 1 × 109 vg/eye of the vector when they were 4–6 weeks old, with the vehicle injected to the contralateral eyes as controls. The number of mice tested in each group is indicated by white lettering inside each bar. The stimulus intensities for dark- and light-adapted ERGs were −4.0 to 3.0 and −1.0 to 2.0 log cd s/m2, respectively. Only the amplitudes with the highest flash stimuli are shown. The ERG response with a full range of flash stimuli is shown in the Supplementary Material, Figure S11. ERG amplitudes from vector- and vehicle- injected eyes were compared by two-tailed paired t-test and represented as mean ± SEM. *P < 0.05, **P < 0.01, ****P < 0.0001. Lower dark-adapted ERG response in the vector-treated eyes indicates retinal toxicity likely caused by high RP2 expression. (B) RP2 staining of retinal sections from 18-month-old Rp2-KO mice that received injections of 1 × 109 or 1 × 108 vg/eye of the vector. Thinning of ONL was observed at multiple regions in the retina treated with 1 × 109 vg/eye, while relatively normal ONL thickness was maintained in the 1 × 108 vg-treated retina. The magnified images of the marked areas are shown. Scale bar 50 μm. IS, inner segments; ONL, outer nuclear layer; INL, inner nuclear layer.
Prevention of cone dysfunction and loss in Rp2-KO mice by RP2 gene delivery at late stages
To assess whether Rp2-KO mice with more advanced cone dysfunction and degeneration would still benefit from the treatment, we administered the vector to 10-month-old Rp2-KO mice at a dose of 3 × 108 vg/eye and examined their retinal function and structure at 18 months of age. Although the therapeutic effect was not as remarkable as in the case of early intervention (Fig. 4A), the vector-treated eyes still displayed significantly higher light-adapted b-wave amplitude than vehicle-treated eyes (Fig. 9A and B and Supplementary Material, Fig. S10). Consistently, substantial M- and S-opsin and cone PDE6 expressing cells were preserved in the vector-treated retina, in contrast to the vehicle-treated retina (Fig. 9C).
Figure 9.

Preservation of cone function and cone proteins in Rp2-KO mice following vector treatment at an older age. (A) Light-adapted ERG b-wave amplitude of 18-month-old Rp2-KO mice that received subretinal injection of AAV8-scRK-hRP2 vector at 10 months of age. Mice were injected unilaterally with the vector at a dose of 3 × 108 vg/eye, with the vehicle injected to the contralateral eyes as controls. The stimulus intensities were from −1.0 to 2.0 log cd s/m2. Only the amplitude with the highest flash stimulus is shown. The ERG response with a full range of flash stimuli is shown in the Supplementary Material, Figure S10. The number of mice tested in each group is indicated by white lettering inside each bar. ERG amplitudes from vector- and vehicle-injected eyes were compared by two-tailed paired t-test and represented as mean ± SEM. **P < 0.01. The vector-injected eyes exhibited significantly higher amplitude than control eyes. (B) Representative light-adapted and 10 Hz flicker ERG traces. (C) Immunofluorescence of retinal sections. Higher numbers of S- and M-opsin expressing cells were maintained in the vector-treated retinal sections. Cone PDE6 (cPDE6) expression was also restored in the OS of the vector-treated retina. Scale bar 50 μm. OS, outer segments; IS, inner segments; ONL, outer nuclear layer; INL, inner nuclear layer.
Discussion
Gene replacement therapy is an attractive approach for treating monogenic retinal diseases because of relatively easy access to target cells for gene delivery. To date, almost 250 genes have been associated with clinical phenotypes that include retinal dysfunction (RetNet, https://sph.uth.edu/Retnet/sum-dis.htm); many of these are potential targets for therapy by replacement of the normal gene. AAV and lentivirus vectors are efficient in transducing retinal cells and are being tested in human clinical trials (6). With the initial success in RPE65-LCA patients, gene replacement therapy for several retinal degenerative diseases, including choroideremia, Usher syndrome type 1B, Stargart's disease and X-linked retinoschisis, has rapidly entered clinical stage (https://clinicaltrials.gov/). Like most inherited retinal degeneration, RP2-XLRP afflicts a small patient population but gained our attention for developing gene therapy because of early onset of disease and relatively severe vision impairment in affected individuals. The disease is a good candidate for gene replacement since the RP2-coding sequence is small enough to be delivered by an AAV vector and RP2 mutations primarily affect photoreceptors, which can be efficiently transduced by AAV. In the present study, we report long-term dose efficacy profile for the treatment of RP2 disease by an AAV vector in Rp2-KO mouse model, establishing the framework for future clinical development.
A therapeutic vector should provide effective treatment with minimal safety concerns. Compared with the other viral vectors, AAV exhibits a much better safety profile due to its low integration frequency and immunogenicity and non-pathogenicity of its parental form (41). However, a large amount of vector payload could be toxic to the retina due to limited capacity of retinal cells for processing the capsid protein and vector DNA, as suggested in our recent efficacy/toxicity study for RPGR-XLRP disease in mice (20). An ideal vector should be able to provide adequate transgene expression at a relatively low dose, with minimal adverse effects owing to gene delivery. To meet this requirement, we adopted a combinatorial strategy in which optimal AAV serotype, vector genome form (scAAV instead of ssAAV) and transcriptional elements for photoreceptor transduction were incorporated into the RP2 vector design. AAV8 appears to be better than the other commonly used serotypes (such as AAV2 and AAV5) in transducing photoreceptors of mouse and/or non-human primates as shown in a few previous studies (37,39,40) and was therefore chosen in the present study. A 20-fold vector dose range (5 × 107 to 109 vg/eye) was tested for its efficacy and toxicity, and except for the highest dose, administration of 3 × 108, 1 × 108 or 5 × 107 vg/eye was able to provide long-term preservation of cone function and viability in the Rp2-KO mice. The lowest dose, 5 × 107 vg/eye, if converted to human application, will be 3.5 × 109 vg/eye, considering about 70-fold difference between mouse and human retinal surface areas. Although the transgene and the AAV serotype are different, this dose is far below the lowest AAV vector dose that has been tested in the RPE65 clinical trial (1.5 × 1010 vg/eye) (42). Our results suggest that the vector could be efficacious and well tolerated if applied to XLRP-RP2 patients. Not surprisingly, 1 × 109 vg/eye dose lead to high level of human RP2 expression and exhibited retinal toxicity, especially to rod photoreceptors. This toxicity is less likely caused by the vector capsid and genome since a similar dose of AAV8 was well tolerated in mice (20,43,44).
In our recent (27) and current studies, mis-trafficking of M-cone opsin was observed in Rp2-KO retina, likely due to defect in cilia trafficking caused by the loss of RP2 protein (27). Consistent with previous observations (28), expression of cone PDE6 was diminished in cone OS of the KO retina. As a consequence of the lack of RP2-GAP function, the resulting high concentration of hyperactive ARL3-GTP could have impeded the interaction of PDE6D with its prenylated cargo, such as GRK1 and PDE6. In addition, we replicated the finding of significant delay in the falling phase of photopic b-wave in the KO mice (28). Similar patterns of prolonged falling phase of photopic b-wave have been observed in Pde9a-KO mouse (45) and in tetrodotoxin (TTX) or cis-2,3-piperidine-dicarboxylic acid (PDA)-treated mice (46). Mechanism underlying this altered kinetics of photopic ERG by RP2 ablation is unclear and is beyond the scope of our current study. The diminished GRK1 in cone OS, which delays the deactivation of opsin-mediated phototransduction cascade, could partially account for this observation (28), but the involvement of cells in inner retinal layers cannot be ruled out. These pathological processes, including mis-trafficking or loss of cone protein and the alteration in ERG kinetics, were either prevented or reversed in mice receiving RP2 gene delivery.
A sufficient number of photoreceptors should remain in the degenerating retina for gene replacement therapy to be successful, thus requiring an early intervention. However, such a scenario is often difficult in patients with early-onset and rapidly progressing retinal degeneration. Administration of vectors in model animals with substantial retinal degeneration may simulate the paradigm for the majority of patients who have already presented severe clinical symptoms at the time of diagnosis or treatment. Although not as pronounced as early intervention, our studies demonstrate that gene delivery even at a late stage of disease might partially rescue RP2 loss-of-function defects, thus providing hope for older XLRP-RP2 patients.
Our current vector dose efficacy/toxicity profile obtained in the Rp2-KO mice can be extrapolated to patients with predicted null alleles. It will be of interest to determine whether vector dose responses are different in mouse models with other Rp2 mutations (e.g. missense, truncation or splicing mutations). In addition, gain-of-function RP2 mutations, though not identified as yet, would likely require other treatment strategies including gene knock-down. Whether gene replacement would work in the presence of a mutant protein is worth investigation.
The RP2-KO mice used in our current study displayed cone-dominant retinal phenotype. Although rod dysfunction was also observed, the cone impairment was far more severe as reported previously (27,28). The mechanism underlying this discrepancy between rod and cone dysfunction in response to loss of RP2 remains to be elucidated. Since rod-related symptoms are presented in a majority of RP2 patients, additional animal models might be required to evaluate the efficacy of the vector for treating both rod and cone impairments. Regardless, the therapeutic AAV8-RP2 vector developed in the present study is clearly efficacious in preserving cone function. As the first report of gene replacement therapy for RP2 disease, our study is attractive for clinical development because of the wide effective vector dose range, a broad treatment window and long-lasting therapeutic effects that are obvious in the treated Rp2-KO mice.
Materials and Methods
Generation of the Rp2-KO mouse line and animal husbandry
Studies conform to the ARVO statement for the Use of Animals in Ophthalmic and Vision Research. NEI Animal Care and Use Committee approved all animal protocols. Rp2-KO mouse line was created by crossing Rp2flox/flox mice (27) with a Cre-expressing line (CAG-Cre or Zp3-Cre line) (31,32). All mice were maintained in NIH animal care facility in controlled ambient illumination on a 12 h light/12 h dark cycle.
AAV vector construction and production
To generate the therapeutic AAV vector, a 1073 bp DNA fragment containing the human RP2-coding sequence (NCBI Ref. Seq. No.: NM_006915.2) with flanking Cla I and Xho I sites was synthesized and sequence-verified (DNA2.0, Menlo Park, CA, USA). The fragment also contains GCCACC Kozak sequence just upstream of the start codon. This fragment was digested with Cla I and Xho I restriction enzymes and cloned into an existing AAV shuttle plasmid maintained in laboratory. It was inserted in between an upstream RK promoter and a chimeric (β-globin/CMV) intron and a downstream β-globin polyadenylation signal. AAV type 2 ITRs were used in the AAV vector plasmid construction. To make a sc AAV vector, the left ITR (ITR near the promoter region) was modified to eliminate the terminal resolution site and AAV D sequence. The resultant plasmid was named as pV4.7scRK-RP2.
Triple-plasmid transfection to HEK293 cells was used to produce the AAV-RP2 vector as described previously (47). The sc AAV human RP2 construct was packaged into AAV8 capsid. The vector was purified by polyethylene glycol precipitation followed by cesium chloride density gradient fractionation, as described earlier (47). The purified vector was maintained in solution containing 10 mm Tris–HCl, 180 mm NaCl and pH 7.4. The virus was titered by real-time polymerase chain reaction using the following primers and fluorescent-labeled probes:
Forward primer: 5′ GCACCTTCTTGCCACTCCTA 3′
Reverse primer: 5′ GACACAGCACCAGGCTAAATCC 3′
Probe: 5′ FAM-CGTCCTCCGTGACCCCGGC-TAMRA 3′
Linearized plasmid DNA was used as standard to quantify the virus.
Subretinal injection
Subretinal injections were performed as previously described (48) with some modifications. Mice were anesthetized with an intra-peritoneal injection of ketamine (80 mg/Kg) and xylazine (8 mg/Kg). The pupils were dilated with topical atropine (1%) and tropicamide (0.5%). Proparacaine (0.5%) was used as topical anesthesia. Surgery was performed under an ophthalmic surgical microscope. An 18-gauge sharp needle was used to make a small incision in the cornea adjacent to the limbus. A 33-gauge blunt needle fitted to a Hamilton syringe was inserted through the incision while avoiding the lens and pushed through the retina. One microliter of either therapeutic AAV vector or vehicle (10 mm Tris–HCl, 180 mm NaCl, pH 7.4) was delivered subretinally to the eye. The vector was given in the right eyes, and the vehicle was given in the left eyes. Visualization during injection was aided by addition of fluorescein (100 mg/ml AK-FLUOR, Alcon, Fort Worth, TX, USA) to the vector suspension at 0.1% by volume.
Electroretinogram (ERG)
ERGs were performed using Espione E2 system (E2, Diagnosys LLC, Lowell, MA, USA). Mice were dark-adapted overnight. Pupils were dilated with topical phenylephrine (2.5%) and tropicamide (0.5%). Mice were anesthetized with an intra-peritoneal injection of ketamine (80 mg/Kg) and xylazine (8 mg/Kg). All the above procedures were conducted in dim red light. ERGs were recorded from both the eyes using gold wire loops with 0.5% proparacaine topical anesthesia and a drop of 2.5% hypromellose ophthalmic demulcent solution for corneal hydration. A gold wire loop placed in the mouth was used as reference, and a ground electrode was placed on the tail. Flash intensities for dark-adapted ERG ranged from −4.0 to +3.0 log cd s m−2/flash. Light-adapted ERGs were recorded after 2 min of adaptation to a white 32 cd m−2 rod-suppressing background. ERG recording was conducted with a brief white flash intensity ranging from −0.53 to +2.0 log cd s m–2/flash. The flicker response was taken with 10 Hz light flicks. The responses were computer averaged and recorded at 3–60 s intervals depending on flash intensity. For recording M- and S-cone-mediated ERG response, the mice were first light adapted for 2 min in green light with 20 cd m−2 intensity. ERG was recorded by alternating green and UV flashes. The intensities ranged from 0.1671 × 104 to 55.7 × 104 photon/µm2 for green flashes and from 0.0113 × 104 to 33.9 × 104 photon/µm2 for UV flashes, with a background green illumination of 20 cd m−2. ERG analyzer that employs a zero-phase low-pass digital filter and a program built in MatLab by NEI Visual Function Core was used to remove oscillatory potentials in recorded ERG waveforms.
Optomotor test
Visual behavior of the mice was determined by optomotor test using OptoMotry developed by Cerebral Mechanics (http://www.cerebralmechanics.com/) following the protocol described elsewhere (49,50). Briefly, the mouse was placed at the center of closed OptoMotry surrounded by four computer screens. A camera was place on the top to monitor the movement of the mouse. The computer screens created a virtual image of rotating drum with sine-wave grating. The tracking of the rotating gratings by the mouse was scored by its head and neck movements. The spatial frequency of the grating was controlled and monitored by OptoMotry© software (Ver14). The maximum spatial frequency in 100% background contrast that generated a tracking movement of the animal was recorded for each eye.
Immunoblotting
Whole retinal lysate was prepared in RIPA buffer containing protease inhibitor cocktail by sonication. The lysate was cleared by brief spinning, and the protein concentration is estimated using the Bradford reagent. Approximately 20 µg of protein was loaded in each lane of 10% sodium dodecyl sulphate-polyacrylamide gel (BioRad USA, Cat. No. 456–8033). Immunoblot was performed using a protocol described previously (20). The primary antibodies used included a rabbit polyclonal antibody against polyhistidine-conjugated full-length mouse RP2 protein (Geneway Biotech, San Diego, CA) and a mouse monoclonal anti-β actin antibody (Sigma, Cat. No. A5316). The proteins were visualized with peroxidase-conjugated goat anti-rabbit or goat anti-mouse secondary antibody using SuperSignal West Pico Chemiluminescent substrate (Thermo Fisher Scientific, Inc., Rockford, IL, Cat. No. 34087).
Immunofluorescence
Mice were euthanized, and the eyes were enucleated and fixed in 4% paraformaldehyde (PFA) solution for 1–2 h and passed through a series of sucrose solution for cryo-protection and flash frozen in OCT solution. A series of 12 µm sections was cut through the eyes in superior–inferior pole orientation by cryostat. The sections were stained with specific antibodies using the following protocol. Sections were blocked in 5% goat serum in phosphate-buffered saline (PBS) containing 0.1% Triton X-100 (PBST) for 1 h, followed by incubation with primary antibodies diluted in 2% goat serum at 4°C overnight. Sections were washed three times in PBST and incubated with fluorochrome-conjugated secondary antibodies and 0.2 µg/ml DAPI for 1 h. Sections were washed again with PBST and mounted in Fluoromount-G (SouthernBiotech, Birmingham, AL). Images were captured on a confocal scanning microscope LSM700 (Zeiss, Germany).
To prepare flat mount of retina, enucleated eyes from euthanized mice were first incubated in chilled PBS solution for 15 min over ice. The eye balls were squeezed gently several times to detach the retina. The eye balls were then fixed in 4% PFA for 1 h, and the retinas were separated from other parts of the eye, washed with PBST and blocked in 5% goat serum in PBST for 4 h. The retina whole mounts were then incubated with primary antibodies diluted in 2% goat serum at 4°C overnight with slow agitation. The whole mounts were washed three times (twice for 45 min each and once for 1 h) in PBST, incubated with fluorochrome-conjugated secondary antibodies for 4 h. They were then washed again in PBST and mounted in Fluoromount-G, with photoreceptor layers facing up. Imaging was conducted on confocal scanning microscope LSM700.
The primary antibodies used for immunofluorescence included rabbit polyclonal antibody against mouse RP2 protein as described earlier, mouse monoclonal antibody against rhodopsin (1D4, Santa Cruz Biotechnology, Dallas, TX, Cat. No. sc-57432), rabbit polyclonal antibody against M-opsin (Millipore, Billerica, MA, Cat. No. AB5404), chicken polyclonal antibody against S-opsin (48), rabbit polyclonal antibody against cone PDE6 (48) and rabbit polyclonal antibody against rod PDE (rod PDE β subunit; Affinity BioReagent, Golden, CO). Secondary antibodies included goat anti-rabbit, anti-mouse or anti-chicken antibodies conjugated with Alexa Fluor 488,568 or 647 (Life Technologies, Grand Island, NY, USA). DAPI and Alexa Fluor 594-conjugated peanut hemagglutinin (PNA) (Life Technologies) were used to stain the nuclei and cone photoreceptors, respectively.
Quantification of cones in mouse retina
For quantification of cone cells in vector- and vehicle-treated eyes, flat mounts of retina stained with PNA were used. PNA-stained cells from 10 randomly selected non-overlapping 0.01 mm2 areas (5 from superior and 5 from inferior retina) were counted. From the numbers obtained, total projected numbers of cone cells in 1 mm2 area were calculated. Cone cell numbers from vector- and vehicle-treated eyes were compared by two-tailed paired t-test.
Statistical analysis
Two-tailed unpaired t-test was performed to compare the results from Rp2-KO and WT mice. Two-tailed paired t-test was performed to compare the results from vector- and vehicle-injected eyes. GraphPad Prism 6 (GraphPad Software, La Jolla, CA) was used for statistical analysis.
Supplementary Material
Funding
This work was supported by the intramural research program of the National Eye Institute, National Institutes of Health (NIH), NIH grant EY022372 and Foundation Fighting Blindness.
Supplementary Material
Acknowledgements
We thank Yi-Sheng Chang, Yide Mi and Jacob Nellissery for their assistance. We would also like to thank Biological Imaging Core of National Eye Institute for their assistance with tissue preparation and immunofluorescence.
Conflict of Interest statement. None declared.
References
- 1.Collins F.S., Varmus H. (2015) A new initiative on precision medicine. N. Engl. J. Med., 372, 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collins F.S. (2015) Exceptional opportunities in medical science: a view from the National Institutes of Health. JAMA, 313, 131–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Reilly M., Federoff H.J., Fong Y., Kohn D.B., Patterson A.P., Ahmed N., Asokan A., Boye S.E., Crystal R.G., De Oliveira S., et al. (2014) Gene therapy: charting a future course—summary of a National Institutes of Health Workshop, April 12, 2013. Hum. Gene. Ther., 25, 488–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nathwani A.C., Reiss U.M., Tuddenham E.G., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D., et al. (2014) Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med., 371, 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scott L.J. (2015) Alipogene tiparvovec: a review of its use in adults with familial lipoprotein lipase deficiency. Drugs, 75, 175–182. [DOI] [PubMed] [Google Scholar]
- 6.Boye S.E., Boye S.L., Lewin A.S., Hauswirth W.W. (2013) A comprehensive review of retinal gene therapy. Mol. Ther., 21, 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hartong D.T., Berson E.L., Dryja T.P. (2006) Retinitis pigmentosa. Lancet, 368, 1795–1809. [DOI] [PubMed] [Google Scholar]
- 8.Estrada-Cuzcano A., Roepman R., Cremers F.P., den Hollander A.I., Mans D.A. (2012) Non-syndromic retinal ciliopathies: translating gene discovery into therapy. Hum. Mol. Genet., 21, R111–R124. [DOI] [PubMed] [Google Scholar]
- 9.Daiger S.P., Sullivan L.S., Bowne S.J. (2013) Genes and mutations causing retinitis pigmentosa. Clin. Genet., 84, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daiger S.P., Rossiter B.J.F., Greenberg J., Christoffels A., Hide W. (1998) Data services and software for identifying genes and mutations causing retinal degeneration. Invest. Ophthalmol.Vis. Sci., 39:S295. [Google Scholar]
- 11.Vervoort R., Lennon A., Bird A.C., Tulloch B., Axton R., Miano M.G., Meindl A., Meitinger T., Ciccodicola A., Wright A.F. (2000) Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat. Genet., 25, 462–466. [DOI] [PubMed] [Google Scholar]
- 12.Sharon D., Sandberg M.A., Rabe V.W., Stillberger M., Dryja T.P., Berson E.L. (2003) RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am. J. Hum. Genet., 73, 1131–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meindl A., Dry K., Herrmann K., Manson F., Ciccodicola A., Edgar A., Carvalho M.R., Achatz H., Hellebrand H., Lennon A., et al. (1996) A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat. Genet., 13, 35–42. [DOI] [PubMed] [Google Scholar]
- 14.Schwahn U., Lenzner S., Dong J., Feil S., Hinzmann B., van Duijnhoven G., Kirschner R., Hemberger M., Bergen A.A., Rosenberg T., et al. (1998) Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat. Genet., 19, 327–332. [DOI] [PubMed] [Google Scholar]
- 15.Webb T.R., Parfitt D.A., Gardner J.C., Martinez A., Bevilacqua D., Davidson A.E., Zito I., Thiselton D.L., Ressa J.H., Apergi M., et al. (2012) Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum. Mol. Genet., 21, 3647–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Breuer D.K., Yashar B.M., Filippova E., Hiriyanna S., Lyons R.H., Mears A.J., Asaye B., Acar C., Vervoort R., Wright A.F., et al. (2002) A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am. J. Hum. Genet., 70, 1545–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Branham K., Othman M., Brumm M., Karoukis A.J., Atmaca-Sonmez P., Yashar B.M., Schwartz S.B., Stover N.B., Trzupek K., Wheaton D., et al. (2012) Mutations in RPGR and RP2 account for 15% of males with simplex retinal degenerative disease. Invest. Ophthalmol. Vis. Sci., 53, 8232–8237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jayasundera T., Branham K.E., Othman M., Rhoades W.R., Karoukis A.J., Khanna H., Swaroop A., Heckenlively J.R. (2010) RP2 phenotype and pathogenetic correlations in X-linked retinitis pigmentosa. Arch. Ophthalmol., 128, 915–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beltran W.A., Cideciyan A.V., Lewin A.S., Iwabe S., Khanna H., Sumaroka A., Chiodo V.A., Fajardo D.S., Roman A.J., Deng W.T., et al. (2012) Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc. Natl. Acad. Sci. USA., 109, 2132–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu Z., Hiriyanna S., Qian H., Mookherjee S., Campos M.M., Gao C., Fariss R., Sieving P.A., Li T., Colosi P., et al. (2015) A long-term efficacy study of gene replacement therapy for RPGR-associated retinal degeneration. Hum. Mol. Genet., 24, 3956–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans R.J., Hardcastle A.J., Cheetham M.E. (2006) Focus on molecules: X-linked retinitis pigmentosa 2 protein, RP2. Exp. Eye Res., 82, 543–544. [DOI] [PubMed] [Google Scholar]
- 22.Grayson C., Bartolini F., Chapple J.P., Willison K.R., Bhamidipati A., Lewis S.A., Luthert P.J., Hardcastle A.J., Cowan N.J., Cheetham M.E. (2002) Localization in the human retina of the X-linked retinitis pigmentosa protein RP2, its homologue cofactor C and the RP2 interacting protein Arl3. Hum. Mol. Genet., 11, 3065–3074. [DOI] [PubMed] [Google Scholar]
- 23.Chapple J.P., Hardcastle A.J., Grayson C., Spackman L.A., Willison K.R., Cheetham M.E. (2000) Mutations in the N-terminus of the X-linked retinitis pigmentosa protein RP2 interfere with the normal targeting of the protein to the plasma membrane. Hum. Mol. Genet., 9, 1919–1926. [DOI] [PubMed] [Google Scholar]
- 24.Chapple J.P., Hardcastle A.J., Grayson C., Willison K.R., Cheetham M.E. (2002) Delineation of the plasma membrane targeting domain of the X-linked retinitis pigmentosa protein RP2. Invest. Ophthalmol. Vis. Sci., 43, 2015–2020. [PubMed] [Google Scholar]
- 25.Veltel S., Gasper R., Eisenacher E., Wittinghofer A. (2008) The retinitis pigmentosa 2 gene product is a GTPase-activating protein for Arf-like 3. Nat. Struct. Mol. Biol., 15, 373–380. [DOI] [PubMed] [Google Scholar]
- 26.Schwarz N., Hardcastle A.J., Cheetham M.E. (2012) Arl3 and RP2 mediated assembly and traffic of membrane associated cilia proteins. Vision Res., 75, 2–4. [DOI] [PubMed] [Google Scholar]
- 27.Li L., Khan N., Hurd T., Ghosh A.K., Cheng C., Molday R., Heckenlively J.R., Swaroop A., Khanna H. (2013) Ablation of the X-linked retinitis pigmentosa 2 (Rp2) gene in mice results in opsin mislocalization and photoreceptor degeneration. Invest. Ophthalmol. Vis. Sci., 54, 4503–4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang H., Hanke-Gogokhia C., Jiang L., Li X., Wang P., Gerstner C.D., Frederick J.M., Yang Z., Baehr W. (2014) Mistrafficking of prenylated proteins causes retinitis pigmentosa 2. FASEB J., 29, 932–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu F., Chen J., Yu S., Raghupathy R.K., Liu X., Qin Y., Li C., Huang M., Liao S., Wang J., et al. (2015) Knockout of RP2 decreases GRK1 and rod transducin subunits and leads to photoreceptor degeneration in zebrafish. Hum. Mol. Genet., 24, 4648–4659. [DOI] [PubMed] [Google Scholar]
- 30.Veltel S., Wittinghofer A. (2009) RPGR and RP2: targets for the treatment of X-linked retinitis pigmentosa? Expert Opin Ther. Targets, 13, 1239–1251. [DOI] [PubMed] [Google Scholar]
- 31.Lewandoski M., Wassarman K.M., Martin G.R. (1997) Zp3-cre, a transgenic mouse line for the activation or inactivation of loxP-flanked target genes specifically in the female germ line. Curr. Biol., 7, 148–151. [DOI] [PubMed] [Google Scholar]
- 32.Sakai K., Miyazaki J. (1997) A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem. Biophys. Res. Commun., 237, 318–324. [DOI] [PubMed] [Google Scholar]
- 33.Khani S.C., Pawlyk B.S., Bulgakov O.V., Kasperek E., Young J.E., Adamian M., Sun X., Smith A.J., Ali R.R., Li T. (2007) AAV-mediated expression targeting of rod and cone photoreceptors with a human rhodopsin kinase promoter. Invest. Ophthalmol. Vis. Sci., 48, 3954–3961. [DOI] [PubMed] [Google Scholar]
- 34.Wang Z., Ma H.I., Li J., Sun L., Zhang J., Xiao X. (2003) Rapid and highly efficient transduction by double-stranded adeno-associated virus vectors in vitro and in vivo. Gene Ther., 10, 2105–2111. [DOI] [PubMed] [Google Scholar]
- 35.McCarty D.M., Fu H., Monahan P.E., Toulson C.E., Naik P., Samulski R.J. (2003) Adeno-associated virus terminal repeat (TR) mutant generates self-complementary vectors to overcome the rate-limiting step to transduction in vivo. Gene Ther., 10, 2112–2118. [DOI] [PubMed] [Google Scholar]
- 36.Yokoi K., Kachi S., Zhang H.S., Gregory P.D., Spratt S.K., Samulski R.J., Campochiaro P.A. (2007) Ocular gene transfer with self-complementary AAV vectors. Invest. Ophthalmol. Vis. Sci., 48, 3324–3328. [DOI] [PubMed] [Google Scholar]
- 37.Natkunarajah M., Trittibach P., McIntosh J., Duran Y., Barker S.E., Smith A.J., Nathwani A.C., Ali R.R. (2008) Assessment of ocular transduction using single-stranded and self-complementary recombinant adeno-associated virus serotype 2/8. Gene Ther., 15, 463–467. [DOI] [PubMed] [Google Scholar]
- 38.Kong F., Li W., Li X., Zheng Q., Dai X., Zhou X., Boye S.L., Hauswirth W.W., Qu J., Pang J.J. (2010) Self-complementary AAV5 vector facilitates quicker transgene expression in photoreceptor and retinal pigment epithelial cells of normal mouse. Exp. Eye Res., 90, 546–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Allocca M., Mussolino C., Garcia-Hoyos M., Sanges D., Iodice C., Petrillo M., Vandenberghe L.H., Wilson J.M., Marigo V., Surace E.M., et al. (2007) Novel adeno-associated virus serotypes efficiently transduce murine photoreceptors. J. Virol., 81, 11372–11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vandenberghe L.H., Bell P., Maguire A.M., Cearley C.N., Xiao R., Calcedo R., Wang L., Castle M.J., Maguire A.C., Grant R., et al. (2011) Dosage thresholds for AAV2 and AAV8 photoreceptor gene therapy in monkey. Sci. Transl. Med., 3, 88ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dismuke D.J., Tenenbaum L., Samulski R.J. (2013) Biosafety of recombinant adeno-associated virus vectors. Curr. Gene Ther., 13, 434–452. [DOI] [PubMed] [Google Scholar]
- 42.Maguire A.M., Simonelli F., Pierce E.A., Pugh E.N., Jr, Mingozzi F., Bennicelli J., Banfi S., Marshall K.A., Testa F., Surace E.M., et al. (2008) Safety and efficacy of gene transfer for Leber's congenital amaurosis. N. Engl. J. Med., 358, 2240–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pawlyk B.S., Bulgakov O.V., Liu X., Xu X., Adamian M., Sun X., Khani S.C., Berson E.L., Sandberg M.A., Li T. (2010) Replacement gene therapy with a human RPGRIP1 sequence slows photoreceptor degeneration in a murine model of Leber congenital amaurosis. Hum. Gene Ther., 21, 993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Black A., Vasireddy V., Chung D.C., Maguire A.M., Gaddameedi R., Tolmachova T., Seabra M., Bennett J. (2014) Adeno-associated virus 8-mediated gene therapy for choroideremia: preclinical studies in in vitro and in vivo models. J. Gene Med., 16, 122–130. [DOI] [PubMed] [Google Scholar]
- 45.Dhingra A., Tummala S.R., Lyubarsky A., Vardi N. (2014) PDE9A is expressed in the inner retina and contributes to the normal shape of the photopic ERG waveform. Front. Mol. Neurosci., 7, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miura G., Wang M.H., Ivers K.M., Frishman L.J. (2009) Retinal pathway origins of the pattern ERG of the mouse. Exp. Eye Res., 89, 49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grimm D., Zhou S., Nakai H., Thomas C.E., Storm T.A., Fuess S., Matsushita T., Allen J., Surosky R., Lochrie M., et al. (2003) Preclinical in vivo evaluation of pseudotyped adeno-associated virus vectors for liver gene therapy. Blood, 102, 2412–2419. [DOI] [PubMed] [Google Scholar]
- 48.Sun X., Pawlyk B., Xu X., Liu X., Bulgakov O.V., Adamian M., Sandberg M.A., Khani S.C., Tan M.H., Smith A.J., et al. (2010) Gene therapy with a promoter targeting both rods and cones rescues retinal degeneration caused by AIPL1 mutations. Gene Ther., 17, 117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prusky G.T., Alam N.M., Beekman S., Douglas R.M. (2004) Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Invest. Ophthalmol. Vis. Sci., 45, 4611–4616. [DOI] [PubMed] [Google Scholar]
- 50.Douglas R.M., Alam N.M., Silver B.D., McGill T.J., Tschetter W.W., Prusky G.T. (2005) Independent visual threshold measurements in the two eyes of freely moving rats and mice using a virtual-reality optokinetic system. Vis. Neurosci., 22, 677–684. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.