

Abstract
Purpose
To identify the underlying genetic defect in Korean patients with macular corneal dystrophy (MCD).
Methods
Genomic DNA was isolated from peripheral blood leukocytes of seven patients from six unrelated families with MCD (three men and four women). Polymerase chain reaction was performed for coding regions of the carbohydrate sulfotransferase (CHST6), gene followed by bidirectional sequencing. Targeted mutational analysis (exons 4, 11–12, 14) of the transforming growth factor, beta-induced (TGFBI) gene was performed for all patients.
Results
All seven patients were found to have compound heterozygous mutations in the CHST6 gene. In addition to six previously reported mutations, c.95C>A (p.Ser32*), c.521A>G (p.Lys174Arg), c.557C>G (p.Pro186Arg), c.613C>T (p.Arg205Trp), c.820G>A (p.Glu274Lys), and c.1072T>C (p.Tyr358His), three novel mutations were identified in this study, including two missense mutations, c.353C>T (p.Ser118Phe) and c.922C>T (p.His308Tyr), and one frameshift mutation, c.786delC (p.L264Cfs*117). Among the three novel mutations, only the c.353C>T mutation had been reported in the Exon Aggregation Consortium database at an extremely low frequency of 0.00005072. In addition, these three novel mutations were absent from controls in 1,000 genomes, dbSNP, and the TIARA genome database, which is a Korean personal genome database. The most frequent mutation was c.613C>T (p.Arg205Trp), revealed in four unrelated Korean families, which has not previously been reported in other populations. No mutations were detected in the TGFBI gene.
Discussion
This is the first report on genetic analysis of Korean MCD patients. Three novel and six previously reported disease-causing CHST6 mutations were identified, which expands the mutational spectrum of MCD.
Introduction
Macular corneal dystrophy (MCD, OMIM 217800) is an autosomal recessive corneal disorder characterized by bilateral, progressive, diffuse stromal haze; irregular corneal whitish opacity; and central corneal thinning [1,2]. Over time, the non-transparent areas progressively merge as the entire corneal stroma gradually becomes cloudy, leading to severe visual impairment between 10 and 30 years of age [1,2]. As a far-famed corneal disorder, MCD has been reported in a wide range of prevalence in many different countries [3-12]. In particular, MCD accounts for one-third of every penetrating keratoplasty in Iceland [8]. MCD is also a common type of corneal dystrophy leading to penetrating keratoplasty in Japan, India, and Saudi Arabia [9,10,12].
Histologically, MCD is characterized by the accumulation of glycosaminoglycans (GAGs) between the stromal lamellae, underneath the epithelium, and within keratocytes and endothelial cells [13,14]. Explants from MCD-affected corneas have been reported to synthesize low-sulfated keratan sulfate (KS), suggesting that the sulfate groups in KS may play critical roles in maintaining corneal transparency [15-17]. N-acetylglucosamine-6-O-sulfotransferase (GlcNAc6ST) activity in the extracts from MCD-affected corneas was much lower than in those in corneas with keratoconus and in normal control corneas [15]. This decrease in GlcNAc6ST activity in corneas with MCD is associated with the occurrence of low- or non-sulfated KS, and this has been proposed as an underlying mechanism for the observed corneal opacity [15,18,19]. Since the carbohydrate sulfotransferase 6 (CHST6) gene, located on chromosome 16q22, which encodes the corneal (C)-GlcNAc6ST comprising 395 amino acids, was first identified in 2000 as a candidate gene for MCD in the Japanese population [6], numerous mutations have been reported in MCD patients with different ethnic backgrounds [2-7,9,10]. The CHST6 gene contains four exons; the coding region is contained only within exon 3 [20]. C-GlcNAc6ST catalyzes the transfer of a sulfate group to N-acetylglucosamine in KS, a common component of corneal proteoglycans [20].
Jee et al. reported that MCD accounts for 12.9% of corneal dystrophy encountered in Koreans, following granular dystrophy (29.2%) and Fuchs’ endothelial dystrophy (23.6%) [21]. However, little is known about the molecular characterization of MCD in Koreans. Recently, only one homozygous missense mutation (c.613C>T, p.Arg205Trp) has been reported in a 59-year-old Korean woman with MCD [22]. Here, we analyzed the CHST6 gene in seven Korean MCD patients and identified three novel and six previously reported disease-causing mutations.
Methods
This study was performed according to the principles of the Declaration of Helsinki, after approval by the Institutional Review Board of Seoul St. Mary’s Hospital, College of Medicine, Catholic University of Korea, Seoul, Korea (KC14RISI0419). Informed consent was obtained from all patients.
Patient selection and clinical evaluation
The medical records of seven patients from six unrelated families with MCD in Seoul St. Mary’s Hospital were investigated. Seven patients were diagnosed as having MCD based on typical clinical features after evaluation by a corneal specialist (M.S.K.) and referred for molecular confirmation. Thorough ocular examinations were performed, including best-corrected visual acuity (BCVA), intraocular pressure (IOP), refractive measurement, central corneal thickness, and slit lamp biomicroscopy of the anterior segment and fundus. The age, gender, visual acuity, family history, and previous ocular records of the patients were reviewed. The obtained pedigrees were consistent with an autosomal recessive inheritance pattern, and there was no known consanguinity in all six families.
Mutational analysis
After receiving informed consent, blood samples were collected from all patients for DNA extraction and CHST6 gene analysis. In addition, targeted mutational analysis (exons, 4, 11–12, 14) of the transforming growth factor, beta-induced (TGFBI) gene was performed for all patients. Genomic DNA was extracted from the peripheral blood leukocytes using the QIAmp DNA Mini Kit (Qiagen, Hamburg, Germany). Polymerase chain reaction (PCR) was performed using previously published primer sets for CHST6 [6]. All the coding exons and the flanking intron/exon boundaries of CHST6 were amplified. The PCR amplicons were bidirectionally sequenced using the Big Dye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems). The chromatograms were analyzed with the Sequencher software version 5.0 (Gene Codes, Ann Arbor, MI). Mutations were confirmed by sequencing two or more independent PCR reactions. When a novel missense mutation was identified, in silico analysis was performed using Polyphen-2 and SIFT. In cases of missense mutations, conservation of the involved amino acids among several sulfotransferases of human and mouse origin was investigated using Clustal Omega. DNA from available family members was sequenced to determine whether the mutation cosegregated with the phenotype within the pedigree. All mutations were described according to the Human Genome Variation Society nomenclature, and GenBank accession numbers NM_021615.4 and NM_000358.2 were used for CHST6 and TGFBI alignment, respectively.
Results
Molecular analysis of the CHST6 gene
The patients’ molecular findings in the present study are summarized in Table 1. Nine different mutations within the coding region of the CHST6 gene were identified in seven patients from the six unrelated families (Table 1). In addition to six previously reported mutations (c.95C>A, c.521A>G, c.557C>G, c.613C>T, c.820G>A, and c.1072T>C), three novel mutations, c.353C>T, c.786delC, and c.922C>T, were identified. There were seven missense mutations, one nonsense mutation, and one frameshift mutation. The c.613C>T (Arg205Trp) mutation was the most frequently detected mutation in our study. No mutations were detected in the TGFBI gene.
Table 1. Mutation alleles of the CHST6 gene identified in 7 Korean patients with MCD.
| Gene | Nucleotide change | Amino acid change | Mutation effect | Number of families | Number of individuals | Polyphen | SIFT | Reference |
|---|---|---|---|---|---|---|---|---|
| |
c.95C>A |
p.Ser32* |
Nonsense mutation |
1 |
1 |
- |
- |
Chinese7 |
| |
c.353C>T |
p.Ser118Phe |
Missense mutation |
1 |
2 |
Probably damaging |
Not tolerated |
Novel |
| |
c.521A>G |
p.Lys174Arg |
Missense mutation |
1 |
1 |
Probably damaging |
Not tolerated |
Japanese6 |
| |
c.557C>G |
p.Pro186Arg |
Missense mutation |
1 |
1 |
Probably damaging |
Not tolerated |
African American5 |
| CHST6 |
c.613C>T |
p.Arg205Trp |
Missense mutation |
4 |
4 |
Probably damaging |
Not tolerated |
Korean24 |
| |
c.786delC |
p.L264Cfs*117 |
Frameshift mutation |
1 |
1 |
- |
- |
Novel |
| |
c.820G>A |
p.Glu274Lys |
Missense mutation |
1 |
1 |
Probably damaging |
Not tolerated |
Japanese6, Indian9 |
| |
c.922C>T |
p.His308Tyr |
Missense mutation |
1 |
1 |
Probably damaging |
Not tolerated |
Novel |
| c.1072T>C | p.Tyr358His | Missense mutation | 1 | 2 | Probably damaging | Not tolerated | British4, Chinese3 |
Figure 1 shows the nucleotide sequences in the three novel mutations, c.353C>T, c.786delC, and c.922C>T. The c.922C>T mutation detected in a 41-year-old female (patient 2) was a missense mutation resulting in a histidine-to-tyrosine substitution at codon 308. Another missense c.353C>T mutation, detected in one family (patients 5–1 and 5–2), led to a serine-to-phenylalanine substitution at codon 118. Patient 6 was a 15-year-old female and had a novel mutation (c.786delC, p.L264Cfs*117), which resulted in premature termination due to a frameshift. The clinically unaffected father and brother carried either one of the two mutations, which implied MCD as a recessive inherited disorder. Among the three novel mutations, only the c.353C>T mutation had been reported in the Exon Aggregation Consortium (ExAC) database at an extremely low frequency of 0.00005072. In addition, these three novel mutations were absent from controls in 1,000 genomes, dbSNP, and the TIARA genome database, a Korean personal genome database.
Figure 1.
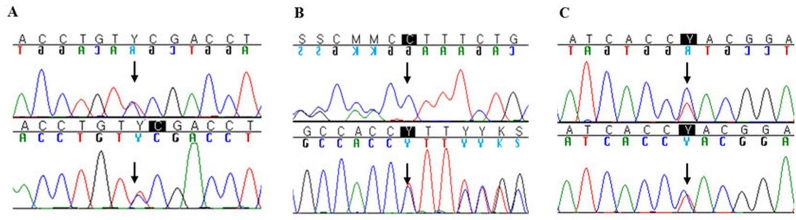
Sequencing chromatograms of the three novel CHST6 mutations identified in this study. A: c.353C>T (p.Ser118Phe); B: c.786delC (p.L264Cfs*117); C: c.922C>T (p.His308Tyr).
All seven missense mutations identified in Korean MCD patients, including two novel mutations, were predicted to have a pathogenic effect by Polyphen-2 and SIFT software, which produced results designated as “probably damaging” and “not tolerated,” respectively (Table 1). In addition, multiple protein sequence alignment analyses between several sulfotransferases of human and mouse origin demonstrated that the majority of the amino acids substituted in the missense mutations identified in Korean MCD patients were highly conserved residues, while the amino acids substituted in the newly identified p.Ser118Phe and p.His308Tyr mutations showed a lesser degree of conservation (Table 2).
Table 2. Multiple sequence alignment of sulfotransferases showing conservation of amino acid residues mutated in Korean patients with MCD.
| Sulfotransferase | S118F | K174R | P186R | R205W | E274K | H308Y | Y358H | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Human carbohydrate sulfotransferase 6 precursor (GI:11055976) | L | S | D | L | L | K | E | V | L | Y | P | L | D | P | R | A | R | F | E | D | T | H | G | S | L | G | Y | R | |||||||
| Human carbohydrate sulfotransferase 5 (GI:21362052) | L | S | A | F | L | K | E | V | L | Y | P | L | D | P | R | A | R | F | E | D | T | H | G | S | L | G | Y | R | |||||||
| Human N-acetylglucosamine 6-O-sulfotransferase (GI:4927114) | Q | S | S | L | L | K | E | V | L | Y | P | L | D | P | R | A | R | Y | E | D | T | R | G | K | L | G | Y | R | |||||||
| Human L-selectin ligand sulfotransferase (GI:13897504) | Q | S | S | L | L | K | E | V | L | Y | P | L | D | P | R | A | R | Y | E | D | T | R | G | K | L | G | Y | R | |||||||
| Human chondroitin 6-sulfotransferase-2 (GI:930944) | T | A | A | L | I | K | D | V | L | V | P | L | D | P | R | A | R | Y | E | D | T | R | G | A | L | A | Y | P | |||||||
| Human keratan sulfate Gal-6-sulfotransferase (GI:2887403) | T | D | R | I | I | K | T | V | L | R | A | L | D | P | R | G | R | Y | E | D | T | R | G | D | L | G | Y | K | |||||||
| Mouse L-selectin ligand sulfotransferase (GI:5596406) | Q | S | S | L | L | K | E | V | L | Y | P | L | D | P | R | A | R | Y | E | D | T | R | G | K | L | G | Y | L | |||||||
| Mouse carbohydrate sulfotransferase 5 precursor (GI:9910284) | I | S | D | L | L | K | E | V | L | Y | P | L | D | P | R | A | R | Y | E | D | T | H | G | S | L | G | Y | R | |||||||
| Mouse chondroitin 6-sulfotransferase (GI:3253091) | T | Q | F | L | L | K | A | V | L | Q | P | L | D | P | R | A | R | Y | E | D | T | Q | A | T | F | G | Y | K | |||||||
Ocular phenotypes associated with mutations
Table 3 shows the ocular phenotypes of seven patients from six unrelated families tested (three males and four females). All seven patients were found to have compound heterozygous mutations in the CHST6 gene. Slit lamp examination of all affected patients revealed bilateral stromal corneal cloudiness. Multiple irregular-shaped, gray-white, poorly delineated opacities were found involving the center of the cornea (Figure 2A–C) and extending to the peripheral limbus in some patients (Figure 2D). The two chief complaints were progressive visual disturbance and photophobia. Participants from a wide range of age at detection revealed various visual acuity. Central corneal thickness was decreased in all 12 eyes that were examined at initial visit. The mean values of central corneal thickness measured by ultrasound pachymetry were 450.3±32.8 μm and 446.0±31.2 μm in the right and left eyes, respectively.
Table 3. Clinical findings of Korean MCD patients carrying CHST6 mutations.
| Case No. | Age/Gender | Mutation | Age at detection (years) | Family History |
Initial |
Therapeutic procedures | Final VA | ||
|---|---|---|---|---|---|---|---|---|---|
| VA (Rt / Lt) | Pachy (Rt / Lt) | ECD (Rt / Lt) | (Rt / Lt) | ||||||
| 1 | 50/Male | c.521A>G / c.613C>T | 15 | + | NA | NA | NA | Keratoplasty, Both eyes | 0.4 / 0.25 |
| 2 | 41/Female | c.820G>A / c.922C>T | 41 | - | 1.0 /1.0 | 441 / 432 | 1377 /2237 | - | NA |
| 3 | 44/Male | c.557 C>G / c.613 C>T | 42 | - | 0.4 / 0.32 | 492 / 498 | - | - | NA |
| 4 | 35/Male | c.95C>A / c.613 C>T | 15 | - | 0.16 / 0.125 | 482 / 460 | - | Keratoplasty, Right eye | 0.5 / 0.8 |
| 5-1 | 15/Female | c.353 C>T / c.1072 T>C | 11 | + | 0.8 / 0.8 | 407 / 405 | - | - | 0.4 / 0.4 |
| 5-2 | 19/Female | c.353 C>T / c.1072 T>C | 15 | + | 0.63 / 0.32 | 425 / 445 | - | - | 0.32 / 0.2 |
| 6 | 15/Female | c.613C>T / c.786delC | 10 | - | 0.4 / 0.4 | 455 / 436 | - | Keratoplasty, Left eye | 0.1 / 0.5 |
Figure 2.

Slit lamp photography of patients with MCD. A, B: A 15-year-old female carrying p.Arg205Trp/p.L264Cfs*117 in the CHST6 gene (patient 6) revealed diffuse ground-glass-like stromal haze and multiple gray-white opacities with irregular borders. Arrows (B) indicate newly developed opacities three years after initial diagnosis (A). The patient complained of severe photophobia, which required penetrating keratoplasty in her left eye. C: A 19-year-old female carrying p.Ser118Phe/p.Tyr358His in the CHST6 gene (patient 5–2) shows coalescence of individual stromal opacities. The absence of clear areas between corneal opacities is a characteristic finding, unlike granular dystrophy. D: A 44-year-old male carrying p.Pro186Arg/p.Arg205Trp in the CHST6 gene (patient 3) has multiple, irregular, gray-white opacities extending to the limbus in both eyes.
Five eyes from three patients received therapeutic penetrating keratoplasty. In patient 1, carrying the c.[521A>G];[613C>T] mutations, penetrating keratoplasty was performed in both eyes at the age of 38 in another hospital. This patient is currently waiting for a repeat penetrating keratoplasty in the left eye due to the recurrence of corneal opacity. Patient 5–1, carrying the c.[95C>A];[613C>T] mutations, received penetrating keratoplasty in both eyes. Patient 6, carrying the c.[613C>T];[786delC] mutations, received penetrating keratoplasty only in the left eye. Basophilic deposits between the stromal lamellae and within keratocytes and endothelial cells were positive to Alcian blue and periodic acid-Schiff stains, but negative to Congo red and Masson’s trichrome stain, which are consistent with the accumulation of GAGs (Figure 3). The BCVA improved after surgery in these three eyes without any postoperative complications. The clarity of grafted corneas was maintained throughout the follow-up period, which ranged from 14 to 48 months.
Figure 3.
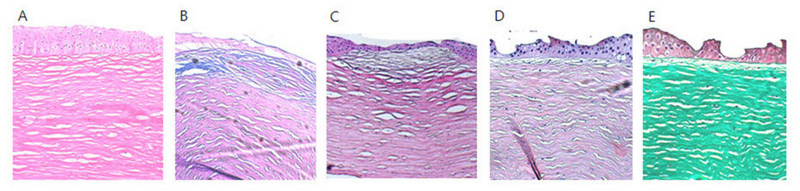
Histopathological findings in a 15-year-old female patient (patient 6). A: Hematoxylin and eosin stain. B: Alcian blue stain. C: Periodic acid-Schiff stain. D: Congo red stain. E: Masson’s trichrome stain. Basophilic deposits between the stromal lamellae and within keratocytes and endothelial cells were positive for Alcian blue and periodic acid-Schiff stain, but negative for Congo red and Masson’s trichrome stain, which are consistent with the accumulation of glycosaminoglycans.
Discussion
In this report, we describe CHST6 mutations in Korean MCD patients. Three novel mutations were identified in this study, including two missense mutations, c.353C>T (p.Ser118Phe) and c.922C>T (p.His308Tyr), and one frameshift mutation c.786delC (p.L264Cfs*117). The most frequent mutation was the c.613C>T (p.Arg205Trp) mutation revealed in four unrelated Korean families, which has not previously been reported in other populations.
CHSTs are a family of related enzymes that catalyze the sulfation of specific carbohydrates [20]. The N-acetylglucosamine-6-O-sulfotransferase (GlcNAc6ST) enzyme encoded by the CHST6 gene transfers sulfate to N-acetylglucosamine on KS GAG chains [17,20]. Abnormalities in the synthesis of this enzyme have been identified as a cause of MCD [6,15,18,19]. Unsulfated polylactosamine chains are likely to be less water soluble than the fully sulfated KS because of the decrease in GAG chain polarity in the absence of sulfate esters [20]. The degradation of unsulfated KS proteoglycans may also be inhibited by its insolubility [20]. The intracellular and extracellular accumulation of poorly or non-sulfated KS in the corneal stroma, corneal endothelium, and Descemet membrane resulted in corneal opacity in patients with MCD [1,2,13,14].
The present study revealed the mutational heterogeneity of the CHST6 gene in Korean MCD patients (Table 1 and Table 3). It is noteworthy that the most frequent abnormality found in this study was the c.613C>T mutation, leading to an arginine-to-tryptophan substitution at codon 205. Recently, this mutation was reported in a 59-year-old Korean woman with MCD, but was not previously reported in other populations [22]. The amino acid substitutions at codon 205 have been previously identified in Japanese (p.Arg205Leu from c.614G>T) and Indian (p.Arg205Gln from c.614G>A) MCD patients, but they are different from the mutation detected in our study [9,23]. The identified c.613C>T (p.Arg205Trp) mutation was located in the RX7S sequence in the 3′-phosphate binding domain, which was one of the two binding sites for the high-energy sulfate donor in the C-GlcNAc6S protein [23-25]. The sequence between the two binding sites is thought to contribute to a binding pocket that interacts with an acceptor to bring it into apposition with the sulfate donor [24,25]. It is known that mutations of these binding motif sequences abolish HKN-1 sulfotransferase activity [23].
Among seven missense mutations of the CHST6 gene, the c.521A>G (p.Lys174Arg), c.557C>G (p.Pro186Arg), c.820G>A (p.Glu274Lys), and c.1072T>C (p.Tyr358His) mutations have been previously identified to be responsible for MCD in Japanese [6], African-American [5], Indian [9], British [4], and Chinese [3] patients (Table 1). The c.353C>T (p.Ser118Phe) and c.922C>T (p.His308Tyr) mutations were detected for the first time in the present study. These two novel missense mutations were predicted to have a pathogenic effect by Polyphen-2 and SIFT software, which produced results designated as “probably damaging” and “not tolerated,” respectively. We also searched the genome databases, including ExAC, 1,000 genomes, dbSNP, and an ethnic-specific Korean personal genome database, TIARA. Only the p.Ser118Phe mutation had been reported in ExAC, though at an extremely low frequency of 0.00005072. A mutation of this low frequency, especially in a recessive disorder, could be considered to have moderate evidence of pathogenicity, according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines for the interpretation of sequence variants. Additionally, the multiple protein sequence alignment analysis between several sulfotransferases of human and mouse origin demonstrated that the novel mutations substitute relatively well-conserved amino acid residues, which can also be regarded as a supporting evidence of pathogenicity of these novel missense mutations (Table 2).
Generally, nonsense or frameshift mutations in the CHST6 gene occur less frequently in MCD [1,2,26]. The nonsense mutation of the CHST6 gene was first identified in three patients in one French family, who experienced rapid visual deterioration at an early age and all required keratoplasty in the second decade of life [27]. A genetic analysis of German patients with MCD demonstrated that frameshift mutations in the CHST6 gene have a tendency to be associated with more severe clinical phenotypes with much deeper corneal deposits [28]. Gruenauer-Kloevekorn et al. suggested that the application of therapeutical options such as primary phototherapeutic keratectomy or primary penetrating keratoplasty could be based on the molecular genetic findings in each patient [28]. However, Sultana et al. reported that there were no consistent differences in phenotype between patients with the various amino acid substitutions and truncating mutations [29]. In our study, two different kinds of nonsense or frameshift mutations were detected in two families (patients 4 and 6).
All of the study participants carried compound heterozygous mutations in the CHST6 gene, which may be related to the non-consanguinity of the study participants. They had bilateral corneal cloudiness and corneal thinning, which are characteristic features of MCD [1,2]. The reduced corneal thickness is expected to result from the dysregulation of KS proteoglycan synthesis or catabolism [30]. Although different mutations of the CHST6 gene were confirmed in each patient, we did not notice any phenotypic differences according to the identified genotype (Table 3). Further studies with larger numbers of patients are required to delineate the genotype-phenotype correlation of MCD.
This study had some limitations. Relatively small numbers of patients were analyzed, and the parents and/or siblings of patients were not thoroughly examined. The serum levels of KS and the immunohistochemical reaction to the corneal tissue were not sufficiently evaluated to characterize the immunophenotype of MCD. However, recently there has been increased awareness of and interest in genetic diseases in Korea. Genetic analysis of Koreans with MCD could provide valuable information for correct diagnosis, carrier detection, and genetic counseling in patients and their families.
In conclusion, this is the first report of a genetic analysis of Korean MCD patients. Three novel and six previously reported disease-causing CHST6 mutations were identified, which expands the mutational spectrum of MCD.
Acknowledgments
The authors thank all the patients for participating in this study. This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2013R1A1A2006801). The authors declare no conflict of interest. Man Soo Kim (mskim@catholic.ac.kr) and Myungshin Kim (microkim@catholic.ac.kr) contributed equally.
References
- 1.Klintworth GK. Corneal dystrophies. Orphanet J Rare Dis. 2009;4:7. doi: 10.1186/1750-1172-4-7. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19236704&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weiss JS, Møller HU, Lisch W, Kinoshita S, Aldave AJ, Belin MW, Kivela T, Busin M, Munier FL, Seitz B, Sutphin J, Bredrup C, Mannis MJ, Rapuano CJ, Van Rij G, Kim EK, Klintworth GK. The IC3D classification of the corneal dystrophies. Cornea. 2008;27(Suppl 2):S1–83. doi: 10.1097/ICO.0b013e31817780fb. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19337156&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dang X, Zhu Q, Wang L, Su H, Lin H, Zhou N, Liang T, Wang Z, Huang S, Ren Q, Qi Y. Macular corneal dystrophy in a Chinese family related with novel mutations of CHST6. Mol Vis. 2009;15:700–5. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19365571&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 4.El-Ashry MF, Abd El-Aziz MM, Shalaby O, Wilkins S, Poopalasundaram S, Cheetham M, Tuft SJ, Hardcastle AJ, Bhattacharya SS, Ebenezer ND. Novel CHST6 nonsense and missense mutations responsible for macular corneal dystrophy. Am J Ophthalmol. 2005;139:192–3. doi: 10.1016/j.ajo.2004.07.001. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15652851&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 5.Patel DA, Harocopos GJ, Chang SH, Vora SC, Lubniewski AJ, Huang AJ. Novel CHST6 gene mutations in 2 unrelated cases of macular corneal dystrophy. Cornea. 2011;30:664–9. doi: 10.1097/ICO.0b013e3182012888. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=21242781&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akama TO, Nishida K, Nakayama J, Watanabe H, Ozaki K, Nakamura T, Dota A, Kawasaki S, Inoue Y, Maeda N, Yamamoto S, Fujiwara T, Thonar EJ, Shimomura Y, Kinoshita S, Tanigami A, Fukuda MN. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nat Genet. 2000;26:237–41. doi: 10.1038/79987. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11017086&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 7.Liu Z, Tian X, Iida N, Fujiki K, Xie P, Wang W, Ma Z, Kanai A, Murakami A. Mutation analysis of CHST6 gene in Chinese patients with macular corneal dystrophy. Cornea. 2010;29:883–8. doi: 10.1097/ICO.0b013e3181ca2e74. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20539220&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 8.Jonasson F, Oshima E, Thonar EJ, Smith CF, Johannsson JH, Klintworth GK. Macular corneal dystrophy in Iceland. A clinical, genealogic, and immunohistochemical study of 28 patients. Ophthalmology. 1996;103:1111–7. doi: 10.1016/s0161-6420(96)30559-9. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=8684802&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 9.Warren JF, Aldave AJ, Srinivasan M, Thonar EJ, Kumar AB, Cevallos V, Whitcher JP, Margolis TP. Novel mutations in the CHST6 gene associated with macular corneal dystrophy in southern India. Arch Ophthalmol. 2003;121:1608–12. doi: 10.1001/archopht.121.11.1608. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=14609920&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 10.Malbran ES. Macular corneal dystrophy in Saudi Arabia: a study of 56 cases and recognition of a new immunophenotype. Am J Ophthalmol. 1998;125:417–8. doi: 10.1016/s0002-9394(99)80167-6. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9512173&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 11.Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrating keratoplasty for macular corneal dystrophy. Ophthalmology. 2005;112:220–4. doi: 10.1016/j.ophtha.2004.08.017. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15691554&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 12.Santo RM, Yamaguchi T, Kanai A, Okisaka S, Nakajima A. Clinical and histopathologic features of corneal dystrophies in Japan. Ophthalmology. 1995;102:557–67. doi: 10.1016/s0161-6420(95)30982-7. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=7724173&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 13.Klintworth GK, Vogel FS. Macular corneal dystrophy. An inherited acid mucopolysaccharide storage disease of the corneal fibroblast. Am J Pathol. 1964;45:565–86. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=14217673&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 14.Snip RC, Kenyon KR, Green WR. Macular corneal dystrophy: ultrastructural pathology of corneal endothelium and Descemet's membrane. Invest Ophthalmol. 1973;12:88–97. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=4265065&dopt=Abstract [PubMed] [Google Scholar]
- 15.Hasegawa N, Torii T, Kato T, Miyajima H, Furuhata A, Nakayasu K, Kanai A, Habuchi O. Decreased GlcNAc 6-O-sulfotransferase activity in the cornea with macular corneal dystrophy. Invest Ophthalmol Vis Sci. 2000;41:3670–7. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11053262&dopt=Abstract [PubMed] [Google Scholar]
- 16.Klintworth GK, Smith CF. Macular corneal dystrophy. Studies of sulfated glycosaminoglycans in corneal explant and confluent stromal cell cultures. Am J Pathol. 1977;89:167–82. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=143892&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 17.Funderburgh JL. Keratan sulfate: structure, biosynthesis, and function. Glycobiology. 2000;10:951–8. doi: 10.1093/glycob/10.10.951. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11030741&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 18.Hasegawa N, Torii T, Nagaoka I, Nakayasu K, Miyajima H, Habuchi O. Measurement of activities of human serum sulfotransferases which transfer sulfate to the galactose residues of keratan sulfate and to the nonreducing end N-acetylglucosamine residues of N-acetyllactosamine trisaccharide: comparison between normal controls and patients with macular corneal dystrophy. J Biochem. 1999;125:245–52. doi: 10.1093/oxfordjournals.jbchem.a022280. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9990120&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 19.Akama TO, Nakayama J, Nishida K, Hiraoka N, Suzuki M, McAuliffe J, Hindsgaul O, Fukuda M, Fukuda MN. Human corneal GlcNac 6-O-sulfotransferase and mouse intestinal GlcNac 6-O-sulfotransferase both produce keratan sulfate. J Biol Chem. 2001;276:16271–8. doi: 10.1074/jbc.M009995200. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11278593&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 20.Musselmann K, Hassell JR. Focus on molecules: CHST6 (carbohydrate sulfotransferase 6; corneal N-acetylglucosamine-6-sulfotransferase). Exp Eye Res. 2006;83:707–8. doi: 10.1016/j.exer.2005.11.020. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=16549065&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 21.Jee DH, Lee YD, Kim MS. Epidemiology of corneal dystrophy in Korea. J Korean Ophthalmol Soc. 2003;44:581–7. [Google Scholar]
- 22.Lee YK, Chang DJ, Chung SK. A case of Korean patient with macular corneal dystrophy associated with novel mutation in the CHST6 gene. Korean J Ophthalmol. 2013;27:454–8. doi: 10.3341/kjo.2013.27.6.454. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24311932&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iida-Hasegawa N, Furuhata A, Hayatsu H, Murakami A, Fujiki K, Nakayasu K, Kanai A. Mutations in the CHST6 gene in patients with macular corneal dystrophy: immunohistochemical evidence of heterogeneity. Invest Ophthalmol Vis Sci. 2003;44:3272–7. doi: 10.1167/iovs.02-0910. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=12882769&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 24.Ong E, Yeh JC, Ding Y, Hindsgaul O, Pedersen LC, Negishi M, Fukuda M. Structure and function of HNK-1 sulfotransferase. Identification of donor and acceptor binding sites by site-directed mutagenesis. J Biol Chem. 1999;274:25608–12. doi: 10.1074/jbc.274.36.25608. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10464296&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 25.Kakuta Y, Pedersen LG, Pedersen LC, Negishi M. Conserved structural motifs in the sulfotransferase family. Trends Biochem Sci. 1998;23:129–30. doi: 10.1016/s0968-0004(98)01182-7. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9584614&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 26.Klintworth GK, Smith CF, Bowling BL. CHST6 mutations in North American subjects with macular corneal dystrophy: a comprehensive molecular genetic review. Mol Vis. 2006;12:159–76. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=16568029&dopt=Abstract [PubMed] [Google Scholar]
- 27.Niel F, Ellies P, Dighiero P, Soria J, Sabbagh C, San C, Renard G, Delpech M, Valleix S. Truncating mutations in the carbohydrate sulfotransferase 6 gene (CHST6) result in macular corneal dystrophy. Invest Ophthalmol Vis Sci. 2003;44:2949–53. doi: 10.1167/iovs.02-0740. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=12824236&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 28.Gruenauer-Kloevekorn C, Braeutigam S, Heinritz W, Froster UG, Duncker GI. Macular corneal dystrophy: mutational spectrum in German patients, novel mutations and therapeutic options. Graefes Arch Clin Exp Ophthalmol. 2008;246:1441–7. doi: 10.1007/s00417-008-0836-1. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18500531&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 29.Sultana A, Sridhar MS, Jagannathan A, Balasubramanian D, Kannabiran C, Klintworth GK. Novel mutations of the carbohydrate sulfotransferase-6 (CHST6) gene causing macular corneal dystrophy in India. Mol Vis. 2003;9:730–4. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=14735064&dopt=Abstract [PubMed] [Google Scholar]
- 30.Dudakova L, Palos M, Svobodova M, Bydzovsky J, Huna L, Jirsova K, Hardcastle AJ, Tuft SJ, Liskova P. Macular corneal dystrophy and associated corneal thinning. Eye (Lond) 2014;28:1201–5. doi: 10.1038/eye.2014.164. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25081284&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]