

Abstract

Glutamate racemase (GR) catalyzes the cofactor independent stereoinversion of l- to d-glutamate for biosynthesis of bacterial cell walls. Because of its essential nature, this enzyme is under intense scrutiny as a drug target for the design of novel antimicrobial agents. However, the flexibility of the enzyme has made inhibitor design challenging. Previous steered molecular dynamics (MD), docking, and experimental studies have suggested that the enzyme forms highly varied complexes with different competitive inhibitor scaffolds. The current study employs a mutant orthogonal tRNA/aminoacyl-tRNA synthetase pair to genetically encode a non-natural fluorescent amino acid, l-(7-hydroxycoumarin-4-yl) ethylglycine (7HC), into a region (Tyr53) remote from the active site (previously identified by MD studies as undergoing ligand-associated changes) to generate an active mutant enzyme (GRY53/7HC). The GRY53/7HC enzyme is an active racemase, which permitted us to examine the nature of these idiosyncratic ligand-associated phenomena. One type of competitive inhibitor resulted in a dose-dependent quenching of the fluorescence of GRY53/7HC, while another type of competitive inhibitor resulted in a dose-dependent increase in fluorescence of GRY53/7HC. In order to investigate the environmental changes of the 7HC ring system that are distinctly associated with each of the GRY53/7HC–ligand complexes, and thus the source of the disparate quenching phenomena, a parallel computational study is described, which includes essential dynamics, ensemble docking and MD simulations of the relevant GRY53/7HC–ligand complexes. The changes in the solvent exposure of the 7HC ring system due to ligand-associated GR changes are consistent with the experimentally observed quenching phenomena. This study describes an approach for rationally predicting global protein allostery resulting from enzyme ligation to distinctive inhibitor scaffolds. The implications for fragment-based drug discovery and high throughput screening are discussed.
Short abstract
A mutant glutamate racemase enzyme possessing a genetically encoded non-natural fluorescent amino acid is developed and studied as a reporter of global enzyme changes occurring upon ligand binding.
Introduction
Many protein and enzyme drug targets are intractable to the most sophisticated methods in structure-based drug design and discovery (SBDDD) in large part because a high degree of flexibility negates the use of one, or even a few, crystal structures in virtual screening or de novo design. Recent years have seen the development of a myriad of tools for obtaining ensembles, which can be used in SBDDD approaches, with mixed results. There are many mechanistic options to consider when attempting to model a flexible enzyme drug target. One of the most critical parameters is whether a “selection” or an “induced-fit” model will be followed. Although it is exceedingly rare for any protein–ligand binding study to contain a sufficient level of physical detail to elucidate selection vs induced-fit, a number of exhaustive studies (both experimental and computational) have recently shown them both to take place in particular systems.1−3 Indeed, it is possible for a variety of complicated phenomena to be operational in a single enzyme–ligand association process.4 Nevertheless, from an SBDDD point of view, a related question is can we employ experimental and computational methods that give us insight into the ligand-associated enzyme changes, independent of exactly how they are manifested?
In the current study, experimental and computational insights into protein–ligand complexation are obtained for a highly flexible enzyme, glutamate racemase (GR), which exhibits very puzzling and idiosyncratic ligand-associated changes. These ligand-associated changes have previously been described using computational selection models, in which a very flexible apoenzyme produces a diverse ensemble of conformations possessing ligand-binding pockets of variable solvent accessible surface areas and protein solvation energies, which has significant implications on the quality of ligand binding to each of these distinct enzyme conformations.5 The current study employs an approach that allows for both selection and induced-fit changes in the computational workflow, concomitant with parallel experimental studies.
GR is a bacterial enzyme responsible for the essential task of the stereoisomerization of l-glutamate to d-glutamate, a member of the cross-linking peptide side chain component of all bacterial peptidoglycan cell walls.6d-Glutamate is not readily available in the environment, and no other viable biosynthetic pathways exists for generation intracellularly, making GR a vital enzyme to bacterial growth and survival.7 The lack of any human isozyme makes GR a valuable target for potential antibacterial therapeutics.
Several isozymes of GR have been the subject of X-ray crystallography campaigns, providing a suite of cocrystal structures for use in SBDDD.8−10 One of the first structures solved belonged to the nonpathogenic, mesophilic bacteria Bacillus subtilis.8 Thus, this isozyme has served as a model system for an assortment of functional studies and drug discovery campaigns.5,11−13 In general, GR has proven to be a flexible enzyme capable of binding a variety of small molecules both in the active site and at allosteric binding pockets,10,14 some of which have included cyclic substrate-product analogues.15 Further complications of GR–ligand complexation are derived from the important role that interstitial waters play in recognizing various classes of competitive inhibitors.13
Inherent flexibility is notoriously a bane to traditional SBDDD, largely due to the unknown nature of the relationship between ligand binding and enzyme conformation. This limits the representative nature of single cocrystal structures and requires the acquisition of additional structural information concurrent with hit discovery and lead optimization. The current study addresses these difficulties in the case of GR, by employing an integrated experimental and computational approach to gain insight into the nature of ligand-dependent changes in complexation. Site-specific incorporation of a genetically encoded fluorescent non-natural amino acid is used to generate a GR which substitutes Tyr53, located in a dynamic region remote from the active site, with l-(7-hydroxycoumarin-4-yl) ethylglycine (7HC) (using the approach developed by Wang et al.,16 as described below), to make an active enzyme (GRY53/7HC). GRY53/7HC is a sensitive reporter for ligand-dependent changes and is studied with two differing competitive inhibitors (e.g., croconate and glucuronate), which have been previously identified as selecting distinct forms of GR.5 A parallel computational study of ligand-GRY53/7HC selection and dynamics is employed to elucidate the source of the ligand-associated allosteric changes. The use of GRY53/7HC offers insight into ligand-associated changes taking place within the active site, which is reflected by global changes occurring remote from the active site and is reported on by the fluorescent probe. The implications of this study are highly relevant to improving approaches to fragment-based drug discovery, virtual screening, and high throughput screening against flexible enzymes and proteins (vide infra).
Results
Selection of Probe and Locus for Site-Specific Incorporation into GR
Molecular dynamics (MD) simulations on GR from previous studies have suggested particular regions of GR that undergo relatively large changes, both in terms of substrate unbinding (via steered MD studies), as well as equilibrated GR–ligand complexes that show movement relative to one another (i.e., GR complexes with different types of active site small molecules equilibrated to distinct conformers).5 A salient residue associated with these altered regions, which could likely be a locus of incorporation for a non-natural fluorescent amino acid, is Tyr53. Incorporation of a site-specific genetically encoded non-natural amino acid 7HC by an orthogonal tRNA/aminoacyl-tRNA synthetase pair, pioneered by Wang et al.,16 is an ideal solution in the case of GR. This system allows targeted inclusion of a single coumarin-based fluorophore with high quantum yield and high environmental sensitivity to pH and solvent polarity.16,17 This is particularly attractive in the case of GR in that a number of MD studies have suggested that Tyr53, although not directly involved in ligand binding and not part of the active site, nevertheless has a dynamic pattern highly associated with distinctive ligand conformers. Previous MD studies have revealed that binding of a variety of ligands causes obvious conformational changes in two regions: a helix and loop/helix that form the entrance to the ligand cleft.5 The loop/helix itself contains Tyr42, which according to in silico docking is seen interacting with specific ligands. The loop/helix is immediately followed by a turn and helix which contains a tyrosine residue, Tyr53. Tyr53 is distinctly outside of the active site binding cleft and does not interact directly with ligands bound to the active site. Thus, the Tyr53 mutated to the 7HC functional group should provide ligand-associated fluorescent sensitivity to changes in the local environment (and thus serve as an allosteric reporter), without sacrificing particular contacts with ligands. The placement of the 7HC moiety at the surface of the enzyme, remote from any ligand pockets, places it in a microenvironment where dielectric values are significantly larger,18 which should restrict fluorescence changes to largely water polarization effects.
Site-Directed Mutagenesis and Expression Conditions
Site-directed mutagenesis was carried out to make two mutations to B. subtilis GR (also known as RacE) , the first being the alteration of the endogenous stop-codon from amber (TAG) to ochre (TAA), in order to prevent probe placement at this location and a read-through mutation. Using this altered gene, mutagenesis was again utilized to mutate the Tyr53 codon to the amber stop codon. This mutation was confirmed by sequencing. The RacE gene (B. subtilis) was placed in a pET15b plasmid, which codes for placement of a cleavable hexahistidine-tag at the N-terminal of the recombinant protein. The pEB-JYRS-courRS plasmid containing the non-natural tRNA and corresponding tRNA synthetase was transformed first into BL21(DE3) E. coli cells. Cells were made chemically competent and transformed with the pET15b plasmid. The resulting cells are resistant to both tetracycline and ampicillin.
The GRY53/7HC mutant protein was expressed and purified (Figure S1) as described in Method S2. It should be noted that cell growth was attenuated upon induction with IPTG, implicating protein expression of these mutants as being toxic. The expression of both the GR pET15b and pEB-JYRS plasmids within E. coli BL21(DE3) appears to reduce the overall fitness of the strain, as demonstrated by a reduced growth rate in liquid culture.
A circular dichroism spectrum of the wild-type and mutant protein was performed to assess proper folding (Figure 1). The two CD spectra are nearly identical, yielding the characteristic profile seen in previous studies.11,14
Figure 1.
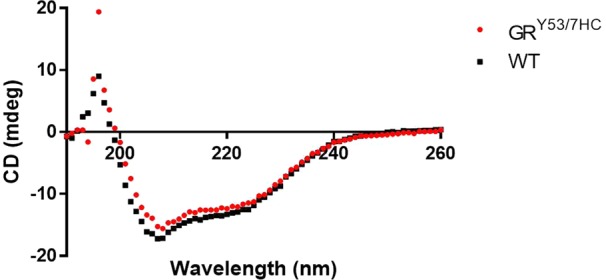
Circular dichroism spectra confirms retained secondary structure of equal concentrations of the GRY53/7HC mutant (red) relative to WT (black).
Fluorescence Wavelength Profile for GRY53/7HC
The intrinsic fluorescence of the GRY53/7HC mutant protein was compared to purified wild-type GR to confirm incorporation of the non-natural amino acid (Figure 2). Wild-type GR has no fluorescence signal when excited at 340 nm, even at substantially higher concentrations than the GRY53/7HC mutant. On the contrary, the mutant has substantial fluorescence signal in this range, which increases with protein concentration. Fluorescence at 455 nm was monitored for 60 min without any notable fluctuations in signal intensity (Figure S2).
Figure 2.
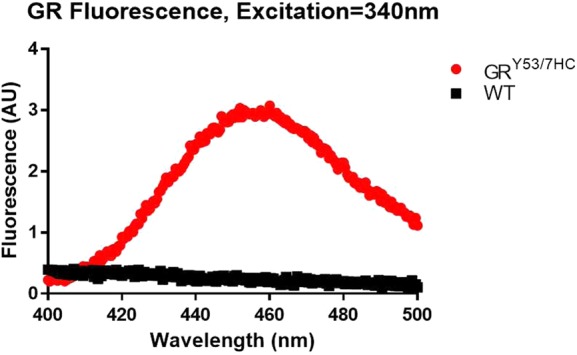
Fluorescence scans using an excitation wavelength of 340 nm confirms the presence of 7HC (λmax = 455 nm) in the GRY53/7HC mutant (red), but not in a similar purification of wild-type GR (black). The concentration of wild-type GR is 4-fold more than that of GRY53/7HC.
GRY53/7HC Is an Active Racemase
To ensure the fluorescent mutant could in fact be used as a representative model of wild-type GR, activity of the GRY53/7HC mutant was assessed in vitro using a coupled-enzyme assay measuring the turnover of l- to d-glutamate (Figure 3). The observed KM value of the mutant was 0.27 ± 0.03 mM, within error of the published value of 0.25 mM.11 The observed kcat value was 0.031 ± 0.001 s–1, a ∼40-fold reduction relative to the published value of 1.3 s–1.11
Figure 3.
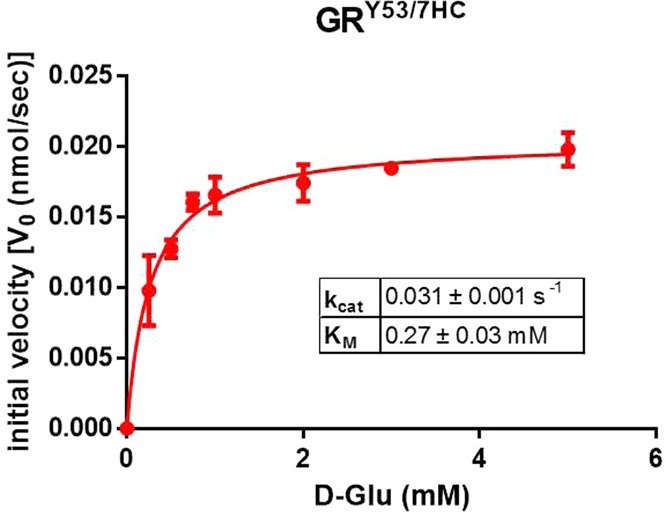
Racemase activity of GRY53/7HC mutant has a KM value within error of the published values (0.25 mM)11 of wild-type GR and a kcat reduced by ∼40-fold relative to the published wild-type GR value (1.3 s–1).11
Titration of Ligands into GRY53/7HC
The fluorescence wavelength profiles of both croconate and glucuronate were obtained at saturating concentrations to determine their specific contribution to fluorescence intensity at the measured wavelength. Croconate had little to no fluorescence at a saturating concentration, while glucuronate did show minimal signal contribution at a concentration of 50 mM. The specific methodology of inhibitor titration is described in detail in Method S3. Titration results are presented in Figure 4. Fitting to a one-site binding model results in KD values that are within experimental error of KI values determined previously via fitting of the Michaelis–Menten expression for competitive inhibition to the steady state kinetic data. This reaffirms that the fluorescent event being measured is directly related to inhibitor binding to GRY53/7HC. Most striking is the dramatic difference in the titration profiles between the two inhibitors. Croconate results in a quenching of the fluorescence intensity, while glucuronate binding results in fluorescence intensity enhancement. It is also encouraging that the incorporation of the probe has not altered the GR structure significantly enough to alter inhibitor binding affinities. These results provide strong experimental support for hypotheses formed from the computational studies by Whalen et al.,5 that ascribed dramatically different receptor conformations to the croconate- and glucuronate-bound GR. Importantly, it is also known that both of these inhibitors are competitive with the glutamate substrate, implicating distinct allosteric linkages between the Y53 position and the small buried active site of GR.
Figure 4.

Binding of (A) croconate and (B) glucuronate to GRY53/7HC elicits distinct changes in the fluorescent signature of GRY53/7HC. The fluorescent titrations were fit to a one-site binding equation giving a KD of 35 ± 1 μM and 1.6 ± 0.3 mM for croconate and glucuronate, respectively. In both cases, the KD is within error of the KI, 42 ± 10 μM for croconate12 and 1.5 ± 0.3 mM for glucuronate. The similarity of the KI and KD values suggests that the fluorescent titration is reporting on actual binding events.
Given the properties of the fluorescent probe, changes to fluorescence intensity can be explained in several ways: conformational changes associated with ligand binding to GRY53/7HC may be altering solvent exposure of the 7HC ring; significant changes in the microenvironment of the 7HC ring may be perturbing the pKa of the 7HC ring hydroxyl; and/or there may be changes in a simple stacking interaction with several local Tyr residues. In order to examine which of these scenarios is most likely, we employed a computational study on the ligand-GRY53/7HC systems, as described below.
Study of GRY53/7HC–Ligand Complexes in silico
In order to gain insight into the types of ligand-associated conformational changes that are responsible for the observed in vitro differential fluorescence pattern outlined above, an MD- and ensemble-docking-based in silico study of croconate- and glucuronate-bound GRY53/7HC was performed. Figure 5 provides a broad overview of the computational workflow.
Figure 5.

Computational workflow used to rationalize the differential fluorescence pattern of croconate and glucuronate binding to GR. (A) A ∼20 ns MD simulation using the YAMBER3 force field,19 a derivative of AMBER,20 was performed on the unliganded form of GR to relax the structure. The low energy structure from the MD simulation was then subjected to the CONCOORD algorithm, as described by de Groot et al.,21 to sample conformational space of the GR structure. (B) Croconate and glucuronate were docked to an ensemble of the original unliganded GR crystal structure and eight CONCOORD generated structures using AutoDock VINA.22 These two ligands selected distinctly different conformations for the top-docked form. (C) The 7HC ring was built into the croconate and glucuronate associated GR structures to generate the GRY53/7HC mutant in silico. A ∼15 ns MD simulation using the YAMBER3 force field19 was then performed on each of the GRY53/7HC–ligand complexes. (D) Clustering of the MD snapshots was performed using the method of Pettersen et al.23 (Method S5), which generated representative forms of the enzyme–ligand complexes from the MD simulation. (E) The low energy structure from the MD simulation, the time averaged structure from the MD simulation, and top representative clustered forms of the MD snapshots were used in a variety of surface area analyses. Molecular graphics created with YASARA (www.yasara.org) and POVRay (www.povray.org).
The unliganded GR crystal structure was relaxed in a ∼20 ns atomistic MD simulation using the YAMBER3 knowledge-based force field,19 employing explicit solvent and physiological conditions. The low energy structure from the production portion of the simulation was used as a seed structure for the CONCOORD (from CONstraints to COORDinates) method of de Groot et al.,21 in which an empirical restraint system is used to attempt to capture the few dominant motions that govern a protein’s flexibility (a simplified form of essential dynamics, ED, which is used to calculate eigenvectors of the covariance matrix of atomic fluctuations that occur in long atomistic MD simulations). The CONCOORD approach is extremely computationally efficient and has yielded results comparable to long MD simulations and experimental NMR data on a number of proteins.21 One of the advantages of this approach, especially for ensemble docking, is that a covariance analysis of randomly generated structures, which satisfy a set of distance constraints, is employed. We elected to generate eight mutually distinct CONCOORD structures using this utility (henceforth referred to as ‘CONCOORD 1–8’). We employed the Global Distance Test (GDT) as a metric for assessing structural uniqueness, which is summarized in the matrix in Table 1. This method is fully described in Method S10.
Table 1. Matrix of Global Distance Test (GDT) Results Showing % Similarity of the Unliganded GR Crystal Structure and CONCOORD Generated Structures Using a 1 Å Cutoff.
| structure | unliganded GRa | CONCOORD 1 | CONCOORD 2 | CONCOORD 3 | CONCOORD 4 | CONCOORD 5 | CONCOORD 6 | CONCOORD 7 | CONCOORD 8 |
|---|---|---|---|---|---|---|---|---|---|
| unliganded GRa | 100 | 28.15 | 29.577 | 20.079 | 17.667 | 26.427 | 19.193 | 16.683 | 23.228 |
| CONCOORD 1 | 28.15 | 100 | 20.472 | 16.093 | 32.677 | 14.124 | 14.665 | 17.224 | 20.276 |
| CONCOORD 2 | 29.577 | 20.471 | 100 | 23.278 | 21.555 | 28.593 | 11.86 | 10.433 | 22.195 |
| CONCOORD 3 | 20.079 | 16.093 | 23.278 | 100 | 12.894 | 10.531 | 17.372 | 13.829 | 13.484 |
| CONCOORD 4 | 17.667 | 32.677 | 21.555 | 12.894 | 100 | 17.52 | 8.563 | 14.075 | 16.978 |
| CONCOORD 5 | 26.427 | 14.124 | 28.593 | 10.531 | 17.52 | 100 | 11.811 | 7.136 | 33.612 |
| CONCOORD 6 | 19.193 | 14.665 | 11.86 | 17.372 | 8.563 | 11.811 | 100 | 14.764 | 10.974 |
| CONCOORD 7 | 16.683 | 17.224 | 10.433 | 13.829 | 14.075 | 7.136 | 14.764 | 100 | 11.614 |
| CONCOORD 8 | 23.228 | 20.276 | 22.195 | 13.484 | 16.978 | 33.612 | 10.974 | 11.614 | 100 |
The crystal structure (PDB 1ZUW)8 of GR bound to d-glutamate was used as the initial structure. d-Glutamate was deleted from the structure, and the unliganded GR form was subjected to a 20 ns MD simulation. The low energy structure from the production phase of the MD simulation was used as the seed structure for the CONCOORD method of structural sampling. The aggregate of the structures in Table 1 represent a crude model for the apo GR ensemble. YASARA’s CONCOORD utility was used to generate structures which sample the conformational space of the GR structure.21 A GDT using a 1 Å cutoff was then performed on a structural superpose of the ensemble of the unliganded GR crystal structure and the CONCOORD generated structures. The GDT assesses the quality of superpose of two structures within a defined distance, providing the percentage of superposed atoms (Method S10).
Next, an ensemble docking screen of croconate and glucuronate against the unliganded GR low energy structure of the 20 ns MD simulation (the seed structure used in the CONCOORD calculations) and the eight CONCOORD-generated structures was performed using AutoDock VINA.22 As previously discussed, croconate and glucuronate were found to select unique forms of GR for their top-docked pose, suggesting these small molecule scaffolds may be selecting distinct forms of GR in solution.5 Glucuronate prefers a conformer with a more solvent-exposed and opened active site cleft, which is unsurprising given the previous studies of Whalen et al. using the FERM-SMD approach.5 On the other hand, croconate is predicted to have the most binding affinity to a conformer with a more closed active site cleft, with less solvent exposure, as well as a smaller active site volume (vide infra). Again this is consistent with previous computational studies.5,12
The GR–ligand complexes were then used as starting points for 15 ns of production atomistic MD simulations of GRY53/7HC–ligand complexes in physiological conditions, using the YAMBER3 force field.19 Note that the 7HC moiety was built into the respective structures and subjected to a refinement before the production phase of the respective simulations (as described in Method S4). A variety of structures from the respective trajectories, including time-averaged, low energy, and major forms from a clustering protocol (henceforth referred to as ‘croconate 1–3’ and ‘glucuronate 1–4’ for representative clustered forms of croconate- and glucuronate-bound GR, respectively), were all subjected to further analysis, which is summarized below. The clustering protocol that was employed, the method of Pettersen et al.,23 is based on the approach of Kelley et al.24 (Method S5). The resulting GRY53/7HC–ligand representative clustered structures showed sufficient structural variation among themselves, based on the matrix of their respective GDT values, that all were subjected to further analysis (Table 2). Through visual inspection, surface area analyses, and analysis of correlated motions of the 7HC ring structure with residues throughout the protein, we studied ligand-associated binding events to gain insight into the varied fluorescence pattern upon croconate and glucuronate binding, respectively.
Table 2. Matrix of Global Distance Test (GDT) Results Showing % Similarity of Representative Clustered Forms of Enzyme–Ligand Complexes from MD Simulation Using a 1 Å Cutoff.
| representative structure | glucuronate 1a | glucuronate 2a | glucuronate 3a | glucuronate 4a |
|---|---|---|---|---|
| croconate 1a | 69.721 | 69.206 | 69.598 | 70.823 |
| croconate 2a | 67.32 | 64.919 | 66.438 | 69.255 |
| croconate 3a | 66.291 | 62.788 | 69.892 | 66.879 |
Representative clustered forms of GRY53/7HC–ligand complexes. Clustering of the MD snapshots was performed based on the method of Pettersen et al.,23 which generated representative forms of the enzyme–ligand complexes from the MD simulations. A GDT using a 1 Å cutoff was then performed from a structural superpose of the top representative enzyme-croconate and enzyme-glucuronate clustered forms (Method S10).
Ligand-Binding Associated Structural Changes
Analysis of the clustered, time-averaged, and low energy structures of the respective GRY53/7HC–ligand complexes all show the general trend of a greater volume active site in the glucuronate complex and a smaller volume active site in the croconate complex (Table 3). These results demonstrate consistency between this work and previous studies in which glucuronate was shown to prefer a more open, solvent-exposed active site, while croconate was shown to prefer a more closed, less solvent-exposed active site.5,12
Table 3. Volume of Active Site (A3) of Key GRY53/7HC–Ligand Structures.
| GRY53/7HC–croconate complexes |
GRY53/7HC–glucuronate complexes |
||
|---|---|---|---|
| structure | volume of active site (Å3) | structure | volume of active site (Å3) |
| LowEa | 124 | LowEa | 142 |
| time avgb | 104 | time avgb | 241 |
| croconate 1c | 99 | glucuronate 1c | 154 |
| croconate 2c | 96 | glucuronate 2c | 124 |
| croconate 3c | 135 | glucuronate 3c | 198 |
| glucuronate 4c | 101 | ||
Low energy structure from the MD simulation.
Time averaged structure from the MD simulation.
Representative clustered forms of GRY53/7HC–ligand complexes. The smaller volume of the GRY53/7HC–croconate complexes suggests a more closed active site, while the greater volume of the GRY53/7HC–glucuronate complexes suggests a more open active site.
There was no substantial difference in interaction of the 7HC ring with any other neighboring Tyr residues between the GRY53/7HC–croconate and GRY53/7HC–glucuronate complexes. Therefore, the differential fluorescence pattern exhibited in the in vitro studies cannot be explained by a simple stacking interaction with nearby Tyr residues. Additionally, no significant changes within the local environment of the 7HC moiety seemed to be occurring that may have altered the pKa of the 7HC hydroxyl group. There have been several reported cases where pKa shifts in a non-natural 7HC moiety are ascribed as partial contributors to the fluorescence changes upon binding of a ligand.25,26 However, in both of these cases, the 7HC moiety was placed in buried regions proximal to the binding of highly negatively charged ligands, which is not analogous to the environment of the probe in the GRY53/7HC system. However, a difference in solvent exposure of the 7HC ring between the croconate and glucuronate complexes was observed. Therefore, it is most probable that this is the cause of the differential fluorescence pattern exhibited in the in vitro studies. Surface area analysis of the 7HC ring in the GRY53/7HC–croconate and GRY53/7HC–glucuronate complexes showed that the 7HC ring is more solvent exposed in the GRY53/7HC–croconate complex than in the GRY53/7HC–glucuronate complex (Table 4 and Figure 6). That this trend was evident looking at the low energy and time averaged structures from the MD as well as the representative cluster structures strongly suggests that this represents a real difference between GRY53/7HC–croconate and GRY53/7HC-glucuronate. The increased solvent exposure of the 7HC ring in the GRY53/7HC– croconate complex and relative decreased solvent exposure of the 7HC ring in the GRY53/7HC–glucuronate complex is consistent with the experimental results, which show fluorescence quenching due to complexation with croconate and an increase in fluorescence intensity due to complexation with glucuronate. It is not possible to predict the magnitude of the fluorescence changes that would result from the current ensemble using molecular mechanics methods, as performed in the current study. Nevertheless, there is a consensus among the representative forms of the individual trajectories, low energy, time averaged, and structurally clustered snapshots, that the GRY53/7HC–croconate complex has a 7HC moiety that is more solvent exposed than the GRY53/7HC–glucuronate 7HC moiety. The magnitude of change in the solvent accessible surface area of the 7HC ring between the two complexes (ranging from 70 to 107 Å2) represents a change that is ∼13% of the total potential solvent accessible surface of the 7HC ring. Although it is challenging to extrapolate the changes observed in the GRY53/7HC system to early model studies that were performed on free 7HC (due to the uncertainty of knowing the dielectric of the protein environment), the work of Zinsli,17 in which the effect of fractional solutions of ethanol and water on 7HC fluorescence was studied, showed very large changes in the fluorescence when going from no water to 14% mole fraction of water (nearly half of the fluoresce signal was lost).
Table 4. Solvent-Accessible Surface Area (Å2) of 7HC Residue of Key GRY53/7HC–Ligand Structures.
| GRY53/7HC–croconate complexes |
GRY53/7HC–glucuronate complexes |
||
|---|---|---|---|
| structure | 7HC solvent accessible surface area (Å2) | structure | 7HC solvent accessible surface area (Å2) |
| LowEa | 91.68 | LowEa | 76.08 |
| time avgb | 97.43 | time avgb | 70.34 |
| croconate 1c | 104.5 | glucuronate 1c | 76.42 |
| croconate 2c | 107.08 | glucuronate 2c | 76.36 |
| croconate 3c | 107.27 | glucuronate 3c | 80.06 |
| glucuronate 4c | 92.62 | ||
Low energy structure from the MD simulation.
Time-averaged structure from the MD simulation.
Representative clustered forms of GRY53/7HC–ligand complexes. Greater solvent exposure of the 7HC moiety is observed in the GRY53/7HC–croconate complexes than in the GRY53/7HC–glucuronate complexes. The magnitude of the change in solvent accessible surface area, ranging from 70 to 107 A2, represents ∼13% of the total potential solvent accessible surface of the 7HC ring system.
Figure 6.

Superpose of most representative clustered GRY53/7HC structures selected by croconate (solvent accessible surface area of this complex is indicated by yellow surface; croconate and 7HC moiety of this complex is indicated by green stick structures) and glucuronate (solvent accessible surface area of this complex is indicated by blue surface; glucuronate and 7HC moiety of this complex is indicated by magenta stick structures). (A) Conformational changes at the active site correspond to changes at the 7HC position. The closing of the active site in the GRY53/7HC–croconate complex corresponds to greater solvent accessible surface area of the 7HC ring, while opening of the active site in the GRY53/7HC–glucuronate complex corresponds to reduced solvent accessible surface area of the 7HC ring. (B) Ligand associated shift in plane of 7HC rings, concomitant with changes in solvent associated surface areas. Molecular graphics created with YASARA (www.yasara.org) and POVRay (www.povray.org).
Correlated Movements of the 7HC Ring with Helix System (res 74–87) behind the Active Site
In order to investigate the dynamic relationship between the Y53/7HC position and the rest of the enzyme, especially the region of the active site, a dynamic cross-correlation matrix was calculated (DCCM) (Method S9) for the GRY53/7HC–croconate and GRY53/7HC–glucuronate complexes, which is represented graphically and numerically in Figure 7 and Table 5, respectively. This matrix indicates how movements of all residue pairs correlate. These values are normalized (as described in Method S9) and range from −1 (perfectly anticorrelated, graphically represented by shades of blue) to +1 (perfectly correlated, graphically represented by shades of yellow). The values on the diagonal must be +1 (because the motion of any atom is perfectly autocorrelated). Figure 7 shows regions of strong cross-correlation of residues highlighted within the white box, which links residues 50–60 (which comprise the helix on which the 7HC ring is located) to residues 74–87 (which comprise two helices connected by a short loop region located behind the croconate/glucuronate binding site). Table 5 lists the normalized cross-correlation values between the position with the 7HC ring and this key region. Behind the active site, there is a helix (residues 74–79) with which both croconate and glucuronate interact. This helix is connected to a second helix (residues 82–87) by a short loop region. The environment surrounding the 7HC ring is formed in part by this second helix (residues 82–87). Visual inspection suggested that the interaction of croconate and glucuronate with the first helix causes significant movement of the second helix, altering the 7HC environment and the solvent exposure of the 7HC ring between the GRY53/7HC–croconate and GRY53/7HC–glucuronate complexes, potentially contributing to the fluorescence changes observed experimentally. Figure 8 depicts the proposed interaction profile. These data provide a structural and dynamic link to the observed changes seen in the experimental fluorescence titration data described above.
Figure 7.

DCCM of (A) GRY53/7HC–croconate and (B) GRY53/7HC–glucuronate. Yellow regions are indicative of positively correlated residue movements, while blue regions are indicative of anticorrelated residue movements. Correlated movements between residues 50–60 (which comprise the helix on which the 7HC is located) to residues 74–87 (which comprise two helices connected by a short loop region located behind the ligand binding site) is shown by the raised yellow regions highlighted by the white boxes. Table 5 provides the normalized values for the DCCM. Molecular graphics created with YASARA (www.yasara.org) and POVRay (www.povray.org).
Table 5. Normalized Cross-Correlation Values between the 7HC Ring and Selected Regions of GRY53/7HC–Ligand Complexesa.
| GRY53/7HC–croconate complex |
GRY53/7HC–glucuronate complex |
||
|---|---|---|---|
| region | averaged correlated movement with 7HC | region | averaged correlated movement with 7HC |
| helix 1 (Res 74–79) | 0.10 ± 0.06 | helix 1 (Res 74–79) | 0.13 ± 0.04 |
| helix 2 (Res 82–87) | 0.2 ± 0.1 | helix 2 (Res 82–87) | 0.2 ± 0.1 |
| entire protein (Res 1–262) | 0.0 ± 0.2 | entire protein (Res 1–262) | 0.0 ± 0.2 |
The normalized values of the DCCM are given where a value of −1 indicates residue movements are perfectly anticorrelated, and a value of +1 indicates residue movements are perfectly correlated. A strong correlation is seen in the movement of the 7HC ring with helix 1 (residues 74–79) and helix 2 (residues 82–87) in both the GRY53/7HC–croconate and GRY53/7HC–glucuronate complexes. There is no statistically significant correlation in the movement of the 7HC ring with all residues, as expected.
Figure 8.

Results of the DCCM suggest that interaction of croconate/glucuronate with helix 1 (residues 74–79) alters the movement of helix 2 (residues 82–87), which in turn causes significant movement of the helix possessing the 7HC ring and alters its environment in the (A) GRY53/7HC-croconate and (B) GRY53/7HC–glucuronate complexes. Molecular graphics created with YASARA (www.yasara.org) and POVRay (www.povray.org).
Discussion
Previous MD simulation studies on GR have implicated the region in the helix comprised of residues 50–60 as undergoing large movements, depending on the type of ligand bound to the active site of GR, and thus represented a reasonable position for incorporation of the fluorescent reporter moiety l-(7-hydroxycoumarin-4-yl) ethylglycine by use of a mutant orthogonal tRNA/aminoacyl-tRNA synthetase approach developed by Wang et al.16 GRY53/7HC, an active glutamate racemase with the 7HC moiety inserted into the Y53 position, was subsequently biosynthesized and utilized as an experimental probe to study conformational changes occurring upon ligand binding. Although the GRY53/7HC mutant has maintained the same KM value as the wild-type GR, it exhibits a ∼40 fold decrease in kcat. This represents a ∼2.2 kcal/mol increase in the kinetic barrier for racemization in the d → l direction. It is not possible to determine the source of this kinetic difference from the current MD studies. The small magnitude of this change may be due to any number of factors, such as an additional hydrogen bond in the ground state of the GRY53/7HC–substrate complex, subtle repositioning of the catalytic base, or perhaps dynamic events in the domain opening that leads to product release. A complicating factor in addressing such subtle kinetic changes is that it is reasonable that product release is at least partially rate determining, based on the primary hydrogen substrate kinetic isotope effects performed on the related L. bacillus GR.27
A significant body of structural, biochemical, and computational work on GR has characterized it as a very plastic enzyme, which has been necessary to rationalize both its structure, function, and nature of inhibition by small molecules.8,10−12,14 Nevertheless, direct experimental feedback about the dynamics and selectivity of allostery employed by this enzyme has been difficult to obtain. A major application of GRY53/7HC is to examine how different competitive inhibitor chemotypes, which remarkably share the same active site pocket, seem to be associated with different conformations of the enzyme. The results from this study provide strong support for the hypotheses from earlier steered MD simulations that point toward very large changes in enzyme structure upon binding and unbinding of ligands to the active site.5 From a ligand- or drug-design viewpoint, one is not dealing with a small variability in the “druggability” of a single pocket, but rather an ensemble-docking of radically different active sites, with a range of druggabilities. Indeed, accurately predicting the binding affinities for these two compounds, croconate and glucuronate, involved assessing not just a range of structures from steered MD simulations of GR, but also examining relative changes in the protein solvation energy of the target.5 The current studies with GRY53/7HC complexation with these two competitive inhibitors showed that croconate binding results in a 57% reduction in the volume of the, already very buried, active site cavity; this may be an important contributor to its significantly higher ligand efficiency (−0.6 kcal/mol for croconate vs −0.3 kcal/mol for glucuronate). Indeed, recent free energy of binding calculations, using the extended linear response (ELR) method, performed on a series of GR-competitive inhibitors showed that the collection of polar ligands, which are preferred by GR, necessitate either a very narrowly tailored van der Waals contact surface or the ability to form a network of interstitial waters deep within the active site.13 In addition to glucuronate, GR binds weakly to a number of other carboxylate-containing ligands, which tend to remain partially solvated within the active site, and lack the enhanced van der Waals contribution observed for croconate binding.13 The two ligands examined in detail in the current study are archetypal scaffolds selected based on a previous extensive steered MD and binding free energy study,5 which encompassed 17 different GR inhibitors. Indeed, these two scaffolds were selected because they typified radically different ligand-associated complexation events with the GR receptor (one with an extraordinarily good and one with a very poor ligand efficiency value). Indeed, many problematic scaffolds described in Whalen et al., 2011,5 including glucuronate, were hypothesized to associate with conformations of GR that have much more solvated subpockets and lead to much poorer than expected binding free energies. These previous findings are supported by the differences in the ligand binding cavities of the respective GRY53/7HC complexes, as described in Table 3. It will be an important next step to determine if the nature of the GRY53/7HC ligand associated changes in fluorescence can be used to identify poor ligand efficiencies in future experimental fragment screening campaigns.
The extraordinary sensitivity of the 7HC moiety within GRY53/7HC, and its incorporation into the dynamic region, provides a valuable experimental probe, quickly identifying a more “open” and solvated form, which should be avoided for high quality complexation; indeed, use of GRY53/7HC could be used in high throughput screening to quickly identify attractive candidates by examining ligand-associated changes in fluorescence. The dynamic cross-correlation matrix, calculated from the MD studies of the two respective GR-complexes, shows strong coupling between regions behind the active site (a two helix system comprised of res 74–87) and the position that incorporates the 7HC moiety. The results from our computational studies indicate that this coupled motion provides a reporter for these active site forms, which provides a nexus between ensemble structures and ligand binding preferences.
Fragment-based drug discovery has been a useful and productive new approach for exploiting novel chemical space in challenging protein and enzyme targets.28,29 The most successful campaigns in optimizing fragment hits usually employ numerous biophysical approaches (X-ray crystallography, NMR, SPR, and ITC) to predict how a fragment’s ligand efficiency (as well as other metrics, such as LogP) will be maintained or improved, while potency (i.e., molecular weight) is increased. Structural knowledge about the complexation is absolutely essential to optimization, yet the very nature of many plastic enzymes means that this comes with a host of potential difficulties, particularly related to different fragments associating with different forms of the target protein, which may or may not be amenable to crystallization or even prediction about these novel structural forms. Indeed, the results of the current study underscore an even more serious challenge: small molecules which bind to the same active site, but to substantially different forms of the target enzyme! Furthermore, it is not at all apparent that attempting to optimize a fragment hit in the usual rational synthetic “growth” strategy would result in this same mode of selection (i.e., fragment modification may have nontrivial effects on selection of the target conformer). One may envisage the use of MD-informed 7HC-labeling strategies, as outlined in the current study, as a means of improving fragment optimization protocols in many other flexible protein and enzyme drug targets.
Future studies will be focused on employing GRY53/7HC in fragment-based screens, in order to determine a broader profile of compounds and their respective allosteric signatures with respect to fluorescence changes. It would be particularly interesting to make GRY53/7HC analogues of H. pylori and B. anthracis GRs, as these have well characterized allosteric pockets remote from the active site. It is quite possible that the unifying theme in the allosteric inhibition of these enzymes may be accurately described by the type of extended selection model employed in these studies (i.e., the nature of noncompetitive, small molecule regulation of GR enzymes resides in an extended selection model, as presented in this study). The generation of GRY53/7HC analogues for these species would yield potentially powerful insight into how small molecules associate with particular conformers of a flexible protein target. Indeed, as our understanding about the linkage between in vitro and in silico studies grows, such knowledge may allow future campaigns to presage how large libraries of fragments will dynamically interact with flexible protein targets.
Acknowledgments
This work was supported by the NIH in the forms of Grant R01-GM097373 to M.A.S., T32-GM067795 to S.F.D., as well as T32-GM704217 to K.L.W. The authors thank Dr. Peter Schultz for the use of the pEB-JYRS(couRS) plasmid.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.5b00211.
Experimental methods, materials, and computational methods, and additional figures (PDF)
Author Contributions
# S.F.D and K.L.W. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Lange O. F.; Lakomek N. A.; Fares C.; Schroder G. F.; Walter K. F.; Becker S.; Meiler J.; Grubmuller H.; Griesinger C.; de Groot B. L. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science 2008, 320, 1471–1475. 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- Gsponer J.; Christodoulou J.; Cavalli A.; Bui J. M.; Richter B.; Dobson C. M.; Vendruscolo M. A coupled equilibrium shift mechanism in calmodulin-mediated signal transduction. Structure 2008, 16, 736–746. 10.1016/j.str.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant B. J.; McCammon J. A.; Gorfe A. A. Conformational selection in G-proteins: lessons from Ras and Rho. Biophys. J. 2010, 99, L87–89. 10.1016/j.bpj.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csermely P.; Palotai R.; Nussinov R. Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem. Sci. 2010, 35, 539–546. 10.1016/j.tibs.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen K. L.; Chang K. M.; Spies M. A. Hybrid Steered Molecular Dynamics-Docking: An Efficient Solution to the Problem of Ranking Inhibitor Affinities Against a Flexible Drug Target. Mol. Inf. 2011, 30, 459–471. 10.1002/minf.201100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. T. Enzymes in the d-alanine branch of bacterial cell wall peptidoglycan assembly. J. Biol. Chem. 1989, 264, 2393–2396. [PubMed] [Google Scholar]
- Kobayashi K.; Ehrlich S. D.; Albertini A.; Amati G.; Andersen K. K.; Arnaud M.; Asai K.; Ashikaga S.; Aymerich S.; Bessieres P.; Boland F.; Brignell S. C.; Bron S.; Bunai K.; Chapuis J.; Christiansen L. C.; Danchin A.; Debarbouille M.; Dervyn E.; Deuerling E.; Devine K.; Devine S. K.; Dreesen O.; Errington J.; Fillinger S.; Foster S. J.; Fujita Y.; Galizzi A.; Gardan R.; Eschevins C.; Fukushima T.; Haga K.; Harwood C. R.; Hecker M.; Hosoya D.; Hullo M. F.; Kakeshita H.; Karamata D.; Kasahara Y.; Kawamura F.; Koga K.; Koski P.; Kuwana R.; Imamura D.; Ishimaru M.; Ishikawa S.; Ishio I.; Le Coq D.; Masson A.; Mauel C.; Meima R.; Mellado R. P.; Moir A.; Moriya S.; Nagakawa E.; Nanamiya H.; Nakai S.; Nygaard P.; Ogura M.; Ohanan T.; O’Reilly M.; O’Rourke M.; Pragai Z.; Pooley H. M.; Rapoport G.; Rawlins J. P.; Rivas L. A.; Rivolta C.; Sadaie A.; Sadaie Y.; Sarvas M.; Sato T.; Saxild H. H.; Scanlan E.; Schumann W.; Seegers J. F.; Sekiguchi J.; Sekowska A.; Seror S. J.; Simon M.; Stragier P.; Studer R.; Takamatsu H.; Tanaka T.; Takeuchi M.; Thomaides H. B.; Vagner V.; van Dijl J. M.; Watabe K.; Wipat A.; Yamamoto H.; Yamamoto M.; Yamamoto Y.; Yamane K.; Yata K.; Yoshida K.; Yoshikawa H.; Zuber U.; Ogasawara N. Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 4678–4683. 10.1073/pnas.0730515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzheinikov S. N.; Taal M. A.; Sedelnikova S. E.; Baker P. J.; Rice D. W. Substrate-induced conformational changes in Bacillus subtilis glutamate racemase and their implications for drug discovery. Structure 2005, 13, 1707–1713. 10.1016/j.str.2005.07.024. [DOI] [PubMed] [Google Scholar]
- May M.; Mehboob S.; Mulhearn D. C.; Wang Z.; Yu H.; Thatcher G. R.; Santarsiero B. D.; Johnson M. E.; Mesecar A. D. Structural and functional analysis of two glutamate racemase isozymes from Bacillus anthracis and implications for inhibitor design. J. Mol. Biol. 2007, 371, 1219–1237. 10.1016/j.jmb.2007.05.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundqvist T.; Fisher S. L.; Kern G.; Folmer R. H.; Xue Y.; Newton D. T.; Keating T. A.; Alm R. A.; de Jonge B. L. Exploitation of structural and regulatory diversity in glutamate racemases. Nature 2007, 447, 817–822. 10.1038/nature05689. [DOI] [PubMed] [Google Scholar]
- Spies M. A.; Reese J. G.; Dodd D.; Pankow K. L.; Blanke S. R.; Baudry J. Determinants of catalytic power and ligand binding in glutamate racemase. J. Am. Chem. Soc. 2009, 131, 5274–5284. 10.1021/ja809660g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen K. L.; Pankow K. L.; Blanke S. R.; Spies M. A. Exploiting Enzyme Plasticity in Virtual Screening: High Efficiency Inhibitors of Glutamate Racemase. ACS Med. Chem. Lett. 2010, 1, 9–13. 10.1021/ml900005b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen K. L.; Spies M. A. Flooding enzymes: quantifying the contributions of interstitial water and cavity shape to ligand binding using extended linear response free energy calculations. J. Chem. Inf. Model. 2013, 53, 2349–2359. 10.1021/ci400244x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen K. L.; Tussey K. B.; Blanke S. R.; Spies M. A. Nature of allosteric inhibition in glutamate racemase: discovery and characterization of a cryptic inhibitory pocket using atomistic MD simulations and pKa calculations. J. Phys. Chem. B 2011, 115, 3416–3424. 10.1021/jp201037t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal M.; Bearne S. L. Inhibition of glutamate racemase by substrate-product analogues. Bioorg. Med. Chem. Lett. 2014, 24, 1432–1436. 10.1016/j.bmcl.2013.12.114. [DOI] [PubMed] [Google Scholar]
- Wang J.; Xie J.; Schultz P. G. A genetically encoded fluorescent amino acid. J. Am. Chem. Soc. 2006, 128, 8738–8739. 10.1021/ja062666k. [DOI] [PubMed] [Google Scholar]
- Zinsli P. E. Investigation of rate parameters in chemical reactions of excited hydroxycoumarins in different solvents. J. Photochem. 1974, 3, 55–69. 10.1016/0047-2670(74)80006-7. [DOI] [Google Scholar]
- Li L.; Li C.; Zhang Z.; Alexov E. On the Dieletric ″Constant″ of Proteins: Smooth Dielectric Function for Macromolecular Modeling and Its Implementation in DelPhi. J. Chem. Theory Comput. 2013, 9, 2126–2136. 10.1021/ct400065j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger E.; Darden T.; Nabuurs S. B.; Finkelstein A.; Vriend G. Making optimal use of empirical energy functions: force-field parameterization in crystal space. Proteins: Struct., Funct., Genet. 2004, 57, 678–683. 10.1002/prot.20251. [DOI] [PubMed] [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M. Jr.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. 10.1021/ja00124a002. [DOI] [Google Scholar]
- de Groot B. L.; van Aalten D. M.; Scheek R. M.; Amadei A.; Vriend G.; Berendsen H. J. Prediction of protein conformational freedom from distance constraints. Proteins: Struct., Funct., Genet. 1997, 29, 240–251. . [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Kelley L. A.; Gardner S. P.; Sutcliffe M. J. An automated approach for clustering an ensemble of NMR-derived protein structures into conformationally related subfamilies. Protein Eng., Des. Sel. 1996, 9, 1063–1065. 10.1093/protein/9.11.1063. [DOI] [PubMed] [Google Scholar]
- Mendes K. R.; Martinez J. A.; Kantrowitz E. R. Asymmetric allosteric signaling in aspartate transcarbamoylase. ACS Chem. Biol. 2010, 5, 499–506. 10.1021/cb9003207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey V. K.; Parrish A. R.; Han S.; Shen Z.; Briggs S. P.; Ma Y.; Wang L. A Fluorescent Reporter of the Phosphorylation Status of the Substrate Protein STAT3. Angew. Chem., Int. Ed. 2011, 50, 8692–8696. 10.1002/anie.201102923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner M. E.; Gallo K. A.; Knowles J. R. Isotope effects and the identifiation of catalytic residues in the reaction catalyzed by glutamate racemase. Biochemistry 1993, 32, 3998–4006. 10.1021/bi00066a021. [DOI] [PubMed] [Google Scholar]
- Murray C. W.; Blundell T. L. Structural biology in fragment-based drug design. Curr. Opin. Struct. Biol. 2010, 20, 497–507. 10.1016/j.sbi.2010.04.003. [DOI] [PubMed] [Google Scholar]
- Murray C. W.; Verdonk M. L.; Rees D. C. Experiences in fragment-based drug discovery. Trends Pharmacol. Sci. 2012, 33, 224–232. 10.1016/j.tips.2012.02.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.