

Abstract
Extracellular signaling is commonly mediated through post-translational protein modifications that propagate messages from membrane-bound receptors to ultimately regulate gene expression. Signaling cascades are ubiquitously intertwined, and a full understanding of function can only be gleaned by observing dynamics across multiple key signaling nodes. Importantly, targets within signaling cascades often represent opportunities for therapeutic development or can serve as diagnostic biomarkers. Protein phosphorylation is a particularly important post-translational modification that controls many essential cellular signaling pathways. Not surprisingly, aberrant phosphorylation is found in many human diseases, including cancer, and phosphoprotein-based biomarker signatures hold unrealized promise for disease monitoring. Moreover, phosphoprotein analysis has wide-ranging applications across fundamental chemical biology, as many drug discovery efforts seek to target nodes within kinase signaling pathways. For both fundamental and translational applications, the analysis of phosphoprotein biomarker targets is limited by a reliance on labor-intensive and/or technically challenging methods, particularly when considering the simultaneous monitoring of multiplexed panels of phosphoprotein biomarkers. We have developed a technology based upon arrays of silicon photonic microring resonator sensors that fills this void, facilitating the rapid and automated analysis of multiple phosphoprotein levels from both cell lines and primary human tumor samples requiring only minimal sample preparation.
Short abstract
Requiring minimal sample prep, silicon photonic microring resonator technology enables rapid, multiplexed, automated analysis of phosphoprotein targets from cell lines and primary human tumor samples.
Introduction
The post-translational modification of proteins is an essential process through which extracellular recognition events can be communicated from receptor activation through signaling cascades to ultimately control transcription.1 Phosphorylation-driven kinase signaling is perhaps the most common post-translational modification utilized in extracellular signaling.2 Not surprisingly, aberrant regulation of phosphorylation is implicated in many diseases,3−5 including cancer, yet a thorough understanding of phosphorylation dynamics can reveal interventional opportunities. Disease altered signaling cascades provide important targets for both current and emerging therapeutic agents,6−8 while also representing diagnostic or prognostic biomarker signatures that are predictive of patient response to particular treatment regimens. However, the interconnectivity and redundancy between and within kinase signaling cascades often gives rise to resistance against many chemotherapeutic strategies.9−13 This crosstalk also limits the diagnostic utility of any single phosphoprotein-based biomarker. Importantly, a more comprehensive survey of disease-altered kinase signaling can only be achieved by simultaneously analyzing multiple phosphoprotein signatures, effectively probing across multiple intersecting cascades to reveal the functional significance of aberrant pathway activation.
Despite clear applications in both fundamental chemical biology and translational clinical diagnostics, robust multiplexed phosphoprotein analysis remains an unmet analytical challenge. In spite of notorious shortcomings in terms of throughput, plexity, and quantitative capability, electrophoretic methods (e.g., Western blot) remain the gold standard for phosphoprotein expression analysis.14,15 A number of impressive advances have been proposed to increase throughput and reduce reagent and sample consumption,16−22 but these methods are still at relatively early stages of development and have yet to find widespread adoption. Reverse phase protein arrays (RPPA), a miniaturized dot-blot immunoassay with low sample input requirements, allow for many samples to be simultaneously interrogated for the presence of a single protein target, and the method has found utility in clinical trials.9,23,24 However, this approach is not well-suited for molecular diagnostic applications requiring simultaneous assaying of a single sample for multiple phosphoprotein targets. A handful of new antibody-based technologies have also emerged in recent years for multiplex protein analysis, a number of which have taken advantage of the spatial and/or PCR-based multiplexing capacity of DNA–antibody complexes.25−29 These technologies, while requiring the synthesis of a DNA–antibody conjugate, have shown impressive limits of detection and multiplexing capacity, though it is worth pointing out that antibody cross-reactivity typically limits multiplexing to ≤20 protein targets within a single sample volume. Higher levels of multiplexing require the sample to be partitioned into separate reaction volumes. A notable exception is an 88-plex assay for cell surface proteins.30 However, this analysis only targeted the outside of an intact cell and was not subjected to the complex milieu intracellular content, and therefore did not require the application of sandwich pairs for higher specificity.
As an alternative analysis to current methods for multiplexed protein detection, we have developed a silicon photonic detection technology that allows for the routine and robust analysis of biomarker targets from single samples.30−32 Chip-integrated arrays of silicon photonic microring resonators are refractive index sensitive devices that have optical properties that can be monitored to reveal the binding of biomolecules to target-specific capture agents (Figure 1). Microring resonators support optical resonances at specific wavelengths, as defined by
where λ is the wavelength of light, m is an integer, r is the radius of the microring, and neff is the effective refractive index of the propagating optical mode. When functionalized with target-specific capture agents (e.g., antibodies, cDNA, aptamers), analyte binding-induced changes in neff are detected as shifts in resonance wavelength. Importantly, this silicon-based technology allows for high levels of multiplexing, is cost-effective and highly scalable, and supports enhanced assays with demonstrated limits of protein detection as low as 500 fM.32
Figure 1.
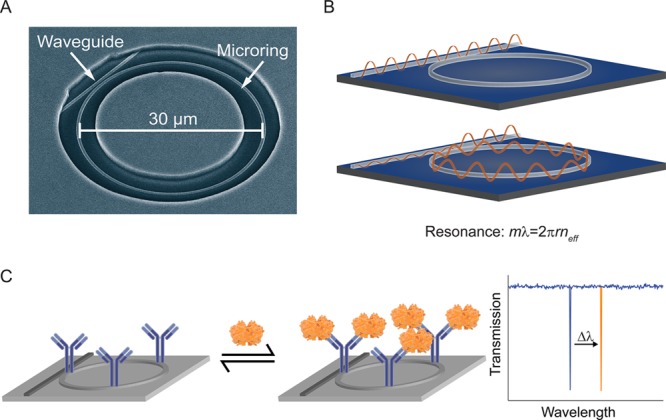
Operating principles for microring resonator sensors. (A) A scanning electron microscope (SEM) image shows a 30 μm active microring with an adjacent linear waveguide. (B) Light is coupled onto the chip via a grating coupler and propagates down the linear waveguide via total internal reflectance. Under resonance conditions, light couples into the adjacent microring, resulting in a narrow dip in the transmittance past the microring, which is measured by a photodetector after coupled off-chip by a second grating coupler. (C) Shifts in the resonant wavelength occur due to changes in the refractive index near the surface of the ring. The schematic example depicts a target protein binding to a capture antibody, resulting in a shift of the resonance to a longer wavelength.
The chief advantages of this technology are the ease of use, rapid analysis times, scalable chip fabrication and functionalization, and robust and reproducible sensor operation. In contrast to our previous efforts, this work reports the highest ever levels of multiplexing using a silicon photonic sensor, and the first demonstration of detection from cell lysate and resected tumor samples. Importantly, sensor substrates are prepared in batch using well-established microspotting techniques. Chips are then simply loaded into a cartridge and fitted with a precut and automatically aligned gasket. This is in contrast to many other reports that require custom microfluidic device fabrication and alignment. The assay is also completely automated using an integrated pumping system so that all that is required is minimal sample preparation (e.g., cell lysis following standard methods). Overall, this type of rapid, scalable, and high throughput method for probing levels of multiple phosphoproteins from single samples could provide new insights into functional and coordinated aspects of disease altered signaling, revealing underlying drivers of cancer progression and informative therapeutic opportunities.
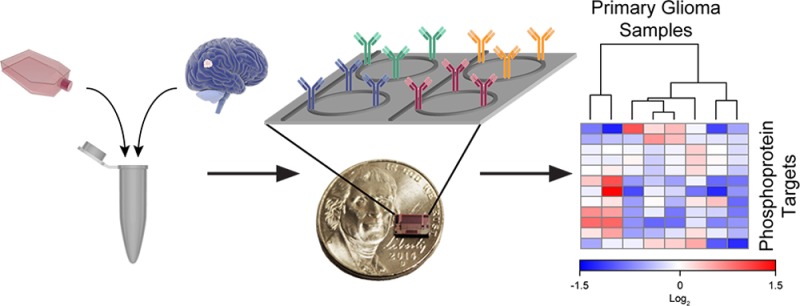
Given the aforementioned needs in both chemical biology and clinical diagnostics, we report the first application of this silicon photonic technology to multiplexed phosphoprotein analysis. Sensor arrays functionalized with monoclonal antibodies specific for 12 kinase cascade-related targets were utilized to rapidly (<2 h) obtain a multiplexed phosphoprotein profile from both glioblastoma cell line models and primary surgical glioma specimens. Spatial multiplexing is achieved by selective immobilization of individually addressable microring sensors with capture antibodies using a commercial microspotter. The method is reproducible, requires low sample input (<104 cells), and was used to monitor dynamic phosphorylation in cell lines responding to stimulatory and inhibitory treatments. Applying this technology to primary surgical glioma specimens, the resulting multiplexed phosphoprotein signature allowed for the rapid discrimination of growing tumor from necrotic tissue: actionable information that, given the rapidity of the assay, could potentially be integrated in near real-time within the clinical decision making process. In general, this multiplexed detection platform, which can be customized to any set of protein targets to which capture agents can be directed, will find broad utility in the monitoring of dynamic cell signaling processes in both fundamental biological and translational diagnostic applications.
Results and Discussion
General Detection Scheme
The general workflow for multiplexed phosphoprotein profiling consists of sample processing (i.e., tissue homogenization and cell lysis), rapid data acquisition, and semiautomated data analysis (Figure 2). Sensor chips were covalently functionalized via microspotting with 12 monoclonal antibodies specific for protein epitopes within targets of interest (Figure S1), including 10 phosphorylation sites. Each antibody was immobilized onto eight independently addressable sensors, providing on-chip technical replicates, and phosphoprotein detection was accomplished using an enzymatically enhanced sandwich immunoassay format (Figure S2).32 After loading the chips into the sensor scanner, whole cell lysates from either cultured glioblastoma cell lines or surgically resected glioma tissue specimens were flowed across the sensor chips. The sandwich immunocomplex assembled on each sensor surface consisted of the capture antibody and antigen, biotinylated tracer antibody, and a streptavidin–horseradish peroxidase conjugate. Protein detection was achieved by monitoring the resonance wavelength shift associated with the enzymatic conversion of 4-chloro-1-naphthol (4CN) to the insoluble 4-chloro-1-naphthon (4CNP) product, which was deposited onto the sensor surface.
Figure 2.

Protein detection scheme for multiplexed phosphoprotein profile. (A) Cells harvested either from human glioma cell lines in culture or from primary resected tumor specimens were homogenized and lysed. (B) Samples were then flowed across a 4 × 6 mm silicon sensor chip containing a 132-element microring sensor array functionalized with target-specific capture antibodies. (C) Resonance wavelength shifts observed from an enhanced sandwich immunoassay correspond to the concentration of each target in the sample. Error bars represent ± SD (n = 8 technical replicates). (D) Resonance wavelength shifts can be displayed as a heat map to reveal heterogeneity between samples.
Targets to be detected (Table S1) were selected due to their key roles in the PI3K/AKT/mTOR and MAPK/ERK pathways, which are aberrantly regulated in many cancers. For multiplex protein detection in cell lysate, relevant antibody pairs (Table S2) were validated and covalently immobilized onto sensor chips using standard bioconjugate chemistry and piezoelectric microspotting. The panel of proteins was then simultaneously detected using the described enzyme-enhanced sandwich immunoassay format. The reproducibility of the assay was demonstrated across multiple biological replicates, and notable differences in phosphoprotein expression were observed between different cell lines.
Sensor Characterization Using a Model Protein
Recombinant β-catenin prepared in running buffer was used as an initial model system to characterize sensor performance (Figure 3A). These results, fit to a logistic function, indicated a ∼3.5-order of magnitude linear dynamic range and a limit of detection and quantitation of 0.6 pM and 1.3 pM, respectively (Table S3). Analysis of β-catenin from within cell lysate showed that this protein could be reliably detected with a sample input of fewer than 10,000 cells (Figure 3B). For reference, fine needle aspirate biopsies consistently yield >500,000 cells,33 and typical cell culture protocols yield >106 cells. While not a focus of this study, which centers on phosphoprotein analysis, absolute quantitation using this platform is achievable for targets that have readily available standards.
Figure 3.

Sensor characterization using a protein standard. (A) Serial dilutions of recombinant β-catenin standard were used to assess sensor performance across 5 orders of magnitude in 10 mM PBST/B buffer. The net shift from the enzymatic signal enhancement step with the net shift from off target mouse IgG isotype control was used as readout. The sensor has a linear dynamic range of 3.55-log with a limit of detection (LOD) of 0.6 pM (solid horizontal black line) and limit of quantification (LOQ) of 1.3 pM (dashed horizontal black line). The data was fit to a logistic function (red trace, adjusted r-squared >0.99). Error bars, SD (n = 3). (B) The minimum sample input for detection of β-catenin cell lysate was determined. The minimum input is between 1000 and 10,000 cells, whereas typical yield of most biopsy methods is >100,000 cells, including fine needle aspirates. The data was fit to a logistic function with adjusted r-squared of 0.985. Error bars represent ± SD (n = 3).
Validation of Antibody Pairs
Identification and screening of antibody sandwich pairs with sufficient specificity and affinity for targets of interest is a major limitation for all immunoassay detection platforms, and there have recently been public calls for dramatic improvements and standardization of antibodies used for research applications (http://www.nature.com/news/1.16827). Without such standardization, rigorous antibody screening procedures are necessary to identify suitable antibody targets. Major advantages of the microring resonator platform are the ability to monitor real-time binding events and the fact that antibody validation can be carried out directly on the same platform on which the final assay will be performed. In contrast to end-point assays, where operators are blind until the conclusion of the multistep assay, the real-time signal observed in microring detection experiments can reveal problematic assay steps. An example of binding events for each step of the protein expression profiling assay is provided in Figure S3. See Materials and Methods for a detailed description of antibody validation procedures.
Phosphoprotein Profiling of Glioblastoma Cell Lines
The ability to discriminate heterogeneities across samples of similar composition is a vital tool for translation of the platform into a workable tool both for research and clinical settings. To test the performance of the multiplexed assay, five model glioblastoma cell lines were analyzed: U-87 MG, U-343 MG-a, U-251 MG, LN-229, and T98-G. Prior to analysis, cell lysate samples were diluted to 50 μg/mL total protein content as determined by a bicinchoninic acid (BCA) assay to ensure a constant sample input across multiple samples. These cell lines were grown using standard tissue culture, lysed under non-denaturing conditions, and analyzed on the microring resonator platform. Within a single 2 h experiment, the 12-plex phosphoprotein analysis can be performed (Figure 4A). Of all the examined targets, Ser780 and Ser807/811 retinoblastoma (Rb) phosphorylation sites showed the greatest variance between individual cell lines (Figure 4B). Rb is a tumor suppressor protein known to be functionally inactive in many cancer types, such as osteosarcoma and small-cell carcinoma.34 Activated forms of Rb block the progression of cell cycle from G1 to S phase by inhibiting E2F transcription factors.35 Cyclin dependent kinase (Cdk) phosphorylation inactivates Rb, typically causing the cell to advance into S phase DNA replication.36 Inactivation of Rb, as appears to be the case in the T-98-G cell lines (Figure 4A), can induce quiescent cells to re-enter the cell cycle, initiating cancerous growth.37 However, Rb is neither necessary nor sufficient for cancer development, as is seen by the low levels of phosphorylation in both the U87 and U343 cell lines.
Figure 4.

Twelve-plex phosphoprotein analysis of glioma cell lines. (A) The multiplex phosphoprotein analysis of 5 model glioma cell lines grown in standard tissue culture reveals difference in protein phosphorylation. The data is normalized across rows and fit to a log-2 scale (n = 3). The bar graph represents a single multiplexed detection experiment on the microring resonator platform for 12-plex phosphoprotein analysis of the U87 MG cell line. (B) Differences in protein phosphorylation for four targets (β-catenin, phospho-Akt Ser 473, phospho-Rb Ser 807/811, phospho-S6 Ser 240/244) across five cell lines. Error bars represent ± SD (n = 3 different samples per cell line). * significant at p < 0.05, ** significant at p < 0.01, and *** significant at p < 0.001.
When presented in the form of a heat map, data normalization is performed for each target of interest, and this method allows for facile comparison between model glioma cell lines. Categorization of samples based on protein profiles has the potential for downstream applications in a clinical setting for subclassification of glioma in patients, as was previously accomplished using miRNA expression levels.38 In general, the assay is highly reproducible (Figure S4) with a technical variance <3% for analyses of cell lysate samples. The biological variance, determined by analysis of lysate from three cultures of cells from across different passages of the same cell line, was determined to be <18%.
In addition to detecting basal phosphoprotein levels, we also utilized this multiplexed platform to monitor dynamic changes in expression in response to four different cell treatments. Cells were cultured in the presence of vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), rapamycin, or wortmannin. Cells cultured in media both with and without 10% fetal bovine serum (FBS) were analyzed as references. Interestingly, LN-229 and U-87 MG cell lines demonstrated differential responses to treatment in culture (Figure 5A). For instance, rapamycin treatment results in a dramatic shift in the phospo-S6 ribosomal protein kinase levels for the Ser235/236 site in both cell lines. However, for that same treatment, U87 cells showed a mild upregulation in phospho-Akt and phospho-mTOR levels in comparison to LN-229 cells. These examples of heterogeneous cell line responses to treatment suggest the potential application of this platform for assessing tumor status and monitoring in vitro therapeutic response.
Figure 5.

Multiplexed phosphoprotein profiling of treated cell lines and primary surgical glioma specimens. (A) Dynamic phosphoprotein expression profiling of U87 MG and LN-229 cell lines in response to 4 different treatment regimens (VEGF, EGF, rapamycin, and wortmannin) with (−)-serum and (+)-serum serving as references. (B) Isolated primary samples from six glioma patients were analyzed for the 12 protein targets and sorted via unsupervised hierarchical clustering based on Euclidean distance. The dashed regions highlight two samples that clustered together that, in contrast to the other samples, were identified in pathology reports as being composed of >50% necrotic tissue.
Analysis of Primary Surgical Glioma Specimens
Cell lysates derived from established cell lines in culture provided a proof-of-concept for the phosphoprotein profiling using the microring resonator platform, but the method should also be applicable to tumor tissue homogenates if it is to serve as a useful diagnostic platform. To that end, primary surgical glioma specimens were homogenized and lysed following the same protocol as the cell culture samples. For the analysis of tumor tissue homogenates, the technical variance was <6% (Figure S4). In general, the surgical glioma samples had reduced phosphoprotein expression levels compared to cultured cell lines,39 so a higher total protein concentration was used for all tumor samples (150 μg/mL). The multiplexed phosphoprotein analysis was performed as described, and the resulting expression levels were used as input for clustering via Euclidean distance. Notably, glioma tissue homogenates were clearly distinguished from isolated primary glioma stemlike cell samples (Figure S5). Furthermore, two samples (21225 and 43096) showed widespread reduction in protein phosphorylation, and clustered together separate from the other glioma samples (Figure 5B). A review of pathology reports indicated that these two samples uniquely had extensive necrosis (>50% necrotic tissue). Necrosis can occur via several mechanisms, including both p53-dependent and independent pathways.40−43 In the context of GBM, necrosis via radiation injury is commonly encountered in recurrent disease and is often difficult to distinguish from actively growing tumor using many noninvasive imaging approaches,40,44 complicating surgical intervention. While beyond the scope of this paper, it is important to note that the 2 h time frame of our multiplexed assay might provide useful information to pathologists regarding molecular tissue/tumor heterogeneity, which in turn might help guide the surgical decision making process intraoperatively.
Conclusion
We have utilized silicon photonic microring resonator arrays to dynamically monitor phosphoprotein levels using both cultured glioblastoma cell lines and surgical human glioma specimens. This multiplexed technology can simultaneously determine expression levels of multiple targets in less than 2 h using sample inputs compatible with many basic science and clinical detection applications. In addition to monitoring differences in phosphoprotein levels from glioblastoma cell lines cultured in the presence or absence of stimulatory and inhibitory factors, this technology revealed differences between surgical glioma specimens. Because phosphorylation-dependent pathway activation plays an important role in tumor growth and resistance, the rapid and broad-based analysis of phosphoprotein patterns as outlined here could provide useful diagnostic and therapeutic information beyond that which can be inferred from genomic or transcriptomic studies.
Using phosphoprotein levels, we could stratify samples that had extensive tissue necrosis, identified as having reduced levels of phosphoprotein targets. Importantly, this rapid discrimination of growing tumor from necrotic tissue provides actionable information that could potentially be integrated in near real-time within the clinical decision making process. In this report we describe the simultaneous detection of 12 protein targets; however, the plexity of the panel can be further increased and, for example, could include comprehensive profiling of both phosphorylated and nonphosphorylated forms of selected targets. In general, this multiplexed detection technology, which can be applied to any panel of protein biomarkers to which high affinity capture agents can be directed, will find utility throughout both fundamental biological and clinical applications that seek to simultaneously probe expression levels of multiple protein targets, including those focusing on dynamic processes such as pathway activation and therapeutic regulation.
Materials and Methods
Reagents
Unless indicated otherwise, all reagents were purchased from Sigma-Aldrich and were used as received according to the manufacturer’s protocol. The following reagents were purchased from Thermo Scientific: (bis[sulfosuccinimidyl]) suberate (BS3) homobifunctional amine-to-amine cross-linking agent (#21585), streptavidin–horseradish peroxidase (SA–HRP) conjugate (#21130), 1-step chloronaphthol solution containing 4-chloro-1-naphthol (4-CN) and proprietary peroxide-containing buffer (#34012 or NC0544546), and StartingBlock (PBS) blocking buffer (#37548). 3-Aminopropyltriethoxysilane (APTES) was from Gelest (#SIA0610.1). DryCoat assay stabilizer was from Virusys (#AG066-1). Recombinant β-catenin was from abcam (#ab63175). PBS buffer (10 mM) with 0.1% BSA and 0.05% Tween-20 (PBST/B) was used as a running buffer for all experiments on the microring resonator platform. All antibodies were purchased from Cell Signaling Technology (Table S2) as custom formulations in 10 mM PBS. Biotinylation of tracer antibodies was performed using the EZ-Link NHS-PEG4-Biotin reagent from Thermo Scientific (21329).
Microring Resonator Instrumentation, Chip Fabrication, and Operation
Maverick M1 optical scanning instrumentation, control software, and sensor array chips were purchased from Genalyte Inc. (San Diego, CA). Sensor fabrication and operation have been previously described.30,45 Briefly, sensor chips are batch fabricated on a silicon-on-insulator (SOI) wafer, and sub-micrometer features (e.g., grating couplers, waveguides, and microrings) are generated via deep UV lithography and reactive ion etching. A perfluoropolymer cladding layer is added to the entire chip surface via spin coating, and annular windows are etched over 132 of 136 rings. The exposed rings are termed active, and the remaining 4 rings are used as controls to correct for thermal drift. The chips come coated with a protective 1 μm thick photoresist layer, which is removed prior to use by immersing first in an acetone bath immediately followed by rinsing the chip in isopropanol.
Sensor interrogation is performed using an external cavity tunable diode laser centered at 1550 nm coupled via fiber optics to a free-space optical scanner.45 The scanner focuses and steers the optical beam to serially probe each of the 136 rings while sweeping the laser wavelength through a suitable spectral window. Light is coupled into microrings via chip-integrated grating couplers. A 30 μm microring is adjacent to the waveguide, and light couples from the waveguide into the ring only under resonance conditions (described above). When functionalized with an appropriate capture agent, binding-induced changes in neff lead to shifts in the resonance wavelength, which can be used to quantitatively detect biomolecular targets. Resonance wavelength shifts are monitored as a function of time and reported in units of Δpm. Additional instrumental specifications and a more exhaustive description of operation have been previously reported.45
Array Surface Functionalization
Capture biomolecules are covalently immobilized onto the ring surface into a spatially multiplexed array using robotic microspotting. After removal of the protective photoresist layer, chips are silanized in a 1% APTES solution in acetone for 2 min, followed by 2 min rinses in acetone and then isopropanol. The chips are then briefly (<10 s) rinsed in water and dried under a N2 gas stream. Chips are subsequently loaded into the microarrayer. Each cluster of 4 active microrings was spotted with a 2 mM acetic acid solution containing 5 mM BS3 cross-linker. Antibody solutions of 400 μg/mL in 10 mM PBS with 5% glycerol were then spatially arrayed onto individual clusters of four microrings on the chip surface. Spotting locations were randomly assigned across each channel of the sensor array (Figure S1). Spotted chips were then transferred to a humidity chamber for 1 h. The chips were next coated with DryCoat by gently pipetting the solution across the chips’ surface. The chips were then transferred to a desiccator at 4 °C for storage.
Lower plexity chips utilized for antibody validation, cross reactivity screening, and β-catenin detection standardization were fabricated using identical attachment chemistries; however, solutions were spotted by hand rather than using the robotic microspotter.
Antibody Validation Protocol
Selection of antibody targets for screening was guided by recommendations from commercial vendors in order to ensure that epitopes for each component of the antibody sandwich did not overlap. In addition to vendor recommendations, various research facilities (e.g., RPPA Core Facility—Functional Proteomics, MD Anderson Cancer Center, http://www.mdanderson.org) provide databases containing antibodies that have undergone extensive screening procedures that were useful in selecting successful antibody clones.
In order to validate recommended antibody pairs for protein detection in cell lysate on the microring resonator platform, antibodies were purchased from vendors in a custom formulation in a 10 mM PBS buffer. Custom formulations were necessary to ensure that no species were present that would interfere with either surface conjugation or biotinylated tracer antibodies. On-target capture antibodies and an off-target control antibody (e.g., mouse IgG isotype control) were functionalized onto a chip following the methodology described above. When available, reference protein standards were used as positive controls. When no standard was available, as is the case for the phosphoprotein targets, cell lysates isolated from the 5 model glioma cell lines were used as reference samples. An antibody hit was considered valid when the following were observed: (1) on-target signal significantly above off-target control, (2) concentration-dependent signal response (typically undiluted, 1:10, and 1:100 cell lysate dilutions), (3) statistically insignificant signal for on-target and off-target antibodies for negative control experiments, and (4) Western blot experiments indicated a single band at the appropriate molecular weight using the same antibody clone.
The absolute minimum signal for a validation target is the limit of quantitation (LOQ), which was experimentally determined to be an 11.5 pm shift based off of the signal and standard deviation of a blank response (Figure S3). Practically, all qualifying targets provided a net shift of at least 300 pm above background. Two types of negative control experiments were used: (1) removal of on-target tracer antibodies from the standard assay and (2) removal of diluted whole lysate form the standard assay. The combination of the two negative control runs ensured that tracer antibodies would only form the antibody sandwich when target analyte is present. Additionally, the off-target control capture antibody ensured that the tracer antibodies did not bind to cell lysate proteins that nonspecifically adsorb to the sensor surface. Nonspecific adsorption is also prevented by blocking the sensor surface with a carrier protein (see below). The Western blot validation ensured that the antibody targets correctly bound the target of interest without cross reactivity (Figure S6). A major benefit of using a sandwich immunoassay is that cross-reactivity will be observed only if there is off-target or nonspecific binding during both the capture and tracer steps in the assay.
For all cases, antibodies were tested in both sandwich configurations to determine which provided the best performance (e.g., run 1, A as capture, B as tracer; run 2, B as capture, A as tracer). Additionally, antibodies were validated using Western blotting for lysate samples from at least 3 cell lines.
Protein Expression Profiling
Twelve-plex protein expression profile chips were spotted as described above. For each run, flow was coupled to the sensor surface via a fluidic cartridge assembly (Figure S7). Sensor chips were sandwiched between an aluminum cartridge holder, a 0.007 in. laser cut biaxially oriented polyethylene terephthalate (Mylar) gasket, and a polytetrafluorethylene (Teflon) cartridge top. Solvent was delivered to the cartridge assembly from via a 0.25 mm flangeless 1/4-28 fitting screwed directly into the cartridge top. PEEK plugs were used to cap unused fluidic ports. The entire cartridge assembly was then loaded into the Maverick system.
For all steps in the assay, the flow rate was 30 μL/min. The assay consisted of (1) StartingBlock protein blocking step (10 min), (2) rinse with 10 mM PBST/B (10 min), (3) 20 min cell lysate (50 μg/mL in 10 mM PBST/B, 20 min), (4) protein blocking with StartingBlock (10 min), (5) rinse with 10 mM PBST/B (2 min), (6) tracer antibodies (1 μg/mL in PBST/B, 10 min), (7) rinse with PBST/B (2 min), (8) streptavidin–HRP conjugate (2 μg/mL in 10 mM PBST/B, 10 min), (9) 1-step chloronaphthol solution (25 min), and (10) rinse with 10 mM PBST/B (10 min). The total assay time was 117 min; however, the assay can be shortened further through optimization of blocking and reagent loading steps. For example, a nonoptimized 45 min assay was successfully carried out, but with <25% loss in signal.
Data Analysis
All data analysis was performed using Origin 9.1 (OriginLab Corporation, Northampton, MA) as well as a custom program for semiautomated data analysis. For analysis of protein and phosphoproteins in cell lysate, the net shift resulting from the enzymatic signal enhancement during the 25 min chloronaphthol oxidation step was used as assay readout. For each target, the data represents averaged responses from 4 to 8 replicate measurements on a single chip, corresponding to either 1 or 2 clusters of 4 microrings responding to a given target. Sensor calibration data, determined using β-catenin in PBST/B buffer, was plotted as sensor response (measure in Δpm) versus time and fit to the logistic function (Figure 3 and Table S3):
where A1 is the initial value (Δpm), A2 is the final value (Δpm), x is the analyte concentration (pM), x0 is the center value (inflection point, pM), and p is the power parameter affecting the slope of the linear portion of the fit surround the inflection point.
To generate expression heat maps, the data from the 12-plex protein expression profile was normalized by row to the average response for that target across the samples of interest according to the equation
where x is target signal for a single sample and x̅ is the mean target signal for a sample set. Data visualization was performed using Gene-E (http://broadinstitute.org/cancer/software/GENE-E/). The heat map data is also presented in Tables S4–S6 as the standard score (i.e., z-score) to indicate the deviation from the mean for each.
Cell Culture and Treatments
Model glioma cell lines were obtained from ATCC (U-87 MG [HTB-14], LN-229 [CRL-2611], T98-G [CRL-1690]) or Cell Line Service (U-251 MG [300385], U-343 MGa [300365]). For comparison of untreated samples, cells were cultured in Dulbecco’s modification of Eagle’s medium (DMEM, Corning, #10-013-CV) supplemented with 10% FBS (VWR, #1400-500) and 1% penicillin–streptomycin (Life Technologies, #15070-063). Cells were subcultured at ∼80% confluency using 0.05% Trypsin-EDTA (Life Technologies, #25300-062) for cell detachment and reseeded at 2 × 106 cells/mL.
For treatments, cells were cultured using the same method as untreated cells for one passage cycle. Cells were then starved in serum-free medium for 6 h immediately prior to treatment. One of the following reagents was supplemented into serum-free DMEM medium for each of the four treatments: VEGF (Cell Signaling Technology, #8065SC) at 200 ng/mL, EGF (Cell Signaling Technology, #8916SC) at 200 ng/mL, wortmannin (Cell Signaling Technology, #9951S) at 200 nM, or rapamycin (Cell Signaling Technology, #9904S) at 100 nM. Two control treatments were also performed consisting of serum-free DMEM with 200 μL of DMSO added as a loading control and DMEM supplemented as described for untreated samples. Cells were harvested and lysed after 18 h of treatment.
Cell lysis was performed following the manufacturer’s protocol using the cell lysis buffer containing protease and phosphatase inhibitors (Cell Signaling Technology, #9803S), supplemented with phenylmethanesulfonyl fluoride (PMSF) as a protease inhibitor (Cell Signaling Technology, #8553S). The buffer contains 1% Triton X-100 (Fisher, #BP151-100), a nonionic detergent, for cell lysis. The use of nonionic detergent ensures that the capture antibodies do not denature upon exposure to cell lysate solution, as has been observed when using lysis buffers containing sodium dodecyl sulfate (SDS) or other ionic detergents. Ionic detergents could be used though; however, an additional step of detergent removal would need to be incorporated into the workflow.
During the lysis protocol, a small fraction (50 μL) was set aside to determine the total protein concentration using a bicinchoninic acid (BCA) assay (Fisher, #PI-23227) following the manufacturer’s protocol. All lysate from cell culture was analyzed at 50 μg/mL total protein concentration.
Preparation of Primary Glioma Samples
All primary surgical glioma samples were collected at Brigham and Women’s Hospital under authorized IRB protocols. Samples were homogenized and lysed according to the same protocol as the cell culture samples. Total protein levels were determined using a BCA assay (see above), and all samples were analyzed at 150 μg/mL.
Western Blot
Cell lysate samples were quantified with a BCA assay (see above) and brought to similar final concentrations. Samples were electrophorectically separated via SDS–PAGE with 4–20% MINI PROTEAN TGX gels (Bio-Rad, #4561093). Gels were loaded with a visible protein ladder for transfer visualization (Bio-Rad, #161-0374) and biotinylated protein ladder (Cell Signaling Technology, #7727) for chemiluminescent signaling. Proteins were transferred to nitrocellulose membranes (Cell Signaling Technology, #12369) via standard methods using the Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad, #1703930). The blots were blocked with 5% w/v nonfat dry milk in 50 mM tris-buffered saline (TBS) with 1% Tween-20 (TBST) for 1 h. Membranes were incubated with primary antibody overnight at 4 °C following the manufacturer’s protocols. All antibodies were used at a 1:1000 dilution unless otherwise specified by the manufacturer. Blots were washed three times in 15 mL of TBST prior to an incubation step (1 h, rt) with secondary anti-rabbit or anti-mouse IgG and anti-biotin, all HRP-linked antibody (1:1000 in 5% w/v nonfat dry milk in TBST, Cell Signaling Technology, #7074, 7075, and 7076). Blots were rinsed again with 15 mL of TBST three times prior to imaging with chemiluminescence using either LumiGlo chemiluminescent reagent or peroxide SignalFire ECL Reagent (Cell Signaling Technology, #7003, 6883).
Acknowledgments
The authors gratefully acknowledge financial support from the National Institutes of Health (NIH) through the National Cancer Institute (CA177462-01). J.H.W. was supported by the National Science Foundation Graduate Research Fellowship Program. The content is solely the responsibility of the authors and does not represent the official views of any funding agencies. Credit to somersault18:24 (somersault1824.com) for figure components, shared under a Creative Commons license (CC BY-NC-SA 4.0).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.5b00250.
List of protein targets and antibody pairs, fitting parameters, standard scores for heat maps, sensor spotting map, assay schematic, real-time binding demonstration, additional glioma sample clustering, antibody validation with Western blots, and fluidic interface with cartridge assembly (PDF)
The authors declare the following competing financial interest(s): R.C.B. has a financial interest in Genalyte, Inc.
Supplementary Material
References
- Gomperts B. D.; Kramer I. M.; Tatham P. E. R.. Prologue: Signal Transduction, Origins, and Ancestors. In Signal Transduction, 2nd ed.; Academic Press: San Diego, 2009; Chapter 1, pp 1–20. [Google Scholar]
- Gomperts B. D.; Kramer I. M.; Tatham P. E. R.. Phosphorylation and Dephosphorylation: Protein Kinases A and C. In Signal Transduction, 2nd ed.; Academic Press: San Diego, 2009; Chapter 9, pp 243–272. [Google Scholar]
- Krook A.; Björnholm M.; Galuska D.; Jiang X. J.; Fahlman R.; Myers M. G.; Wallberg-Henriksson H.; Zierath J. R. Characterization of signal transduction and glucose transport in skeletal muscle from type 2 diabetic patients. Diabetes 2000, 49 (2), 284–292. 10.2337/diabetes.49.2.284. [DOI] [PubMed] [Google Scholar]
- Steeg P. S. Metastasis suppressors alter the signal transduction of cancer cells. Nat. Rev. Cancer 2003, 3 (1), 55–63. 10.1038/nrc967. [DOI] [PubMed] [Google Scholar]
- Hoyer S. Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur. J. Pharmacol. 2004, 490 (1–3), 115–125. 10.1016/j.ejphar.2004.02.049. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Yang P. L.; Gray N. S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9 (1), 28–39. 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivanco I. Targeting molecular addictions in cancer. Br. J. Cancer 2014, 111 (11), 2033–2038. 10.1038/bjc.2014.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliwkowski M. X.; Mellman I. Antibody Therapeutics in Cancer. Science 2013, 341 (6151), 1192–1198. 10.1126/science.1241145. [DOI] [PubMed] [Google Scholar]
- Pierobon M.; Wulfkuhle J.; Liotta L.; Petricoin E. Application of molecular technologies for phosphoproteomic analysis of clinical samples. Oncogene 2015, 34, 805. 10.1038/onc.2014.16. [DOI] [PubMed] [Google Scholar]
- Quaranta V.; Tyson D. R. What Lies Beneath: Looking Beyond Tumor Genetics Shows the Complexity of Signaling Networks Underlying Drug Sensitivity. Sci. Signaling 2013, 6 (294), pe32–pe32. 10.1126/scisignal.2004715. [DOI] [PubMed] [Google Scholar]
- Niederst M. J.; Engelman J. A. Bypass Mechanisms of Resistance to Receptor Tyrosine Kinase Inhibition in Lung Cancer. Sci. Signaling 2013, 6 (294), re6. 10.1126/scisignal.2004652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klempner S. J.; Myers A. P.; Cantley L. C. What a Tangled Web We Weave: Emerging Resistance Mechanisms to Inhibition of the Phosphoinositide 3-Kinase Pathway. Cancer Discovery 2013, 3 (12), 1345–1354. 10.1158/2159-8290.CD-13-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz L. A. Jr; Williams R. T.; Wu J.; Kinde I.; Hecht J. R.; Berlin J.; Allen B.; Bozic I.; Reiter J. G.; Nowak M. A.; Kinzler K. W.; Oliner K. S.; Vogelstein B. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486 (7404), 537–540. 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnette W. N. Western Blotting”: Electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal. Biochem. 1981, 112 (2), 195–203. 10.1016/0003-2697(81)90281-5. [DOI] [PubMed] [Google Scholar]
- Corley R. B.Detection and Analysis of Proteins. In A Guide to Methods in the Biomedical Sciences; Springer US: 2005; pp 1–24. [Google Scholar]
- Yen Y. K.; Jiang Y. W.; Chang S. C.; Wang A. B. Western blotting by thin-film direct coating. Anal. Chem. 2014, 86 (10), 5164–70. 10.1021/ac5010162. [DOI] [PubMed] [Google Scholar]
- Hughes A. J.; Spelke D. P.; Xu Z.; Kang C.-C.; Schaffer D. V.; Herr A. E. Single-cell western blotting. Nat. Methods 2014, 11 (7), 749–755. 10.1038/nmeth.2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S.; Anderson G. J.; Kennedy R. T. Western blotting using microchip electrophoresis interfaced to a protein capture membrane. Anal. Chem. 2013, 85 (12), 6073–9. 10.1021/ac400940x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W.; Chen W.; Jiang X. Microfluidic Western Blot. Anal. Chem. 2010, 82 (10), 3974–3976. 10.1021/ac1000493. [DOI] [PubMed] [Google Scholar]
- Hughes A. J.; Herr A. E. Microfluidic Western blotting. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 21450–21455. 10.1073/pnas.1207754110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson G. J.; M. Cipolla C.; Kennedy R. T. Western Blotting Using Capillary Electrophoresis. Anal. Chem. 2011, 83 (4), 1350–1355. 10.1021/ac102671n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung M.; Kim D.; Herr A. E. Microchamber Western blotting using poly-L-lysine conjugated polyacrylamide gel for blotting of sodium dodecyl sulfate coated proteins. Anal. Chem. 2013, 85 (16), 7753–61. 10.1021/ac401012j. [DOI] [PubMed] [Google Scholar]
- Akbani R.; Becker K.-F.; Carragher N.; Goldstein T.; de Koning L.; Korf U.; Liotta L.; Mills G. B.; Nishizuka S. S.; Pawlak M.; Petricoin E. F.; Pollard H. B.; Serrels B.; Zhu J. Realizing the Promise of Reverse Phase Protein Arrays for Clinical, Translational, and Basic Research: A Workshop Report: The RPPA (Reverse Phase Protein Array) Society. Mol. Cell. Proteomics 2014, 13 (7), 1625–1643. 10.1074/mcp.O113.034918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibes R.; Qiu Y.; Lu Y.; Hennessy B.; Andreeff M.; Mills G. B.; Kornblau S. M. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol. Cancer Ther. 2006, 5 (10), 2512–2521. 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- Schweitzer B.; Roberts S.; Grimwade B.; Shao W.; Wang M.; Fu Q.; Shu Q.; Laroche I.; Zhou Z.; Tchernev V. T.; Christiansen J.; Velleca M.; Kingsmore S. F. Multiplexed protein profiling on microarrays by rolling-circle amplification. Nat. Biotechnol. 2002, 20 (4), 359–365. 10.1038/nbt0402-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey R. C.; Kwong G. A.; Radu C. G.; Witte O. N.; Heath J. R. DNA-Encoded Antibody Libraries: A Unified Platform for Multiplexed Cell Sorting and Detection of Genes and Proteins. J. Am. Chem. Soc. 2007, 129 (7), 1959–1967. 10.1021/ja065930i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan R.; Vermesh O.; Srivastava A.; Yen B. K. H.; Qin L.; Ahmad H.; Kwong G. A.; Liu C.-C.; Gould J.; Hood L.; Heath J. R. Integrated barcode chips for rapid, multiplexed analysis of proteins in microliter quantities of blood. Nat. Biotechnol. 2008, 26 (12), 1373–1378. 10.1038/nbt.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M.; Wei W.; Su Y.; Kim J.; Shin Y. S.; Mai W. X.; Nathanson D. A.; Heath J. R. Chemical Methods for the Simultaneous Quantitation of Metabolites and Proteins from Single Cells. J. Am. Chem. Soc. 2015, 137 (12), 4066–4069. 10.1021/jacs.5b00944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullal A. V.; Peterson V.; Agasti S. S.; Tuang S.; Juric D.; Castro C. M.; Weissleder R. Cancer Cell Profiling by Barcoding Allows Multiplexed Protein Analysis in Fine-Needle Aspirates. Sci. Transl. Med. 2014, 6 (219), 219ra9. 10.1126/scitranslmed.3007361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn A. L.; Luchansky M. S.; Bowman A. L.; Bailey R. C. Quantitative, Label-Free Detection of Five Protein Biomarkers Using Multiplexed Arrays of Silicon Photonic Microring Resonators. Anal. Chem. 2010, 82 (1), 69–72. 10.1021/ac902451b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchansky M. S.; Bailey R. C. Rapid, multiparameter profiling of cellular secretion using silicon photonic microring resonator arrays. J. Am. Chem. Soc. 2011, 133 (50), 20500–6. 10.1021/ja2087618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindt J. T.; Luchansky M. S.; Qavi A. J.; Lee S.-H.; Bailey R. C. Subpicogram Per Milliliter Detection of Interleukins Using Silicon Photonic Microring Resonators and an Enzymatic Signal Enhancement Strategy. Anal. Chem. 2013, 85 (22), 10653–10657. 10.1021/ac402972d. [DOI] [PubMed] [Google Scholar]
- Rajer M.; Kmet M. Quantitative analysis of fine needle aspiration biopsy samples. Radiol. Oncol. 2005, 39 (4), 269–272. [Google Scholar]
- Burkhart D. L.; Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 2008, 8 (9), 671–682. 10.1038/nrc2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg R. A. The retinoblastoma protein and cell cycle control. Cell 1995, 81 (3), 323–330. 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Rubin S. M. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem. Sci. 2013, 38 (1), 12–19. 10.1016/j.tibs.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajioka I.; Martins R. A. P.; Bayazitov I. T.; Donovan S.; Johnson D. A.; Frase S.; Cicero S. A.; Boyd K.; Zakharenko S. S.; Dyer M. A. Differentiated Horizontal Interneurons Clonally Expand to Form Metastatic Retinoblastoma in Mice. Cell 2007, 131 (2), 378–390. 10.1016/j.cell.2007.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T.-M.; Huang W.; Park R.; Park P. J.; Johnson M. D. A Developmental Taxonomy of Glioblastoma Defined and Maintained by MicroRNAs. Cancer Res. 2011, 71 (9), 3387–3399. 10.1158/0008-5472.CAN-10-4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J.; Zhou J.; Sutherland A.; Wei W.; Shin Y. S.; Xue M.; Heath J. R. Microfluidics-Based Single-Cell Functional Proteomics for Fundamental and Applied Biomedical Applications. Annu. Rev. Anal. Chem. 2014, 7 (1), 275–295. 10.1146/annurev-anchem-071213-020323. [DOI] [PubMed] [Google Scholar]
- Kim J.; Brown S.; Jenrow K.; Ryu S. Mechanisms of radiation-induced brain toxicity and implications for future clinical trials. J. Neuro-Oncol. 2008, 87 (3), 279–286. 10.1007/s11060-008-9520-x. [DOI] [PubMed] [Google Scholar]
- Peña L. A.; Fuks Z.; Kolesnick R. N. Radiation-induced Apoptosis of Endothelial Cells in the Murine Central Nervous System: Protection by Fibroblast Growth Factor and Sphingomyelinase Deficiency. Cancer Res. 2000, 60 (2), 321–327. [PubMed] [Google Scholar]
- Santana P.; Peña L. A.; Haimovitz-Friedman A.; Martin S.; Green D.; McLoughlin M.; Cordon-Cardo C.; Schuchman E. H.; Fuks Z.; Kolesnick R. Acid Sphingomyelinase–Deficient Human Lymphoblasts and Mice Are Defective in Radiation-Induced Apoptosis. Cell 1996, 86 (2), 189–199. 10.1016/S0092-8674(00)80091-4. [DOI] [PubMed] [Google Scholar]
- Wong C. S.; Van der Kogel A. J. Mechanisms of radiation injury to the central nervous system: implications for neuroprotection. Mol. Interventions 2004, 4 (5), 273–284. 10.1124/mi.4.5.7. [DOI] [PubMed] [Google Scholar]
- Siu A.; Wind J.; Iorgulescu J. B.; Chan T.; Yamada Y.; Sherman J. Radiation necrosis following treatment of high grade glioma—a review of the literature and current understanding. Acta Neurochir. 2012, 154 (2), 191–201. 10.1007/s00701-011-1228-6. [DOI] [PubMed] [Google Scholar]
- Iqbal M.; Gleeson M. A.; Spaugh B.; Tybor F.; Gunn W. G.; Hochberg M.; Baehr-Jones T.; Bailey R. C.; Gunn L. C. Label-Free Biosensor Arrays Based on Silicon Ring Resonators and High-Speed Optical Scanning Instrumentation. IEEE J. Sel. Top. Quantum Electron. 2010, 16 (3), 654–661. 10.1109/JSTQE.2009.2032510. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.