

ABSTRACT
Herpetic stromal keratitis (HSK) is a blinding ocular disease that is initiated by HSV-1 and characterized by chronic inflammation in the cornea. Although HSK immunopathology of the cornea is well documented in animal models, events preceding this abnormal inflammatory cascade are poorly understood. In this study, we have examined the activation of pathological CD4+ T cells in the development of HSK. Dendritic cell autophagy (DC-autophagy) is an important pathway regulating major histocompatibility complex class II (MHCII)-dependent antigen presentation and proper CD4+ T cell activation during infectious diseases. Using DC-autophagy-deficient mice, we found that DC-autophagy significantly and specifically contributes to HSK disease without impacting early innate immune infiltration, viral clearance, or host survival. Instead, the observed phenotype was attributable to the abrogated activation of CD4+ T cells and reduced inflammation in HSK lesions. We conclude that DC-autophagy is an important contributor to primary HSK immunopathology upstream of CD4+ T cell activation.
IMPORTANCE
Herpetic stromal keratitis (HSK) is the leading cause of infectious blindness in the United States and a rising cause worldwide. HSK is induced by herpes simplex virus 1 but is considered a disease of inappropriately sustained inflammation driven by CD4+ T cells. In this study, we investigated whether pathways preceding CD4+ T cell activation affect disease outcome. We found that autophagy in dendritic cells significantly contributed to the incidence of HSK. Dendritic cell autophagy did not alter immune control of the virus or neurological disease but specifically augmented CD4+ T cell activation and pathological corneal inflammation. This study broadens our understanding of the immunopathology that drives HSK and implicates the autophagy pathway as a new target for therapeutic intervention against this incurable form of infectious blindness.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is the leading cause of infectious blindness in developed countries and a rising cause of visual impairment worldwide (1, 2). One form of HSV ocular disease, herpetic stromal keratitis (HSK), is an inflammatory disease of the cornea characterized by corneal opacity and neovascularization. Due to the incurable and reactivating nature of HSV, recurrence of HSK approaches 40% after 5 years from initial presentation (1, 3–6). While HSK is induced by HSV infection, the inflammatory response is sustained long after clearance of replicating virus (7–10) and is ultimately responsible for the tissue damage that leads to loss of visual acuity. As a result, supplementing antivirals with topical corticosteroids significantly shortens the duration of HSK and is the current standard of care (11). Topical steroids, however, nonspecifically suppress the immune response and have significant ocular side effects such as cataracts and glaucoma (12). Development of future therapies depends on continued study of HSK pathogenesis and exposing targetable immunomodulatory pathways specific to this disease.
Studies in a variety of animal models have elucidated the sequence of events that lead to HSK. During the preclinical phase, HSV-1 infection of the corneal epithelium leads to a productive initial influx of myeloid cells that limit viral growth (13, 14). After clearance of infectious virus, however, there is a second chronic leukocytic infiltration driven by CD4+ T cells that coincides with clinical disease (7, 15). The persistent immune cells, inflammatory cytokines, lymphangiogenesis (16, 17), and angiogenesis perpetuate the chronic tissue damage, compromising corneal integrity and function. Canonically, the destructive inflammation characterizing HSK is regulated by CD4+ T cells (18–21). These pathological CD4+ T cells are skewed toward inflammatory Th1 (22) and Th17 (23) subsets. Consequently, corneal interleukin-2 (IL-2) (9, 24), gamma interferon (IFN-γ) (9, 25), and IL-17 (26) are important mediators of disease. Anti-inflammatory T-regulatory cells (Tregs), however, are also present on the ocular surface and serve a protective role against HSK (27–29). In addition, it has been suggested that virus-specific CD4+ T cells are important for the control of acute infection and maintenance of HSV latency in neurological tissue (30, 31). Therefore, reducing the levels of CD4+ T cells to alleviate HSK may exacerbate the overall pathogenesis of HSV (32). An alternative strategy, therefore, might be to modulate the activation of CD4+ T cells to specifically ameliorate HSK.
Upstream of CD4+ T cell activation, dendritic cells (DCs) are the most potent professional antigen-presenting cell. Present in most tissues, including the cornea (33, 34), DCs are important for both innate and adaptive immunity, and manipulating their abilities may coordinate significant changes in the immune response and disease pathogenesis. Previous studies show that autophagy in DCs (DC-autophagy) is a nonclassical pathway for antigen processing and presentation on major histocompatibility complex class II (MHCII) (35, 36). Consistent with these findings, manipulating DC-autophagy alters CD4+ T cell activation and the outcome of HSV-2 genital disease (37), respiratory syncytial virus (RSV)-induced respiratory disease (38), and experimental autoimmune encephalomyelitis (EAE) (39). In this study, we examine the impact of DC-autophagy on HSV-1 pathogenesis. We show that mice lacking DC-autophagy exhibit abrogated activation of CD4+ T cells, limited corneal inflammation in HSK lesions, and reduced clinical disease. This phenotype is independent of early corneal immune infiltration and viral growth, and there are no changes in overall survival following ocular challenge. Taken together, our data provide in vivo evidence for the pathological role of DC-autophagy in orchestrating primary HSK immunopathology and point toward a new possible avenue of therapeutic intervention for this blinding disease.
RESULTS
DC-autophagy contributes to HSK disease.
In dendritic cells, autophagy is important for delivery and processing of antigens in the MHCII antigen presentation pathway and thereby promoting the activation of CD4+ T cells (36, 37). Since HSK is driven by CD4+ T cells, we hypothesized that DC-autophagy would contribute to HSK. To address this, we generated atg5fl/fl CD11c-cre (referred to in this text as DC-autophagy−/−) mice. atg5 is considered an essential autophagy and is necessary for the conversion of LC3, which forms the membrane of the autophagosome (40–42). Offspring were fully viable, and we validated the tissue-specific deletion of autophagy in genotyped mice by immunoblotting. CD11c+ bone marrow-derived DCs from DC-autophagy−/− mice showed reduced levels of atg5, reduced conversion of LC3-I to LC3-II, and increased accumulation of p62 relative to cells from atg5fl/fl control mice (see Fig. S1A in the supplemental material). In comparison, levels of these autophagy proteins were unaltered in CD11c− peritoneal macrophages. In addition, the numbers and percentages of DCs in the spleen were unaltered in DC-autophagy−/− mice relative to controls (see Fig. S1B). Using these mice, we addressed whether the pathogenesis of HSK was altered. We infected DC-autophagy−/− mice unilaterally with HSV-1, and disease was imaged and scored by a masked observer for corneal opacity and neovascularization. While DC-autophagy−/− mice consistently showed reduced corneal opacity (Fig. 1A) and neovascularization (Fig. 1B), neither parameter achieved statistical significance. However, these mice exhibited a significantly lower incidence of disease at the end of the 2nd week, the peak of HSK severity, than did the atg5fl/fl control mice (Fig. 1C, P = 0.0286; Fig. 1D, P = 0.0017).
FIG 1 .
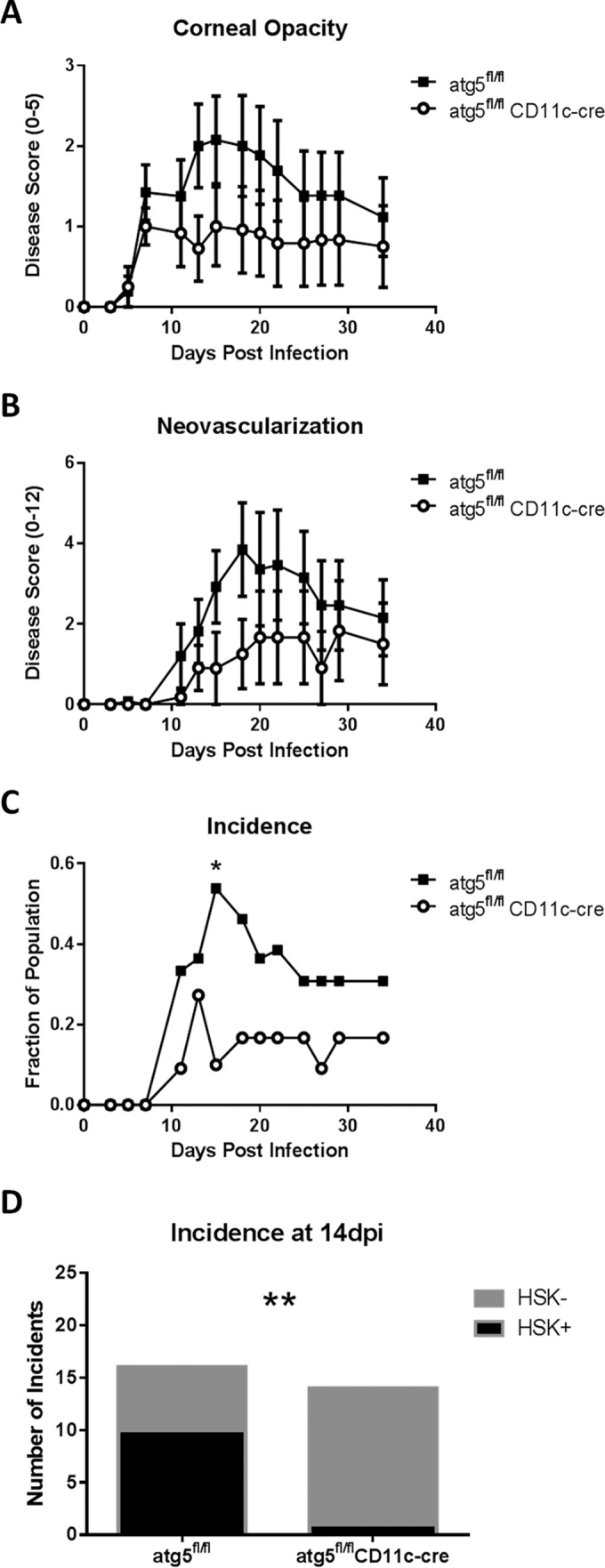
HSK disease is attenuated in the absence of DC-autophagy. atg5fl/fl (n = 13) and atg5fl/fl CD11c-cre (n = 12) female mice were challenged with 2 × 106 PFU of HSV-1 (strain 17) unilaterally. (A) Corneal opacity was scored on a scale of 0 to 5. (B) Neovascularization was scored on a scale of 0 to 12. (C) Positive HSK disease was defined as a corneal opacity score of ≥1.5 and a neovascularization score of ≥2. (D) Summation of 2 other independent experiments showing incidences at 14 dpi of positive (black) and negative (gray) disease. Error bars represent standard errors of the means. Statistical significance was determined by unpaired parametric t test (A and B) and chi-square test (C and D). *, P < 0.05; **, P < 0.005.
Since MHCII is downstream of DC-autophagy, we further confirmed our findings in MHCII−/− mice. Compared to controls, the MHCII−/− mice had reduced corneal opacity (see Fig. S2A in the supplemental material) and neovascularization (see Fig. S2B), but again, neither parameter achieved statistical significance. However, the MHCII−/− mice had significantly lower disease incidence (see Fig. S2C) at 11 and 32 days postinfection (dpi) (11 dpi, P = 0.0427; 32 dpi, P = 0.0241). This deficiency in antigen presentation was not accompanied by changes in the viral replication on the corneal surface (see Fig. S2D). Overall, these data suggest that MHCII-dependent antigen presentation and DC-autophagy contribute to the incidence of HSK.
DC-autophagy does not impact viral replication or host survival following peripheral infection with neuroinvasive HSV-1.
Previous work by Lee et al. has shown that mice with defects in DC-autophagy succumb more rapidly to genital HSV-2 infection (37). We therefore investigated whether DC-autophagy played a role in determining survival of ocular HSV-1 infection. After corneal challenge with a neurovirulent strain of HSV-1 (McKrae), DC-autophagy−/− mice and control mice exhibited comparable neurologic symptoms (see Fig. S3 in the supplemental material) and rates of mortality within a 21-day experimental period (Fig. 2A; P < 0.45). This phenotype was consistent with the similar viral titers observed in trigeminal ganglia and brains (Fig. 2B to D). These data suggest that DC-autophagy has only a minimal role in the control of neurological spread and growth of HSV-1 or survival of central nervous system (CNS) infection. Therefore, DC-autophagy seems to play a specific role in contributing to HSK pathogenesis.
FIG 2 .

DC-autophagy does not alter survival or neurological viral spread after acute corneal challenge with HSV-1. (A) Attainment of endpoint survival criteria of atg5fl/fl CD11-cre/DC-autophagy−/− (n = 49) and atg5fl/fl (n = 31) mice after bilateral corneal infection with 2 × 106 PFU of HSV-1 (McKrae) per eye. Data are a summation of 5 independent experiments. (B to D) Viral titers in trigeminal ganglia (B), brainstem and cerebellum (C), and cerebrum (D) of C57BL/6 (n = 7) and atg5fl/fl CD11-cre (n = 7) mice after HSV-1 (McKrae) corneal infection. Data represent 2 to 3 independent experiments. The dotted line marks the limit of detection. Statistical significance was determined by log rank test (A) and unpaired parametric t test (B to D).
DC-autophagy does not alter early immune infiltration and control of viral replication in the cornea.
To investigate further how DC-autophagy contributes to HSK disease, we evaluated parameters that have been shown to be involved in HSK pathogenesis. Since early immune control of viral burden in the cornea is protective against HSK (43, 44), we hypothesized that the attenuated HSK phenotype observed in DC-autophagy−/− mice may be due to greater early immune infiltration and suppression of viral growth. Three days post-corneal infection with HSV-1, we examined early myeloid infiltration by flow cytometry (Fig. 3A). Surprisingly, we saw no differences in neutrophil (CD11b+ Ly6G+ Ly6Clow), macrophage (CD11b+ Ly6G− Ly6C+ or CD11b+ Ly6G+ Ly6Chigh) (45), or overall myeloid (CD11b+) infiltration into the cornea (Fig. 3B). This was consistent, however, with the comparable levels of viral replication in corneas at 3 and 5 dpi in DC-autophagy−/− and control mice (Fig. 3C). Furthermore, infectious particles in both DC-autophagy−/− and control mice became undetectable after 7 dpi, prior to the onset of HSK. Taken together, these data further support the idea that antigen presentation pathways do not modulate acute control of viral growth and that DC-autophagy does not alter early preclinical events in the cornea leading to HSK disease.
FIG 3 .

DC-autophagy does not alter preclinical myeloid infiltration or viral growth in the cornea. atg5fl/fl CD11-cre (n = 10) and atg5fl/fl (n = 7) mice were infected bilaterally with 2 × 106 PFU per eye of HSV-1 (strain 17). (A and B) Corneas were harvested at 3 dpi and analyzed using flow cytometry for all myeloid cells (CD11b+), neutrophils (CD11b+ Ly6G+ Ly6Clow), or macrophages (CD11b+ Ly6G− Ly6C+ or CD11b+ Ly6G+ Ly6Chigh) (A) and quantified as percentage of cells (left) and total number of cells per cornea (right) (B). Data represent 3 independent experiments. (C) Viral titers of ocular tear film after challenge with 2 × 106 PFU of HSV-1 (strain 17) in one eye. Data represent 5 similar experiments. Statistical significance was determined by unpaired parametric t test (B) and analysis of variance (C).
DC-autophagy is important for activation of CD4+ T cells in vivo.
Previous reports show that CD4+ T cell activation is enhanced by DC-autophagy (37, 38, 46, 47). In turn, activated CD4+ T cells and their associated cytokines IFN-γ and IL-2 contribute to HSK incidence and persistence (9, 32). Together, these observations provide a likely explanation for the reduced HSK observed in the DC-autophagy−/− mice (Fig. 2). To assess this, we evaluated expression of the early activation marker CD69 on CD4+ T cells in the draining lymph nodes of DC-autophagy−/− and control mice 3 days after corneal HSV-1 challenge (Fig. 4A). We found a significant reduction in the percentage of CD69+ cells in the CD4+ T cell compartment of DC-autophagy−/− mice relative to control mice (Fig. 4B; P = 0.0053). This is consistent with the hypothesis that DC-autophagy is important for maximal activation of CD4+ T cells and for the promotion of HSK.
FIG 4 .
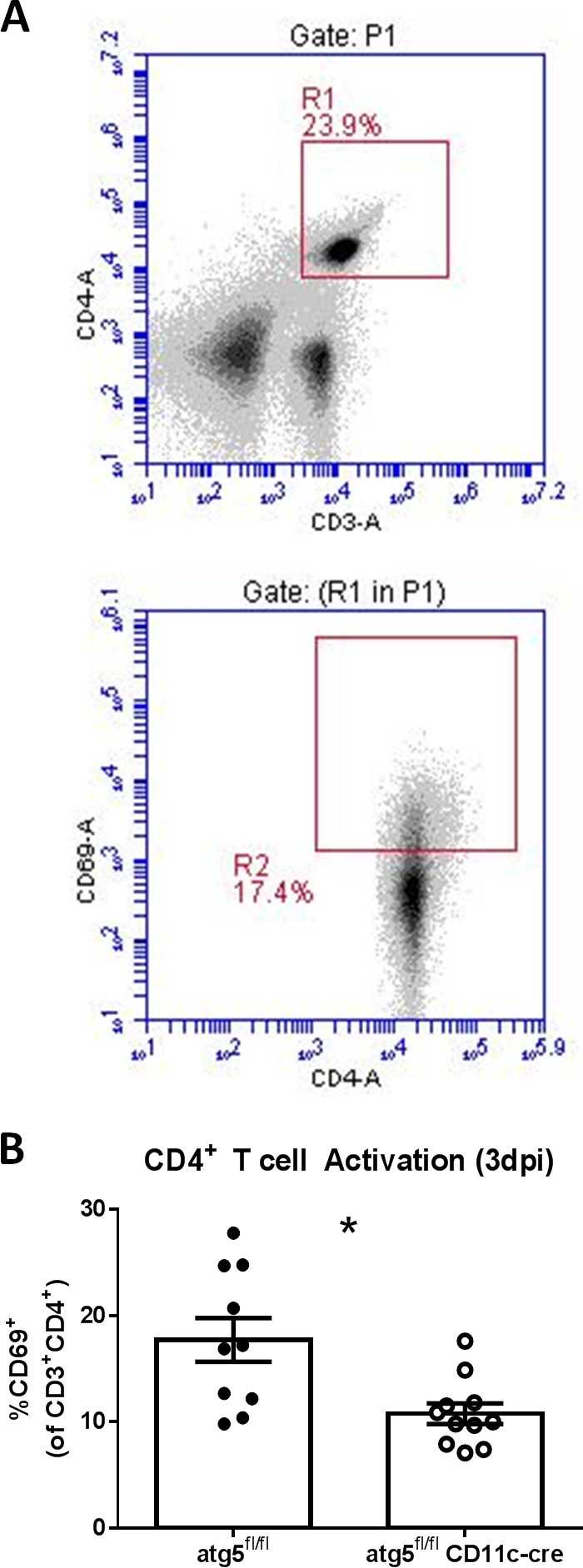
DC-autophagy promotes early activation of CD4+ T cells after HSV-1 infection. atg5fl/fl CD11-cre (n = 11) and atg5fl/fl (n = 10) mice were infected bilaterally with 2 × 106 PFU per eye, and draining cervical lymph nodes were harvested at 3 days postinfection and analyzed using flow cytometry. Using the gating scheme (A), the percentage of CD69+ cells in the CD4+ T cell compartment was quantified (B). Data represent 2 independent experiments. Error bars represent standard errors of the means. Significance was determined by unpaired parametric t test. *, P < 0.05.
DC-autophagy promotes immunopathology in HSK lesions.
Based on the attenuated CD4+ T cell activation in DC-autophagy−/− mice, we hypothesized that the differences in clinical HSK were due to reduced CD4+ T cell infiltration into the cornea. To test this, we evaluated immune infiltration into corneas at 14 dpi and 38 dpi using flow cytometry (Fig. 5). Although no differences in the relative proportions of the analyzed cells infiltrating the cornea were observed at 14 dpi, we found a significant reduction in the number of CD45+ cells (Fig. 5A; P = 0.037) and CD4+ T cells (Fig. 5E; P = 0.044) in the corneas of DC-autophagy−/− mice relative to control mice. In DC-autophagy−/− mice, we also observed a reproducible but statistically nonsignificant decrease in the numbers of CD11b+ GR1+ neutrophils and macrophages (Fig. 5C; P = 0.08), which made up a large proportion of the CD45+ immune infiltrate. In contrast, we found no differences in the numbers of CD11b+ GR1− myeloid cells (Fig. 5B; P = 0.20), CD11c+ dendritic cells (Fig. 5D; P = 0.75), or CD8+ T cells (Fig. 5F; P = 0.32). Any differences in immune infiltration were transient and became blunted by 38 dpi (Fig. 5G to L), which mirrors disease recovery after 15 dpi (Fig. 1). At 38 dpi, there were no significant differences in numbers of immune cells between the floxed control and DC-autophagy−/− mice, even though we observed a reduction in the frequency of CD4+ (Fig. 5K; P = 0.021) and CD8+ (Fig. 5L; P = 0.026) T cells in the cornea. This discrepancy in percentage but not absolute number might indicate corneal thinning, a well-documented sequela of HSK (48, 49), which would cause a greater loss of total corneal cell number in the more severely diseased control mice.
FIG 5 .

DC-autophagy contributes to immune infiltration into the cornea during HSK. atg5fl/fl and atg5fl/fl CD11c-cre female mice were challenged with 2 × 106 PFU of HSV-1 (strain 17) unilaterally. Corneas were analyzed by flow cytometry at 14 dpi (A to F; n ≥ 7 per group) and 38 dpi (G to L; n ≥ 11 per group). Samples were first gated on CD45+ cells before gating on subpopulations. In each panel, quantification of each cell subset is presented as percentage (left) and total number (right) in the cornea. Cell populations analyzed include CD45+ immune cells (A and G), CD11b+ GR1+ neutrophils and macrophages (C and I), CD11b+ GR1− myeloid cells (B and H), CD11c+ dendritic cells (D and J), CD4+ T cells (E and K), and CD8+ T cells (F and L). Statistical significance was determined by unpaired parametric t test. *, P < 0.05.
To further analyze these immunological differences at 14 dpi, we measured cytokine levels in the cornea with a Luminex cytokine array. Notably, we observed significantly reduced levels of IL-2 and skewing away from Th1 cytokines in the cornea of DC-autophagy−/− mice relative to control mice (Fig. 6A), consistent with the reduced number of CD4+ T cells (Fig. 5E). We also observed significant reductions in proinflammatory cytokines IL-1β and IL-6 (Fig. 6B); chemokines monocyte chemoattractant protein 1 (MCP-1)/CCL2, LIX/CXCL5, and IP-10/CXCL10 (Fig. 6C); and leukemia inhibitory factor (LIF) (Fig. 6D). Additionally, we also observed nonsignificant but trending decreases (P < 0.10) in IL-12p40 (Fig. 6B); KC/CXCL1, MIP-1β/CCL4, and RANTES/CCL5 (Fig. 6C); and granulocyte colony-stimulating factor (G-CSF) and vascular endothelial growth factor (VEGF) (Fig. 6D). Overall, the pathological cytokine signature produced in the cornea of DC-autophagy−/− mice after HSK onset was significantly attenuated. Taken together, we conclude that DC-autophagy mediates CD4+ T cell activation and infiltration into HSK lesions, driving pathological inflammation and exacerbating corneal disease.
FIG 6 .

DC-autophagy contributes to cytokine production in cornea during HSK. Control atg5fl/fl (n = 5) and CD11c-cre (n = 2) and atg5fl/fl CD11c-cre (n = 7) female mice were challenged with 2 × 106 PFU of HSV-1 (strain 17) unilaterally. Two or three corneas were pooled to make 3 samples per group, which were analyzed by multiplex protein assay. Cytokines were stratified as T cell cytokines (A), proinflammatory cytokines (B), chemokines (C), and growth factors (D). We found no detectable amounts of IL-3, IL-4, IL-5, and IL-7 in any of the corneas. Error bars represent standard errors of the means. Statistical significance was determined by unpaired parametric t test. *, P < 0.05; ND, not detected.
DISCUSSION
Dendritic cells are important determinants of HSV-1 pathogenesis (43, 50–52). Ablation of the CD11c+ cells prior to HSV-1 infection promotes greater neurological spread (50) and corneal replication of the virus (43), while also influencing subsequent adaptive immune responses (50, 53). Here, we show that the innate and adaptive functions of dendritic cells have differential requirements for autophagy, thereby specifically limiting HSK pathogenesis.
Our study shows that DC-autophagy has limited or no effect on innate immunity and viral growth, which directly impact host mortality. While previous work showed that DCs control viral growth through natural killer cell activation (50) and recruitment (43), we observed no changes in myeloid cell recruitment or viral titers in the cornea in the DC-autophagy−/− mice. Our findings, however, are consistent with the previous observation that autophagy does not alter the ability of DCs to mature, phagocytize, migrate, or produce innate cytokines (37). Therefore, we reason that mortality following ocular challenge is unaltered in the absence of DC-autophagy because innate immune control of viral replication, a key determinant of neuropathogenesis (54–58), remains intact.
Consistent with previous reports (37–39, 47), we found that DC-autophagy has a significant impact on the development of adaptive immunity, specifically CD4+ T cell responses. DC-autophagy was also reported to promote host survival following intravaginal infection with HSV-2 (37). In our study, however, we saw no significant changes in viral titers or survival following ocular infection with neurovirulent HSV-1. This contrast in survival data is most likely due to altered viral spread and tropism following use of disparate viruses and routes of infection. Intravaginal HSV-2 affects the genital mucosal and epidermis, spreading through the dorsal root ganglia to the spinal cord and pelvic autonomic ganglia, leading to encephalitis and urinary/fecal retention, respectively (59, 60). Conversely, ocular HSV-1 infection is limited to the orofacial region and its innervating trigeminal and autonomic pathways (61), leading to mortality from CNS pathology (62). Differences at these mucosal sites and specific routes of neurological spread likely require different immune components for control of the virus and downstream pathology. Additionally, HSV-2 is generally more virulent than HSV-1 regardless of the route of infection (63). Taken together, the impact of DC-autophagy on pathogenesis is likely pathogen and disease specific.
The major finding that we report in this study is that DC-autophagy is a pathological contributor to HSK disease. This finding is consistent with HSK being a chronic human disease with known pathological involvement of host immunity (11), and the immunological findings are consistent with the well-supported paradigm that HSK is a CD4+ T cell-driven disease (7–10). Robust viral replication in the cornea is necessary for adaptive immune priming and subsequent induction of HSK (64). HSV replication and HSK are therefore positively correlated. In this study, we showed modest but reproducibly increased corneal viral titers in DC-autophagy−/− mice and yet a reduced incidence of HSK. This strongly supports the idea that ablation of DC-autophagy can specifically reduce HSK immunopathology, even with increased viral loads. Furthermore, our findings recapitulate the attenuated HSK seen in CD4−/− and CD4-depleted mice (32, 65), but we did not see the reported transient corneal pathology mediated by the compensatory infiltration of CD8+ T cells (32). This difference could simply result from different virus and mouse strains used in this study (10). Alternatively, these disparate findings could be due to the role of DC-autophagy in cross-presentation to CD8+ T cells, which is dependent on both the type of dendritic cell and the type of antigen (66, 67). It could also be due to anti-inflammatory functions of Tregs (27, 68), which have been shown to reduce HSK severity (29) and would not be depleted in our study. Furthermore, the pattern and concentrations of IL-10 in the cornea do not suggest that DC-autophagy exerts its inflammatory effects through suppression of Tregs. That said, unlike CD4-ablated mice, the baseline presence of Tregs in DC-autophagy−/− mice likely exerts overall immune suppression. Given these reasons, our data suggest that DC-autophagy can impact CD4+ T cell activation and HSK pathogenesis without compensatory CD8+ T cell infiltration into the cornea.
As a result of reduced CD4+ T cell activation, DC-autophagy−/− mice showed overall dampening of immune infiltration and cytokine production in the cornea. Notably, we saw a significant reduction in the amount of CD4+ T cells and IL-2 in the cornea. This is consistent with the report that IL-2 promotes HSK (9) and that anti-IL-2 antibody complexes alleviated HSK severity (69). We also observed significant differences in proinflammatory cytokines (IL-6, IL-1β, and MCP-1) previously reported in HSK lesions (17, 70–72) and an overall reduction in infiltrating immune cells. Neutrophils are the predominant immune cell in HSK lesions (73, 74), and we saw a trending reduction in the number of CD11b+ GR1+ cells in the HSK lesions of DC-autophagy−/− mice. DC-autophagy is most likely acting on neutrophils indirectly through activation of CD4+ T cells, which are known to produce cytokines that potentiate neutrophil chemotaxis and survival in the cornea (24, 73, 75). Considering that neutrophils are responsible for much of the pathology in the cornea (73, 74, 76), the reduction in clinical disease seen in DC-autophagy−/− mice is most directly attributable to the diminished neutrophil numbers in HSK lesions. We also observed a reduction in CXCL10, which was previously reported to augment HSK (44). Those authors showed that CXCL10−/− mice had reduced infiltration of neutrophils at the preclinical stage, thereby interfering with viral clearance and aggravating HSK. Since we observed no differences in early myeloid infiltration or viral clearance, we speculate that the absence of DC-autophagy alters CXCL10 and neutrophil infiltration only during the later time points following viral clearance. Given that we see immunological differences in the cornea only during clinical but not at preclinical time points, we conclude that the immunomodulatory effects of DC-autophagy have a temporal specificity that is evident in the cornea only during HSK.
An important caveat of our approach is that we studied HSK following primary HSV-1 infection. While many other studies have used this approach (reviewed in reference 53), it is important to note that most human cases of HSK are recurrences caused by reactivating virus (5, 77). That said, it is notable that depletion of DCs after induction of HSK does not alter disease outcome (53), suggesting that the signature of a pathological immune response determined by dendritic cells occurs early during infection, likely prior to the onset of disease. This may have utility for vaccine design, since modulating DC-autophagy during HSV vaccination, either by engineering the virus or by modulating the host response, may fine-tune the CD4+ T cell response away from promoting HSK immunopathology while retaining the immune control of viral growth.
We have previously reported that HSV-1 γ34.5 can limit maturation of autophagosomes in dendritic cells, thereby attenuating activation of CD4+ T cells (46). In fact, this is one of three known mechanisms by which HSV-1 hinders MHCII antigen presentation (36, 78). This study reveals the intriguing possibility that such viral blockade of antigen presentation actually leads to reduced HSK. Immune modulation has largely been viewed as a pathway for pathogens to cause increased levels of disease. Teleologically, however, such modulation may also be advantageous to the pathogen in minimizing immune system-mediated damage and promoting the success of its host, thereby maximizing the probability of spread within the host population.
In conclusion, we have shown in this study that autophagy in dendritic cells is a pathological host factor that contributes to CD4+ T cell-driven HSK without perturbing innate immunity or control of viral replication. The specificity of this effect and the expanding repertoire of autophagy modulating drugs (reviewed in reference 57) make DC-autophagy an attractive candidate for therapeutic intervention, warranting further investigation.
MATERIALS AND METHODS
Cells and virus.
Viruses used in this study were strain 17syn+ and McKrae. Their titers were determined, and they were propagated on African green monkey kidney (Vero) cells (79–81).
Mice.
In all experiments, 6- to 10-week-old mice were used. atg5fl/fl mice (82) were a generous gift from Skip Virgin (Washington University, St Louis, MO), and the CD11c-cre mice [strain B6.Cg-Tg(Itgax-cre)1-1Reiz/J] (83) and MHCII-knockout mice (strain B6.129S2-H2dlAb1-Ea/J) (84) were purchased from the Jackson Laboratory (Bar Harbor, ME). The atg5fl/fl and CD11c-cre mice were crossed and bred in-house. The genetic backgrounds of the progeny mice were assessed at the Dartmouth Speed Congenic Core Facility at the Geisel School of Medicine at Dartmouth (DartMouse). DartMouse uses the Illumina, Inc. (San Diego, CA), GoldenGate genotyping assay to interrogate 1,449 single-nucleotide polymorphisms (SNPs) spread throughout the genome. The raw SNP data were analyzed using DartMouse’s SNaP-Map and Map-Synth software, allowing the determination of genetic background at each SNP location. Mice were housed in the barrier facility in the Center for Comparative Medicine and Research at the Geisel School of Medicine at Dartmouth, and all procedures were performed in accordance with federal and university policies.
Animal procedures.
For corneal infections, corneas were scarified with a 25-gauge needle and inoculated with 2 × 106 PFU of virus in a volume of 5 µl as previously described (79). For the HSK model, strain 17syn+ was used unilaterally. Microscopic images of the cornea were obtained using a Celestron (Torrance, CA) handheld digital microscope, and images were scored by a masked observer. Corneas were clinically scored for opacity and neovascularization. As previously described (85), corneal opacity was evaluated on a scale of 0 to 5, where 0 indicates clear stroma, 1 indicates mild stromal opacification, 2 indicates moderate opacity, 3 indicates dense opacity, 4 indicates total opacity, and 5 indicates corneal melt. Corneal neovascularization was evaluated using a scale of 0 to 12, where each of the four quadrants of the eye was evaluated for the density of new vasculature, also previously described (85). Eye swab material was collected, and titers were determined by standard plaque assay as previously described (86). Brains and trigeminal ganglia were placed in 1.5-ml tubes containing 1-mm-diameter glass beads and 1 ml of medium. Sample homogenates were prepared by mechanically disrupting them in a Mini-Beadbeater-8 (BioSpec Products) and then sonicated. The titers of homogenates were determined by plaque assay on Vero cells, and the amount of virus was expressed as PFU per milliliter of tissue homogenate. For analysis of survival, corneas were infected bilaterally with strain McKrae. Endpoint criteria for sacrificing mice included loss of ≥15% of starting body weight, body temperature loss of ≥10%, altered locomotion, and labored breathing.
Tissue preparation for flow cytometry.
Eyes were harvested, and corneas were isolated under a dissecting microscope at various times postinfection. Corneas were digested in 400 U/ml collagenase II (Life Technologies) for 1.5 h at 37°C, homogenized with an Eppendorf micropestle, triturated with a pipettor, and filtered through a 70-µm mesh filter before flow cytometry staining. Cervical lymph nodes were dissected, similarly homogenized, and filtered before flow cytometry staining. Blood was harvested by submandibular bleed, and erythrocytes (RBCs) were lysed using Gey’s solution. The following antibodies were used for flow cytometric analysis: Ly6G-fluorescein isothiocyanate (FITC) (1A6), CD4-FITC (GK1.5), CD69-phycoerythrin (PE) (H1.2F3), CD8α-PE (53-6.7), CD11c-FITC (HL3), and GR1-peridinin chlorophyll protein (PerCP)/Cy5.5 (RB6-8C5), all purchased from BD Biosciences (San Jose, CA). Ly6C-PerCP/Cy5.5 (HK1.4), CD11b-allophycocyanin (APC) (M1/70), CD3-PerCP/Cy5.5 (17A2), CD45-APC (30-F11), and CD19-PE (6D5) were purchased from BioLegend (San Diego, CA), and CD3-APC (17A2) was purchased from eBioscience (San Diego, CA). Samples were analyzed using the BD Accuri C6 flow cytometer, and the data were analyzed using CFlow software (Accuri).
BioPlex multiple cytokine assay.
Corneas were harvested from mice and processed as previously described (87). Corneas were quartered in 150 µl of phosphate-buffered saline (PBS) plus protease inhibitor (Roche cOmplete, Mini) and sonicated 6 times for 15 s. After rigorous vortexing, debris was spun down and supernatant was collected for analysis. A mouse 32-plex assay (Millipore) was performed per the manufacturer’s instructions.
SUPPLEMENTAL MATERIAL
Validation of DC-autophagy−/− mice. (A) Bone marrow-derived dendritic cells and peritoneal macrophages were harvested as previously described (88, 89). Immunoblot showing expression of atg5, LC3, and p62 in CD11c+ bone marrow-derived dendritic cells (BMDCs) and CD11c− peritoneal macrophages of atg5fl/fl mice and atg5fl/fl CD11c-cre mice. Proteins were resolved by electrophoresis on denaturing 12% polyacrylamide gels, transferred to polyvinylidene difluoride membranes, blocked with 5% skim milk, and reacted with antibodies at indicated concentrations: atg5, 1:500 (Novus); LC3, 1:1,000 (MBL); p62/SQSTM1, 1:1,000 (Novus); and β-actin, 1:1,000 (BioLegend). Data represent 3 independent experiments. (B) Flow cytometry analysis of CD11c+ dendritic cells in the spleen showing percentage of splenocytes (top) and total number of splenocytes (bottom). Data represent 3 similar experiments. Statistical significance was determined by analysis of variance. Download
HSK disease is attenuated in the absence of MHCII. C57BL/6 (n = 10) and MHCII−/− (n = 10) female mice were challenged with 2 × 106 PFU of HSV-1 (strain 17) unilaterally. (A) Corneal opacity was scored on a scale of 0 to 5. (B) Neovascularization was scored on a scale of 0 to 12. (C) Positive HSK disease was defined as a corneal opacity score of ≥1.5 and a neovascularization score of ≥2. (D) Viral titers of ocular tear film after challenge with 2 × 106 PFU of HSV-1 (strain 17) in one eye. Error bars represent standard errors of the means. Statistical significance was determined by unpaired parametric t test (A, B, and D) and chi-square test (C). *, P < 0.05. Download
DC-autophagy does not alter neurological disease after acute corneal challenge with HSV-1. atg5fl/fl and atg5fl/fl CD11c-cre mice (n = 12 in each group) were challenged with 2 × 106 PFU of HSV-1 (McKrae) bilaterally. Neurological disease was scored using the following scale: 0, normal behavior; 1, ruffled fur and/or hunched posture; 2, ruffled fur/hunched posture and repetitive movements and/or jumpy behavior; 3, motor deficits on one side; 4, moribund. Data represent 4 independent experiments. Error bars represent standard errors of the means. Statistical significance was determined by unpaired parametric t test. Download
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health grants to D.A.L. (EY09083) and to P.M.S. (EY21247 and unrestricted funds from the SLU Department of Ophthalmology). The project was also supported by a Geisel School of Medicine Molecular and Cellular Biology Program training grant (2T32GM008704-16 and 5T32GM008704-17) to Y.J. The DartLab immunoassay and Flow Cytometry Shared Resource at the Geisel School of Medicine at Dartmouth are supported by 5P30CA023108-36 from the National Cancer Institute and 8P30GM103415-14 from the National Institute of General Medical Sciences. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We acknowledge Philipe Gobeil for help breeding the atg5fl/fl CD11c-cre mice; Brian North for mouse colony breeding and genotyping; Edward Usherwood for the MHCII−/− mice; Richard Manivanh for help with the statistical analysis; and Zhuting Hu, Sarah Katzenell, and Brent Berwin for reagents and/or helpful discussions.
Footnotes
Citation Jiang Y, Yin X, Stuart PM, Leib DA. 2015. Dendritic cell autophagy contributes to herpes simplex virus-driven stromal keratitis and immunopathology. mBio 6(6):e01426-15. doi:10.1128/mBio.01426-15.
REFERENCES
- 1.Liesegang TJ. 2001. Herpes simplex virus epidemiology and ocular importance. Cornea 20:1–13. doi: 10.1097/00003226-200101000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Farooq AV, Shukla D. 2012. Herpes simplex epithelial and stromal keratitis: an epidemiologic update. Surv Ophthalmol 57:448–462. doi: 10.1016/j.survophthal.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shuster JJ, Kaufman HE, Nesburn AB. 1981. Statistical analysis of the rate of recurrence of herpesvirus ocular epithelial disease. Am J Ophthalmol 91:328–331. doi: 10.1016/0002-9394(81)90285-3. [DOI] [PubMed] [Google Scholar]
- 4.Stanzel TP, Diaz JD, Mather R, Wong IG, Margolis TP, Gritz DC. 2014. The epidemiology of herpes simplex virus eye disease in Northern California. Ophthal Epidemiol 21:370–377. doi: 10.3109/09286586.2014.966848. [DOI] [PubMed] [Google Scholar]
- 5.Liesegang TJ, Melton LJ, Daly PJ, Ilstrup DM. 1989. Epidemiology of ocular herpes simplex. Incidence in Rochester, Minn, 1950 through 1982. Arch Ophthalmol 107:1155–1159. doi: 10.1001/archopht.1989.01070020221029. [DOI] [PubMed] [Google Scholar]
- 6.Young RC, Hodge DO, Liesegang TJ, Baratz KH. 2010. Incidence, recurrence, and outcomes of herpes simplex virus eye disease in Olmsted County, Minnesota, 1976–2007: the effect of oral antiviral prophylaxis. Arch Ophthalmol 128:1178–1183. doi: 10.1001/archophthalmol.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Metcalf JF, Hamilton DS, Reichert RW. 1979. Herpetic keratitis in athymic (nude) mice. Infect Immun 26:1164–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russell RG, Nasisse MP, Larsen HS, Rouse BT. 1984. Role of T-lymphocytes in the pathogenesis of herpetic stromal keratitis. Invest Ophthalmol Vis Sci 25:938–944. [PubMed] [Google Scholar]
- 9.Hendricks RL, Tumpey TM, Finnegan A. 1992. IFN-gamma and IL-2 are protective in the skin but pathologic in the corneas of HSV-1-infected mice. J Immunol 149:3023–3028. [PubMed] [Google Scholar]
- 10.Hendricks RL, Tumpey TM. 1990. Contribution of virus and immune factors to herpes simplex virus type I-induced corneal pathology. Invest Ophthalmol Vis Sci 31:1929–1939. [PubMed] [Google Scholar]
- 11.Wilhelmus KR, Gee L, Hauck WW, Kurinij N, Dawson CR, Jones DB, Barron BA, Kaufman HE, Sugar J, Hyndiuk RA. 1994. Herpetic eye disease study. A controlled trial of topical corticosteroids for herpes simplex stromal keratitis. Ophthalmology 101:1883–1895. [DOI] [PubMed] [Google Scholar]
- 12.Renfro L, Snow JS. 1992. Ocular effects of topical and systemic steroids. Dermatol Clin 10:505–512. [PubMed] [Google Scholar]
- 13.Tumpey TM, Chen SH, Oakes JE, Lausch RN. 1996. Neutrophil-mediated suppression of virus replication after herpes simplex virus type 1 infection of the murine cornea. J Virol 70:898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biswas PS, Rouse BT. 2005. Early events in HSV keratitis—setting the stage for a blinding disease. Microbes Infect 7:799–810. doi: 10.1016/j.micinf.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Hendricks RL, Tumpey TM. 1991. Concurrent regeneration of T lymphocytes and susceptibility to HSV-1 corneal stromal disease. Curr Eye Res 10(Suppl):47–53. doi: 10.3109/02713689109020357. [DOI] [PubMed] [Google Scholar]
- 16.Wuest TR, Carr DJJ. 2010. VEGF-A expression by HSV-1-infected cells drives corneal lymphangiogenesis. J Exp Med 207:101–115. doi: 10.1084/jem.20091385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bryant-Hudson KM, Gurung HR, Zheng M, Carr DJJ. 2014. Tumor necrosis factor alpha and interleukin-6 facilitate corneal lymphangiogenesis in response to herpes simplex virus 1 infection. J Virol 88:14451–14457. doi: 10.1128/JVI.01841-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Newell CK, Sendele D, Rouse BT. 1989. Effects of CD4+ and CD8+ T-lymphocyte depletion on the induction and expression of herpes simplex stromal keratitis. Reg Immunol 2:366–369. [PubMed] [Google Scholar]
- 19.Newell CK, Martin S, Sendele D, Mercadal CM, Rouse BT. 1989. Herpes simplex virus-induced stromal keratitis: role of T-lymphocyte subsets in immunopathology. J Virol 63:769–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doymaz MZ, Rouse BT. 1992. Herpetic stromal keratitis: an immunopathologic disease mediated by CD4+ T lymphocytes. Invest Ophthalmol Vis Sci 33:2165–2173. [PubMed] [Google Scholar]
- 21.Mercadal CM, Bouley DM, DeStephano D, Rouse BT. 1993. Herpetic stromal keratitis in the reconstituted scid mouse model. J Virol 67:3404–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niemialtowski MG, Rouse BT. 1992. Predominance of Th1 cells in ocular tissues during herpetic stromal keratitis. J Immunol 149:3035–3039. [PubMed] [Google Scholar]
- 23.Xia L, Zhang S, Cao Z, Hu Y, Yang H, Wang D. 2013. Interleukin-17 enhanced immunoinflammatory lesions in a mouse model of recurrent herpetic keratitis. Microbes Infect 15:126–139. doi: 10.1016/j.micinf.2012.10.017. [DOI] [PubMed] [Google Scholar]
- 24.Tang Q, Chen W, Hendricks RL. 1997. Proinflammatory functions of IL-2 in herpes simplex virus corneal infection. J Immunol 158:1275–1283. [PubMed] [Google Scholar]
- 25.Minami M, Kita M, Yan X, Yamamoto T, Iida T, Sekikawa K, Iwakura Y, Imanishi J. 2002. Role of IFN-gamma and tumor necrosis factor-alpha in herpes simplex virus type 1 infection. J Interferon Cytokine Res 22:671–676. doi: 10.1089/10799900260100150. [DOI] [PubMed] [Google Scholar]
- 26.Maertzdorf J, Osterhaus AD, Verjans GM. 2002. IL-17 expression in human herpetic stromal keratitis: modulatory effects on chemokine production by corneal fibroblasts. J Immunol 169:5897–5903. doi: 10.4049/jimmunol.169.10.5897. [DOI] [PubMed] [Google Scholar]
- 27.Suvas S, Azkur AK, Kim BS, Kumaraguru U, Rouse BT. 2004. CD4+CD25+ regulatory T cells control the severity of viral immunoinflammatory lesions. J Immunol 172:4123–4132. doi: 10.4049/jimmunol.172.7.4123. [DOI] [PubMed] [Google Scholar]
- 28.Nesburn AB, Bettahi I, Dasgupta G, Chentoufi AA, Zhang X, You S, Morishige N, Wahlert AJ, Brown DJ, Jester JV, Wechsler SL, BenMohamed L. 2007. Functional Foxp3+ CD4+ CD25(Bright+) “natural” regulatory T cells are abundant in rabbit conjunctiva and suppress virus-specific CD4+ and CD8+ effector T cells during ocular herpes infection. J Virol 81:7647–7661. doi: 10.1128/JVI.00294-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Veiga-Parga T, Suryawanshi A, Mulik S, Giménez F, Sharma S, Sparwasser T, Rouse BT. 2012. On the role of regulatory T cells during viral-induced inflammatory lesions. J Immunol 189:5924–5933. doi: 10.4049/jimmunol.1202322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson AJ, Chu C-F, Milligan GN. 2008. Effector CD4+ T-cell involvement in clearance of infectious herpes simplex virus type 1 from sensory ganglia and spinal cords. J Virol 82:9678–9688. doi: 10.1128/JVI.01159-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Velzen M, Jing L, Osterhaus ADME, Sette A, Koelle DM, Verjans GMGM. 2013. Local CD4 and CD8 T-cell reactivity to HSV-1 antigens documents broad viral protein expression and immune competence in latently infected human trigeminal ganglia. PLoS Pathog 9:e1003547. doi: 10.1371/journal.ppat.1003547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lepisto AJ, Frank GM, Xu M, Stuart PM, Hendricks RL. 2006. CD8 T cells mediate transient herpes stromal keratitis in CD4-deficient mice. Invest Ophthalmol Vis Sci 47:3400–3409. doi: 10.1167/iovs.05-0898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mastropasqua L, Nubile M, Lanzini M, Carpineto P, Ciancaglini M, Pannellini T, Di Nicola M, Dua HS. 2006. Epithelial dendritic cell distribution in normal and inflamed human cornea: in vivo confocal microscopy study. Am J Ophthalmol 142:736–744. doi: 10.1016/j.ajo.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 34.Mayer WJ, Irschick UM, Moser P, Wurm M, Huemer HP, Romani N, Irschick EU. 2007. Characterization of antigen-presenting cells in fresh and cultured human corneas using novel dendritic cell markers. Invest Ophthalmol Vis Sci 48:4459–4467. doi: 10.1167/iovs.06-1184. [DOI] [PubMed] [Google Scholar]
- 35.Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Münz C. 2005. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 307:593–596. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 36.Schmid D, Pypaert M, Münz C. 2007. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, Mizushima N, Grinstein S, Iwasaki A. 2010. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 32:227–239. doi: 10.1016/j.immuni.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reed M, Morris SH, Jang S, Mukherjee S, Yue Z, Lukacs NW. 2013. Autophagy-inducing protein beclin-1 in dendritic cells regulates CD4 T cell responses and disease severity during respiratory syncytial virus infection. J Immunol 191:2526–2537. doi: 10.4049/jimmunol.1300477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhattacharya A, Parillon X, Zeng S, Han S, Eissa NT. 2014. Deficiency of autophagy in dendritic cells protects against experimental autoimmune encephalomyelitis. J Biol Chem 289:26525–26532. doi: 10.1074/jbc.M114.575860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuma A, Mizushima N, Ishihara N, Ohsumi Y. 2002. Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J Biol Chem 277:18619–18625. doi: 10.1074/jbc.M111889200. [DOI] [PubMed] [Google Scholar]
- 41.Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. 1998. A protein conjugation system essential for autophagy. Nature 395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 42.Mizushima N, Ohsumi Y, Yoshimori T. 2002. Autophagosome formation in mammalian cells. Cell Struct Funct 27:421–429. doi: 10.1247/csf.27.421. [DOI] [PubMed] [Google Scholar]
- 43.Frank GM, Buela K-G, Maker DM, Harvey SAK, Hendricks RL. 2012. Early responding dendritic cells direct the local NK response to control herpes simplex virus 1 infection within the cornea. J Immunol 188:1350–1359. doi: 10.4049/jimmunol.1101968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen FH, Wang SW, Yeh TM, Tung YY, Hsu SM, Chen SH. 2013. Absence of CXCL10 aggravates herpes stromal keratitis with reduced primary neutrophil influx in mice. J Virol 87:8502–8510. doi: 10.1128/JVI.01198-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fischer MA, Davies ML, Reider IE, Heipertz EL, Epler MR, Sei JJ, Ingersoll MA, Van Rooijen N, Randolph GJ, Norbury CC. 2011. CD11b+, Ly6G+ cells produce type I interferon and exhibit tissue protective properties following peripheral virus infection. PLoS Pathog 7:e1002374. doi: 10.1371/journal.ppat.1002374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gobeil PAM, Leib DA. 2012. Herpes simplex virus γ34.5 interferes with autophagosome maturation and antigen presentation in dendritic cells. mBio 3:e00267–12. doi: 10.1128/mBio.00267-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu E, Van Grol J, Subauste CS. 2015. ATG5 but not ATG7 in dendritic cells enhances IL-2 and IFN-γ production by Toxoplasma gondii-reactive CD4(+) T cells. Microbes Infect 17:275–284. doi: 10.1016/j.micinf.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaye S, Choudhary A. 2006. Herpes simplex keratitis. Prog Retin Eye Res 25:355–380. doi: 10.1016/j.preteyeres.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 49.Sugar A. 2014. Herpes simplex keratitis. UpToDate http://www.uptodate.com/contents/herpes-simplex-keratitis.
- 50.Kassim SH, Rajasagi NK, Zhao X, Chervenak R, Jennings SR. 2006. In vivo ablation of CD11c-positive dendritic cells increases susceptibility to herpes simplex virus type 1 infection and diminishes NK and T-cell responses. J Virol 80:3985–3993. doi: 10.1128/JVI.80.8.3985-3993.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hendricks RL, Janowicz M, Tumpey TM. 1992. Critical role of corneal Langerhans cells in the CD4- but not CD8-mediated immunopathology in herpes simplex virus-1-infected mouse corneas. J Immunol 148:2522–2529. [PubMed] [Google Scholar]
- 52.Knickelbein JE, Buela K, Hendricks RL. 2014. Antigen-presenting cells are stratified within normal human corneas and are rapidly mobilized during ex vivo viral infection. Invest Ophthalmol Vis Sci 55:1118–1123. doi: 10.1167/iovs.13-13523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buela K-G, Hendricks RL. 2015. Cornea-infiltrating and lymph node dendritic cells contribute to CD4+ T cell expansion after herpes simplex virus-1 ocular infection. J Immunol 194:379–387. doi: 10.4049/jimmunol.1402326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mansur DS, Kroon EG, Nogueira ML, Arantes RME, Rodrigues SCO, Akira S, Gazzinelli RT, Campos MA. 2005. Lethal encephalitis in myeloid differentiation factor 88-deficient mice infected with herpes simplex virus 1. Am J Pathol 166:1419–1426. doi: 10.1016/S0002-9440(10)62359-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vollstedt S, Arnold S, Schwerdel C, Franchini M, Alber G, Di Santo JP, Ackermann M, Suter M. 2004. Interplay between alpha/beta and gamma interferons with B, T, and natural killer cells in the defense against herpes simplex virus type 1. J Virol 78:3846–3850. doi: 10.1128/JVI.78.8.3846-3850.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang S-Y, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, Picard C, Chapgier A, Plancoulaine S, Titeux M, Cognet C, von Bernuth H, Ku C-L, Casrouge A, Zhang X-X, Barreiro L, Leonard J, Hamilton C, Lebon P, Héron B, Vallée L, Quintana-Murci L, Hovnanian A, Rozenberg F, Vivier E, Geissmann F, Tardieu M, Abel L, Casanova JL. 2007. TLR3 deficiency in patients with herpes simplex encephalitis. Science 317:1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 57.Lafaille FG, Pessach IM, Zhang S, Ciancanelli MJ, Herman M, Abhyankar A, Ying S, Keros S, Goldstein PA, Mostoslavsky G, Ordovas-Montanes J, Jouanguy E, Plancoulaine S, Tu E, Elkabetz Y, Al-Muhsen S, Tardieu M, Schlaeger TM, Daley GQ, Abel L, Casanova J-L, Studer L, Notarangelo LD. 2012. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 491:769–773. doi: 10.1038/nature11583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Casrouge A, Zhang S-Y, Eidenschenk C, Jouanguy E, Puel A, Yang K, Alcais A, Picard C, Mahfoufi N, Nicolas N, Lorenzo L, Plancoulaine S, Sénéchal B, Geissmann F, Tabeta K, Hoebe K, Du X, Miller RL, Héron B, Mignot C, de Villemeur TB, Lebon P, Dulac O, Rozenberg F, Beutler B, Tardieu M, Abel L, Casanova J-L. 2006. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 314:308–312. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- 59.Parr MB, Parr EL. 2003. Intravaginal administration of herpes simplex virus type 2 to mice leads to infection of several neural and extraneural sites. J Neurovirol 9:594–602. doi: 10.1080/jnv.9.6.594.602. [DOI] [PubMed] [Google Scholar]
- 60.Morahan PS, Breinig MC, McGeorge MB. 1977. Immune responses to vaginal or systemic infection of BALB/c mice with herpes simplex virus type 2. J Immunol 119:2030–2036. [PubMed] [Google Scholar]
- 61.Lee S, Ives AM, Bertke AS. 2015. HSV-1 reactivates from autonomic ciliary ganglia independently from sensory trigeminal ganglia to cause recurrent ocular disease. J Virol 89:8383–8391. doi: 10.1128/JVI.00468-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kintner RL, Brandt CR. 1995. The effect of viral inoculum level and host age on disease incidence, disease severity, and mortality in a murine model of ocular HSV-1 infection. Curr Eye Res 14:145–152. doi: 10.3109/02713689508999926. [DOI] [PubMed] [Google Scholar]
- 63.Richards JT, Kern ER, Overall JC, Glasgow LA. 1981. Differences in neurovirulence among isolates of herpes simplex virus types 1 and 2 in mice using four routes of infection. J Infect Dis 144:464–471. doi: 10.1093/infdis/144.5.464. [DOI] [PubMed] [Google Scholar]
- 64.Babu JS, Thomas J, Kanangat S, Morrison LA, Knipe DM, Rouse BT. 1996. Viral replication is required for induction of ocular immunopathology by herpes simplex virus. J Virol 70:101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stuart PM, Summers B, Morris JE, Morrison LA, Leib DA. 2004. CD8(+) T cells control corneal disease following ocular infection with herpes simplex virus type 1. J Gen Virol 85:2055–2063. doi: 10.1099/vir.0.80049-0. [DOI] [PubMed] [Google Scholar]
- 66.Ravindran R, Khan N, Nakaya HI, Li S, Loebbermann J, Maddur MS, Park Y, Jones DP, Chappert P, Davoust J, Weiss DS, Virgin HW, Ron D, Pulendran B. 2014. Vaccine activation of the nutrient sensor GCN2 in dendritic cells enhances antigen presentation. Science 343:313–317. doi: 10.1126/science.1246829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mintern JD, Macri C, Chin WJ, Panozza SE, Segura E, Patterson NL, Zeller P, Bourges D, Bedoui S, McMillan PJ, Idris A, Nowell CJ, Brown A, Radford KJ, Johnston AP, Villadangos JA. 2015. Differential use of autophagy by primary dendritic cells specialized in cross-presentation. Autophagy 11:906–917. doi: 10.1080/15548627.2015.1045178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sehrawat S, Suvas S, Sarangi PP, Suryawanshi A, Rouse BT. 2008. In vitro-generated antigen-specific CD4+ CD25+ Foxp3+ regulatory T cells control the severity of herpes simplex virus-induced ocular immunoinflammatory lesions. J Virol 82:6838–6851. doi: 10.1128/JVI.00697-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gaddipati S, Estrada K, Rao P, Jerome AD, Suvas S. 2015. IL-2/anti-IL-2 antibody complex treatment inhibits the development but not the progression of herpetic stromal keratitis. J Immunol 194:273–282. doi: 10.4049/jimmunol.1401285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Staats HF, Lausch RN. 1993. Cytokine expression in vivo during murine herpetic stromal keratitis. Effect of protective antibody therapy. J Immunol 151:277–283. [PubMed] [Google Scholar]
- 71.Arrunategui-Correa V, Baltatzis S, Foster CS. 1999. The role of cytokines in experimental herpes simplex keratitis. Acta Virol 43:325–329. [PubMed] [Google Scholar]
- 72.Lee SK, Choi BK, Kang WJ, Kim YH, Park HY, Kim KH, Kwon BS. 2008. MCP-1 derived from stromal keratocyte induces corneal infiltration of CD4+ T cells in herpetic stromal keratitis. Mol Cells 26:67–73. [PubMed] [Google Scholar]
- 73.Heiligenhaus A, Foster CS. 1994. Histological and immunopathological analysis of T-cells mediating murine HSV-1 keratitis. Graefes Arch Clin Exp Ophthalmol 232:628–634. doi: 10.1007/BF00193124. [DOI] [PubMed] [Google Scholar]
- 74.Thomas J, Gangappa S, Kanangat S, Rouse BT. 1997. On the essential involvement of neutrophils in the immunopathologic disease: herpetic stromal keratitis. J Immunol 158:1383–1391. [PubMed] [Google Scholar]
- 75.Tang Q, Hendricks RL. 1996. Interferon gamma regulates platelet endothelial cell adhesion molecule 1 expression and neutrophil infiltration into herpes simplex virus-infected mouse corneas. J Exp Med 184:1435–1447. doi: 10.1084/jem.184.4.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Opremcak EM, Rice BA, Wells PA, Foster CS. 1990. Histology and immunohistology of Igh-1-restricted herpes simplex keratitis in BALB/c congenic mice. Invest Ophthalmol Vis Sci 31:305–312. [PubMed] [Google Scholar]
- 77.Labetoulle M, Auquier P, Conrad H, Crochard A, Daniloski M, Bouée S, Elhasnaoui A, Colin J. 2005. Incidence of herpes simplex virus keratitis in France. Ophthalmology 112:888–895. doi: 10.1016/j.ophtha.2004.11.052. [DOI] [PubMed] [Google Scholar]
- 78.Neumann J, Eis-Hübinger AM, Koch N. 2003. Herpes simplex virus type 1 targets the MHC class II processing pathway for immune evasion. J Immunol 171:3075–3083. doi: 10.4049/jimmunol.171.6.3075. [DOI] [PubMed] [Google Scholar]
- 79.Rader KA, Ackland-Berglund CE, Miller JK, Pepose JS, Leib DA. 1993. In vivo characterization of site-directed mutations in the promoter of the herpes simplex virus type 1 latency-associated transcripts. J Gen Virol 74:1859–1869. doi: 10.1099/0022-1317-74-9-1859. [DOI] [PubMed] [Google Scholar]
- 80.Brown SM, Ritchie DA, Subak-Sharpe JH. 1973. Genetic studies with herpes simplex virus type 1. The isolation of temperature-sensitive mutants, their arrangement into complementation groups and recombination analysis leading to a linkage map. J Gen Virol 18:329–346. doi: 10.1099/0022-1317-18-3-329. [DOI] [PubMed] [Google Scholar]
- 81.Williams LE, Nesburn AB, Kaufman HE. 1965. Experimental induction of disciform keratitis. Arch Ophthalmol 73:112–114. doi: 10.1001/archopht.1965.00970030114023. [DOI] [PubMed] [Google Scholar]
- 82.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. 2006. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 83.Caton ML, Smith-Raska MR, Reizis B. 2007. Notch-RBP-J signaling controls the homeostasis of CD8− dendritic cells in the spleen. J Exp Med 204:1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Madsen L, Labrecque N, Engberg J, Dierich A, Svejgaard A, Benoist C, Mathis D, Fugger L. 1999. Mice lacking all conventional MHC class II genes. Proc Natl Acad Sci U S A 96:10338–10343. doi: 10.1073/pnas.96.18.10338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.West DM, Del Rosso CR, Yin X-, Stuart PM. 2014. CXCL1 but not IL-6 is required for recurrent herpetic stromal keratitis. J Immunol 192:1762–1767. doi: 10.4049/jimmunol.1302957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leib DA, Coen DM, Bogard CL, Hicks KA, Yager DR, Knipe DM, Tyler KL, Schaffer PA. 1989. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J Virol 63:759–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Divito SJ, Hendricks RL. 2008. Activated inflammatory infiltrate in HSV-1-infected corneas without herpes stromal keratitis. Invest Ophthalmol Vis Sci 49:1488–1495. doi: 10.1167/iovs.07-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Patankar YR, Lovewell RR, Poynter ME, Jyot J, Kazmierczak BI, Berwin B. 2013. Flagellar motility is a key determinant of the magnitude of the inflammasome response to Pseudomonas aeruginosa. Infect Immun 81:2043–2052. doi: 10.1128/IAI.00054-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Amiel E, Alonso A, Uematsu S, Akira S, Poynter ME, Berwin B. 2009. Pivotal advance: Toll-like receptor regulation of scavenger receptor-A-mediated phagocytosis. J Leukoc Biol 85:595–605. doi: 10.1189/jlb.1008631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rowe AM, St. Leger AJ, Jeon S, Dhaliwal DK, Knickelbein JE, Hendricks RL. 2013. Herpes keratitis. Prog Retin Eye Res 32:88–101. doi: 10.1016/j.preteyeres.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rubinsztein DC, Codogno P, Levine B. 2012. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Validation of DC-autophagy−/− mice. (A) Bone marrow-derived dendritic cells and peritoneal macrophages were harvested as previously described (88, 89). Immunoblot showing expression of atg5, LC3, and p62 in CD11c+ bone marrow-derived dendritic cells (BMDCs) and CD11c− peritoneal macrophages of atg5fl/fl mice and atg5fl/fl CD11c-cre mice. Proteins were resolved by electrophoresis on denaturing 12% polyacrylamide gels, transferred to polyvinylidene difluoride membranes, blocked with 5% skim milk, and reacted with antibodies at indicated concentrations: atg5, 1:500 (Novus); LC3, 1:1,000 (MBL); p62/SQSTM1, 1:1,000 (Novus); and β-actin, 1:1,000 (BioLegend). Data represent 3 independent experiments. (B) Flow cytometry analysis of CD11c+ dendritic cells in the spleen showing percentage of splenocytes (top) and total number of splenocytes (bottom). Data represent 3 similar experiments. Statistical significance was determined by analysis of variance. Download
HSK disease is attenuated in the absence of MHCII. C57BL/6 (n = 10) and MHCII−/− (n = 10) female mice were challenged with 2 × 106 PFU of HSV-1 (strain 17) unilaterally. (A) Corneal opacity was scored on a scale of 0 to 5. (B) Neovascularization was scored on a scale of 0 to 12. (C) Positive HSK disease was defined as a corneal opacity score of ≥1.5 and a neovascularization score of ≥2. (D) Viral titers of ocular tear film after challenge with 2 × 106 PFU of HSV-1 (strain 17) in one eye. Error bars represent standard errors of the means. Statistical significance was determined by unpaired parametric t test (A, B, and D) and chi-square test (C). *, P < 0.05. Download
DC-autophagy does not alter neurological disease after acute corneal challenge with HSV-1. atg5fl/fl and atg5fl/fl CD11c-cre mice (n = 12 in each group) were challenged with 2 × 106 PFU of HSV-1 (McKrae) bilaterally. Neurological disease was scored using the following scale: 0, normal behavior; 1, ruffled fur and/or hunched posture; 2, ruffled fur/hunched posture and repetitive movements and/or jumpy behavior; 3, motor deficits on one side; 4, moribund. Data represent 4 independent experiments. Error bars represent standard errors of the means. Statistical significance was determined by unpaired parametric t test. Download